

Abstract
The organization of the extracellular matrix has a profound impact on cancer development and progression. The matrix becomes aligned throughout tumor progression, providing “highways” for tumor cell invasion. Aligned matrix is associated with breast density and is a negative prognostic factor in several cancers; however, the underlying mechanisms regulating this reorganization remain poorly understood. Deletion of the tumor suppressor Pten in the stroma was previously shown to promote extracellular matrix expansion and tumor progression. However, it was unknown if PTEN also regulated matrix organization. To address this question, a murine model with fibroblast-specific Pten deletion was used to examine how PTEN regulates matrix remodeling. Using second harmonic generation microscopy, Pten deletion was found to promote collagen alignment parallel to the mammary duct in the normal gland and further remodeling perpendicular to the tumor edge in tumor-bearing mice. Increased alignment was observed with Pten deletion in vitro using fibroblast-derived matrices. PTEN loss was associated with fibroblast activation and increased cellular contractility, as determined by traction force microscopy. Inhibition of contractility abrogated the increased matrix alignment observed with PTEN loss. Murine mammary adenocarcinoma cells cultured on aligned matrices derived from Pten−/− fibroblasts migrated faster than on matrices from wild-type fibroblasts. Combined, these data demonstrate that PTEN loss in fibroblasts promotes extracellular matrix deposition and alignment independently from cancer cell presence, and this reorganization regulates cancer cell behavior. Importantly, stromal PTEN negatively correlated with collagen alignment and high mammographic density in human breast tissue, suggesting parallel function for PTEN in patients.
Abbreviations: ECM, extracellular matrix; FDM, fibroblast-derived matrix; MMF, murine mammary fibroblast; MMP, matrix metalloproteinase; PTEN, phosphatase and tensin homolog; SHG, second harmonic generation; TACS, tumor-associated collagen signature; WT, wild-type
Introduction
Mammographic density is a significant risk factor for both ductal carcinoma in situ and invasive breast cancer [1], representing one of the largest independent risk factors for breast cancer development [2]. High mammographic density in more than 50% of breast tissue is associated with a 3.5-fold higher risk of invasive breast cancer as compared to breasts with less than 10% density [1]. Although high mammographic density poses a lower relative risk for cancer initiation than mutations such as BRCA1/2 [3], it contributes greatly to this etiology due to its frequent occurrence. It is estimated that breast density contributes to the development of 30%-65% of breast cancers [2], [3]. Mammographic density is attributable to a number of structural changes within the breast, including increased collagen deposition [4], [5], higher proteoglycan expression [6], and a greater area of glandular structures [5]. Of these factors, collagen deposition is most strongly associated with mammographic density [5]. Increased collagen density has been shown to directly promote tumorigenesis in a mouse model [7], underscoring the importance of the extracellular matrix (ECM) in cancer initiation. In cases of high mammographic density, not only do changes in collagen deposition occur but also changes in collagen organization. Collagen coherency, a measure of local fiber alignment, and increased fibrillar organization surrounding mammary ducts were recently observed to correlate with mammographic density and local tissue stiffness [8]. However, the changes in ECM organization that occur in cases of high mammographic density is understudied, and it remains unclear how these changes affect ECM organization in the tumor microenvironment.
ECM reorganization occurs extensively throughout tumor development and progression, and this remodeling regulates cancer cell behavior. Throughout tumor progression, collagen fibers surrounding the tumor become linearized and aligned, first parallel to the tumor edge (termed tumor-associated collagen signature 2 or TACS-2) then locally perpendicular (TACS-3) [9]. High collagen density has been shown to promote early formation of the TACS-3 phenotype in the tumor microenvironment in a mouse model [7]. The presence of these perpendicularly aligned fibers promotes cancer cell invasion away from the primary tumor [9], [10]. In the peritumoral stroma of the breast, there is a strong correlation between collagen linearization and cancer invasion, as well as tissue stiffness [11]. Indeed, collagen alignment is an independent prognostic factor in breast carcinoma patients [12]. The effects of fiber alignment on cancer cell behavior have been extensively studied in vitro using a variety of methods, including 2D studies on micropatterned surfaces and 3D studies using collagen gels or fibroblast-derived matrices (FDMs). Cancer-associated fibroblasts (CAFs) have been found to produce highly aligned extracellular matrices as compared to normal fibroblasts [13], [14], and matrix alignment promotes elongation [15] and directional migration [14] of cancer cells in CAF-derived matrices. Similarly, cancer cells migrate with higher directionality [16] and persistence [17] in aligned collagen gels.
Fibroblasts play a central role in ECM deposition and organization. Downregulation of the tumor suppressor phosphatase and tensin homolog (PTEN) is a common feature of the activated stroma surrounding tumors, with nearly half of patients with invasive breast carcinoma showing low stromal PTEN expression [18]. Pten deletion in mammary fibroblasts greatly increases collagen deposition surrounding mammary ducts, promotes gelatinase activity, and increases macrophage infiltration into the mammary gland [18]. Furthermore, Pten deletion in fibroblasts modifies the adjacent epithelium, increasing the mammary stem cell–enriched myoepithelial cell population [19]. Stromal Pten deletion additionally increases tumorigenesis and tumor burden in the presence of the Neu oncogene [18], acting in part through downregulation of miR320 [20]. As stromal PTEN loss dramatically modifies the mammary environment, it is not possible to separate these factors in vivo to determine how changes to the ECM contribute to tumor growth.
PTEN has been shown to function as a negative regulator of fibroblast activation in a pulmonary fibrosis model, and deletion of Pten exacerbates fibrosis [21]. Therefore, we hypothesized that loss of PTEN in stromal fibroblasts could stimulate activation (similar to a CAF phenotype) and increase matrix alignment even in the absence of a tumor. To test this hypothesis, we used a previously developed mouse model in which Pten is specifically deleted in fibroblasts to investigate how loss of PTEN affects ECM reorganization in vivo [18]. Fibroblasts from this model were used to examine fibroblast activation, matrix organization, and the effects of matrix alignment on cancer cell migration in vitro. The role of PTEN in regulating cellular contractility—and the subsequent effect on matrix alignment—was also investigated. Finally, the relationship of PTEN loss and human breast density was explored.
Materials and Methods
Mouse Studies
PtenloxP/loxP, Fsp-cre;PtenloxP/loxP, MMTV-Neu;PtenloxP/loxP, and MMTV-Neu;Fsp-cre;PtenloxP/loxP FVB/N mice were generated and maintained as previously described [18], [22] in accordance with NIH regulations and approved by the Ohio State University Institutional Animal Care and Use Committee.
Collagen Imaging and Orientation Analysis
Tissues from the upper mammary gland of 8-week-old mice were fixed using 4% paraformaldehyde or formalin, then paraffin-embedded and sectioned at 5-μm thickness. The sections were deparaffinized, soaked for 8 minutes in Weigert's hematoxylin (Sigma-Aldrich), washed for 10 minutes in deionized water, and stained with picrosirius red (Abcam) according to the manufacturer's protocol. The sections were imaged using an Olympus FV1000 MPE microscope equipped with a 25× XLPlan water immersion objective lens (N.A. 1.05) and a Mai Tai DeepSee Ti:Sapphire laser (Spectra-Physics, Newport Corp.) tuned to a wavelength of 950 nm. Images were taken through the sample depth in 1-μm intervals, and a maximum intensity projection of these images was analyzed using CurveAlign software (version 2.3; http://loci.wisc.edu/software/curvealign). Using the picrosirius red images, a border was drawn manually at the duct edge or at the tumor-stroma boundary. This boundary was superimposed onto second harmonic generation images, and collagen fiber orientation was analyzed within 25 μm of the boundary. Collagen fiber orientation was analyzed relative to the duct edge for mice without tumors and relative to the tumor-stroma boundary in MMTV-Neu mice. Overall, 8478 points were analyzed for orientation in WT samples, 8763 in Pten−/− samples, 1081 in WT;MMTV-Neu samples, and 2151 in Pten−/−;MMTV-Neu samples.
Cell Lines and Culture Conditions
Wild-type and Pten−/− murine mammary fibroblasts (MMFs) were isolated from mice as previously described [20] and used at passages 30-50. They were maintained in high-glucose DMEM (Invitrogen) with 10% heat-inactivated fetal bovine serum (FBS; VWR), 2 mM L-glutamine, 10 U/ml penicillin, and 10 μg/ml streptomycin. FBS was heat-inactivated at 55°C for 30 minutes. DB7 murine mammary adenocarcinoma cells [23] were maintained in the same growth media except the FBS was not heat inactivated, and used at passage <30. Cells were tested for mycoplasma upon receipt and maintained in a humidified 37°C incubator with 5% CO2.
Fibroblast-Derived Matrix Production
FDMs were produced over a period of 5 days according to previously published protocols [24], with the exception that media changes occurred daily. In brief, fibroblasts were plated on gelatin-coated plates at 50,000 cells/cm2. Medium was changed daily with the addition of 50 μg/ml L-ascorbic acid (Sigma-Aldrich). Matrices were fixed or decellularized 5 days after plating. Cells were extracted from the matrices using 0.5% Triton X-100 (Sigma-Aldrich) and 20 mM NH4OH for 3-5 minutes, at which point three times the volume of phosphate-buffered saline (PBS) was added to each well and the matrices were allowed to equilibrate at 4°C for a minimum of 12 hours. The matrices were washed in PBS three times and then treated with 10 U/ml DNAse (Sigma-Aldrich) in PBS supplemented with CaCl2 and MgCl2 for 30 minutes at 37°C. WT matrices were extracted three times and Pten−/− matrices were extracted once due to differences in matrix thickness. Matrices were stored in PBS at 4°C for a maximum of 1 week before use.
Immunofluorescence and Cell Morphology Analysis
To fix samples, half of the media was removed from each sample, and an equivalent volume of fixative (4% paraformaldehyde and 5% sucrose in PBS) was added for 20 minutes at room temperature. Samples were permeabilized in 0.5% Triton X-100 and blocked in 10% normal goat serum (Invitrogen) before incubating with primary antibodies in blocking solution. Samples were washed and incubated with fluorescent secondary antibodies, 1:2000 Hoechst 33342 (Invitrogen, Thermo Fisher Scientific) and, if applicable, 1:100 AlexaFluor 555-conjugated phalloidin (Invitrogen, Thermo Fisher Scientific), and then washed before mounting on slides using ProLong Gold Antifade solution (Life Technologies, Thermo Fisher Scientific). To quantify cell morphology, phalloidin staining was analyzed using CellProfiler software [25]. In CellProfiler, Hoechst staining was used to identify nuclei, with a minimum diameter of 10 pixels, and shape was used to distinguish clumped objects. The phalloidin staining for each cell was propagated from the identified nuclei, and the area and axis lengths of each cell were determined.
Antibodies and Dilutions
The following antibodies were used in these studies: α-smooth muscle actin (Sigma-Aldrich A2547, clone 1A4, used 1:1000 for Western blotting and 1:300 for immunofluorescence), fibronectin (Abcam ab23750, used 1:200 for immunofluorescence), GAPDH (Thermo Fisher Scientific NB600502, clone 6C5, used 1:1000 for Western blotting), PTEN (Cell Signaling 9552, used 1:1000 for Western blotting, or Cell Signaling 9559, used for immunohistochemistry), AlexaFluor-conjugated goat anti-rabbit or anti-mouse (Thermo Fisher Scientific A11034 and A21422, used 1:500 for immunofluorescence), and horseradish peroxidase–conjugated goat anti-rabbit or anti-mouse (Thermo Fisher Scientific 32460 and 32430, used 1:1000 for Western blotting).
Fibroblast-Derived Matrix Imaging and Orientation Analysis
An Olympus FV1000 confocal microscope equipped with a 40× (N.A. 1.3) oil-immersion objective lens was used to image the fibronectin structure in the top 12 μm of each matrix in 1-μm sections. The maximum intensity projection of these images was analyzed with the OrientationJ plugin for ImageJ [26], which analyzes fiber orientation on a pixel-by-pixel basis, using previously published settings [24]. The mode of the orientation distribution of the fibers was set to 0°, and matrix alignment was quantified by determining the fraction of fibers falling within 10° or 20° of the mode.
Western Blotting
Cells were lysed in RIPA buffer containing HALT protease/phosphatase inhibitor (Thermo Fisher Scientific) and frozen at −70°C until use. Samples were thawed and centrifuged at 12,000 RPM for 15 minutes. Protein in the supernatant was quantified using the μBCA assay (Thermo Fisher Scientific), and 20 μg of protein were loaded into each well. Samples were separated using SDS-PAGE and transferred onto a PVDF membrane, then incubated with primary and secondary HRP-conjugated antibodies and imaged using chemiluminescent substrate. Densitometry was performed using ImageJ software (version 1.43u, National Institutes of Health).
Encapsulation of Cells for Determination of Matrix Metalloproteinase (MMP) Activity
This assay was performed as previously described [27], with the exception that MMP activity was normalized to Hoechst 33342 fluorescence to control for the number of cells per sample. In brief, a single cell suspension of MMF was encapsulated at 2×106 cells/ml in a solution of 6 wt% 4-arm 20 kDa polyethylene glycol (PEG) (JenKem) to form 50-μl hydrogels. PEG-norbornene was synthesized as previously described [27] and cross-linked with a MMP-degradable cross-linker (KCGPQG↓IWGQCK, where the arrow denotes the site of cleavage; American Peptide Company), 5 mM CRGDS peptide (American Peptide Company), and a fluorogenic MMP-sensitive peptide (GGPQG↓IWGQKDdeAEEAcC). The mixture was polymerized using 1.7 mM lithium phenyl-2,4,6-trimethylbenzoyl-phosphinate and exposure to UV light at 4 mW/cm2 for 3 minutes. One hour before reading, Hoechst 33342 was added to each well at a dilution of 1:1000. Hoechst fluorescence was measured (excitation/emission 350/461 nm) along with collagenase fluorescence (excitation/emission 494/521 nm) at 24 hours after encapsulation using area scans on a SpectraMax M2 plate reader (Molecular Devices). Background fluorescence was subtracted using readings from corresponding no-cell conditions. Collagenase fluorescence was normalized to Hoechst fluorescence to control for the number of cells in each sample.
PCR
Cells were plated at 50,000/cm2 and cultured for 24 hours in growth medium, at which point the medium was changed and supplemented with 50 μg/ml ascorbic acid. Cells were cultured for another 24 hours before lysis. RNA was isolated using Tri-Reagent (Sigma-Aldrich) according to the manufacturer's protocol, reverse transcribed, and used for qRT-PCR. Primers were obtained from the Harvard Primer Bank (https://pga.mgh.harvard.edu/primerbank/) and verified using Primer BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). qRT-PCR was run using SYBR Green (Thermo Fisher Scientific) for detection. Primers were used at 900 nM, and expression levels were normalized to the endogenous control, 18S (Thermo Fisher Scientific). Primer sequences were as follows:
Col1a1: sense (s), GCTCCTCTTAGGGGCCACT; antisense (as), CCACGTCTCACCATTGGGG; Col4a1: s, TCCGGGAGAGATTGGTTTCC; as, CTGGCCTATAAGCCCTGGT; Fn1: s, ATGTGGACCCCTCCTGATAGT; as, GCCCAGTGATTTCAGCAAAGG; Tnc: s, GCATCCGTACCAAAACCATCA; as, AACCCGTAGGGATTAGTGTCG.
Traction Force Microscopy
Traction force microscopy was performed as previously described [28]. In brief, polyacrylamide gels containing 0.01% 0.5 μm red fluorescent carboxylate modified microspheres (Invitrogen, Thermo Fisher Scientific) were prepared by mixing 8% acrylamide and 0.04% bis-acrylamide (Bio-Rad) with 0.05% ammonium persulfate and 0.05% N,N,N′,N′-tetramethylethylenediamine to polymerize on 22-mm square glass coverslips. The gel surface was activated twice with 50 mM sulfo-SANPAH (Thermo Fisher Scientific) for 5 minutes under a 254-nm UV lamp and then coated with 200 μg/ml type I bovine collagen (Advanced BioMatrix) for 30 minutes at room temperature. Cells were seeded onto the surface at 200 cells/cm2 and grown overnight. Cells were imaged under phase contrast to obtain the location of the cell boundary. Fluorescence images of the beads were taken before and after trypsinization of the cells to obtain the positional changes of the beads with cell detachment. Using MATLAB (MathWorks) image registration, the bead positions were aligned and were used to calculate displacement field by correlation based particle image velocimetry. The 3D finite element software COMSOL Multiphysics was used to compute traction stresses, Ti(r), by setting the displacement values as the boundary conditions on cell surface. Maximum traction force, Tmax = max[Ti(r)], and net contractile moment, Mnet, were analyzed as follows:
where r is the position on the gel surface and x is the distance from center of mass of the cell to the gel surface.
Migration Studies
Spheroids were produced via the hanging drop method, using 1000 cells per 10-μl drop. Cells were suspended for 3 days to form spheroids before plating on TCPS or decellularized FDM; drops were transferred using a 1000-μl pipette. The spheroids were allowed to adhere for 7 hours before imaging. Images were taken under transmitted light every 10 minutes for 12 hours. The cells were maintained at 37°C and 5% CO2. To quantify cell migration, the area of the spheroid was determined by manual tracing in ImageJ. To control for variation in initial spheroid size, the final area covered by the cells after 12 hours of imaging was normalized to the initial area covered by the spheroid. The aspect ratio of the spheroids after 12 hours of imaging was determined using the FitEllipse function in ImageJ.
Human Breast Density, PTEN, and Collagen Analysis
Fibroglandular breast tissue >2 cm from the primary tumor site was dissected from the breast specimens of women undergoing definitive surgery for breast cancer at the James Cancer Hospital of the Ohio State University. Tissue collection was performed in accordance with institutional guidelines under IRB protocol #1999C0262. Fibroglandular tissue was dissected within 1 hour of surgical excision and placed immediately into 10% buffered formalin for 24 hours. Samples were then changed to 70% ethanol and paraffin embedded. Mammographic density was evaluated in accordance with the fifth edition of BIRADS of the American College of Radiology, with four categories for breast density. Patients were classified as high density (heterogeneously dense or dense) or low density (fatty or scattered fibroglandular density) based on presurgery mammography. A total of 13 patients' samples were used for this study, 5 of which were classified as low density, while 8 were high density.
Tissue sections were stained using the Bond RX autostainer (Leica Biosystems Inc.). Slides were baked at 65°C for 15 minutes, and the automated system performed immunohistochemistry using Bond reagents (Leica), a PTEN primary antibody (Cell Signaling), and DAB for detection. Slides were counterstained with hematoxylin. Samples were then dehydrated through a series of ethanol and xylenes and mounted. The VECTRA Automated Quantitative Pathology Imaging system (PerkinElmer) was used to quantify PTEN immunostaining using the pattern recognition algorithm in inForm 2.0 software (PerkinElmer). The slides were scanned in their entirety, and the region with the lowest visual DAB staining in the periductal stroma in each sample was imaged. Multispectral image cubes were prepared using inForm software, followed by trainable tissue segmentation of images into epithelium and stroma and further segmentation into nuclei and cytoplasm. The spectrally unmixed DAB signal was scored based on a user-defined threshold into four categories (0+, 1+, 2+, and 3+). The percent of cells within each scoring category was determined based on cell segmentation with the hematoxylin counterstain. An H-Score was then calculated using following formula: [1×(% cells 1+) + 2×(% cells 2+) + 3×(% cells 3+)].
Collagen imaging was performed analogously to the murine samples. Samples were blinded to the experimenter with regards to mammographic density. Epithelial PTEN staining was used to manually draw a border at the duct edge, and collagen orientation was determined relative to this border.
Cell Proliferation Assays
Cell proliferation was determined using the CyQUANT assay (ThermoFisher) according to the manufacturer's instructions. +MMFs were seeded at 50,000/cm2 in 96-well plates. At 4, 24, or 48 hours after seeding, the medium was aspirated, and the cells were frozen at −70°C prior to using CyQUANT.
Data Analysis
Statistical analyses were done using GraphPad Prism 7 software. Comparisons between two groups were done using an unpaired one-sided t test, except for traction force microscopy and patient data, which were analyzed using a Mann-Whitney test. Comparisons between more conditions were done using a one-way ANOVA followed by Tukey's multiple comparisons test. Data were considered significant at P<.05. Each experiment was repeated a minimum of three times with two technical replicates. For experiments using tissue sections, each tissue section was obtained from a different mouse or patient. A minimum of four images was taken per section, with the exception of PTEN quantification, and one MMTV-Neu;PtenloxP/loxP section where only two images were taken due to a lack of stroma in the section. Fiber orientation distributions were compared using a two-sample Kolmogorov-Smirnov test.
Results
Stromal Pten Loss Alters Collagen Organization
While PTEN has been identified as a tumor suppressor in many cancers for its role in cancer cells, PTEN expression in the stromal compartment also critically regulates disease progression. Pten deletion specifically in fibroblasts using a murine model (Fsp-Cre;PtenloxP/loxP) has been shown to greatly promote collagen deposition within the mammary gland [18]. However, it remains unclear if PTEN controls other key characteristics of the ECM that regulate tumor invasion and metastasis, such as matrix alignment [7], [9], [29]. To investigate how collagen organization changes with loss of stromal PTEN, second harmonic generation (SHG) microscopy was used to observe fibrillar collagen organization surrounding ducts in mammary gland tissue sections from wild-type (WT, PtenloxP/loxP) and Pten−/− (Fsp-Cre;PtenloxP/loxP) mice. As previously reported [18], stromal Pten−/− mammary glands exhibited remarkably enhanced ECM deposition surrounding mammary ducts (Figure 1A). The orientation of each fiber in the SHG imaging was quantified, and the distribution of fiber orientation was plotted as a histogram (Figure 1B). The cumulative probability distribution of fiber orientation was significantly different between the WT and Pten−/− samples. The fraction of fibers oriented parallel to the duct edge was quantified for each duct imaged, and this fraction was significantly higher with Pten knockout (where parallel was defined as <10° from the tangent line of the duct edge; Figure 1C). The number of fibers oriented perpendicular to the duct edge in Pten−/− samples also significantly decreased (where perpendicular is defined as >80° from the tangent line of the duct edge; Figure 1C).
Figure 1.

Pten knockout affects collagen alignment in vivo. (A) H&E staining, second harmonic generation, and picrosirius red staining of representative ducts within murine mammary gland tissue sections. SHG images are shown above their corresponding picrosirius red image. Solid lines represent the lumen edge. (B) Histogram of collagen fiber angle relative to the duct, quantified using CurveAlign software. Bin size = 10°. P value represents the probability of the WT and Pten−/− histograms originating from the same distribution. (C) Plot of the fraction of fibers oriented at <10° or >80° relative to the duct edge. Each dot represents the fraction of fibers surrounding an individual duct. WT: n=32. Pten−/−: n=23, where n represents the number of ducts imaged from 7 and 5 mice, respectively. *P<.05, **P<.01.
Stromal Pten Loss Promotes Perpendicular Collagen Orientation Surrounding Tumors
Collagen alignment changes throughout tumor progression [9]; therefore, we also examined the effect of stromal Pten knockout on collagen organization in the tumor microenvironment. PtenloxP/loxP;MMTV-Neu mice (WT;MMTV-Neu) and Fsp-Cre;PtenloxP/loxP;MMTV-Neu (Pten−/−;MMTV-Neu) mice were generated as previously described [18], and collagen fiber alignment at the tumor-stroma boundary was examined (Figure 2A). Collagen fiber orientation distribution relative to the tumor edge, graphed as a histogram in Figure 2B, was significantly different between two groups. The proportion of fibers oriented parallel to the tumor edge (Figure 2C) did not change with stromal loss of PTEN; however, significantly more fibers were oriented perpendicular to the tumor edge in Pten−/−;MMTV-Neu mice (Figure 2C).
Figure 2.

Pten knockout affects collagen alignment surrounding tumors. (A) H&E staining, second harmonic generation, and picrosirius red staining of murine mammary gland tissue sections containing tumors. SHG images are shown above their corresponding picrosirius red image. Solid lines represent the tumor-stroma boundary. (B) Histogram of collagen fiber angle relative to the tumor edge, quantified using CurveAlign software. Bin size=10°. P value represents the probability of the WT and Pten−/− histograms originating from the same distribution. (C) Graph of the fraction of fibers oriented at <10° or >80° relative to the tumor edge, shown as mean±SEM. WT;MMTV-Neu: n=12. Pten−/−;MMTV-Neu: n=15, where n represents the number of tumor regions imaged from 3 separate mice. *P<.05.
Pten Deletion in Fibroblasts Enhances Matrix Alignment In Vitro
To investigate the mechanism by which Pten deletion regulates ECM alignment, we used an in vitro system in which fibroblasts are treated with ascorbic acid to stabilize their natural matrix production. These FDMs can then be subsequently decellularized and reseeded with cancer cells, allowing isolation of the effects of matrix alignment on cancer cell behavior. Pten−/− MMFs produced highly aligned matrices compared to matrices from WT MMF, both when the matrices were fixed immediately after matrix production and following decellularization of the matrix (Figure 3A; quantified in Supplemental Figure 1). Fibronectin is the most prevalent protein expressed in FDM [13]; therefore, fibronectin fibers were analyzed for orientation. Fibronectin is necessary for collagen network formation by cells [30], and the two proteins have been found to orient equivalently and colocalize in FDM [30], [31]. Matrix fiber orientation was graphed as a histogram (Figure 3B), and the fraction of fibers falling within 10° or 20° of the mode orientation was subsequently determined. The fraction of fibers falling within these ranges in the Pten−/− matrices was nearly twice that of the WT matrices, indicating that loss of PTEN greatly increases matrix alignment (Figure 3C). This is consistent with the enhanced collagen alignment observed in vivo, where Pten knockout promoted collagen organization parallel to the edge of the duct. The matrices produced by the WT fibroblasts were significantly thicker than those produced by the Pten−/− fibroblasts (Figure 3D). However, when the matrix was solubilized, the Pten−/− matrices contained significantly more protein relative to the DNA content (Figure 3E), indicating that the Pten−/− fibroblasts produce more protein on a “per cell” basis. As such, the increased thickness of the WT matrices is likely due to the higher proliferation rate of the WT cells in vitro (Supplemental Figure 2).
Figure 3.

Pten knockout in fibroblasts alters matrix structure. (A) Fibronectin immunostaining of fixed or decellularized FDMs, color-coded by fiber orientation. (B) Histogram of fibronectin fiber orientation from fixed matrices; bin size=1°. (C) Graph of the fraction of fibers in the matrix falling within 10° or 20° of the mode orientation. (D) Height of unextracted matrices. (E) Protein content of solubilized matrices normalized to DNA content. n=3±SEM. *P<.05, **P<.01, ***P<.001.
Supplemental Figure 1.

Graph of the fraction of fibers in the matrix falling within 10° or 20° of the mode orientation, before and after the extraction process. n=2-3±SEM.
Supplemental Figure 2.
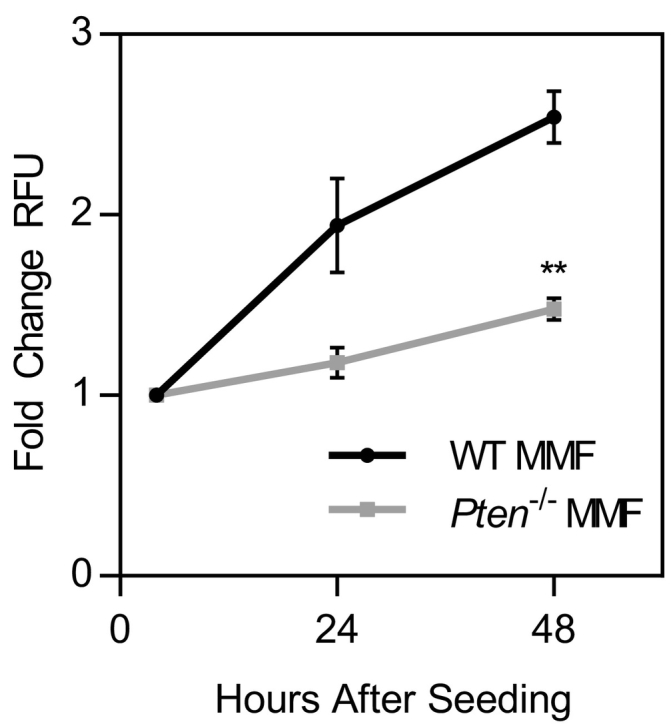
MMF proliferation measured by the CyQUANT assay. n=3±SEM. **P<.01 as compared to WT MMF at the same time point.
Fibroblast Activation with Pten Deletion
CAFs are often activated to a myofibroblast-like state, with heightened cellular contractility, MMP secretion, and ECM synthesis [32]. CAFs produce significantly more aligned matrices in vitro as compared to normal fibroblasts [13], [14]. Because we observed changes in matrix alignment in vivo and in vitro with loss of stromal PTEN, we hypothesized that Pten deletion may result in an activated phenotype, similar to CAFs. To evaluate the activation state of the MMFs, we examined the myofibroblast marker α-smooth muscle actin (αSMA). Immunofluorescence of αSMA revealed pronounced stress fiber formation in Pten−/− MMF compared to WT MMF (Figure 4A). Moreover, αSMA protein expression was significantly higher in Pten−/− cells (as normalized to GAPDH) (Figure 4B), indicating that Pten knockout regulates αSMA in terms of both protein expression and organization.
Figure 4.

Pten deletion increases fibroblast activation. (A) Immunofluorescence images of WT or Pten−/− MMF cultured for 5 days with ascorbic acid, then fixed and stained for αSMA and nuclei. (B) Western blotting and quantification of PTEN (54 kDa), αSMA (42 kDa), and GAPDH loading control (37 kDa), n=5±SEM. (C) MMP activity of MMF encapsulated in PEG hydrogels, n=3±SEM. (D) Normalized mRNA expression of matrix components as determined by RT-PCR, n=3±SEM. *P<.05, ****P<.0001.
We additionally hypothesized that MMP activity would be upregulated in the Pten−/− fibroblasts. MMPs are a family of zinc-dependent enzymes widely involved in matrix reorganization, and their activity is typically upregulated in CAFs [32]. To assess MMP activity, fibroblasts were encapsulated in PEG-based hydrogels with an MMP-sensitive fluorescent peptide [27], which is cleaved by MMP-1, -2, -3, -7, -8, and -9, among others [33]. MMP activity of Pten−/− MMF was found to be approximately two-fold higher than that of WT MMF (Figure 4C).
The matrix produced by CAFs is different than that of quiescent fibroblasts, including increased assembly of collagen I and tenascin C [34], [35], [36]. Given the CAF-like phenotype of the Pten−/− fibroblasts, we hypothesized that Pten−/− fibroblasts would produce higher quantities of these proteins. mRNA levels for several ECM components in ascorbic acid–stimulated MMF were measured by qRT-PCR. When normalized to the loading control 18S, Pten−/− MMF expressed greater than 10-fold more tenascin C (Tnc) mRNA as compared to WT cells (Figure 4D). No significant changes were detected in mRNA levels of collagen I (Col1a1), collagen IV (Col4a1), or fibronectin (Fn).
Regulation of Matrix Alignment by Cellular Contractility
Changes in ECM organization during cancer progression have been attributed to a variety of factors, including increases in lysyl oxidase expression [37] and increases in cellular contractility [37], [38]. Decreases in contractility have been observed to correlate with lower collagen alignment in vivo as well as decreased FDM alignment in vitro in a caveolin-1 knockout model [29]. In addition, the direction of fibronectin fiber assembly by fibroblasts closely correlates with the direction of cell-generated traction forces [39] and the orientation of F-actin stress fibers in the cell [31]. Because Pten deletion promoted fibroblast activation, we hypothesized that loss of PTEN increases cellular contractility to regulate matrix alignment. Traction force microscopy was used to determine how PTEN loss affects cellular contractility. Traction forces generated by the Pten−/− cells were higher than those from WT fibroblasts, as shown by color maps of the traction stress fields (Figure 5A). The maximum traction stress generated by each cell was significantly higher with Pten knockout (Figure 5B). The net contractile moment, a measure of the directionality of the traction forces, was also significantly higher in Pten−/− cells (Figure 5C).
Figure 5.

Increased contractility in Pten−/− fibroblasts regulates matrix alignment. (A) Color maps of the traction stress field in WT and Pten−/− fibroblasts. (B) Maximum traction stress was quantified and plotted as median±95% confidence interval, n=17-22. (C) The net contractile moment of each cell was quantified and plotted as median±95% confidence interval, n=17-22. (D) Fibronectin staining of Pten−/− matrices treated with contractility inhibitors, pseudocolored by fiber orientation. (E) Alignment of Pten−/− matrices treated with Blebbistatin (myosin inhibitor) or Y-27632 (ROCK inhibitor) as compared to a DMSO vehicle control. n=3±SEM. *P<.05, **P<.01, ***P<.001, ****P<.0001 relative to control. ^^P<.01 relative to 1-μM concentration of the same inhibitor.
To determine if the increased contractility in Pten−/− fibroblasts was necessary for the enhanced matrix alignment, contractility was reduced using Blebbistatin, a nonmuscle myosin inhibitor, as well as Y-27632, a Rho-associated protein kinase (ROCK) inhibitor. ROCK signaling has been shown to contribute to myosin light chain phosphorylation and is necessary for fibroblast contractility [40]. Cell-generated traction forces are important for fibronectin fibrillogenesis, and previous studies have shown that complete ablation of cellular contractility, such as through high concentrations of myosin or ROCK inhibitors, can inhibit this process [14], [39], [41]. Therefore, relatively low concentrations of Blebbistatin and Y-27632 (1 and 10 μM) were used here to investigate the effect of reduced contractility on Pten−/− FDM alignment. Previous work has shown that these concentrations of Blebbistatin are insufficient to cause changes in matrix assembly [14]. Treatment with either Blebbistatin or Y-27632 resulted in significant decreases in matrix alignment (Figure 5 D and E). Treatment with 10 μM Blebbistatin resulted in a significant decrease in alignment as compared with a 1-μM dose of the same inhibitor. Collectively, these results demonstrate that the increased cellular contractility observed with Pten knockout was necessary for the changes in matrix alignment.
Effect of Matrix Organization on −−Cancer Cell Elongation and Migration
Changes in ECM composition and organization can influence cancer cell behavior. To examine the effect of the observed changes in matrix structure on cancer cell function, we used DB7 murine mammary adenocarcinoma cells, a cell line with low metastatic potential derived from MMTV-PyMT mice with a PyMTY315F/Y322F mutation [23]. FDMs were decellularized in order to isolate the effects of the ECM from any effects due to fibroblast cell signaling, and subsequently reseeded with DB7 cells. DB7 cells were stained with phalloidin and Hoechst to visualize cell morphology, and fibronectin staining was used to visualize the matrix (Figure 6A). Because the Pten−/− FDMs were thinner than the WT FDMs (Figure 3D), it is possible that cells seeded on Pten−/− FDMs could respond to stiffness of the glass underneath the matrices. Previous work has yielded varying estimates of the minimum height necessary to prevent cells from sensing the glass substrate underneath thin coatings, ranging from as small as 3.4 μm [42], well below the thickness of the matrices used here, to several tens of microns [43]. DB7 cells had a significantly higher cell area when plated on glass coverslips as compared to either of the FDM, while no difference in cell area was observed between cells on WT or Pten−/− FDM (Figure 6B). This suggests that the cells were not responding to the rigidity of the glass through the matrix. Furthermore, previous work has shown that cells plated in FDMs form matrix adhesions similar to those seen in vivo, both of which are morphologically and compositionally distinct from cells plated on stiff, 2D substrates [44]. Interestingly, the DB7 cells were significantly more elongated on the Pten−/− FDM as compared to those plated on glass or WT FDM, as determined by the aspect ratio of the cells (the ratio of the long axis of each cell divided by its short axis; Figure 6C). The area of the nuclei was not significantly different between any of the substrates. However, nuclei were significantly more elongated in the cells plated on the Pten−/− FDM as compared to those plated on glass or the WT FDM (Figure 6C), a phenomenon which has been previously connected to cell migration [45].
Figure 6.

Pten FDMs promote DB7 cancer cell elongation and migration. (A) Staining of DB7 cells plated on glass or FDMs. (B) Quantification of DB7 cell and nuclear area from phalloidin and Hoechst staining using CellProfiler. (C) Quantification of DB7 cell and nuclear aspect ratio using CellProfiler. n=3±SEM. (D) Representative images of DB7 spheroids at time 0 or after 12 hours of imaging. (E) DB7 spheroid spreading as determined by the final area of the spheroid divided by its initial area. (F) Aspect ratio of the spheroids at 12 hours. n=13-16 spheroids across 4 experiments. *P<.05, ***P<.001, *** P<.001, ****P<.0001.
Matrix reorganization in vivo has previously been implicated in providing “highways” for cancer migration [9]. We hypothesized that the increased alignment found in the Pten−/− matrices would therefore increase cancer cell migration. However, very little single cell random migration occurred on any substrate (Supplemental Figure 3). To provide impetus for the cells to migrate, DB7 cells were formed into spheroids and plated on FDM or on tissue culture polystyrene (TCPS). Over 12 hours, the spheroids plated on TCPS spread the most, followed by those plated on Pten−/− matrices (Figure 6, D-E). In comparison, the WT spheroids showed little change over time. The aspect ratio of the spheroids was significantly higher on Pten−/− matrices than on TCPS (Figure 6F), although there was no significant difference between the spheroids on WT or Pten−/− FDM. In agreement with the increase in spheroid aspect ratio, the spheroids on Pten−/− matrices often spread preferentially in one direction, whereas cells on TCPS spread radially (Figure 6D). This is likely due to local matrix alignment within the Pten−/− matrices, as the contact guidance cues provided by matrix alignment have been shown to increase the directionality and persistence of cancer cell migration [14], [17], [46]. Overall, these results indicate that matrix reorganization due to Pten deletion in fibroblasts promotes cancer cell migration, even in the absence of fibroblast cell signaling.
Supplemental Figure 3.
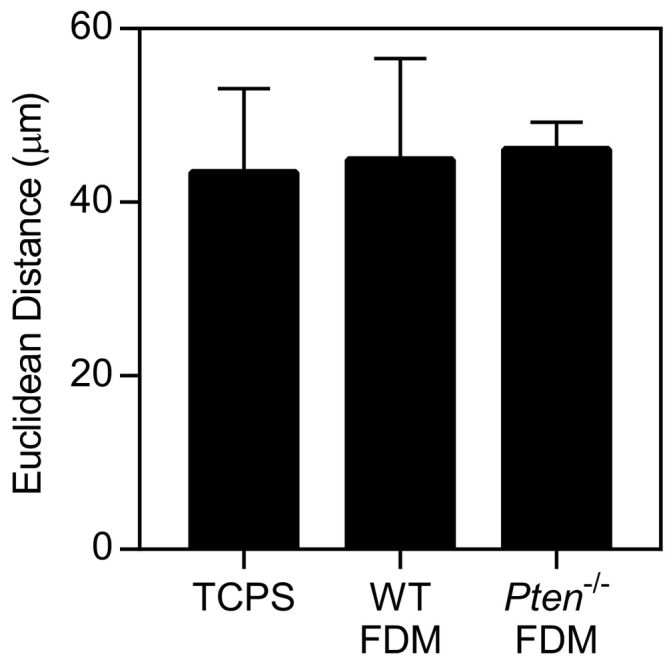
Single-cell random migration with DB7 cells resulted in minimal movement regardless of the substrate. Cells were imaged for 12 hours and tracked using ImageJ manual tracking plugin. n=2 ±SEM.
Correlation of High Mammographic Density with Low Stromal PTEN and Collagen Alignment
Although our data support a role for stromal PTEN in ECM deposition and reorganization, the relevance of the murine model to human disease remained unclear. To begin to test the hypothesis that stromal PTEN levels contribute to mammographic density, we performed a small pilot study in which we evaluated stromal PTEN expression and matrix alignment in fibroglandular breast tissue obtained from breast cancer patients determined to have high or low mammographic density (Figure 7A). Paraffin-embedded tissue sections were immunostained for PTEN (Figure 7B). To quantitatively assess the expression of PTEN in the stroma, the PTEN H-score was calculated where the stromal PTEN expression surrounding mammary ducts was lowest throughout each tissue section. The stromal PTEN H-score was significantly lower in samples from women with high mammographic density (Figure 7C).
Figure 7.

Low stromal PTEN expression and collagen alignment correlate with high mammographic density. (A) Representative mammography for breasts classified as high or low density. (B) PTEN immunohistochemistry (brown) surrounding mammary ducts in low- and high-density breasts. (C) Stromal PTEN H score in low- and high-density breasts. (D) PTEN and hematoxylin staining (top row) and corresponding SHG images (bottom row) surrounding mammary ducts in low- and high-density breasts. (E) Histogram of fiber alignment relative to the duct edge. (F) Quantification of fiber alignment by the percent of fibers falling within 10° of the duct edge in low- and high-density breasts. (G) Correlation of collagen fiber alignment relative to the duct edge and stromal PTEN H score, P<.05 by Pearson's correlation. n=5 sections from low-density breasts, 8 high-density breasts, with images of 5 ducts taken per sample. *P<.05.
Fibrillar collagen organization surrounding the epithelial ducts was then evaluated using SHG microscopy (Figure 7D). The orientation distribution of fibers relative to the duct in high-density breasts was significantly different from the distribution in low-density breasts (Figure 7E). The fraction of fibers oriented parallel to the duct edge was significantly higher in samples from high-density breasts (Figure 7F). This observation is consistent with a recent study which demonstrated that collagen coherency is increased in cases of high mammographic density [8].
To determine how collagen organization and PTEN expression were related, we graphed the fraction of fibers parallel to the duct edge versus the stromal PTEN H-score (Figure 7G). A Pearson correlation indicated that the collagen alignment and PTEN expression were significantly negatively correlated. In combination with our data in the Fsp-Cre;PtenloxP/loxP mouse model, this suggests that low PTEN expression in fibroblasts may promote collagen deposition and alignment, both characteristic of high mammographic density.
Discussion
Low stromal PTEN expression likely primes the microenvironment for tumor development through several mechanisms, including ECM reorganization and fibroblast activation. Stromal fibroblasts are activated during tumor progression to a myofibroblast-like state (CAFs) and produce more aligned ECM than quiescent fibroblasts [13], [14]. A similar phenotype was observed here with deletion of Pten, suggesting that PTEN loss promotes CAF-like behaviors. PTEN loss was also associated with greatly increased mRNA expression of the ECM protein tenascin C. Tenascin C deposition is commonly upregulated in the breast tumor microenvironment [47], and expression of tenascin C has been shown to promote lung metastasis in breast cancer [48], [49].
The changes in collagen structure observed with loss of stromal PTEN are reminiscent of the tumor-associated collagen signatures (TACSs) observed in breast tumors, which describe changes in collagen organization throughout tumor progression [9]. TACS-1 is defined as a drastic increase in collagen deposition and correlates with early stages of tumor growth. TACS-2 is characterized by collagen linearization and organization parallel to the tumor edge, while TACS-3 is indicated by local collagen fiber orientation perpendicular to the tumor edge. This perpendicular orientation creates “highways” for cancer cell invasion away from the primary tumor and into the stroma [9]. The presence of TACS-3 is a prognostic factor in breast cancer patients, associated with poor disease-free and overall survival [12]. In tumor-bearing mice with loss of stromal PTEN, we observed a small but statistically significant increase in the number of fibers oriented perpendicular to the tumor edge, similar to TACS-3, as compared to control mice. The presence of TACS-3 correlates with significantly reduced overall survival in breast cancer patients irrespective of its quantity [12], suggesting that small changes in TACS-3 organization have biological significance. This change in matrix alignment may be a contributing factor to the increased tumor size and tumor burden reported during the initial development of this mouse model [18]. Recent work has demonstrated that an antibody to lysyl oxidase homolog 2 notably decreases collagen alignment surrounding mammary tumors in a murine model, thereby reducing tumor growth and lung metastasis [50]. Modulation of ECM alignment in the tumor microenvironment through effectors of PTEN signaling may therefore provide a potential therapeutic target.
A variety of inhibitors for downstream effectors of PTEN signaling are currently in clinical trials. PTEN is best characterized as a lipid phosphatase which antagonizes PI3K/AKT pathway signaling through dephosphorylation of PIP3, although it has additional protein phosphatase activity [51]. The PI3K inhibitors idelalisib and copanlisib, as well as the mTOR inhibitors temsirolimus and everolimus, are FDA-approved for treatment of several cancers, and a variety of other PI3K/AKT pathway inhibitors are currently undergoing clinical trials [52]. The Rho/ROCK contractility pathway provides another potential target to reduce the ability of CAFs to remodel the tumor microenvironment. No ROCK inhibitors are currently approved for cancer treatment [53]. However, the ROCK inhibitor fasudil, which is FDA-approved for treatment of cerebral vasospasms, has been shown to markedly reduce breast cancer metastasis in a mouse model [54]. Cell contractility pathways may therefore prove a useful target for reducing metastasis by abrogating matrix remodeling by CAFs.
Interestingly, even in the absence of oncogene expression, deletion of stromal Pten increased collagen deposition and promoted organization parallel to the edge of ducts in the murine mammary gland. We observed similar increases in parallel collagen orientation surrounding mammary ducts in patients with high mammographic density, and this negatively correlated with stromal PTEN expression. This organization is reminiscent of the TACS-2 signature, despite the lack of tumor presence. This suggests that low PTEN expression in fibroblasts may prime the microenvironment for tumor development, allowing the ECM to be more easily reorganized into a TACS-3 orientation after tumor development. Consistent with this idea, collagen accumulation promotes tumorigenesis and earlier formation of TACS-3 surrounding tumors in a Col1a1TmJae mouse model, in which the sequence of type I collagen is mutated to abrogate MMP-mediated cleavage of collagen I [7].
Although increased collagen deposition has been well established as a contributing factor to breast density, it is unclear how collagen organization may change with breast density and how this might contribute to tumor development. We show here that stromal Pten deletion in a murine model leads to increased ECM deposition and reorganization of fiber alignment parallel to the edge of mammary ducts. We correspondingly demonstrated in a small cohort of patients that stromal PTEN expression negatively correlated with both increased mammographic density and collagen alignment. Taken together, this suggests that low stromal PTEN expression may contribute to high mammographic density. Stromal PTEN loss is associated with metastasis in prostate cancer [55], suggesting that stromal PTEN loss may additionally contribute to cancer progression. Future investigation to extend this pilot study and determine how stromal PTEN expression relates to clinical outcome will be vital in elucidating the role of stromal PTEN in human breast cancer development and progression.
The following are the supplementary data related to this article.
Acknowledgments
Acknowledgements
Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. This facility is supported in part by NIH cancer center grant P30 CA016058 to The Ohio State University, National Cancer Institute, Bethesda, MD.
Author Contributions
This study was conceived and designed by C. E. J., G. M. S., E. C., M. C. O., and J. L. L. Experiments were performed by C. E. J., A. M. H., Y. C., and L. D. Y. The data were analyzed and interpreted by C. E. J., A. M. H., L. D. Y., S. N. G., and J. L. L. The manuscript was drafted by C. E. J., A. M. H., Y. C., and J. L. L. All authors revised and approved the manuscript.
Footnotes
Financial Support: This work was supported by The Ohio State University College of Engineering, Biomedical Engineering Department; the National Institutes of Health cancer center grant to The Ohio State University [grant number P30 CA016058]; and the National Cancer Institute [grant numbers R01 CA113451 and CA06927] in support of the Talbot Library and Tissue Culture facility at Fox Chase Cancer Center, as well as the Commonwealth of Pennsylvania.
References
- 1.Gill JK, Maskarinec G, Pagano I, Kolonel LN. The association of mammographic density with ductal carcinoma in situ of the breast: the Multiethnic Cohort. Breast Cancer Res. 2006;8:R30. doi: 10.1186/bcr1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engmann NJ, Golmakani MK, Miglioretti DL, Sprague BL, Kerlikowske K. Population-attributable risk proportion of clinical risk factors for breast cancer. JAMA Oncol. 2017;3:1228–1236. doi: 10.1001/jamaoncol.2016.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyd NF, Rommens JM, Vogt K, Lee V, Hopper JL, Yaffe MJ. Mammographic breast density as an intermediate phenotype for breast cancer. Lancet Oncol. 2005;6:798–808. doi: 10.1016/S1470-2045(05)70390-9. [DOI] [PubMed] [Google Scholar]
- 4.Huo C.W., Chew G., Hill P., Huang D., Ingman W., Hodson L., Brown K.A., Magenau A., Allam A.H., McGhee E. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 2015;17 doi: 10.1186/s13058-015-0592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li T, Sun L, Miller N, Nicklee T, Woo J, Hulse-Smith L, Tsao M-S, Khokha R, Martin L, Boyd N. The association of measured breast tissue characteristics with mammographic density and other risk factors for breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:343–349. doi: 10.1158/1055-9965.EPI-04-0490. [DOI] [PubMed] [Google Scholar]
- 6.Alowami S, Troup S, Al-Haddad S, Kirkpatrick I, Watson PH. Mammographic density is related to stroma and stromal proteoglycan expression. Breast Cancer Res. 2003;5:R129. doi: 10.1186/bcr622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6 doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McConnell JC, O’Connell OV, Brennan K, Weiping L, Howe M, Joseph L, Knight D, O’Cualain R, Lim Y, Leek A. Increased peri-ductal collagen micro-organization may contribute to raised mammographic density. Breast Cancer Res. 2016;18 doi: 10.1186/s13058-015-0664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4 doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016;35:655–667. doi: 10.1007/s10555-016-9650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conklin M.W., Eickhoff J.C., Riching K.M., Pehlke C.A., Eliceiri K.W., Provenzano P.P., Friedl A., Keely P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amatangelo MD, Bassi DE, Klein-Szanto AJ, Cukierman E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol. 2005;167:475–488. doi: 10.1016/S0002-9440(10)62991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdogan B., Ao M, White LM, Means AL, Brewer BM, Yang L, Washington MK, Shi C, Franco OE, Weaver AM. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799–3816. doi: 10.1083/jcb.201704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castelló-Cros R, Khan DR, Simons J, Valianou M, Cukierman E. Staged stromal extracellular 3D matrices differentially regulate breast cancer cell responses through PI3K and beta1-integrins. BMC Cancer. 2009;9 doi: 10.1186/1471-2407-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carey SP, Goldblatt ZE, Martin KE, Romero B, Williams RM, Reinhart-King CA. Local extracellular matrix alignment directs cellular protrusion dynamics and migration through Rac1 and FAK. Integr Biol. 2016;8:821–835. doi: 10.1039/c6ib00030d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107:2546–2558. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong J-L. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sizemore GM, Balakrishnan S, Hammer AM, Thies KA, Trimboli AJ, Wallace JA, Sizemore ST, Kladney RD, Woelke SA, Yu L. Stromal PTEN inhibits the expansion of mammary epithelial stem cells through Jagged-1. Oncogene. 2017;36:2297–2308. doi: 10.1038/onc.2016.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2011;14:159–167. doi: 10.1038/ncb2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD. Negative regulation of myofibroblast differentiation by PTEN (phosphatase and tensin homolog deleted on chromosome 10) Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- 23.Borowsky AD, Namba R, Young LJT, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff RD, Gregg JP. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22:47–59. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- 24.Franco-Barraza J, Beacham D, Amatangelo MD, Cukierman E. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr Protoc Cell Biol. 2016;71:10.9.1–10.9.34. doi: 10.1002/cpcb.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rezakhaniha R, Agianniotis A, Schrauwen JTC, Griffa A, Sage D, Bouten CVC, van de Vosse FN, Unser M, Stergiopulos N. Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech Model Mechanobiol. 2011;11:461–473. doi: 10.1007/s10237-011-0325-z. [DOI] [PubMed] [Google Scholar]
- 27.Leight JL, Alge DL, Maier AJ, Anseth KS. Direct measurement of matrix metalloproteinase activity in 3D cellular microenvironments using a fluorogenic peptide substrate. Biomaterials. 2013;34:7344–7352. doi: 10.1016/j.biomaterials.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zielinski R, Mihai C, Kniss D, Ghadiali SN. Finite element analysis of traction force microscopy: influence of cell mechanics, adhesion, and morphology. J Biomech Eng. 2013;135 doi: 10.1115/1.4024467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibáñez T, Pellinen T, Echarri A. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1and α2β1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Lin Z, Foolen J, Schoen I, Santoro A, Zenobi-Wong M, Vogel V. Disentangling the multifactorial contributions of fibronectin, collagen and cyclic strain on MMP expression and extracellular matrix remodeling by fibroblasts. Matrix Biol. 2014;40:62–72. doi: 10.1016/j.matbio.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 33.Nagase H, Fields GB. Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers. 1996;40:399–415. doi: 10.1002/(SICI)1097-0282(1996)40:4%3C399::AID-BIP5%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 34.Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–497. [PMC free article] [PubMed] [Google Scholar]
- 35.De Wever O. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 1016-1018;18:1016–1018. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- 36.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18 doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abhilash AS, Baker BM, Trappmann B, Chen CS, Shenoy VB. Remodeling of fibrous extracellular matrices by contractile cells: predictions from discrete fiber network simulations. Biophys J. 2014;107:1829–1840. doi: 10.1016/j.bpj.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemmon CA, Chen CS, Romer LH. Cell traction forces direct fibronectin matrix assembly. Biophys J. 2009;96:729–738. doi: 10.1016/j.bpj.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 41.Yoneda A, Ushakov D, Multhaupt HAB, Couchman JR, Assoian R. Fibronectin matrix assembly requires distinct contributions from rho kinases I and -II. Mol Biol Cell. 2006;18:66–75. doi: 10.1091/mbc.E06-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buxboim A, Rajagopal K, Brown AEX, Discher DE. How deeply cells feel: methods for thin gels. J Phys Condens Matter. 2010;22 doi: 10.1088/0953-8984/22/19/194116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sen S, Engler AJ, Discher DE. Matrix strains induced by cells: computing how far cells can feel. Cell Mol Bioeng. 2009;2:39–48. doi: 10.1007/s12195-009-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 45.Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23:55–64. doi: 10.1016/j.ceb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ray A, Slama ZM, Morford RK, Madden SA, Provenzano PP. Enhanced directional migration of cancer stem cells in 3D aligned collagen matrices. Biophys J. 2017;112:1023–1036. doi: 10.1016/j.bpj.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hancox RA, Allen MD, Holliday DL, Edwards DR, Pennington CJ, Guttery DS, Shaw JA, Walker RA, Pringle JH, Jones J.L. Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009;11:R24. doi: 10.1186/bcr2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oskarsson T, Acharyya S, Zhang XH-F, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Z, Schwenzer A, Rupp T, Murdamoothoo D, Vegliante R, Lefebvre O, Klein A, Hussenet T, Orend G. Tenascin-C promotes tumor cell migration and metastasis through integrin α9β1-mediated YAP inhibition. Cancer Res. 2018;78:950–961. doi: 10.1158/0008-5472.CAN-17-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grossman M, Ben-Chetrit N, Zhuravlev A, Afik R, Bassat E, Solomonov I, Yarden Y, Sagi I. Tumor cell invasion can be blocked by modulators of collagen fibril alignment that control assembly of the extracellular matrix. Cancer Res. 2016;76:4249–4258. doi: 10.1158/0008-5472.CAN-15-2813. [DOI] [PubMed] [Google Scholar]
- 51.Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15:65–79. doi: 10.2174/1389450114666140106100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–291. doi: 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- 53.Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov. 2015;10:991–1010. doi: 10.1517/17460441.2015.1058775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ying H, Biroc SL, Li W, Alicke B, Xuan J-A, Pagila R, Ohashi Y, Okada T, Kamata Y, Dinter H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 2006;5:2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 55.Wartenberg M, Centeno I, Haemmig S, Vassella E, Zlobec I, Galván JA, Neuenschwander M, Schlup C, Gloor B, Lugli A. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur J Cancer. 2016;65:80–90. doi: 10.1016/j.ejca.2016.06.013. [DOI] [PubMed] [Google Scholar]