

Summary
Safeguarding cell function and identity following a genotoxic stress challenge entails a tight coordination of DNA damage signaling and repair with chromatin maintenance. How this coordination is achieved and with what impact on chromatin integrity remains elusive. Here, we address these questions by investigating the mechanisms governing the distribution in mammalian chromatin of the histone variant H2A.X, a central player in damage signaling. We reveal that H2A.X is deposited de novo at sites of DNA damage in a repair-coupled manner, whereas the H2A.Z variant is evicted, thus reshaping the chromatin landscape at repair sites. Our mechanistic studies further identify the histone chaperone FACT (facilitates chromatin transcription) as responsible for the deposition of newly synthesized H2A.X. Functionally, we demonstrate that FACT potentiates H2A.X-dependent signaling of DNA damage. We propose that new H2A.X deposition in chromatin reflects DNA damage experience and may help tailor DNA damage signaling to repair progression.
Keywords: ANP32E, chromatin, DNA damage signaling, DNA repair, FACT, histone chaperones, histone variants, H2A.X, H2A.Z, UV damage
Graphical Abstract
Highlights
-
•
H2A.X is deposited de novo at sites of DNA damage repair, whereas H2A.Z is evicted
-
•
FACT promotes new H2A.X deposition coupled to repair synthesis
-
•
FACT stimulates H2A.X-dependent signaling of DNA damage
-
•
H2A.X is not only a starting point of damage signaling but also an output of repair
Histone variants convey epigenetic information and define functional chromatin states. Piquet et al. describe a reshaping of histone variant patterns at sites of UVC damage repair with H2A.Z removal followed by de novo deposition of H2A.X. The H2A.X deposition machinery provides a means for fine-tuning DNA damage signaling.
Introduction
Cells are constantly exposed to genotoxic stress and respond by activating dedicated DNA damage signaling and repair pathways that safeguard genome stability (Ciccia and Elledge, 2010, Hoeijmakers, 2009, Jackson and Bartek, 2009). DNA damage signaling consists in the activation of checkpoint kinase cascades upon DNA damage detection and/or processing to coordinate cell-cycle progression with DNA repair (Lazzaro et al., 2009). Adding another layer of complexity, damage signaling and repair machineries operate on chromatin substrates in eukaryotic cell nuclei, where DNA wraps around histone proteins (Luger et al., 2012). Chromatin landscapes, defined by specific patterns of histone variants (Buschbeck and Hake, 2017), post-translational modifications (Bannister and Kouzarides, 2011), and various degrees of chromatin folding, convey epigenetic information that instructs cell function and identity through the regulation of gene expression and replication programs (Allis and Jenuwein, 2016). While the importance of maintaining epigenome integrity is widely recognized, it is still poorly understood how this is achieved during the DNA damage response.
DNA damage signaling and repair indeed elicit profound chromatin rearrangements, challenging epigenome maintenance (Dabin et al., 2016), with a transient destabilization of chromatin organization, accompanied by DNA-damage-induced changes in histone modifications (Dantuma and van Attikum, 2016). Chromatin structure is then restored concomitantly with the repair of DNA damage (Polo and Almouzni, 2015, Smerdon, 1991). It remains unclear whether chromatin restoration is an entirely faithful process or if genotoxic stress responses alter the epigenetic landscape, leaving a signature of DNA damage repair. Restoration of chromatin at damage sites involves the deposition of newly synthesized histones, as shown for H2A, H3.1, and H3.3 histone variants at sites of UVA and UVC damage in human cells (Adam et al., 2013, Dinant et al., 2013, Juhász et al., 2018, Luijsterburg et al., 2016, Polo et al., 2006). New histone deposition employs dedicated histone chaperones, including the H3 variant-specific chaperones HIRA (histone regulator A) and CAF-1 (chromatin assembly factor 1) that operate at UVC damage sites (Adam et al., 2013, Polo et al., 2006), while the histone chaperone FACT (facilitates chromatin transcription) controls H2A-H2B turnover (Dinant et al., 2013). Regarding the histone variant H2A.Z, its dynamic exchange at sites of DNA double-strand breaks (DSBs) in human cells involves the concerted action of the histone chaperone ANP32E (acidic nuclear phosphoprotein 32 family member E) and of the chromatin remodeling factors p400 and INO80 (inositol-requiring 80) (Alatwi and Downs, 2015, Gursoy-Yuzugullu et al., 2015, Xu et al., 2012).
Here, we investigate how such repair-coupled chromatin rearrangements may leave an imprint on the epigenetic landscape and cross-talk with damage signaling by focusing on the H2A.X histone variant, which represents the ancestral form of H2A, conserved in all eukaryotes (Talbert and Henikoff, 2010). Making up only 10%–25% of total H2A, H2A.X is nevertheless central to the DNA damage response, owing to a particular C-terminal serine, in position 139 in mammals, targeted by DNA-damage-responsive kinases (Rogakou et al., 1998). H2A.X S139 phosphorylation spreads at a distance from the damage, which is key for amplifying the DNA damage signal through the coordinated recruitment of DNA damage checkpoint mediators (Altmeyer and Lukas, 2013, Smeenk and van Attikum, 2013). Best described in response to DSBs, this signaling cascade also operates following other types of genomic insults, including UV irradiation. UV damage processing triggers checkpoint signaling by activating the ATR (ataxia-telangiectasia-mutated and Rad3-related) kinase, which phosphorylates H2A.X (Hanasoge and Ljungman, 2007). This in turns recruits checkpoint mediators, including MDC1 (mediator of DNA damage checkpoint 1) and downstream factors that control, among others, 53BP1 (p53-binding protein 1) accumulation (Marteijn et al., 2009).
Regardless of which type of DNA insult activates the H2A.X signaling cascade, a salient feature of H2A.X phosphorylation is that it takes place in situ in damaged chromatin (Rogakou et al., 1998). The original distribution of H2A.X in chromatin is thus a critical determinant of the damage response, as it will govern the distribution of the phosphorylated form, known as γH2A.X. A commonly held view is that H2A.X is phosphorylated at DNA damage sites but ubiquitously incorporated in chromatin, independently of DNA damage. However, recent chromatin immunoprecipitation sequencing (ChIP-seq) studies in mammalian cells challenged this view by revealing a nonrandom distribution of H2A.X, with enrichments at active transcription start sites and sub-telomeric regions in activated human lymphocytes (Seo et al., 2012, Seo et al., 2014) and at extra-embryonic genes in mouse pluripotent stem cells (Wu et al., 2014). The mechanisms underpinning the nonrandom distribution of H2A.X in chromatin are unknown, as is their potential connection to the DNA damage response.
In this study, by investigating H2A.X dynamics during UVC damage repair in mammalian cells, we reveal that H2A.X is deposited de novo in damaged chromatin by the histone chaperone FACT, concomitantly with repair synthesis. We also uncover H2A.Z eviction from UV-damaged chromatin by ANP32E, which, together with FACT-mediated H2A.X deposition, reshapes the chromatin landscape by altering histone variant patterns at repair sites. Functionally, both histone chaperones are key for mounting an efficient cellular response to DNA damage, with FACT potentiating H2A.X-dependent damage signaling.
Results
De Novo Deposition of H2A Histone Variants at Repair Sites
To characterize H2A.X deposition pathways, we monitored de novo histone deposition using SNAP-tag technology (Bodor et al., 2012) in human U2OS cells stably expressing SNAP-tagged H2A variants (Figure 1A and S1). Our initial analyses did not reveal any detectable accumulation of new H2A variants at UVC damage sites, contrary to what we had observed with newly synthesized H3.3 (Adam et al., 2013) (Figure 1B). We reasoned that this discrepancy might be due to the higher mobility of outer core histones (H2A-H2B) compared to inner core histones (H3-H4) (Kimura and Cook, 2001, Louters and Chalkley, 1985), which may hinder the detection of their local accumulation. Because outer core histone mobility is partly transcription dependent (Jackson, 1990, Kimura and Cook, 2001), we tracked new histones in the presence of transcription inhibitors, 5,6-Dichlorobenzimidazole 1-beta-D-ribofuranoside (DRB), flavopiridol, or α-amanitin (Bensaude, 2011) (Figures 1C and S2A). Note that short-term transcription inhibition reduces but does not abolish histone neosynthesis because of preexisting mRNAs. Thus, we revealed new H2A.X accumulation at sites of UVC damage in the vast majority of cells (>85%; Figures 1C–1E). We recapitulated our observations in mouse embryonic fibroblasts (Figures S3A–S3D). Importantly, new H2A.X accumulation at UVC damage sites was not an artifact of transcription inhibition, as it was also detectable in the absence of transcription inhibitors upon exposure to higher UVC doses, with a modest but reproducible enrichment at UV sites relative to the whole nucleus approaching 1.2-fold (Figure S2B). No significant enrichment was observed when staining for total H2A.X (Figure 1D), arguing that new H2A.X accumulation most likely reflects histone exchange at damage sites. Noteworthy, de novo accumulation of H2A.X was also observed at sites of UVA laser micro-irradiation (Figure S2C) and thus is not unique to the UVC damage response. We clarified the nature of the DNA damage that was driving new H2A.X deposition upon UVC irradiation by showing that UVC did not elicit DSB signaling (Figure S2D). Thus, the new H2A.X deposition observed at UVC damage sites is unlikely to be driven by DSBs. To test whether it was specific for the damage-responsive histone H2A.X, we extended our analyses to other H2A variants, namely canonical H2A and another replacement variant conserved in all eukaryotes, H2A.Z, considering both H2A.Z.1 and H2A.Z.2 forms, which display different dynamics in response to UVA laser damage in human cells (Nishibuchi et al., 2014). For this, we established U2OS cell lines that stably express comparable levels of SNAP-tagged H2A variants (Figure S1). We detected de novo accumulation of H2A, but not of H2A.Z.1 and H2A.Z.2, at UVC damage sites (Figure 1E). Similar results were obtained without transcription inhibition (Figure S2E), pointing to a specific histone deposition mechanism that is not general to all H2A variants.
Figure 1.

De Novo Accumulation of H2A Variants at UVC Damage Sites
(A) Assay for monitoring the accumulation of newly synthesized histones labeled with tetramethylrhodamine (TMR)-star at UVC damage sites marked by the repair factor XPA (xeroderma pigmentosum, complementation group A) in human cells stably expressing SNAP-tagged histones.
(B–E) New histone accumulation at UVC damage sites marked by XPA analyzed 2 hr after irradiation in U2OS cells stably expressing the indicated SNAP-tagged histone variants, wild-type (B–D) and mutants (E).
Cells were treated without (B) or with (C) RNAPII inhibitors (DRB; FLV, flavopiridol; AMA, α-amanitin; DMSO and PBS, vehicles). Total H2A.X is detected by immunostaining for SNAP or H2A.X (D). S139A/E, phospho-deficient/mimetic mutant; WT, wild-type. Bar chart shows mean ± SD from 2 independent experiments. Scale bars, 10 μm.
ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figures S1–S4.
Collectively, these data reveal a transcription-independent deposition of newly synthesized H2A and H2A.X, but not H2A.Z, histone variants at repair sites.
New H2A.X Deposition at Repair Sites Is Independent of S139 Phosphorylation
We next examined the importance of H2A.X S139 phosphorylation for new H2A.X deposition at repair sites. We verified that the C-terminal SNAP tag did not prevent H2A.X phosphorylation (data not shown). By comparing the dynamics of H2A.X wild-type to phospho-mimetic (S139E) and phospho-deficient (S139A) mutants, we established that new H2A.X accumulation at sites of UVC damage repair occurred irrespective of H2A.X S139 phosphorylation status (Figure 1E). Since SNAP-tagged H2A.X proteins are expressed in the context of wild-type endogenous H2A.X, we also treated cells with an ATR kinase inhibitor, which inhibited UV-induced phosphorylation of all H2A.X forms but did not impair the de novo deposition of H2A.X at UVC-damage sites (Figure S4A). Thus, H2A.X phosphorylation is dispensable for the deposition of new H2A.X at sites of DNA damage repair.
De Novo Deposition of H2A Histone Variants at Replication Foci
During the course of our analyses, we noticed that new H2A.X displayed a punctuate pattern in a subset of undamaged cells upon transcription inhibition. This deposition pattern corresponded to replication foci in S phase cells, as shown by co-staining with the replication-coupled histone chaperone CAF-1 (Figure 2A). We observed new H2A.X accumulation at replication foci throughout S phase in U2OS cells treated with transcription inhibitors (Figure 2B). In mouse embryonic fibroblasts, transcription inhibitors could be omitted to visualize new H2A.X accumulation in replicating heterochromatin domains, which are poorly transcribed by nature (Figures S3E and S3F). As observed at repair sites, the enrichment at replication foci was specific for newly synthesized H2A.X (Figure 2C) and only detected for H2A and H2A.X, but not H2A.Z variants (Figure 2D).
Figure 2.

De Novo Accumulation of H2A Variants at Replication Foci
(A–D) New histone accumulation at replication foci marked by CAF-1 p150 analyzed in U2OS cells stably expressing SNAP-tagged H2A.X, wild-type form (A–C) or different H2A variants (D) and treated with the indicated RNAPII inhibitors (DRB, flavopriridol [FLV], α-amanitin [AMA], and DMSO and PBS [vehicles]). Replication patterns (B) distinguish early, mid-, and late S phase cells. Total H2A.X is detected by immunostaining for SNAP or H2A.X (C). Bar chart, mean ± SD from 2 independent experiments. Scale bars, 10 μm. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figures S1–S3.
These findings demonstrate that newly synthesized H2A and H2A.X are deposited both at repair sites and at replication foci.
New H2A.X Deposition Is Coupled to Replicative and Repair Synthesis
To dissect the mechanisms underlying new H2A.X deposition at repair sites and replication foci, we investigated a possible dependency on DNA synthesis, which is a common feature of both responses. We prevented repair synthesis by downregulating the late repair factor XPG (xeroderma pigmentosum, group G), an endonuclease involved in the excision of the UVC-damaged oligonucleotide, which is a prerequisite for repair synthesis (Figure 3A). XPG knockdown did not impede damage detection by early repair factors but markedly reduced the accumulation of newly synthesized H2A.X at sites of UVC damage (Figure 3B), with no detectable effect on new H2A.X deposition at replication foci (data not shown). We confirmed that new H2A.X deposition at repair sites was dependent on the DNA synthesis machinery by knocking down the DNA polymerase processivity factor PCNA (proliferating cell nuclear antigen; Figures 3C and S4B). We also uncovered a dependency on DNA synthesis for new H2A.X deposition at replication foci by inhibiting replicative synthesis with aphidicolin (Figure 3D). These results indicate that the deposition of newly synthesized H2A.X at replication foci and UVC damage sites is dependent on replicative and repair synthesis machineries.
Figure 3.

New H2A.X Accumulation at UV Sites and Replication Foci Is Coupled to DNA Synthesis
(A) Scheme: XPG’s role in the late steps of UVC damage repair. Percentage of cells showing repair synthesis (EdU) at UVC-damage sites (CPDs [cyclobutane pyrimidine dimers]) in U2OS cells treated with the indicated siRNAs (siLUC, control). At least 200 cells were scored in 2 independent experiments.
(B and C) New H2A.X accumulation at UVC damage sites marked by XPA or XPB analyzed 2 hr after irradiation in the presence of flavopiridol (+FLV) in U2OS H2A.X-SNAP cells treated with siRNAs targeting XPG (B) or PCNA (C) (siLUC, control). Knockdown efficiencies are verified by western blot and by impaired CAF-1 recruitment to damage sites.
(D) New H2A.X accumulation at replication foci (marked by EdU before aphidicolin addition) in U2OS H2A.X-SNAP cells treated with flavopiridol (+FLV) and the replication inhibitor aphidicolin (Aphi). Replication inhibition is shown by impaired CAF-1 recruitment to EdU foci.
Bar charts show mean ± SD from at least 2 independent experiments. Scale bars, 10 μm. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figure S4.
New H2A.X Deposition Is Not Coupled to New H3 Deposition
We next investigated whether new H2A.X deposition was coordinated with the deposition of newly synthesized H3 variants that occurs at replication and repair sites (Adam et al., 2013, Polo et al., 2006, Ray-Gallet et al., 2011). Knocking down the histone chaperone CAF-1, responsible for new H3.1 deposition at UV sites (Polo et al., 2006), did not significantly affect new H2A.X accumulation at sites of UVC damage repair (Figure S4C). Similar results were obtained upon downregulation of the histone chaperone HIRA, which deposits new H3.3 at UV sites (Adam et al., 2013) (Figure S4C) and upon loss of function of both pathways simultaneously (data not shown). These data demonstrate that new H2A.X deposition occurs independently of new H3 deposition by CAF-1 and HIRA at UVC damage sites.
The Histone Chaperone FACT Promotes New H2A.X Deposition at UV Damage Sites
To uncover the molecular determinants of new H2A.X accumulation at sites of UVC damage repair, we examined the effect of knocking down candidate histone chaperones and chromatin remodelers, focusing on FACT and INO80. Indeed, the latter maintains the levels of chromatin-bound H2A.X in human cells (Seo et al., 2014) and promotes H2A deposition in yeast (Papamichos-Chronakis et al., 2006), while FACT incorporates H2A and H2A.X, but not H2A.Z, into chromatin (Heo et al., 2008) and stimulates H2A turnover at UVC damage sites in human cells (Dinant et al., 2013). Moreover, FACT associates with replisome components in human cells (Alabert et al., 2014, Tan et al., 2006), is involved in replication-coupled nucleosome assembly in yeast (Yang et al., 2016), and is critical for replisome progression through chromatin in vitro (Kurat et al., 2017). Interestingly, like new H2A.X deposition, FACT recruitment to replication foci was abrogated upon aphidicolin treatment (Figure S5A), and FACT accumulation at repair sites was impaired by XPG and PCNA knockdowns (Figure 4A–4C). Consistent with a dependency on late repair steps, FACT recruitment to UV damage sites peaked 2–4 hr after UV irradiation (Figure S5B). FACT recruitment was independent of H2A.X phosphorylation by ATR (Figure S5C), similar to new H2A.X deposition. Furthermore, FACT trapping to chromatin by the intercalating agent curaxin CBL0137 (Gasparian et al., 2011) (Figure S5D) impaired FACT accumulation and new H2A.X deposition at UVC damage sites (Figures S5E and S5F). In line with these findings, siRNA-mediated downregulation of both FACT subunits, but not of the remodeler INO80, markedly reduced new H2A.X deposition at UVC damage sites (Figures 4D and S5G, left). This effect was specific for the histone chaperone FACT, as depletion of NAP-1 (Nucleosome Assembly Protein-1) family chaperones, which similarly handle H2A variants (Okuwaki et al., 2010), did not impair new H2A.X deposition at repair sites (Figure S4D). The decrease in new H2A.X deposition upon FACT knockdown was also observed in the whole nucleus, most likely reflecting replication- and transcription-coupled deposition of H2A.X by FACT, and cannot be explained by reduced histone neo-synthesis, as FACT knockdown did not inhibit nascent transcription (Figure S5H). De novo accumulation of H2A was similarly reduced in FACT knockdown cells (Figure S5I). These results establish that FACT promotes both new H2A and H2A.X deposition at repair sites.
Figure 4.

The Histone Chaperone FACT Promotes New H2A.X Deposition at UV Damage Sites
(A) Recruitment of FACT subunits SSRP1 and SPT16 to repair sites 2 hr after local UVC irradiation in U2OS cells. Scheme: FACT complex bound to H2A-H2B.
(B–D) Accumulation of FACT subunit SSRP1 and new H2A.X at repair sites marked by XPA/XPB analyzed 2 hr (B and C) or 1.5 hr (D) after local UVC irradiation in U2OS or U2OS H2A.X-SNAP cells treated with the indicated siRNAs (siLUC, control; siFACT, siSPT16 + siSSRP1). Knockdown efficiencies are verified by western blot.
Bar charts represent mean ± SD from at least 2 independent experiments. Scatterplots show mean ± SD from at least 100 cells (1 representative experiment out of 3). Scale bars, 10 μm. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figures S4 and S5.
H2A.Z Removal Precedes New H2A.X Deposition in UVC-Damaged Chromatin
In contrast to H2A and H2A.X, H2A.Z is not deposited de novo at repair sites (Figure 1E), which may impact histone variant patterns in UVC-damaged chromatin. Supporting this idea, when we stained for total histones, we observed that while H2A and H2A.X total levels were not detectably altered, H2A.Z total levels were reduced by ∼10% in damaged chromatin (Figures 5A, 5B, and S6A). This reduction was detectable with different antibodies targeting H2A.Z (N and C termini) and upon fluorescent labeling of total H2A.Z.1- and H2A.Z.2-SNAP (Figures 5A, 5B, and S6B), ruling out the possibility of impaired detection due to post-translational modification of H2A.Z. Such selective depletion affecting H2A.Z, but not H2A and H2A.X, is unlikely to result from a local decompaction of chromatin at damage sites and is not a mere consequence of transcription inhibition at UV sites, because inhibiting transcription did not significantly diminish the levels of chromatin-bound H2A.Z (Figure S6C). When exploring the molecular determinants of H2A.Z removal, we found that it occurred independently of INO80 and FACT (Figures 5B and S5G, right), showing that new H2A.X deposition by FACT is dispensable for H2A.Z removal. Interestingly however, H2A.Z depletion from UV-damaged chromatin was dependent on the histone chaperone ANP32E (Figures 5B and S5G, right, and S6D), previously characterized for its ability to remove H2A.Z from chromatin (Mao et al., 2014, Obri et al., 2014). To gain further mechanistic insights, we analyzed the relative kinetics of H2A.Z removal by ANP32E and new H2A.X deposition by FACT at repair sites by co-staining for both histone variants. We observed that new H2A.X deposition peaked between 1 and 2 hr after irradiation and returned close to basal levels 6 hr after UVC irradiation (Figure 5C). H2A.Z removal reached a maximum ∼30 min after UVC irradiation, clearly preceding new H2A.X deposition (Figure 5C). Notably however, ANP32E-mediated removal of H2A.Z was dispensable for FACT recruitment and new H2A.X deposition at UVC damage sites (Figures 5D and 5E). Thus, ANP32E and FACT chaperones independently orchestrate histone variant turnover in UVC-damaged chromatin, resulting in the maintenance of H2A.X and the loss of H2A.Z. New H2A.X enrichment at UV sites did not persist long-term after UVC irradiation, most likely due to the gradual incorporation of new H2A.X in the entire nucleus over time (data not shown). Nevertheless, it was detectable in early G1 cells that had been damaged in late G2 of the previous cell cycle (Figure S4E), suggesting that new H2A.X enrichment might be maintained through mitosis and contribute to bookmarking repair sites.
Figure 5.

The Histone Chaperone ANP32E Promotes H2A.Z Removal from UV Damage Sites
(A and B) Distribution of the indicated H2A variants (total levels, TMR pulse or H2A.Z Nt/Ct antibody) analyzed 1 hr after local UVC irradiation in U2OS cells expressing SNAP-tagged histones (A) and treated with the indicated siRNAs (B) (siLUC, control; siFACT, siSPT16 + siSSRP1). Arrowheads indicate UV irradiation sites. Fluorescence intensities are measured along the dotted lines. Scatterplots show mean ± SD from at least 40 cells (1 representative experiment out of 3). The significance of H2A.Z loss or enrichment at UV sites is indicated.
(C) Enrichment of new H2A.X and loss of total H2A.Z at UV sites (marked by CPD) analyzed at the indicated time points post 500 J/m2 local UVC irradiation in U2OS H2A.X-SNAP cells. Scatterplots show mean ± SD from at least 39 UV spots (1 representative experiment out of 2). Significance is given compared to a theoretical mean of 1.
(D and E) Accumulation of FACT subunit SSRP1 (D) and new H2A.X (E) at repair sites analyzed as in Figures 4B–4D. Percentages of cells recruiting FACT to repair sites are shown (from at least 250 cells scored in 2 independent experiments).
Scatterplots show mean ± SD from at least 70 cells (1 representative experiment out of 2). Knockdown efficiencies were verified by western blot. Scale bars, 10 μm. ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figures S5 and S6.
FACT Potentiates H2A.X-Dependent Signaling of DNA Damage
Having identified FACT and ANP32E as key factors for H2A.Z and H2A.X turnover in damaged chromatin, we set out to determine the functional relevance of this mechanism. We observed that both FACT and ANP32E knockdowns conferred increased sensitivity of cells to UVC damage (Figure 6A), suggesting that the histone variant turnover orchestrated by these two chaperones may be functionally important for an efficient DNA damage response. However, downregulation of FACT or ANP32E did not reveal any significant contribution of these chaperones to UVC damage repair, as shown by efficient repair factor recruitment (Figures 4D, 5B, 5D, 5E, S5I, and S6D) and unaltered repair synthesis (Figures S7A and S7B). In addition, FACT depletion did not affect the timely removal of UV photoproducts (Figure S7C), and preventing new H2A.X synthesis by siRNA (Figure S7D) did not impair UVC damage repair synthesis (Figure S7E), arguing that new H2A.X deposition by FACT is dispensable for UVC damage repair.
Figure 6.

FACT Potentiates H2A.X-Dependent Signaling of DNA Damage
(A) Clonogenic survival of U2OS cells treated with the indicated siRNAs (siLUC, control) in response to global UVC irradiation (mean ± SD from 4 independent experiments).
(B–E) Quantification of damage signaling factors γH2A.X (B), MDC1 (C), 53BP1 (D), and ATR (E) at sites of UVC damage repair (EdU spots) 2 hr after local UVC irradiation in U2OS cells treated with the indicated siRNAs (siLUC, control). Scatterplots show mean ± SD from at least 78 UV spots (1 representative experiment out of 3). Knockdown efficiencies and total levels of damage signaling factors are controlled by western blot. Scale bars, 10 μm.
ns, non-significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. See also Figure S7.
We next investigated a potential impact of these chaperones on UVC damage signaling. To avoid misinterpretation of the data due to the interference of FACT knockdown with DNA replication, we restricted our analysis to nonreplicating cells based on EdU (5-ethynyl-2′-deoxyuridine) staining. Thus, we noticed that γH2A.X levels were reduced by 30%–35% at sites of UVC damage repair in FACT-depleted cells (Figure 6B), while H2A.X total levels did not show any measurable decrease as assessed in total cell extracts and at damage sites (Figure 6B; data not shown). The reduction in γH2A.X was not explained by lower damage infliction or diminished repair, because repair synthesis and repair factor recruitment were not significantly affected (data not shown). In contrast to FACT loss of function, ANP32E depletion had no inhibitory effect on γH2A.X levels at UVC damage sites (Figure 6B). We next analyzed the recruitment to damaged chromatin of downstream damage signaling factors, including the checkpoint mediators MDC1 and 53BP1. These factors also showed 30%–40% reduction at UVC damage sites in FACT knockdown cells, while their total levels were not decreased (Figures 6C–6D), indicating that FACT is relevant to not only early but also late damage-signaling events. This function of FACT may rely, at least in part, on its ability to promote new H2A.X deposition at repair sites. However, we do not exclude that other FACT activities and/or associated factors may also contribute to H2A.X-dependent signaling. Supporting this idea, we observed reduced recruitment of the ATR kinase to UV damage sites in FACT-depleted cells (Figure 6E). Collectively, these results establish that FACT and ANP32E histone chaperones are critical for the cells to mount an efficient DNA damage response with FACT stimulating H2A.X-dependent signaling of DNA damage.
Discussion
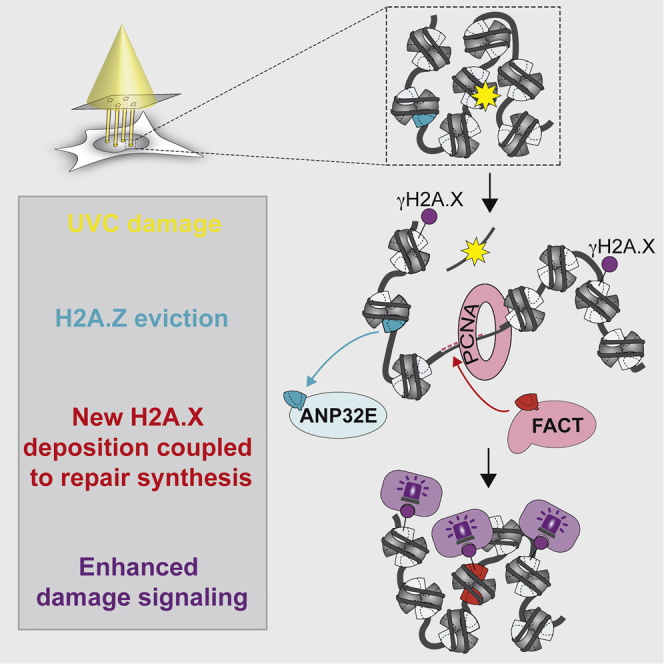
By analyzing the dynamics of H2A histone variants in UV-damaged chromatin, we have uncovered a H2A.Z and H2A.X turnover reaction controlled by two histone chaperones, ANP32E and FACT (Figure 7). Chromatin restoration after repair thus entails at least short-term reshaping of the chromatin landscape with a change in histone variant pattern. We also reveal that H2A.X is deposited de novo at repair sites, which may constitute an extra level of regulation for fine-tuning the DNA damage response in addition to the well-characterized phosphorylation of H2A.X. By ensuring that the target for DNA damage checkpoint kinases is present in sufficient amount and at the right place, new H2A.X deposition may allow an efficient and timely response to genotoxic insults in chromatin regions that are susceptible to DNA damage.
Figure 7.

Model for Chromatin Reshaping during UVC Damage Repair by ANP32E and FACT Histone Chaperones
ANP32E removes H2A.Z from damaged chromatin, which may enhance chromatin accessibility. FACT subsequently promotes new H2A.X deposition coupled to repair synthesis and potentiates DNA damage signaling, thus contributing to coordinate signaling and repair of DNA damage. The resulting distribution of H2A.X in chromatin reflects DNA damage experience.
New H2A.X Deposition at Sites of DNA Synthesis during Replication and Repair
The selective deposition of H2A and H2A.X variants and not H2A.Z at sites of UVC damage repair parallels our previous findings on H3 variants, indicating that H3.1 and H3.3 are deposited de novo in UVC-damaged chromatin while CENP-A is not (Adam et al., 2013, Polo et al., 2006). However, new H2A.X and new H3 deposition at repair sites are uncoupled, suggesting that new H2A.X-H2B associate with parental H3-H4 in repaired chromatin. A selective deposition of H2A variants also occurs on replicating DNA, as previously observed for H2A versus H2A.Z in mouse pericentric heterochromatin (Boyarchuk et al., 2014) and in proteomic studies of human histone variants associated with replicating DNA (Alabert et al., 2015). The coupling between new H2A.X deposition and DNA replication is intriguing given that H2A.X is not a prototypical replicative histone variant (Wu and Bonner, 1981). Nevertheless, the lack of introns and the existence of a non-polyadenylated form of H2A.X mRNA, both characterizing replicative histone transcripts, place H2A.X in a unique position between replicative and replacement histone variants (Mannironi et al., 1989). Functionally, the deposition of newly synthesized H2A.X at replication foci may ensure that H2A.X is not gradually lost during replication, which would happen if only the bona fide replicative variant H2A was deposited. Such replication-coupled deposition of H2A.X could explain the increase in soluble H2A.X in human cells treated with replication inhibitors (Liu et al., 2008). The de novo deposition of H2A.X at repair sites may provide a molecular basis for the H2A.X hotspots found in chromatin regions that are susceptible to endogenous damage (Seo et al., 2014). We speculate that the enrichment of H2A.X may reflect recurrent genomic insults, with H2A.X marking chromatin regions that are prone to damage to facilitate subsequent damage responses. Noteworthy, the H2A.X protein is rapidly stabilized following DNA damage, contributing to a local enrichment at repair sites (Atsumi et al., 2015). However, this stabilization requires H2A.X phosphorylation on Ser139, while new H2A.X deposition at repair sites does not. Thus, these appear to be two distinct and complementary mechanisms controlling the distribution of H2A.X in chromatin, which reflects DNA damage experience.
Role of the Histone Chaperone FACT in New H2A.X Deposition
We have identified FACT as the responsible histone chaperone for new H2A and H2A.X deposition at repair sites. Interestingly, FACT also promotes the deposition of another H2A variant, macroH2A1.2, at sites of replication stress in mammalian cells (Kim et al., 2018). FACT-mediated deposition of new H2A.X at UV damage sites is dependent on PCNA. This is reminiscent of the recruitment of CAF-1 to damaged DNA (Moggs et al., 2000), putting forward PCNA as a platform for histone chaperones on damaged chromatin. FACT may directly associate with PCNA, as the FACT subunit SSRP1 (structure-specific recognition protein 1) harbors a non-canonical PIP (PCNA-interacting protein) box (Mailand et al., 2013). Alternatively, FACT’s interaction with PCNA could be mediated by PCNA-binding factors like XRCC1 (X-ray repair cross-complementing protein 1) (Fan et al., 2004a), which contributes to UVC damage repair (Moser et al., 2007) and was recently reported to associate with FACT (Gao et al., 2017). Other replisome components cooperating with FACT, like MCM (minichromosome maintenance) proteins (Tan et al., 2006), could also contribute to FACT recruitment to sites of UV damage repair.
Impact of New H2A.X Deposition on DNA Damage Signaling and Repair
Although deposited de novo at sites of UVC damage, H2A.X is not required for repair. These findings are in line with previous work showing that H2A.X knockout mouse cells are not particularly sensitive to UV irradiation (Bassing et al., 2002). Similarly, the histone chaperone FACT is dispensable for global genome repair synthesis at UV damage sites. This contrasts with FACT requirement for replicative DNA synthesis (Abe et al., 2011, Kurat et al., 2017, Okuhara et al., 1999), suggesting that FACT activity is more critical for replication fork progression through chromatin than for DNA synthesis per se.
We find that FACT coordinates DNA repair synthesis with damage signaling. While the histone chaperones ASF1 and CAF-1 are required for turning off DSB signaling (Chen et al., 2008, Diao et al., 2017, Kim and Haber, 2009), FACT in contrast enhances γH2A.X levels at sites of UV damage repair, thus potentiating damage signaling. It is not clear at this stage if FACT function in damage signaling governs cell resistance to UV damage or if this also relies on other FACT-dependent activities, including a putative contribution to transcription-coupled repair of UV damage. It is tempting to speculate that FACT stimulates damage signaling, at least in part, through new H2A.X deposition. While this does not lead to a measurable increase in H2A.X total levels at repair sites, it may be enough to amplify the γH2A.X signal by facilitating H2A.X phosphorylation spreading. The repair-coupled deposition of H2A.X may thus contribute to keep damage signaling on while DNA repair is still ongoing. It may also be functionally important for subsequent damage responses by maintaining critical levels of H2A.X in damage-prone chromatin regions.
H2A.Z and H2A.X Turnover at Sites of DNA Damage Repair
New H2A.X deposition at sites of UV damage repair is preceded by the removal of H2A.Z, resulting in a local change in histone variant pattern. It still remains to be determined if the histone variant turnover takes place at the level of the nucleosome, with H2A.X replacing H2A.Z, or at the level of the damaged chromatin domain. H2A.Z is removed from UVC-damaged chromatin by ANP32E, reminiscent of what observed in response to UVA laser damage (Gursoy-Yuzugullu et al., 2015). However, in this case, H2A.Z returns to basal levels in damaged chromatin after a transient accumulation, while H2A.Z is depleted at UVC damage sites. Furthermore, while ANP32E accumulates on laser tracks (Gursoy-Yuzugullu et al., 2015), we did not observe a detectable enrichment of ANP32E at UVC damage sites (data not shown). This histone chaperone may thus be very transiently and/or loosely associated with UVC-damaged chromatin to remove H2A.Z. We did not find any significant role for the remodeler INO80 in H2A.Z removal from UVC-damaged chromatin, contrasting with observations at UVA laser tracks in human cells (Alatwi and Downs, 2015), arguing that different types of DNA lesions may engage distinct chromatin remodeling machineries.
ANP32E promotes cell survival to UVC irradiation, suggesting that ANP32E-mediated removal of H2A.Z from UVC-damaged chromatin may be of functional importance. Given that H2A.Z increases chromatin folding in vitro (Fan et al., 2002, Fan et al., 2004b) and is rapidly removed from UVC-damaged chromatin, the local depletion of H2A.Z could contribute to the early chromatin relaxation observed at UVC damage sites (Adam et al., 2016, Luijsterburg et al., 2012). Future studies will help decipher the functional contribution of H2A.Z displacement to the UV damage response and dissect potential cross-talk with other histone variant dynamics in damaged chromatin.
Dynamics of H2A Variants at Repair Sites and Transcription Regulation
FACT regulates transcription recovery after UVC damage in human cells (Dinant et al., 2013). Thus, FACT-mediated deposition of H2A variants at repair sites could coordinate repair synthesis with transcription restart. Tipping the balance toward H2A and H2A.X as opposed to H2A.Z may also help keeping transcription in check and avoid transcription interference with repair, because H2A.Z poises genes for transcription activation and promotes cryptic transcription when mislocalized in yeast (Jeronimo et al., 2015, Zhang et al., 2005). H2A.Z and H2A.X histone variant turnover at UV sites may thus contribute to mitigate transcription-repair conflicts by maintaining transcription inhibition during repair synthesis. Notably, FACT was shown to limit the formation of RNA-DNA hybrids in yeast and human cells (Herrera-Moyano et al., 2014). It will be of major interest to investigate whether this function of FACT relies on H2A histone variant deposition on newly synthesized DNA. Several recent studies have uncovered a critical role for H2A.X in silencing specific sets of genes during mammalian cell differentiation (Wu et al., 2014, Weyemi et al., 2016). Therefore, alterations in H2A.X distribution following DNA damage may impact cell fate determination via the rewiring of transcriptional programs, thus contributing to the reported effect of DNA damage repair on cell differentiation and reprogramming (Rocha et al., 2013).
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ANP32E | Sigma-Aldrich | Cat# SAB2100124; RRID: AB_10602824 |
| Anti-ANP32E | MyBioSource | Cat# MBS9214243 |
| Anti-CPD | Kamiya Biomedical Company | Cat# MC-062; RRID: AB_1233355 |
| Anti-H2A.X | Abcam | Cat# ab11175; RRID: AB_297814 |
| Anti-γH2A.X | Millipore | Cat# 05-636; RRID: AB_309864 |
| Anti-γH2A.X | Cell Signaling technology | Cat# 2577; RRID: AB_2118010 |
| Anti-H2A.Z Ct | Active motif | Cat# 39113; RRID: AB_2615081 |
| Anti-H2A.Z Nt | GeneTex | Cat# GTX108298; RRID: AB_1950475 |
| Anti-INO80 | Bethyl Laboratories | Cat# A303-371A; RRID: AB_10950580 |
| Anti-SNAP | Pierce Antibodies | Cat# CAB4255; RRID: AB_10710011 |
| Anti-SPT16 (FACT subunit) | Santa Cruz Biotechnology | Cat# sc-28734; RRID: AB_661341 |
| Anti-SSRP1 (FACT subunit) | Ozyme BLE609701 | Cat# BLE609701; RRID: AB_315730 |
| Anti-XPA | BD Biosciences | Cat# 556453; RRID: AB_396423 |
| Anti-XPB | Santa Cruz Biotechnology | Cat# sc-293; RRID: AB_2262177 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5,6-Dichlorobenzimidazole 1-beta-D-ribofuranoside (DRB) | Sigma-Aldrich | Cat# D1916 |
| alpha-Amanitin | Sigma-Aldrich | Cat# A2263 |
| Flavopiridol hydrochloride hydrate | Sigma-Aldrich | Cat# F3055 |
| ATR inhibitor AZ20 | Selleckchem | Cat# S7050 |
| ATM inhibitor KU55933 | Tocris Bioscience | Cat# 3544 |
| Aphidicolin | Sigma-Aldrich | Cat# A0781 |
| SNAP-Cell Block | New England Biolabs | Cat# S9106S |
| SNAP-Cell TMR-Star | New England Biolabs | Cat# S9105S |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Triton X-100 | Euromedex | Cat# 2000 |
| Vectashield medium with DAPI | Vector Laboratories | Cat# H-1200 |
| Critical Commercial Assays | ||
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778075 |
| 4%–20% Mini-PROTEAN TGX gels | Bio-Rad | Cat# 4561096 |
| Click-iT EdU Alexa Fluor 594 Imaging kit | Invitrogen | Cat# C10339 |
| Click-iT RNA Imaging kit | Invitrogen | Cat# C10330 |
| Deposited Data | ||
| Raw data files | This study, Mendeley data | https://doi.org/10.17632/gz5xtcyxzj.1 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | Cat# HTB-96 |
| U2OS H2A.X-SNAP WT form | This study | N/A |
| U2OS H2A.X-SNAP S139A mutant | This study | N/A |
| U2OS H2A.X-SNAP S139E mutant | This study | N/A |
| U2OS H2A.Z.1-SNAP | This study | N/A |
| U2OS H2A.Z.2-SNAP | This study | N/A |
| U2OS H2A-SNAP | This study | N/A |
| U2OS H3.3-SNAP | (Dunleavy et al., 2011) | N/A |
| NIH/3T3 | ATCC | Cat# CRL-1658 |
| NIH/3T3 H2A.X-SNAP + GFP-DDB2 | This study | N/A |
| Oligonucleotides | ||
| siRNAs (Table in Method Details) | Eurofins MWG Operon | N/A (custom sequences) |
| Recombinant DNA | ||
| H2A-SNAP | This study | N/A |
| H2A.X-SNAP (WT) | This study | N/A |
| H2A.X-SNAP (S139A) | This study | N/A |
| H2A.X-SNAP (S139E) | This study | N/A |
| H2A.Z.1-SNAP | This study | N/A |
| H2A.Z.2-SNAP | This study | N/A |
| H3.3-SNAP | (Dunleavy et al., 2011) | N/A |
| GFP-DDB2 | (Adam et al., 2016) | N/A |
| Software and Algorithms | ||
| Metamorph | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy |
| Zen | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html |
| ImageJ | NIH, USA | https://imagej.nih.gov/ij/ |
| ICY | Institut Pasteur, France | http://icy.bioimageanalysis.org/ |
| GraphPad Prism | GraphPad software | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html |
| FlowJo | FlowJo | https://www.flowjo.com |
| Other | ||
| VL-6.LC UVC lamp (254 nm) | Vilber-Lourmat | Cat# 3111 0065 1 |
| VLX-3W dosimeter | Vilber-Lourmat | Cat# 5130 0011 1 |
| CX-254 254 nm sensor | Vilber-Lourmat | Cat# 5190 0001 1 |
| Polycarbonate filter (5 μm pore size) | Millipore | Cat# TMTP01300 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sophie E. Polo (sophie.polo@univ-paris-diderot.fr).
Experimental Model and Subject Details
Cell lines
All U2OS (American Type Culture Collection ATCC HTB-96, human osteosarcoma, female) and NIH/3T3 (ATCC CRL-1658, mouse embryonic fibroblast, male) cell lines were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetal calf serum (EUROBIO), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen) and the appropriate selection antibiotics.
| Stable cell line (reference if not generated in this study) | Selection antibiotics |
|---|---|
| U2OS H2A.X-SNAP WT form | G418 |
| U2OS H2A.X-SNAP S139A mutant | G418 |
| U2OS H2A.X-SNAP S139E mutant | G418 |
| U2OS H2A.Z.1-SNAP | G418 |
| U2OS H2A.Z.2-SNAP | G418 |
| U2OS H2A-SNAP | G418 |
| U2OS H3.3-SNAP (Dunleavy et al., 2011) | G418 |
| NIH/3T3 H2A.X-SNAP + GFP-DDB2 | G418 + Hygromycin |
Antibiotics: G418 (100 μg/mL for U2OS, 500 μg/mL for NIH/3T3, Euromedex), Hygromycin (200 μg/mL, Euromedex).
Method Details
Drug treatments
Transcription inhibition was performed either by adding DRB (100 μM final concentration, Sigma-Aldrich), alpha-Amanitin (AMA, 20 μg/mL final concentration, Sigma-Aldrich) or Flavopiridol hydrochloride hydrate (FLV, 10 μM final concentration, Sigma-Aldrich) to the culture medium at 37°C 4 h before harvesting the cells. Inhibition of replicative synthesis was performed by adding Aphidicolin (Aphi, 1 μg/mL final concentration, Sigma-Aldrich) to the culture medium at 37°C 5 h before harvesting the cells. For ATR and ATM inhibition, cells were incubated with 2 μM ATR inhibitor AZ20 (Selleckchem) or 10 μM ATM inhibitor KU55933 (Tocris Bioscience) 1h before subsequent cell treatment. For FACT trapping, cells were incubated with 2 μM CBL0137 (Bertin Pharma) 15 min before subsequent cell treatment. The DSB-inducing agent neocarzinostatin (NCS, Sigma-Aldrich) was applied for 15 min at 50 ng/ml followed by 1 h recovery in fresh medium. For cell synchronization in late G2, cells were treated for 16 h with 2 mM Thymidine (Sigma-Aldrich), followed by 6 h release in fresh medium and 22 h treatment with the cdk1 inhibitor RO-3306 (10 μM final, Sigma-Aldrich).
SNAP-tag labeling of histone proteins
For specific labeling of newly-synthesized histones, pre-existing SNAP-tagged histones were first quenched by incubating cells with 10 μM non-fluorescent SNAP reagent (SNAP-Cell Block, New England Biolabs) for 30 min (quench) followed by a 30 min-wash in fresh medium and a 2 h-chase. The SNAP-tagged histones neo-synthesized during the chase were then pulse-labeled by incubation with 2 μM red-fluorescent SNAP reagent (SNAP-Cell TMR-Star, New England Biolabs) for 15 min followed by 1h to 1h30-wash in fresh medium. Cells were pre-extracted with Triton detergent before fixation (see Immunofluorescence section for details). If cells were subject to local UVC irradiation, irradiation was performed immediately before the pulse. When transcription inhibitors were used, they were added to the medium at the quench step and kept throughout the experiment. For total labeling of SNAP-tagged histones, the quench step was omitted and cells were pulsed with SNAP-Cell TMR-Star immediately before harvesting. Total H2A.Z is also revealed by immunostaining with antibodies against H2A.Z amino- or carboxy-terminus (Nt/Ct)
siRNA and plasmid transfections
siRNAs purchased from Eurofins MWG Operon were transfected into cells using Lipofectamine RNAiMAX (Invitrogen) following manufacturer’s instructions. The final concentration of siRNA in the culture medium was 50 nM. Cells were analyzed and/or harvested 48 to 72 h post-transfection.
| siRNA | Target sequence |
|---|---|
| siANP32E#1 | 5′AUGGAUUUGAUCAGGAGGAUA3′ |
| siANP32E#2 | 5′UUGGAGCUUAGUGAUAAUAUA3′ |
| siCAF-1 p60 | 5′AAUCUUGCUCGUCAUACCAAA3′ |
| siFACT#1 | 1:1 combination of siSPT16: 5′GGAAUUAAGACAUGGUGUG3′ and siSSRP1: 5′GAUGAGAUCUCCUUUGUCA3′ |
| siFACT#2 | 1:1 combination of siSPT16: 5′ACAUCAGCAUAUGCAUGAC3′ and siSSRP1: 5′UGAGGUGACACUGGAAUUC3′ |
| siFACT#3 | 1:1 combination of siSPT16: 5′ACCGGAGUAAUCCGAAACUGA3′ and siSSRP1: 5′UUCGUUGACUCUGAACAUGAA3′ |
| siH2A.X | 5′CAACAAGAAGACGCGAAUC3′ |
| siHIRA | 5′GGAGAUGACAAACUGAUUA3′ |
| siINO80#1 | 5′GGAGUUAUUUGAACGGCAA3′ |
| siINO80#2 | 5′ACUUGGUCUCCAUUUCAUA3′ |
| siLUC (Luciferase) | 5′CGUACGCGGAAUACUUCGA3′ |
| siNAP1+2 | 1:1 combination of siNAP1: 5′UCACUUUGAACCCAAUG3′ and siNAP2: 5′UGAAAUCAGAACCAGAUAA3′ |
| siPCNA#1 | 5′CCGAGAUCUCAGCCAUAUU3′ |
| siPCNA#2 | 5′GGAGGAAGCUGUUACCAUA3′ |
| siXPG#1 | 5′GAAAGAAGAUGCUAAACGU3′ |
| siXPG#2 | 5′GGAGAAAGAAUUUGAGCUA3′ |
For stable cell line establishment, cells were transfected with plasmid DNA (1 μg/ml final) using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions 48 h before antibiotic selection of clones. All constructs were verified by direct sequencing and/or restriction digests. Cloning details and primer sequences (Sigma-Aldrich) are available upon request.
| Plasmid | Construct details |
|---|---|
| H2A.X-SNAP (WT) | H2AFX coding sequence cloned into pSNAPm (New England Biolabs) |
| H2A.X-SNAP (S139A) | H2AFX coding sequence with insertion of a mutation S139A (TCC to GCG) cloned into pSNAPm (New England Biolabs) |
| H2A.X-SNAP (S139E) | H2AFX coding sequence with insertion of a mutation S139E (TCC to GAG) cloned into pSNAPm (New England Biolabs) |
| H2A.Z.1-SNAP | H2AFZ coding sequence cloned into pSNAPm (New England Biolabs) |
| H2A.Z.2-SNAP | H2AFV coding sequence cloned into pSNAPm (New England Biolabs) |
| H2A-SNAP | HIST1H2AM coding sequence cloned into pSNAPm (New England Biolabs) |
| H3.3-SNAP | H3F3B coding sequence cloned into pSNAPm (New England Biolabs) (Dunleavy et al., 2011) |
| GFP-DDB2 | DDB2 coding sequence (Montpellier Genomic Collections) subcloned into GFP-XPC plasmid (Nishi et al., 2009) replacing XPC (Adam et al., 2016) |
All the coding sequences for histone variants and repair factors are of human origin.
UVC irradiation
Cells grown on glass coverslips (Menzel Gläser) were irradiated with UVC (254 nm) using a low-pressure mercury lamp. Conditions were set using a VLX-3W dosimeter (Vilber-Lourmat). For global UVC irradiation, cells in Phosphate Buffer Saline (PBS) were exposed to UVC doses ranging from 4 to 12 J/m2 by varying the duration of exposure. For local UVC irradiation, cells were covered with a polycarbonate filter (5 μm pore size, Millipore) and irradiated with 150 J/m2 UVC unless indicated otherwise. Only 2% of the UVC light goes through these micropore filters.
UVA laser micro-irradiation
Cells grown on glass coverslips (Menzel Gläser) were presensitized with 10 μM 5-bromo-2′-deoxyuridine (BrdU, Sigma-Aldrich) for 24 hr at 37°C. Damage was introduced with a 405 nm laser diode (3 mW) focused through a Plan-Apochromat 63x /1.4 oil objective to yield a spot size of 0.5-1 μm using a LSM710 NLO confocal microscope (Zeiss) and the following laser settings: 40% power, 50 iterations, scan speed 12.6 μsec/pixel.
Cell extracts and western blot
Total extracts were obtained by scraping cells in Laemmli buffer (50 mM Tris HCl pH 6.8, 1.6% SDS (Sodium Dodecyl Sulfate), 8% glycerol, 4% β-mercaptoethanol, 0.0025% bromophenol blue) followed by 5 min denaturation at 95°C. Cytosolic and nuclear extracts were obtained as previously described (Martini et al., 1998). The chromatin fraction was prepared by addition of benzonase (Novagen) to the pellet after nuclear extraction.
For western blot analysis, extracts were run on 4%–20% Mini-PROTEAN TGX gels (Bio-Rad) in running buffer (200 mM glycine, 25 mM Tris, 0.1% SDS) and transferred onto nitrocellulose membranes (Amersham) with a Trans-Blot SD semidry transfer cell (Bio-Rad). Total proteins were revealed by reversible protein stain (Pierce). Proteins of interest were probed using the appropriate primary and HRP (Horse Radish Peroxidase)-conjugated secondary antibodies (Jackson Immunoresearch), detected using Super-Signal West Pico or Femto chemi-luminescence substrates (Pierce). Alternatively, when fluorescence detection was used instead of chemi-luminescence, total proteins were revealed with REVERT total protein stain, secondary antibodies were conjugated to IRDye 680RD or 800CW and imaging was performed with Odyssey Fc-imager (LI-COR Biosciences).
| Antibody | Species | Dilution | Application | Supplier (reference) | |
|---|---|---|---|---|---|
| Primary | 53BP1 | Rabbit | 1:1000 | WB; IF | Novus Biologicals (NB100-304) |
| ANP32E | Rabbit | 1:100 | WB | Sigma-Aldrich (SAB2100124) | |
| ANP32E | Rabbit | 1:500 | WB | MyBioSource (MBS9214243) | |
| ATR | Goat | 1:300; 1:50 | WB; IF | Santa Cruz Biotechnology (sc-1887) | |
| CAF-1 p60 | Rabbit | 1:500 | IF | Abcam (ab109442) | |
| CAF-1 p150 | Mouse | 1:500 | IF | Abcam (ab24746) | |
| CPD | Mouse | 1:1000 | IF | Kamiya Biomedical Company (MC-062) | |
| DDB2 | Mouse | 1:200 | WB | Abcam (ab51017) | |
| GFP | Mouse | 1:1000 | WB | Roche (11814460001) | |
| H2A | Rabbit | 1:1000 | WB | Millipore (07-146) | |
| H2A.X | Rabbit | 1:4000; 1:1000 | WB; IF | Abcam (ab11175) | |
| γH2A.X | Mouse | 1:1000 | IF | Millipore (05-636) | |
| γH2A.X | Rabbit | 1:1000; 1:250 | WB; IF | Cell Signaling technology (2577) | |
| H2A.Z | Sheep | 1:1000 | WB | Millipore (09-862) | |
| H2A.Z Ct | Rabbit | 1:1000 | IF | Active motif (39113) | |
| H2A.Z Nt | Rabbit | 1:500 | IF | GeneTex (GTX108298) | |
| H3.3 | Rabbit | 1:1000 | WB | Millipore (09-838) | |
| HIRA | Mouse | 1:100 | IF | Active Motif (39557) | |
| INO80 | Rabbit | 1:1000 | WB | Bethyl Laboratories (A303-371A) | |
| MDC1 | Rabbit | 1:1000 1:10 000 | IF; WB | Abcam (ab11171) | |
| NAP1 | Rabbit | 1:4000 | WB | Active Motif (39842) | |
| NAP2 | Rabbit | 1:2000 | WB | Bethyl Laboratories (A304-579A) | |
| PARP1 | Rabbit | 1:1000 | WB | Cell Signaling Technology (9542S) | |
| PCNA | Mouse | 1:1000 | WB; IF | Dako (M0879) | |
| RNAPII Ser2-P | Rabbit | 1:10 000 | WB | Abcam (ab5095) | |
| SNAP | Rabbit | 1:1000 | WB; IF | Pierce Antibodies (CAB4255) | |
| SPT16 (FACT subunit) | Rabbit | 1:500; 1:100 | WB; IF | Santa Cruz Biotechnology (sc-28734) | |
| SSRP1 (FACT subunit) | Mouse | 1:500; 1:1000 | WB; IF | Ozyme (BLE609701) | |
| Tubulin | Mouse | 1:10 000 | WB | Sigma-Aldrich (T9026) | |
| XPA | Mouse | 1:400 | IF | BD Biosciences (556453) | |
| XPB | Rabbit | 1:400 | IF | Santa Cruz Biotechnology (sc-293) | |
| XPG | Rabbit | 1:500 | WB | Bethyl Laboratories (A301-484A) | |
| Secondary | Anti-Mouse HRP | Goat | 1:10 000 | WB | Jackson Immunoresearch (115-035-068) |
| Anti-Rabbit HRP | Donkey | 1:10 000 | WB | Jackson Immunoresearch (711-035-152) | |
| Anti-goat HRP | Donkey | 1:10 000 | WB | Santa Cruz Biotechnology (sc-2020) | |
| Anti-Sheep HRP | Donkey | 1:10 000 | WB | Jackson Immunoresearch (711-035-147) | |
| Anti-Mouse IRDye 680RD | Goat | 1:15 000 | WB | LI-COR Biosciences (926-68070) | |
| Anti-Mouse IRDye 800CW | Goat | 1:15 000 | WB | LI-COR Biosciences (926-32210) | |
| Anti-Rabbit IRDye 680RD | Goat | 1:15 000 | WB | LI-COR Biosciences (926-68071) | |
| Anti-Rabbit IRDye 800CW | Goat | 1:15 000 | WB | LI-COR Biosciences (926-32211) | |
| Anti-Rabbit Alexa Fluor 488 | Goat | 1:1000 | IF | Invitrogen (A11034) | |
| Anti-Rabbit Alexa Fluor 594 | Goat | 1:1000 | IF | Invitrogen (A11037) | |
| Anti-Rabbit Alexa Fluor 647 | Goat | 1:1000 | IF | Invitrogen (A21245) | |
| Anti-Rabbit Alexa Fluor 680 | Goat | 1:1000 | IF | Invitrogen (A21109) | |
| Anti-Mouse Alexa Fluor 488 | Goat | 1:1000 | IF | Invitrogen (A11029) | |
| Anti-Mouse Alexa Fluor 594 | Goat | 1:1000 | IF | Invitrogen (A11032) | |
| Anti-Mouse Alexa Fluor 680 | Goat | 1:1000 | IF | Invitrogen (A21058) | |
| Anti-Goat Alexa Fluor 488 | Donkey | 1:1000 | IF | Invitrogen (A11055) | |
| Anti-Mouse Alexa Fluor 647 | Donkey | 1:1000 | IF | Invitrogen (A31571) |
Immunofluorescence and image acquisition
Cells grown on sterile round glass coverslips 12 mm diameter, thickness No.1.5 (Menzel Gläser) were either fixed directly with 2% paraformaldehyde (PFA, Electron Microscopy Sciences) and permeabilized with 0.5% Triton X-100 in PBS or first pre-extracted with 0.5% Triton X-100 in CSK buffer (Cytoskeletal buffer: 10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) and then fixed with 2% PFA to remove soluble proteins (not bound to chromatin). For CPD staining, DNA was denatured with 0.5 M NaOH for 5 min. For PCNA staining, cells were treated with Methanol (Prolabo) for 15 min at −20°C. Samples were blocked in 5% BSA (Bovine Serum Albumin, Sigma-Aldrich) in PBS supplemented with 0.1% Tween 20 (Euromedex) before incubation with primary antibodies and secondary antibodies conjugated to Alexa Fluor 488, 594, 647 or 680 (Invitrogen). The specificity of H2A.Z Nt and Ct antibodies in immunofluorescence was verified upon H2A.Z knock-down by siRNA (data not shown).
Coverslips were mounted in Vectashield medium with DAPI (Vector Laboratories) and observed with a Leica DMI6000 epifluorescence microscope using a Plan-Apochromat 40x/1.3 or 63x/1.4 oil objective. Images were captured using a CCD camera (Photometrics) and Metamorph software. For confocal images, we used a Zeiss LSM710 confocal microscope with a Plan-Apochromat 63x/1.4 oil objective and Zen software. Images were mounted with Adobe Photoshop applying the same treatment of fluorescence levels to all images from the same experiment except for Figure 5B where intensities in the red channel (TMR) were adjusted on a cell-by-cell basis because of variable levels of SNAP-tagged H2A.Z in the cell population.
Visualization of replicative and repair synthesis
To visualize replicative synthesis, EdU (5-Ethynyl-2′-deoxyUridine) was incorporated into cells during 15 min (10 μM final concentration) and revealed using the Click-iT EdU Alexa Fluor 594 (or 647) Imaging kit (Invitrogen) according to manufacturer’s instructions. To visualize repair synthesis, EdU (10 μM final concentration) was incorporated into cells during 4 h immediately after local UVC irradiation and revealed using the same kit, before CPD labeling by immunofluorescence. For experiments shown in Figures 6B–6E, EdU incorporation was for 2 h post UVC irradiation and replicating cells showing pan-nuclear EdU staining were excluded from the analysis.
Nascent RNA labeling
Cells were incubated in DMEM supplemented with 0.5 mM EU (EthynylUridine) for 45 min, rinsed in cold medium and in PBS before fixation in 2% paraformaldehyde. EU incorporation was revealed with Click-iT RNA Imaging kits (Invitrogen) using Alexa Fluor 594 dye according to manufacturer’s instructions.
Flow cytometry
Cells were fixed in ice-cold 70% ethanol before DNA staining with 50 μg/mL propidium iodide (Sigma-Aldrich) in PBS containing 0.05% Tween 20 and 0.5 mg/mL RNaseA (USB/Affymetrix). DNA content was analyzed by flow cytometry using a FACS Calibur Flow Cytometer (BD Biosciences) and FlowJo Software (TreeStar).
Colony-Forming Assays
Cells were replated 48 h after siRNA transfection and exposed to global UVC irradiation (4, 8 and 12 J/m2) the following day. Colonies were stained 10 days later with 0.5% crystal violet/20% ethanol and counted. Results were normalized to plating efficiencies.
Quantification and Statistical Analysis
Image analyses
ImageJ (U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/) and ICY (Institut Pasteur, Paris, France, http://icy.bioimageanalysis.org/) softwares were used for image analysis. UV damage sites were delineated based on XPA, XPB, CPD or EdU staining (repair synthesis) and nuclei were delineated based on DAPI staining. Replicating cells were excluded based on pan-nuclear EdU staining (Figure 6). CPD levels at UV sites (Figure S5G) were normalized to the average value obtained in siLUC control condition. To calculate the percentage of H2A.Z-SNAP loss at UV damage sites (Figure S5G), total H2A.Z-SNAP levels (TMR-Star labeling) were normalized to DAPI staining and measured at UV sites relative to the entire nucleus. This ratio was subtracted from 1 and the result was then multiplied by 100.
Percentages of positively stained cells (i.e., showing visually detectable enrichment of the signal of interest at UV sites or replication foci) were obtained by scoring at least 100 cells in each experiment unless indicated otherwise. In experiments using transcription inhibitors, only cells showing above background TMR levels were scored.
Statistical analyses
Statistical tests were performed using GraphPad Prism. Loss or enrichment of H2A.X and H2A.Z at UV sites relative to the whole nucleus was compared to a theoretical mean of 1 by one-sample t tests. P values for mean comparisons between two groups were calculated with a Student’s t test with Welch’s correction when necessary. Multiple comparisons were performed by one-way ANOVA with Bonferroni post-tests. Comparisons of clonogenic survival and CPD removal kinetics were based on non-linear regression with a polynomial quadratic model. ns: non-significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Statistical parameters including sample size (n) and dispersion of the data (SD or SEM) are indicated in the figure legends.
Data and Software Availability
Raw data files are deposited on Mendeley Data (https://doi.org/10.17632/gz5xtcyxzj.1).
Acknowledgments
We thank P.-A. Defossez, C. Francastel, V. Mezger, and J. Weitzman for critical reading of the manuscript; E. Dunleavy, A. Hamiche, and S. Jackson for sharing reagents; and the ImagoSeine core facility (Institut Jacques Monod) for assistance with confocal microscopy. This work was supported by the European Research Council (starting grant ERC-2013-StG-336427 “EpIn”), the French National Research Agency (ANR-12-JSV6-0002-01), the “Who am I”? laboratory of excellence (ANR-11-LABX-0071), EDF Radiobiology program (RB 2014-01), the Fondation ARC (SFI20121205449), and France-BioImaging (ANR-10-INSB-04). S.P. is an EMBO Young Investigator. S.A. was the recipient of PhD fellowships from University Paris 6 and La Ligue contre le Cancer.
Author Contributions
S.P., F.L.P., S.-K.B., and S.E.P. designed and performed experiments and analyzed the data. O.C. provided technical assistance and established mouse cell lines. S.A. generated the H2A.X-SNAP plasmids and the H2A-SNAP and H2A.Z.1-SNAP cell lines. S.E.P. supervised the project and wrote the manuscript with input from all authors.
Declaration of Interests
The authors declare no competing interests.
Published: October 18, 2018
Footnotes
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.09.010.
Supplemental Information
References
- Abe T., Sugimura K., Hosono Y., Takami Y., Akita M., Yoshimura A., Tada S., Nakayama T., Murofushi H., Okumura K. The histone chaperone facilitates chromatin transcription (FACT) protein maintains normal replication fork rates. J. Biol. Chem. 2011;286:30504–30512. doi: 10.1074/jbc.M111.264721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S., Polo S.E., Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Adam S., Dabin J., Chevallier O., Leroy O., Baldeyron C., Corpet A., Lomonte P., Renaud O., Almouzni G., Polo S.E. Real-time tracking of parental histones reveals their contribution to chromatin integrity following DNA damage. Mol. Cell. 2016;64:65–78. doi: 10.1016/j.molcel.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Bukowski-Wills J.-C., Lee S.-B., Kustatscher G., Nakamura K., de Lima Alves F., Menard P., Mejlvang J., Rappsilber J., Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Barth T.K., Reverón-Gómez N., Sidoli S., Schmidt A., Jensen O.N., Imhof A., Groth A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alatwi H.E., Downs J.A. Removal of H2A.Z by INO80 promotes homologous recombination. EMBO Rep. 2015;16:986–994. doi: 10.15252/embr.201540330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis C.D., Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Altmeyer M., Lukas J. To spread or not to spread—chromatin modifications in response to DNA damage. Curr. Opin. Genet. Dev. 2013;23:156–165. doi: 10.1016/j.gde.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Atsumi Y., Minakawa Y., Ono M., Dobashi S., Shinohe K., Shinohara A., Takeda S., Takagi M., Takamatsu N., Nakagama H. ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient γH2AX foci formation. Cell Rep. 2015;13:2728–2740. doi: 10.1016/j.celrep.2015.11.054. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing C.H., Chua K.F., Sekiguchi J., Suh H., Whitlow S.R., Fleming J.C., Monroe B.C., Ciccone D.N., Yan C., Vlasakova K. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor D., Rodríguez M., Moreno N. Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol. 2012;Chapter 8 doi: 10.1002/0471143030.cb0808s55. Unit8.8. [DOI] [PubMed] [Google Scholar]
- Boyarchuk E., Filipescu D., Vassias I., Cantaloube S., Almouzni G. The histone variant composition of centromeres is controlled by the pericentric heterochromatin state during the cell cycle. J. Cell Sci. 2014;127:3347–3359. doi: 10.1242/jcs.148189. [DOI] [PubMed] [Google Scholar]
- Buschbeck M., Hake S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017;18:299–314. doi: 10.1038/nrm.2016.166. [DOI] [PubMed] [Google Scholar]
- Chen C.-C., Carson J.J., Feser J., Tamburini B., Zabaronick S., Linger J., Tyler J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabin J., Fortuny A., Polo S.E. Epigenome maintenance in response to DNA damage. Mol. Cell. 2016;62:712–727. doi: 10.1016/j.molcel.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma N.P., van Attikum H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016;35:6–23. doi: 10.15252/embj.201592595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao L.-T., Chen C.-C., Dennehey B., Pal S., Wang P., Shen Z.-J., Deem A., Tyler J.K. Delineation of the role of chromatin assembly and the Rtt101Mms1 E3 ubiquitin ligase in DNA damage checkpoint recovery in budding yeast. PLoS ONE. 2017;12:e0180556. doi: 10.1371/journal.pone.0180556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C., Ampatziadis-Michailidis G., Lans H., Tresini M., Lagarou A., Grosbart M., Theil A.F., van Cappellen W.A., Kimura H., Bartek J. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell. 2013;51:469–479. doi: 10.1016/j.molcel.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Dunleavy E.M., Almouzni G., Karpen G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G1 phase. Nucleus. 2011;2:146–157. doi: 10.4161/nucl.2.2.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.Y., Gordon F., Luger K., Hansen J.C., Tremethick D.J. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 2002;9:172–176. doi: 10.1038/nsb767. [DOI] [PubMed] [Google Scholar]
- Fan J., Otterlei M., Wong H.-K., Tomkinson A.E., Wilson D.M., 3rd XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004;32:2193–2201. doi: 10.1093/nar/gkh556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.Y., Rangasamy D., Luger K., Tremethick D.J. H2A.Z alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol. Cell. 2004;16:655–661. doi: 10.1016/j.molcel.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Gao Y., Li C., Wei L., Teng Y., Nakajima S., Chen X., Xu J., Leger B., Ma H., Spagnol S.T. SSRP1 cooperates with PARP and XRCC1 to facilitate single-strand DNA break repair by chromatin priming. Cancer Res. 2017;77:2674–2685. doi: 10.1158/0008-5472.CAN-16-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparian A.V., Burkhart C.A., Purmal A.A., Brodsky L., Pal M., Saranadasa M., Bosykh D.A., Commane M., Guryanova O.A., Pal S. Curaxins: anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011;3:95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Yuzugullu O., Ayrapetov M.K., Price B.D. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. USA. 2015;112:7507–7512. doi: 10.1073/pnas.1504868112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanasoge S., Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28:2298–2304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- Heo K., Kim H., Choi S.H., Choi J., Kim K., Gu J., Lieber M.R., Yang A.S., An W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Herrera-Moyano E., Mergui X., García-Rubio M.L., Barroso S., Aguilera A. The yeast and human FACT chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014;28:735–748. doi: 10.1101/gad.234070.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Jackson V. In vivo studies on the dynamics of histone-DNA interaction: evidence for nucleosome dissolution during replication and transcription and a low level of dissolution independent of both. Biochemistry. 1990;29:719–731. doi: 10.1021/bi00455a019. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C., Watanabe S., Kaplan C.D., Peterson C.L., Robert F. The histone chaperones FACT and Spt6 restrict H2A.Z from intragenic locations. Mol. Cell. 2015;58:1113–1123. doi: 10.1016/j.molcel.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász S., Elbakry A., Mathes A., Löbrich M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell. 2018;71:11–24.e7. doi: 10.1016/j.molcel.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Kim J.-A., Haber J.E. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. USA. 2009;106:1151–1156. doi: 10.1073/pnas.0812578106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Sturgill D., Sebastian R., Khurana S., Tran A.D., Edwards G.B., Kruswick A., Burkett S., Hosogane E.K., Hannon W.W. Replication stress shapes a protective chromatin environment across fragile genomic regions. Mol. Cell. 2018;69:36–47.e7. doi: 10.1016/j.molcel.2017.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Cook P.R. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001;153:1341–1353. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat C.F., Yeeles J.T.P., Patel H., Early A., Diffley J.F.X. Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol. Cell. 2017;65:117–130. doi: 10.1016/j.molcel.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F., Giannattasio M., Puddu F., Granata M., Pellicioli A., Plevani P., Muzi-Falconi M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst.) 2009;8:1055–1067. doi: 10.1016/j.dnarep.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Liu Y., Parry J.A., Chin A., Duensing S., Duensing A. Soluble histone H2AX is induced by DNA replication stress and sensitizes cells to undergo apoptosis. Mol. Cancer. 2008;7:61. doi: 10.1186/1476-4598-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louters L., Chalkley R. Exchange of histones H1, H2A, and H2B in vivo. Biochemistry. 1985;24:3080–3085. doi: 10.1021/bi00334a002. [DOI] [PubMed] [Google Scholar]
- Luger K., Dechassa M.L., Tremethick D.J. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012;13:436–447. doi: 10.1038/nrm3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M.S., Lindh M., Acs K., Vrouwe M.G., Pines A., van Attikum H., Mullenders L.H., Dantuma N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012;197:267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M.S., de Krijger I., Wiegant W.W., Shah R.G., Smeenk G., de Groot A.J.L., Pines A., Vertegaal A.C.O., Jacobs J.J.L., Shah G.M., van Attikum H. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell. 2016;61:547–562. doi: 10.1016/j.molcel.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Gibbs-Seymour I., Bekker-Jensen S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013;14:269–282. doi: 10.1038/nrm3562. [DOI] [PubMed] [Google Scholar]
- Mannironi C., Bonner W.M., Hatch C.L. H2A.X. a histone isoprotein with a conserved C-terminal sequence, is encoded by a novel mRNA with both DNA replication type and polyA 3′ processing signals. Nucleic Acids Res. 1989;17:9113–9126. doi: 10.1093/nar/17.22.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z., Pan L., Wang W., Sun J., Shan S., Dong Q., Liang X., Dai L., Ding X., Chen S. Anp32e, a higher eukaryotic histone chaperone directs preferential recognition for H2A.Z. Cell Res. 2014;24:389–399. doi: 10.1038/cr.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn J.A., Bekker-Jensen S., Mailand N., Lans H., Schwertman P., Gourdin A.M., Dantuma N.P., Lukas J., Vermeulen W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 2009;186:835–847. doi: 10.1083/jcb.200902150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini E., Roche D.M., Marheineke K., Verreault A., Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J. Cell Biol. 1998;143:563–575. doi: 10.1083/jcb.143.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggs J.G., Grandi P., Quivy J.P., Jónsson Z.O., Hübscher U., Becker P.B., Almouzni G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 2000;20:1206–1218. doi: 10.1128/mcb.20.4.1206-1218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L.H.F., Fousteri M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell. 2007;27:311–323. doi: 10.1016/j.molcel.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Nishi R., Alekseev S., Dinant C., Hoogstraten D., Houtsmuller A.B., Hoeijmakers J.H.J., Vermeulen W., Hanaoka F., Sugasawa K. UV-DDB-dependent regulation of nucleotide excision repair kinetics in living cells. DNA Repair (Amst.) 2009;8:767–776. doi: 10.1016/j.dnarep.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Nishibuchi I., Suzuki H., Kinomura A., Sun J., Liu N.-A., Horikoshi Y., Shima H., Kusakabe M., Harata M., Fukagawa T. Reorganization of damaged chromatin by the exchange of histone variant H2A.Z-2. Int. J. Radiat. Oncol. Biol. Phys. 2014;89:736–744. doi: 10.1016/j.ijrobp.2014.03.031. [DOI] [PubMed] [Google Scholar]
- Obri A., Ouararhni K., Papin C., Diebold M.-L., Padmanabhan K., Marek M., Stoll I., Roy L., Reilly P.T., Mak T.W. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. 2014;505:648–653. doi: 10.1038/nature12922. [DOI] [PubMed] [Google Scholar]
- Okuhara K., Ohta K., Seo H., Shioda M., Yamada T., Tanaka Y., Dohmae N., Seyama Y., Shibata T., Murofushi H. A DNA unwinding factor involved in DNA replication in cell-free extracts of Xenopus eggs. Curr. Biol. 1999;9:341–350. doi: 10.1016/s0960-9822(99)80160-2. [DOI] [PubMed] [Google Scholar]
- Okuwaki M., Kato K., Nagata K. Functional characterization of human nucleosome assembly protein 1-like proteins as histone chaperones. Genes Cells. 2010;15:13–27. doi: 10.1111/j.1365-2443.2009.01361.x. [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M., Krebs J.E., Peterson C.L. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 2006;20:2437–2449. doi: 10.1101/gad.1440206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Almouzni G. Chromatin dynamics after DNA damage: The legacy of the access-repair-restore model. DNA Repair (Amst.) 2015;36:114–121. doi: 10.1016/j.dnarep.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Roche D., Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D., Woolfe A., Vassias I., Pellentz C., Lacoste N., Puri A., Schultz D.C., Pchelintsev N.A., Adams P.D., Jansen L.E.T., Almouzni G. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Rocha C.R.R., Lerner L.K., Okamoto O.K., Marchetto M.C., Menck C.F.M. The role of DNA repair in the pluripotency and differentiation of human stem cells. Mutat. Res. 2013;752:25–35. doi: 10.1016/j.mrrev.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Seo J., Kim S.C., Lee H.-S., Kim J.K., Shon H.J., Salleh N.L.M., Desai K.V., Lee J.H., Kang E.-S., Kim J.S., Choi J.K. Genome-wide profiles of H2AX and γ-H2AX differentiate endogenous and exogenous DNA damage hotspots in human cells. Nucleic Acids Res. 2012;40:5965–5974. doi: 10.1093/nar/gks287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J., Kim K., Chang D.-Y., Kang H.-B., Shin E.-C., Kwon J., Choi J.K. Genome-wide reorganization of histone H2AX toward particular fragile sites on cell activation. Nucleic Acids Res. 2014;42:1016–1025. doi: 10.1093/nar/gkt951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G., van Attikum H. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu. Rev. Biochem. 2013;82:55–80. doi: 10.1146/annurev-biochem-061809-174504. [DOI] [PubMed] [Google Scholar]
- Smerdon M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol. 1991;3:422–428. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- Talbert P.B., Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Tan B.C., Chien C.T., Hirose S., Lee S.C. Functional cooperation between FACT and MCM helicase facilitates initiation of chromatin DNA replication. EMBO J. 2006;25:3975–3985. doi: 10.1038/sj.emboj.7601271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyemi U., Redon C.E., Choudhuri R., Aziz T., Maeda D., Boufraqech M., Parekh P.R., Sethi T.K., Kasoji M., Abrams N. The histone variant H2A.X is a regulator of the epithelial-mesenchymal transition. Nat. Commun. 2016;7:10711. doi: 10.1038/ncomms10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.S., Bonner W.M. Separation of basal histone synthesis from S-phase histone synthesis in dividing cells. Cell. 1981;27:321–330. doi: 10.1016/0092-8674(81)90415-3. [DOI] [PubMed] [Google Scholar]
- Wu T., Liu Y., Wen D., Tseng Z., Tahmasian M., Zhong M., Rafii S., Stadtfeld M., Hochedlinger K., Xiao A. Histone variant H2A.X deposition pattern serves as a functional epigenetic mark for distinguishing the developmental potentials of iPSCs. Cell Stem Cell. 2014;15:281–294. doi: 10.1016/j.stem.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Xu Y., Ayrapetov M.K., Xu C., Gursoy-Yuzugullu O., Hu Y., Price B.D. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell. 2012;48:723–733. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang X., Feng J., Leng H., Li S., Xiao J., Liu S., Xu Z., Xu J., Li D. The histone chaperone FACT contributes to DNA replication-coupled nucleosome assembly. Cell Rep. 2016;14:1128–1141. doi: 10.1016/j.celrep.2015.12.096. [DOI] [PubMed] [Google Scholar]
- Zhang H., Roberts D.N., Cairns B.R. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123:219–231. doi: 10.1016/j.cell.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.