

Abstract
How breast cancer and its treatments affect skeletal muscle is not well defined. To address this question, we assessed skeletal muscle structure and protein expression in 13 women who were diagnosed with breast cancer and receiving adjuvant chemotherapy following tumor resection and 12 nondiseased controls. Breast cancer patients showed reduced single-muscle fiber cross-sectional area and fractional content of subsarcolemmal and intermyofibrillar mitochondria. Drugs commonly used in breast cancer patients (doxorubicin and paclitaxel) caused reductions in myosin expression, mitochondrial loss, and increased reactive oxygen species (ROS) production in C2C12 murine myotube cell cultures, supporting a role for chemotherapeutics in the atrophic and mitochondrial phenotypes. Additionally, concurrent treatment of myotubes with the mitochondrial-targeted antioxidant MitoQ prevented chemotherapy-induced myosin depletion, mitochondrial loss, and ROS production. In patients, reduced mitochondrial content and size and increased expression and oxidation of peroxiredoxin 3, a mitochondrial peroxidase, were associated with reduced muscle fiber cross-sectional area. Our results suggest that chemotherapeutics may adversely affect skeletal muscle in patients and that these effects may be driven through effects of these drugs on mitochondrial content and/or ROS production.
Keywords: cachexia, mitochondria, myotube
INTRODUCTION
Cancer and its treatment can affect skeletal muscle. The most widely studied consequence is cachexia, a syndrome characterized by fat and skeletal muscle tissue depletion that typically occurs in late-stage disease (17). Most studies have evaluated skeletal muscle from patients with cancers that are prone to cachexia and model systems that mimic them. However, patients with other cancer types less prone to cachexia or those with early-stage disease may also experience muscle atrophy (21) and dysfunction. Although these patients comprise the majority of cancer survivors, our understanding of the effect of these cancer types and their treatments on skeletal muscle is limited.
Breast cancer, the most commonly diagnosed cancer in the United States (1), is thought to have minimal effects on skeletal muscle because patients are less prone to cachexia (16) and are typically weight-stable or gain weight during treatment (30). This constancy in body weight, however, belies reciprocal changes in body composition, specifically a gain of fat and a loss of lean tissue (21). Muscle atrophy and the resulting weakness have prognostic importance in breast cancer patients (6, 9, 60), underscoring the need to better understand the effects of this cancer type and its clinical sequelae on skeletal muscle.
In the present study our objective was to examine skeletal muscle in patients receiving adjuvant chemotherapy following tumor resection and in healthy controls without a history of cancer. We hypothesized that cancer therapeutics might promote skeletal muscle maladaptation in patients, as drugs commonly used in breast cancer, such as anthracyclines, cause muscle wasting and dysfunction in preclinical models (22, 25, 43), whereas breast cancer tumors do not (27, 61). More specifically, we hypothesize that chemotherapeutics have myotoxic effects through their ability to provoke mitochondrial dysfunction and oxidative stress, as studies suggest that mitochondrial remodeling and oxidant stress promote skeletal muscle atrophy (48, 51). To begin to address this possibility, and building on preclinical studies showing that chemotherapeutics have effects on mitochondrial biology (3, 43), we also assessed mitochondrial content/morphology and markers of oxidative stress. Finally, we employed the C2C12 murine muscle cell line, as we could not ascribe muscle morphological and mitochondrial phenotypes in humans to chemotherapeutics because of the complex clinical background of these patients and our case-control study design. Cultured muscle cells provide a model where the singular effect of chemotherapeutics can be evaluated independent of clinical factors associated with treatment in patients, as well as effects on other tissue and organ systems. This latter point is particularly relevant to anthracyclines, which cause cardiac damage and heart failure in animal models (26), a syndrome that could affect skeletal muscle (54). Although there are the limitations of C2C12 myotube cultures, results from these experiments can be used as proof-of-principle evidence that chemotherapeutics may have effects on skeletal muscle to provoke phenotypes observed in human patients.
METHODS
Subjects.
Thirteen women with breast cancer were recruited. Patients were included if they had histologically documented stage I, II, or III breast cancer and were receiving or scheduled to receive chemotherapy following surgical resection. We excluded patients with prior history of cancer, other than nonmelanoma skin cancer, prior use of chemotherapeutics, autoimmune, vascular, or neuromuscular disease, or prior knee or hip replacement surgery. All patients received dexamethasone for 1–4 days concurrent with chemotherapy administration to prevent adverse drug reaction and diminish cancer-induced nausea and vomiting. Patients could receive a maximum dose of 12 mg on the day of chemotherapy and 4 mg/day for 3 days thereafter as symptoms necessitate. As dosing was symptom-dependent and not tracked clinically, exact doses for dexamethasone treatment are not available. Three patients were characterized as having experienced weight loss, defined as self-reported, unintentional loss of >5% of body weight within 6 mo before evaluation; other patients who reported little or no (<5%) weight change were characterized as weight-stable. All patients were tested during adjuvant chemotherapy, before radiation therapy. One patient reported current tobacco use (44-pack-year history). None of the patients received nutritional support or anabolic therapy, nor were they taking medications known to influence skeletal muscle size or function aside from chemotherapeutics. One volunteer was on a stable regimen of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor, which our recent work suggests would not affect the variables assessed in this study (50).
Healthy female controls (n = 12) were recruited if they were free from acute or chronic disease, based on medical screening and routine clinical/laboratory tests, and had no prior history of cancer, chronic lung or cardiovascular disease, neurological or orthopedic conditions, or other mobility-limiting ailments. Current or previous smokers were excluded, as were individuals taking medications, with the exception of stable 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor treatment (n = 1). Plasma creatine kinase levels were within the normative range in this volunteer, and she reported no muscle symptoms. Written informed consent was obtained from all volunteers before their participation, and protocols and procedures were approved by the Committees on Human Research at the University of Vermont.
Total and regional body composition.
Body weight was measured on a digital scale (ScaleTronix, Wheaton, IL). Total and regional body composition was measured by dual-energy X-ray absorptiometry, as described elsewhere (57).
Muscle tissue processing.
Percutaneous biopsy of the vastus lateralis was performed at the point midway between the most proximal aspect of the patella and the inguinal fold. The skin was sterilized (2% chlorhexidine-70% isopropyl alcohol), and the site was infiltrated with local anesthetic (1% lidocaine), as described elsewhere (55). A small (~5-mm) incision was made through the skin, fat, and fascia, and the biopsy needle (5-mm Bergstrom) was advanced past the fascia into the muscle at an angle ∼30–45° to the skin surface to position the needle roughly parallel with the muscle fascicles, as described elsewhere (15), to collect long, intact bundles of fibers. Suction was applied and tissue was excised. Tissue for electron microscopy (EM) was tied to a glass rod at a slightly stretched length and placed into 2.5% glutaraldehyde-1% paraformaldehyde for EM. Tissue for immunohistochemistry was embedded in optimal cutting temperature medium and frozen in isopentane cooled in liquid nitrogen. Tissue was also taken for measurement of mechanical properties of skeletal muscle fibers, but those data are not presented. Remaining tissue was frozen in liquid nitrogen and stored at −80°C until analysis. In one breast cancer patient, sufficient tissue was obtained only for EM analysis.
Immunohistochemistry.
Muscle fiber cross-sectional area (CSA) and myonuclear number were quantified by immunohistochemistry using rabbit anti-laminin (Abcam) and mouse anti-myosin heavy chain (MHC) I (Developmental Studies Hybridoma Bank) antibodies, as described elsewhere (11). Nuclei were stained with 4′,6-diamidino-2-phenylindole (Invitrogen). CSA was measured using image analysis software (NIH ImageJ, version 1.51n). Although the myonuclei count will include muscle satellite cells, these are a small fraction (<5%) of the nuclei under the laminin border (46). For healthy controls, immunohistochemistry was available on five volunteers because of limited tissue.
MHC content and isoform distribution.
MHC content and isoform distribution were measured in tissue homogenates via gel electrophoresis, as described elsewhere (58), as a marker of the relative fiber type distribution of muscle.
Electron microscopy.
EM was conducted on intact (i.e., unskinned) skeletal muscle fibers to assess intermyofibrillar and subsarcolemmal mitochondrial area fraction, average area, and number as estimates of mitochondrial content and myofibrillar fractional area, as described elsewhere (10).
Cell culture.
Cultured murine myotubes were used to evaluate the singular effect of chemotherapy agents on skeletal muscle cell biology, primarily, mitochondrial content and oxidant production. Use of this system obviates the possible secondary effects of these drugs on muscle through effects on other organs/tissue systems [e.g., cardiac damage and failure due to doxorubicin (Dox) administration (26)], which could affect skeletal muscle (54). The C2C12 model has been used to characterize the effects of tumor-derived factors on mitochondrial oxidant production (39), effects that have been confirmed in vivo in preclinical models (7).
C2C12 myoblasts (CRL1772, American Type Culture Collection, Manassas, VA) were cultured in low-glucose (1 g/l) DMEM supplemented with 10% fetal bovine serum (FBS; GIBCO Thermo Fisher Scientific, Waltham, MA) and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin). Cells were plated (1.5 × 104 cells/cm2) on Matrigel (40 μg/cm2; Corning, Bedford, MA) and switched to low-serum, high-glucose (4.5 g/l) DMEM (1% heat-inactivated FBS) to induce differentiation, as generally described (34). On day 7 postdifferentiation, myotubes were treated with Dox (Sigma, St. Louis, MO), paclitaxel (Taxol; Sigma), cisplatin (Cis; Tocris, Minneapolis, MN), or respective vehicle control (DMSO for Dox and Taxol and PBS for Cis) for 3 days. Dox dose was based on prior work (24) and serum concentrations (47). Taxol (66) and Cis (56) doses were based on tumor intracellular levels. In all cases, preliminary dose-response studies were undertaken to obtain doses for Dox (0.2 μM), Taxol (40 nM), and Cis (10 µM) that elicited loss of myotube myosin content over the 3-day study period. In Dox and Taxol experiments, where increased oxidant generation was noted (see Figs. 5 and 6), MitoQ (MedKoo Biosciences, Morrisville, NC), a mitochondrial-targeted antioxidant, was added concurrent with chemotherapeutics to determine whether decreasing mitochondrial oxidant stress prevents atrophy. MitoQ has a narrow therapeutic range, a characteristic that we found varied by chemotherapeutic. Accordingly, doses were 0.25 μM for Dox and 0.125 μM for Taxol. On the final day of treatment, myotubes were harvested for analysis of protein expression via Western blotting or underwent mitochondrial measurements.
Fig. 5.

Chemotherapeutic effects on myotube myosin content. A and B: effects of 3 days of doxorubicin (Dox, 0.2 µM) or paclitaxel (Taxol, 40 nM) administration vs. vehicle [DMSO (Ctrl)] on myosin content (A) and reactive oxygen species (ROS) production (B), with representative images of myotubes for each experimental condition in A shown above data. Scale bars = 100 μm. C and D: effects of concomitant treatment with the mitochondrial-targeted antioxidant MitoQ (0.25 µM for Dox and 0.125 µM for Taxol) to prevent myosin loss with Dox or Taxol, with representative images of myotubes for each experimental condition shown above data. Scale bars = 100 µm. Q, MitoQ. Values are means ± SE, with individual data points shown with each bar. *P < 0.05, **P < 0.01 vs. Ctrl.
Fig. 6.

A: colocalization of MitoTracker (green) and MitoSOX (red) in a C2C12 murine myotube. Scale bar = 10 μm. B–E: effect of chemotherapeutics [doxorubicin (Dox, 0.25 µM) and paclitaxel (Taxol, 0.125 µM)] and concomitant treatment with the mitochondrial-targeted antioxidant MitoQ (+MitoQ) on mitochondrial content (B and C) and oxidant production (D and E) compared with vehicle (DMSO) control (Ctrl). Values are means ± SE, with individual data points shown with each bar. *P < 0.05, **P < 0.01 vs. Ctrl; #P < 0.01 vs. Taxol.
No genetic testing was conducted to validate the cell line. However, upon serum withdrawal, these cells fuse to form multinucleated cells (see Fig. 5) and express myofilament proteins, such as myosin, actin, and α-actinin (unpublished data). At day 7 postdifferentiation, when chemotherapy treatment began, myotubes contain myofilaments, contract with electrical stimulation, and demonstrate intracellular calcium release and reuptake (unpublished data). All these anatomic and physiological characteristics have been demonstrated previously in C2C12 myotubes (33, 34, 38). Additionally, we found that electrically stimulated contraction could be mitigated by blocking sodium channels with tetrodotoxin or myosin ATPase activity with N-benzyl-p-toluene sulfonamide, with the former mitigating intracellular calcium currents and contractures and the latter only mitigating contractures (unpublished data). Here again, these characteristics of the contractile response and its sensitivity to specific pharmacological agents have been demonstrated by others (14, 44). Thus, C2C12 myotubes demonstrate many classical phenotypes of in vivo skeletal muscle.
Myotube mitochondrial content and reactive oxygen species production.
Mitochondrial content and reactive oxygen species (ROS) production were measured with fluorometric dyes, as described elsewhere (45, 64) with minor modifications. C2C12 myotubes grown in black-walled, 96-well plates were loaded with fluorescent dyes to assess mitochondrial content (1 μM MitoTracker Green FM, 490/516 nm; Molecular Probes, Eugene, OR) and ROS production (1 μM MitoSOX Red, 510/580 nm; Molecular Probes) 15 min before measurement. Fluorescence was measured on a microplate reader (BioTek, Winooski, VT). The ROS signal (MitoSOX) was expressed relative to the MitoTracker signal to control for any effect of the chemotherapeutic to modify mitochondrial content. MitoTracker Green FM was chosen, because its labeling of mitochondria is insensitive to membrane potential, as this could be affected by chemotherapeutics (23). To confirm colocalization of MitoTracker and MitoSOX signals in myotubes, myoblasts were plated (2.5 × 104 cells/cm2) on Matrigel-coated (60 μg/cm2), 35-mm glass-bottom imaging dishes (MatTek, Ashland, MA) and differentiated following procedures described above, with the exception that the medium was changed daily for the first 3 days following the start of differentiation. Cells were imaged on a Nikon Ti-E inverted microscope in a heated environmental chamber equipped with a Clara charge-coupled device camera (Andor) and Spectra X light engine.
Western blot analysis.
Skeletal muscle tissue was homogenized, as described elsewhere (59), in a subset of patients (n = 9) and controls (n = 11), where tissue was available. In a subset of breast cancer patients (n = 11), mitochondria were isolated, as described elsewhere (35), and treated for peroxiredoxin-3 (Prx 3) assessments (see below). Myotubes were washed with PBS, lysed [50 mM Tris, 150 mM NaCl, 10% (vol/vol) glycerol, 0.5% IGEPAL CA-630, and 1 mM EDTA], incubated on ice for 30 min, and centrifuged. Homogenate, mitochondrial isolate, and lysate protein contents were measured (DC Protein Assay, Bio-Rad, Hercules, CA) and diluted in gel loading buffer. For Prx 3, tissue homogenates and myotube lysates were treated with N-ethylmaleimide (NEM) to prevent oxidation and/or dimerization of Prx 3 during processing and then placed in gel loading buffer with (reducing) or without (nonreducing) DTT. Oxidation of Prx 3 can be used as a marker of intracellular oxidative stress (49), and mitochondria specifically, as Prx 3 is localized to mitochondria. The importance of Prx 3 in muscle is underscored by studies showing that altered expression modulates mitochondrial content, morphology, and function, as well as muscle contractility (36). Proteins separated by SDS-PAGE were transferred to nitrocellulose (Prx 3) or polyvinylidene difluoride (all others), blocked (using BSA, nonfat milk in Tris-buffered saline, or Odyssey blocking buffer), and incubated overnight with Prx 3 (1:6,000 dilution; catalog no. LF-PA0030, Thermo Fisher), p38 (1:1,000 dilution; catalog no. 9112, Cell Signaling Technology), phosphorylated (Thr180/Thr182) p38 (1:500 dilution; catalog no. 4511, Cell Signaling Technology), ERK1/2 (1:1,000 dilution; catalog no. 9102, Cell Signaling Technology), phosphorylated (Thr202/Thr204) ERK1/2 (1:500 dilution; catalog no. 9106, Cell Signaling Technology), α-tubulin (0.5 μg/ml; catalog no. 12G10, Developmental Studies Hybridoma Bank), detyrosinated α-tubulin (1:500 dilution; catalog no. ab48389, Abcam), or myosin fast (1:15,000 dilution; catalog no. M4276, Sigma) antibodies or GAPDH (1:5,000 dilution; catalog no. MCA4739, Bio-Rad). Blots were washed and incubated with secondary antibodies conjugated to horseradish peroxidase and developed by chemiluminescence (Clarity Western ECL substrate, Bio-Rad) or the Odyssey infrared imaging system [for p38, ERK1/2, and tubulin analytes (IRDye 680 or IRDye 800 secondary antibodies, Li-Cor, Lincoln, NE)]. Bands were quantified by densitometry [Quantity One (Bio-Rad) or Odyssey Application Software v3.0 (Li-Cor)]. For analysis that required running more than one gel, data were normalized to an internal loading control to permit comparisons across gels. Technicians were blinded to group assignment or treatment.
Statistics.
Differences between groups were determined using unpaired t-tests. For variables with multiple observations within the same individual (e.g., single-fiber CSA), a linear mixed model, including a between-subject effect of group assignment (control or cancer) and a repeated effect to account for variation in fiber characteristics within each individual, was used. This effect accounts for the fact that each fiber within an individual is not independent and has some relationship to other fibers within that individual. The mixed model is preferable to taking an average value for each individual, which makes the erroneous assumption that within-subject variation is zero. Instead, the mixed model considers this within-subject variation in the derivation of within-group variance to test for fixed effects (i.e., group differences). Least-squared mean and variance estimates are used to test for fixed effects in the mixed model. However, arithmetic means are displayed figuratively and in the text to correspond with individual data (or individual average data for variables with multiple observations per volunteer), the latter of which is plotted to illustrate the spread of data. For cell culture experiments, unpaired t-tests or one-way analysis of variance was used. Finally, relationships between variables were determined by Pearson’s correlation coefficients, with normality confirmed by the Shapiro-Wilk test. For nonnormally distributed variables that remained so following data transformations (e.g., MHC II CSA), Spearman’s rank correlation coefficients were used. SPSS (version 23, IBM, Armonk, NY) was used for all statistical analysis, except mixed-model analysis (SAS version 9.4, SAS Institute, Cary, NC). All data are reported as means ± SE, unless otherwise specified.
RESULTS
Disease and physical characteristics.
Controls were older, but all body size and composition variables were similar between groups (Table 1). Disease and treatment characteristics of patients are shown in Table 1. Five patients reported unintentional weight loss during 6 mo before testing [−4.0 ± 1.1 (range 1–7) kg], with three (1 stage I, 1 stage II, and 1 stage III) reporting >5% loss (−5.7 ± 0.7 kg). Breast cancer patients were studied an average of 59 ± 10 (range 5–114) days into their chemotherapy regimens.
Table 1.
Physical and disease characteristics in controls and breast cancer patients
| Control (n = 12) | Cancer (n = 13) | |
|---|---|---|
| Age, yr | 66 ± 5 | 57 ± 12* |
| Body weight, kg | 67 ± 15 | 70 ± 13 |
| Height, cm | 164 ± 5 | 161 ± 8 |
| Fat mass, kg | 24 ± 12 | 28 ± 11 |
| Fat-free mass, kg | 40 ± 5 | 40 ± 4 |
| Fat-free mass index, kg/m2 | 15.2 ± 1.3 | 15.4 ± 1.6 |
| Leg fat-free mass, kg | 13 ± 2 | 14 ± 2 |
| Cancer stage (I/II/III) | NA | 7/2/4 |
| Histology, n Adenocarcinoma |
NA | 13 |
| Dexamethasone, n | NA | 13 |
| Chemotherapeutics, n Taxanes |
NA | 13 |
| Cyclophosphamide | NA | 8 |
| Doxorubicin | NA | 5 |
| Trastuzumab | NA | 3 |
| History of smoking, n | NA | 1 |
| Radiation, n | NA | 0 |
Values are means ± SD. NA, not applicable to control volunteers.
P < 0.05.
Single-muscle fiber characteristics.
Breast cancer patients showed lower single-muscle fiber CSA than controls (P < 0.05, n = 1,682 and 450 fibers; Fig. 1A), with lower MHC II fiber CSA (P = 0.05, n = 745 and 228 fibers) and a trend toward lower MHC I CSA (P = 0.07, n = 937 and 222 fibers). No differences in CSA were found between patients reporting weight loss and those reporting weight stability in all fibers or in MHC I or II fibers separately (P > 0.40; data not shown). As stated in Statistics, statistical tests for group differences are based on the number of volunteers per group, not the total number of fibers. No differences were found in myonuclear number per fiber in MHC I or II fibers (Fig. 1B). Finally, we found no differences between groups for myofilament fractional area by EM (Fig. 1C) or myosin or actin protein in tissue homogenates (Fig. 1D).
Fig. 1.

Skeletal muscle fiber cross-sectional area (CSA) in all fibers and myosin heavy chain (MHC) I and II fibers (A), myonuclei content in MHC I and II fibers (B), myofilament fractional area (C), and MHC and actin protein content in tissue homogenates (D) in controls and breast cancer patients. Values are means ± SE, with individual data points shown with each bar. For A–C, multiple observations within each individual were averaged to provide a single data point for each volunteer. Representative immunohistochemical images used to measure CSA and myonuclei content for control and cancer patients are shown above A and B, respectively. Scale bars = 100 µm. Representative electron microscopy images used to measure myofilament fractional area (C) for controls and cancer patients are shown above C and D, respectively. Scale bars = 1 µm. AU, arbitrary units. *P ≤ 0.05; †P = 0.07.
Mitochondrial content and structure.
Intermyofibrillar mitochondrial fractional area was lower (P < 0.03) in breast cancer patients than controls (Fig. 2A) due to smaller (P < 0.02) average mitochondrial area (Fig. 2B), with no difference in number of mitochondria (0.24 ± 0.03 and 0.29 ± 0.03 μm−2 in controls and cancer patients, respectively). Similarly, subsarcolemmal mitochondrial fractional area was reduced (P < 0.05; Fig. 2C) due to decreased number of mitochondria per unit area (P < 0.01; Fig. 2D), while average subsarcolemmal mitochondrial area showed a trend (P = 0.07) toward being greater in cancer patients (0.95 ± 0.08 and 1.59 ± 0.26 µm2 in controls and cancer patients, respectively). Subsarcolemmal area did not differ between groups (21.7 ± 2.3 and 21.7 ± 1.6 µm2 in controls and cancer patients, respectively). Finally, in the breast cancer patients, reduced intermyofibrillar mitochondrial size was associated with smaller CSA in both MHC I (r = 0.792, P < 0.01) and MHC II (r = 0.769, P < 0.01) fibers, whereas intermyofibrillar mitochondrial size was not associated with CSA in either fiber type in controls (both P > 0.50). Reduced subsarcolemmal mitochondrial fractional content was associated with smaller MHC II CSA in cancer patients (r = 0.743, P < 0.01), but not controls, in either fiber type (P > 0.60).
Fig. 2.

Skeletal muscle mitochondrial (Mito) content and structure in intermyofibrillar (IMF) and subsarcolemmal (SS) compartments. Representative images for IMF mitochondrial fractional area for controls and cancer patients are shown above A and B, respectively, with average IMF mitochondrial fractional area (A) and average size (B) for controls and breast cancer patients. Representative images for SS mitochondria for controls and cancer patients are shown above C and D, respectively, with average SS fractional area (C) and average number (n) per unit SS area (D). Scale bars = 1 µm for all images. Values are means ± SE, with individual data points shown with each bar. For A–D, multiple observations within each individual were averaged to provide a single data point for each individual. *P < 0.05, **P < 0.01 vs. control.
We examined whether differences in fiber type distribution, a marker of muscle fiber type proportion, might account for variation in mitochondrial parameters. Fractional content of MHC I proteins was lower in breast cancer patients than controls (35.8 ± 0.8 vs. 39.0 ± 1.2%, P < 0.05), although neither MHC IIA (36.3 ± 0.3 vs. 35.8 ± 1.0%) nor MHC IIX (27.9 ± 0.9 vs. 25.2 ± 1.5%) differed. Variation in MHC I fractional content, however, was not associated with mitochondrial fractional content from either compartment in the whole group or in either group separately. Moreover, as slopes of these relationships were shallow (range −0.005 to 0.064), modest differences in MHC I fiber fractional content would likely not explain group differences in mitochondrial content in either compartment.
Prx 3 expression.
To examine whether mitochondrial oxidant stress in cancer patients is associated with muscle fiber size, we evaluated the expression and oxidation state of the mitochondrial-targeted antioxidant enzyme Prx 3 in a subgroup of breast cancer patients (n = 9). We found that Prx 3 expression was negatively related to both MHC I and MHC II CSA (Fig. 3, A and B, respectively). Moreover, in mitochondrial isolates (n = 11), increased amounts of oxidized Prx 3 in breast cancer patients were related to decreased CSA in MHC II fibers (Fig. 3C).
Fig. 3.

Relationship of mitochondrial oxidant stress to myofiber size and chemotherapy administration. A–C: relationship of myosin heavy chain (MHC) I and II cross-sectional area (CSA) to peroxiredoxin 3 (Prx 3) expression and relationship of MHC II CSA to oxidized Prx 3 in breast cancer patients (■). In A, r value is Pearson’s coefficient; in B and C, r values are Spearman’s rank coefficients. For A and B, n = 9; for C, n = 11. AU, arbitrary units. D and E: effects of 3 days of doxorubicin (Dox, 0.2 µM) or paclitaxel (Taxol, 40 nM) treatment vs. control [Ctrl (DMSO)] on C2C12 myotube Prx 3 expression (D, n = 4/condition) and oxidation, with the latter being shown by Prx 3 dimers [oxidized (ox)] under nonreducing conditions (E). F: quantitation of oxidized Prx 3 (n = 4/condition). Data in D and F are means ± SE, with individual data points shown with each bar. For A–C, multiple observations for CSA within each individual were averaged to provide a single data point for each individual. For D, immunoblots are shown above average data with loading control (GAPDH). Loading controls are not shown for E, as oxidized Prx 3 data are expressed as a fraction of total (oxidized + nonoxidized) Prx 3. *P < 0.05 vs. Ctrl.
To further examine whether chemotherapeutics might contribute to variation in Prx 3 expression and/or oxidation, we treated C2C12 myotubes for 3 days with Dox or Taxol, which are commonly used chemotherapeutics in our population (Table 1). Dox did not affect Prx 3 expression, whereas Taxol reduced Prx 3 expression (Fig. 3D; n = 4/group). Note that the two bands shown under reducing conditions are due to addition of NEM to prevent oxidation and/or dimerization during processing, as samples not treated with NEM showed only one band (data not shown). The results above reflect quantification of both bands. Under nonreducing conditions, we found that Dox increased oxidized Prx 3 in myotubes (P < 0.05), as shown by increased Prx 3 dimer (100 ± 8.9, 162.5 ± 11.0, and 106.7 ± 6.1% for control, Dox, and Taxol, respectively; Fig. 3E). Because Taxol reduced Prx 3 expression (Fig. 3D), we calculated the relative amount of dimer in each treatment group as a function of monomer + dimer as a marker of mitochondrial oxidative stress. This did not alter the results, as oxidized Prx 3 in Dox-treated myotubes remained elevated (Fig. 3F; n = 4/group).
Tubulin expression and detyrosination.
Because mitochondrial motility and communication occur via kinesin-dependent movement along microtubules and because taxane chemotherapeutics, which were taken by all the breast cancer patients, alter tubulin expression and posttranslational modification in skeletal muscle, we examined the effects of taxanes on tubulin expression and posttranslational modification. In C2C12 myotubes, treatment with Taxol (40 nM for 3 days, n = 6) increased expression of tubulin and detyrosinated tubulin (Fig. 4A). Similarly, in patients treated with therapeutic doses of taxane chemotherapeutics (n = 9), we found increased tubulin and detyrosinated tubulin expression compared with controls (Fig. 4B; n = 11). Because Taxol can increase ROS production in skeletal muscles, we examined whether variation in tubulin or detyrosinated tubulin correlated with mitochondrial content or morphology. The average area of intermyofibrillar mitochondria was inversely correlated to expression of α-tubulin (r = −0.472, P < 0.05) and detyrosinated tubulin (r = −0.580, P < 0.01). In subsarcolemmal mitochondria, α-tubulin was negatively correlated to fractional content (r = −0.558, P < 0.05) and number per area (r = −0.654, P > 0.01) and positively related to average area (r = 0.582, P < 0.01).
Fig. 4.
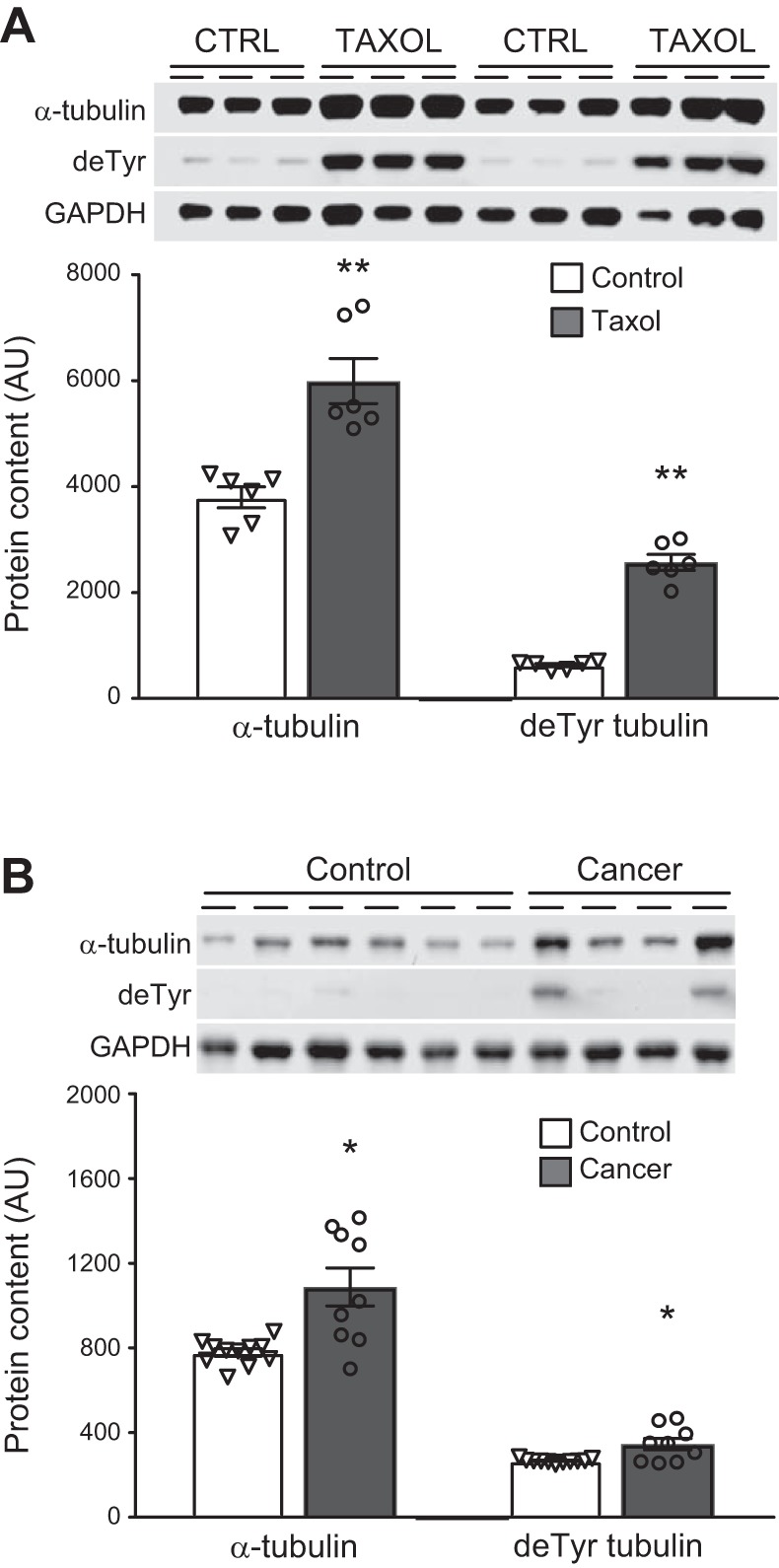
α-Tubulin and detyrosinated (deTyr) tubulin expression. α-Tubulin and detyrosinated tubulin were assessed following 3 days of treatment of C2C12 myotubes (A) with vehicle [Ctrl (DMSO), n = 6] or paclitaxel (Tax, 40 nM, n = 6) and in controls (n = 11) and breast cancer patients (n = 9) (B). Gel images are provided for both experiments. AU, arbitrary units. Values are means ± SE, with individual data points shown with each bar. *P < 0.05, **P < 0.01 vs. control.
Chemotherapy-induced atrophy and oxidant stress.
To further examine the effects of chemotherapeutics on skeletal muscle cell size and mitochondrial content and ROS production, C2C12 myotubes were treated with Dox, a chemotherapeutic with well-characterized myotoxicity, or Taxol, the most common chemotherapeutic in our cohort. Treatment with Dox or Taxol reduced myosin content (Fig. 5A; n = 15 and 9, respectively). The idea that myosin content serves as a proxy of myotube cell size was confirmed by the close tracking of Dox-induced changes in myotube diameter (−22 ± 7% relative to controls, n = 3 wells/condition) with changes in myosin content (−21 ± 2%, n = 10). Accordingly, we utilized myosin protein content as our index of myotube size throughout our studies.
Both Dox and Taxol caused a loss of mitochondrial content [−8 ± 2% (n = 16, P < 0.01) and −38 ± 3% (n = 12, P < 0.001), respectively]. Accordingly, ROS production, as reflected by MitoSOX signal, was expressed per unit MitoTracker signal to control for changes in mitochondrial content. The use of MitoTracker signal to correct MitoSOX signal is based on the mitochondrial targeting of both probes, which we confirmed in our preparations (Fig. 6A). Both chemotherapeutics increased ROS production per unit MitoTracker signal (Fig. 5B; n = 12 and 11, respectively). To examine whether increased ROS production with chemotherapeutics contributes to myotube atrophy, we concurrently treated cells with the mitochondrial-targeted antioxidant MitoQ and observed that it prevented Dox- and Taxol-induced loss of myosin protein content (Fig. 5, C and D; n = 12 and 3, respectively). Additionally, MitoQ application to myotubes prevented the loss of mitochondrial content (Fig. 6, B and C; n = 12 and 12, respectively) and prevented (Dox) or lessened (Taxol) the increase in ROS production (Fig. 6, D and E; n = 12 and 12, respectively).
Our prior work showed mitochondrial rarefaction (−50%) in skeletal muscle from patients with cancers that are prone to cachexia, primarily lung cancer, where platin-based drugs were the most common chemotherapeutic (58). To explore whether they have effects similar to Dox and Taxol, we evaluated the effects of Cis on C2C12 myotubes. Cis caused profound myosin loss [100 ± 17% (control) vs. 19 ± 4% of control (Cis), n = 3, P < 0.01] but did not increase ROS production relative to MitoTracker signal [100 ± 1 (control) vs. 98 ± 1% of control (Cis), n = 12] and, paradoxically, increased MitoTracker signal [100 ± 6 (control) vs. 113 ± 5 of control (Cis), n = 12, P < 0.01].
Signaling pathways.
Recent studies suggest that upregulation of redox-responsive signaling pathways, such as p38 and ERK1/2, may contribute to muscle atrophy with chemotherapeutic administration in preclinical models. However, we found no differences in the expression or phosphorylation of p38 (Fig. 7A) or ERK1/2 (Fig. 7B) or in the ratio of phosphorylated to total content of p38 and ERK1/2, between patients (n = 9) and controls (n = 11). ERK1/2 data represent quantitation of both bands.
Fig. 7.
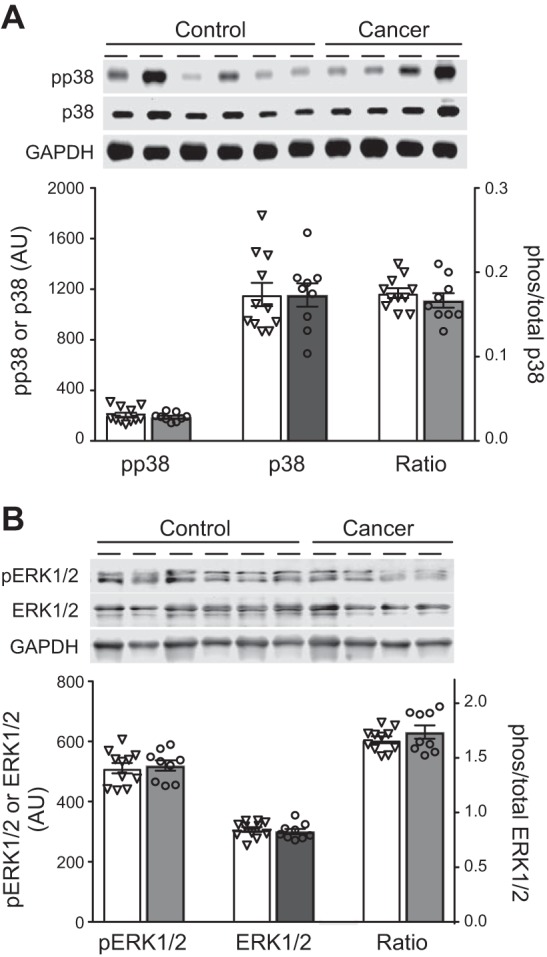
A and B: p38 and ERK1/2 expression and phosphorylation (pp38 and pERK1/2) in tissue homogenates from controls (open bars, n = 11) and breast cancer patients (gray bars, n = 9). Gel images are shown for phosphorylated (phos) and total protein, as well as GAPDH. Ratio, phosphorylated-to-total protein ratio. AU, arbitrary units. Values are means ± SE, with individual data points shown with each bar.
DISCUSSION
Breast cancer patients showed evidence of muscle fiber atrophy and mitochondrial rarefaction, with reduced muscle fiber size tracking with reduced mitochondrial content and increased oxidant stress. Treatment with chemotherapeutics promoted these phenotypes in cultured muscle cells, effects that were mitigated by concomitant treatment with a mitochondrial-targeted antioxidant. These results provide seminal human data that suggest deleterious effects of cancer treatments on muscle secondary to their mitotoxic properties. The idea that chemotherapeutics promote these adaptations provides a potential explanation for muscle atrophy in cancers not prone to cachexia or in early-stage patients, as well as the high prevalence of fatigue and physical disability across different cancer types (12).
Reduced skeletal muscle fiber CSA in breast cancer patients was unlikely due to weight loss, as few patients reported weight loss and fiber CSA did not differ between patients reporting weight loss and those reporting weight stability. Other groups have reported that lean tissue mass is reduced (21), but we use direct measures of muscle cell size in breast cancer patients to provide evidence of muscle loss. We emphasize this point, because tissue level imaging-based measurements may not accurately reflect muscle size in patients because of effects of treatments on tissue hydration (2). Indeed, we found no differences between groups in leg muscle mass by dual-energy X-ray absorptiometry (Table 1). Such discrepancies in humans are not unique to this study, as we (42) and others (4) have shown discordance between measures at these different anatomic levels when tissue hydration may be altered (13). Although we acknowledge that biopsy samples represent a small amount of tissue relative to the amount sampled with imaging techniques, single-fiber CSA measurements agree well with imaging-based whole muscle CSA under stable conditions, where muscle mass and hydration are not changing (11). This potential limitation of imaging techniques is noteworthy, because most studies have used these approaches to define cancer-related muscle atrophy in clinical populations.
This is the first report of skeletal muscle mitochondrial morphological adaptations in breast cancer patients, a cohort with a low prevalence of cachexia, and is consistent with our results in patients with cancers more prone to cachexia (58); however, the reduction was not as pronounced [current study vs. our previous study (58)]. While cancer (62) or chemotherapeutics (3) may reduce mitochondrial content, the facts that our patients’ tumors were removed surgically and that breast cancer tumors generally do not promote cachexia in preclinical models (27, 61) suggest chemotherapeutics as a more likely mediator. Indeed, recent preclinical studies showed that administration of chemotherapeutics used for colorectal tumors to mice reduced intermyofibrillar mitochondrial content secondary to decreased mitochondrion size (3), a morphological phenotype similar to that in our patients. Our results also extend both preclinical and clinical work (58) to show mitochondrial loss in the subsarcolemmal compartment. Interestingly, different structural characteristics accounted for reduced mitochondrial content in subsarcolemmal and intermyofibrillar compartments in breast cancer patients, suggesting different mechanisms for mitochondrial remodeling in the two compartments. Considering the beneficial effect of increasing or maintaining mitochondrial content in other models of atrophy (52), therapeutics or interventions (e.g., exercise) that preserve or increase mitochondria may prevent muscle atrophy and weakness in cancer patients.
Chemotherapeutics may promote mitochondrial loss through their ability to provoke mitochondrial dysfunction and oxidant production, as even modest amounts of oxidant stress can promote mitophagy (20). To address this question, we utilized C2C12 myotubes to examine the singular effect of chemotherapeutics used in breast cancer patients on mitochondrial content and oxidant production. We found that chemotherapeutics caused mitochondrial loss and increased oxidant production in C2C12 myotubes. Moreover, treatment with the mitochondrial-targeted antioxidant MitoQ prevented a loss of mitochondria, further implicating ROS in mitochondrial loss. In contrast, mitochondrial content remained unchanged with Cis, another widely used chemotherapeutic that did not increase ROS production. These data provide proof-of-principle evidence that mitochondrial loss tracks with drug-induced oxidant stress.
Another potential explanation for mitochondrial loss is the effect of taxane chemotherapeutics on microtubules, as all our patients were treated with these drugs. Microtubules form a lattice in muscle fibers and serve as tracks to support kinesin-dependent mitochondrial movement (65) and communication (29). Preclinical studies show that Taxol increases the amount of tubulin and its detyrosinated form in skeletal muscle (31), which we confirmed in C2C12 murine myotubes. Our results further provide seminal data showing that taxanes at therapeutic doses used in human patients upregulate tubulin and detyrosinated tubulin levels in skeletal muscle. The notion that taxanes may have detrimental effects on mitochondria in patients is suggested by the negative correlations of tubulin and detyrosinated tubulin to mitochondrial content and morphology in both compartments. The mechanism whereby Taxol affects mitochondria is not clear, but our data in cultured myotubes and data of others in intact muscle fibers suggest that Taxol increases ROS production (Fig. 4B) (32), which could promote mitophagy (20). In support of a role for ROS in promoting mitochondrial rarefaction, we found that MitoQ prevented Taxol-induced mitochondrial loss. Collectively, our results highlight novel mitotoxic effects of taxane chemotherapeutics in skeletal muscle.
The effect of chemotherapeutics to increase cellular oxidant stress might also promote myofiber atrophy (48). In patients, increased expression and oxidation of Prx 3, which our data and that of others (37) from cultured muscle cells suggest may result from chemotherapeutics, were associated with reduced CSA in MHC I and II fibers. Moreover, in C2C12 myotubes, concomitant treatment with the mitochondrial-targeted antioxidant MitoQ prevented chemotherapy-induced myosin loss. These findings are not confined to cultured muscle cells, as mitochondrial-targeted antioxidants prevent Dox-induced atrophy in rodents (43). Our data show that these effects are also apparent in cultured muscle cells for Taxol, the most commonly used drug among our breast cancer patients. The mechanisms whereby increased cellular or mitochondrial oxidant stress promotes muscle atrophy are not well defined, and we found no evidence in breast cancer patients for activation of p38 and ERK, signaling pathways that have been suggested to be associated with the atrophic effects of chemotherapeutics in preclinical models (3). The effects of chemotherapy may promote muscle atrophy through other signaling pathways that are sensitive to energetic/oxidative stress created by mitochondrial adaptations, such as AMP-activated kinase (63), which can suppress protein synthesis and/or increase proteolysis.
Not all chemotherapeutics derive their myotoxic effects through mitochondrial adaptations, as we found marked myosin depletion in myotubes with Cis in the absence of increased ROS production or mitochondrial rarefaction. The lack of an effect of Cis to induce ROS production may be explained by the concentration we used, as prior work suggests that mitochondrial effects occur at higher concentrations (5). However, the concentrations in our experiments are in line with tumor intracellular levels, and we used PBS as a diluent, which yields greater DNA binding capacity and, in turn, cytotoxicity (18). Despite the lack of effect on mitochondrial content/ROS production, these data reinforce the potential myotoxic effects of chemotherapeutics through multiple pathways.
Several limitations to our studies deserve note. 1) All patients received short courses of dexamethasone concurrent with their chemotherapy (1–4 days for each cycle, depending on symptom severity). Glucocorticoids have well-known effects to cause muscle atrophy (53). Whether the short course of dexamethasone given to these patients would affect muscle structure or function is not known, but longer-term treatment with glucocorticoids combined with bed rest (28 days) led to only modest changes in muscle fiber size (~11% reductions in CSA) (19), and chronic treatment has minimal effects on mitochondrial content (28). Moreover, the atrophic effects of glucocorticoids are generally more apparent in fast-twitch MHC II fibers (53), whereas we found effects in both fiber types. Thus we conclude that it is unlikely that glucocorticoids alone explain the muscle atrophy or mitochondrial rarefaction that we observed in breast cancer patients, but we acknowledge that current data are insufficient to resolve the effects it might have. 2) Because of the cross-sectional nature of our study, we cannot discount the possibility that muscle phenotypes observed in cancer patients are attributable to other factors separate from, or secondary to, cancer and its treatment. For example, acute (e.g., following surgical resection) or chronic physical inactivity (40), which can also induce muscle atrophy and mitochondrial adaptations, may explain these differences. During the review and revision of our manuscript, Mijwel et al. published a study showing declines in skeletal muscle fiber size and citrate synthase activity over 16 wk in breast cancer patients receiving chemotherapy (41), suggesting that muscle atrophy and mitochondrial adaptations occur in concert with cancer treatment. 3) Controls were slightly older than cancer patients. However, most of the phenotypes noted in cancer patients (e.g., fiber atrophy and mitochondrial rarefaction) would worsen with aging, suggesting that the age difference likely lessened group differences in these measures. 4) Myotube cultures are not a perfect model of in vivo skeletal muscle. While they offer a model system to evaluate the singular effects of select agents and obviate off-target effects found in vivo [e.g., cardiac damage/failure associated with Dox administration (26) could affect skeletal muscle size and function (54)], they do not recreate the three-dimensional structural or functional environment of in vivo muscle. Nonetheless, numerous aspects of the atrophic process, such as the effects of cancer to promote oxidant stress, have been studied extensively in myotube cultures and have been shown to correspond with skeletal muscle in vivo (7, 39). 5) We did not include a group of postresection, treatment-naïve patients to further delineate the effects of chemotherapy because of practical difficulties in recruiting and studying these patients. 6) We did not standardize for time on chemotherapeutics among patients, although this factor did not correlate with any outcomes that differed between groups.
In summary, our results provide evidence for skeletal muscle atrophy and mitochondrial rarefaction in breast cancer patients that may be linked to the mitotoxic effects of chemotherapeutics. From a clinical standpoint, these adaptations may contribute to muscle weakness (atrophy) and fatigue (mitochondrial). Accordingly, the notion that chemotherapeutics have such effects on skeletal muscle provides a possible explanation for the high prevalence of fatigue and functional disability across cancer types (12), including those not typically characterized by cachexia, such as breast cancer. Given the contribution of these side effects to a poor quality of life (8) and increased mortality (6, 9, 60), interventions designed to counter these effects on muscle may help alleviate some of the burden of the disease on patients.
GRANTS
This study was funded by a grant from the Vermont Cancer Center/Lake Champlain Cancer Research Organization and by National Institutes of Health Grants R21 CA-191532 and R01 AR-065826. K. Dittus was funded by National Institute of General Medical Sciences Grant P20 GM-103644. B. A. Guigni was funded by Department of Defense SMART Scholarship 2016-85335.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.A.G., K.D., and M.J.T. conceived and designed research; B.A.G., D.M.C., T.W.T., B.F., T.V., B.K.-M., and M.J.T. performed experiments; B.A.G., T.W.T., B.F., T.V., B.K.-M., V.A., and M.J.T. analyzed data; B.A.G., D.M.C., T.W.T., M.S.M., V.A., K.D., and M.J.T. interpreted results of experiments; B.A.G. and M.J.T. prepared figures; B.A.G. and M.J.T. drafted manuscript; B.A.G., D.M.C., M.S.M., V.A., K.D., and M.J.T. edited and revised manuscript; B.A.G., D.M.C., T.W.T., M.S.M., B.F., T.V., B.K.-M., V.A., K.D., and M.J.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank all the volunteers who dedicated their valuable time to these studies. We also thank Dr. Jason Stumpff for assistance with live cell imaging and Dr. Brad Palmer for assistance with electrical field stimulation and assessment of myotube contracture and calcium cycling. The α-tubulin antibody was obtained from the Developmental Studies Hybridoma Bank, created by the National Institute of Child Health and Human Development and maintained at the University of Iowa Department of Biology.
REFERENCES
- 1.American Cancer Society Cancer Facts and Figures 2015 (Online). https://www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf [11 October 2018].
- 2.Aslani A, Smith RC, Allen BJ, Pavlakis N, Levi JA. Changes in body composition during breast cancer chemotherapy with the CMF-regimen. Breast Cancer Res Treat 57: 285–290, 1999. doi: 10.1023/A:1006220510597. [DOI] [PubMed] [Google Scholar]
- 3.Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 7: 43442–43460, 2016. doi: 10.18632/oncotarget.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bechshøft RL, Malmgaard-Clausen NM, Gliese B, Beyer N, Mackey AL, Andersen JL, Kjær M, Holm L. Improved skeletal muscle mass and strength after heavy strength training in very old individuals. Exp Gerontol 92: 96–105, 2017. doi: 10.1016/j.exger.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 5.Berndtsson M, Hägg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer 120: 175–180, 2007. doi: 10.1002/ijc.22132. [DOI] [PubMed] [Google Scholar]
- 6.Braithwaite D, Satariano WA, Sternfeld B, Hiatt RA, Ganz PA, Kerlikowske K, Moore DH, Slattery ML, Tammemagi M, Castillo A, Melisko M, Esserman L, Weltzien EK, Caan BJ. Long-term prognostic role of functional limitations among women with breast cancer. J Natl Cancer Inst 102: 1468–1477, 2010. doi: 10.1093/jnci/djq344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butt Z, Rosenbloom SK, Abernethy AP, Beaumont JL, Paul D, Hampton D, Jacobsen PB, Syrjala KL, Von Roenn JH, Cella D. Fatigue is the most important symptom for advanced cancer patients who have had chemotherapy. J Natl Compr Canc Netw 6: 448–455, 2008. doi: 10.6004/jnccn.2008.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caan BJ, Cespedes Feliciano EM, Prado CM, Alexeeff S, Kroenke CH, Bradshaw P, Quesenberry CP, Weltzien EK, Castillo AL, Olobatuyi TA, Chen WY. Association of muscle and adiposity measured by computed tomography with survival in patients with nonmetastatic breast cancer. JAMA Oncol 4: 798–804, 2018. doi: 10.1001/jamaoncol.2018.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callahan DM, Bedrin NG, Subramanian M, Berking J, Ades PA, Toth MJ, Miller MS. Age-related structural alterations in human skeletal muscle fibers and mitochondria are sex specific: relationship to single-fiber function. J Appl Physiol (1985) 116: 1582–1592, 2014. doi: 10.1152/japplphysiol.01362.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callahan DM, Tourville TW, Miller MS, Hackett SB, Sharma H, Cruickshank NC, Slauterbeck JR, Savage PD, Ades PA, Maughan DW, Beynnon BD, Toth MJ. Chronic disuse and skeletal muscle structure in older adults: sex-specific differences and relationships to contractile function. Am J Physiol Cell Physiol 308: C932–C943, 2015. doi: 10.1152/ajpcell.00014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cella D, Davis K, Breitbart W, Curt G; Fatigue Coalition . Cancer-related fatigue: prevalence of proposed diagnostic criteria in a United States sample of cancer survivors. J Clin Oncol 19: 3385–3391, 2001. doi: 10.1200/JCO.2001.19.14.3385. [DOI] [PubMed] [Google Scholar]
- 13.Damas F, Phillips SM, Lixandrão ME, Vechin FC, Libardi CA, Roschel H, Tricoli V, Ugrinowitsch C. Early resistance training-induced increases in muscle cross-sectional area are concomitant with edema-induced muscle swelling. Eur J Appl Physiol 116: 49–56, 2016. doi: 10.1007/s00421-015-3243-4. [DOI] [PubMed] [Google Scholar]
- 14.De Deyne PG. Formation of sarcomeres in developing myotubes: role of mechanical stretch and contractile activation. Am J Physiol Cell Physiol 279: C1801–C1811, 2000. doi: 10.1152/ajpcell.2000.279.6.C1801. [DOI] [PubMed] [Google Scholar]
- 15.Delbono O, O’Rourke KS, Ettinger WH. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J Membr Biol 148: 211–222, 1995. doi: 10.1007/BF00235039. [DOI] [PubMed] [Google Scholar]
- 16.Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC; Eastern Cooperative Oncology Group . Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am J Med 69: 491–497, 1980. doi: 10.1016/S0149-2918(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 17.Fearon KCH, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166, 2012. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 18.Fischer SJ, Benson LM, Fauq A, Naylor S, Windebank AJ. Cisplatin and dimethyl sulfoxide react to form an adducted compound with reduced cytotoxicity and neurotoxicity. Neurotoxicology 29: 444–452, 2008. doi: 10.1016/j.neuro.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Fitts RH, Romatowski JG, Peters JR, Paddon-Jones D, Wolfe RR, Ferrando AA. The deleterious effects of bed rest on human skeletal muscle fibers are exacerbated by hypercortisolemia and ameliorated by dietary supplementation. Am J Physiol Cell Physiol 293: C313–C320, 2007. doi: 10.1152/ajpcell.00573.2006. [DOI] [PubMed] [Google Scholar]
- 20.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310, 2012. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Freedman RJ, Aziz N, Albanes D, Hartman T, Danforth D, Hill S, Sebring N, Reynolds JC, Yanovski JA. Weight and body composition changes during and after adjuvant chemotherapy in women with breast cancer. J Clin Endocrinol Metab 89: 2248–2253, 2004. doi: 10.1210/jc.2003-031874. [DOI] [PubMed] [Google Scholar]
- 22.Gilliam LA, Ferreira LF, Bruton JD, Moylan JS, Westerblad H, St Clair DK, Reid MB. Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J Appl Physiol (1985) 107: 1935–1942, 2009. doi: 10.1152/japplphysiol.00776.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilliam LAA, Fisher-Wellman KH, Lin C-T, Maples JM, Cathey BL, Neufer PD. The anticancer agent doxorubicin disrupts mitochondrial energy metabolism and redox balance in skeletal muscle. Free Radic Biol Med 65: 988–996, 2013. doi: 10.1016/j.freeradbiomed.2013.08.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilliam LAA, Moylan JS, Patterson EW, Smith JD, Wilson AS, Rabbani Z, Reid MB. Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am J Physiol Cell Physiol 302: C195–C202, 2012. doi: 10.1152/ajpcell.00217.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gouspillou G, Scheede-Bergdahl C, Spendiff S, Vuda M, Meehan B, Mlynarski H, Archer-Lahlou E, Sgarioto N, Purves-Smith FM, Konokhova Y, Rak J, Chevalier S, Taivassalo T, Hepple RT, Jagoe RT. Anthracycline-containing chemotherapy causes long-term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle. Sci Rep 5: 8717, 2015. doi: 10.1038/srep08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res 39: 60–76, 1998. doi: 10.1016/S0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 27.He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci USA 111: 4525–4529, 2014. doi: 10.1073/pnas.1402714111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horber FF, Hoppeler H, Herren D, Claassen H, Howald H, Gerber C, Frey FJ. Altered skeletal muscle ultrastructure in renal transplant patients on prednisone. Kidney Int 30: 411–416, 1986. doi: 10.1038/ki.1986.199. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci USA 110: 2846–2851, 2013. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irwin ML, McTiernan A, Baumgartner RN, Baumgartner KB, Bernstein L, Gilliland FD, Ballard-Barbash R. Changes in body fat and weight after a breast cancer diagnosis: influence of demographic, prognostic, and lifestyle factors. J Clin Oncol 23: 774–782, 2005. doi: 10.1200/JCO.2005.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerr JP, Robison P, Shi G, Bogush AI, Kempema AM, Hexum JK, Becerra N, Harki DA, Martin SS, Raiteri R, Prosser BL, Ward CW. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun 6: 8526, 2015. doi: 10.1038/ncomms9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen Y-W, Raiteri R, Lederer WJ, Dorsey SG, Ward CW. Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal 5: ra56, 2012. doi: 10.1126/scisignal.2002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kontrogianni-Konstantopoulos A, Catino DH, Strong JC, Bloch RJ. De novo myofibrillogenesis in C2C12 cells: evidence for the independent assembly of M bands and Z disks. Am J Physiol Cell Physiol 290: C626–C637, 2006. doi: 10.1152/ajpcell.00442.2005. [DOI] [PubMed] [Google Scholar]
- 34.Langen RCJ, Schols AMWJ, Kelders MCJM, Wouters EFM, Janssen-Heininger YMW. Enhanced myogenic differentiation by extracellular matrix is regulated at the early stages of myogenesis. In Vitro Cell Dev Biol Anim 39: 163–169, 2003. doi:. [DOI] [PubMed] [Google Scholar]
- 35.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol 457: 349–372, 2009. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K-P, Shin YJ, Cho SC, Lee S-M, Bahn YJ, Kim JY, Kwon E-S, Jeong DY, Park SC, Rhee SG, Woo HA, Kwon K-S. Peroxiredoxin 3 has a crucial role in the contractile function of skeletal muscle by regulating mitochondrial homeostasis. Free Radic Biol Med 77: 298–306, 2014. doi: 10.1016/j.freeradbiomed.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Liu MH, Zhang Y, He J, Tan TP, Wu SJ, Fu HY, Chen YD, Liu J, Le QF, Hu HJ, Yuan C, Lin X-L. Upregulation of peroxiredoxin III in doxorubicin-induced cytotoxicity and the FoxO3a-dependent expression in H9c2 cardiac cells. Exp Ther Med 10: 1515–1520, 2015. doi: 10.3892/etm.2015.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manabe Y, Miyatake S, Takagi M, Nakamura M, Okeda A, Nakano T, Hirshman MF, Goodyear LJ, Fujii NL. Characterization of an acute muscle contraction model using cultured C2C12 myotubes. PLoS One 7: e52592, 2012. doi: 10.1371/journal.pone.0052592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLean JB, Moylan JS, Andrade FH. Mitochondria dysfunction in lung cancer-induced muscle wasting in C2C12 myotubes. Front Physiol 5: 503, 2014. doi: 10.3389/fphys.2014.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McTiernan A, Kooperberg C, White E, Wilcox S, Coates R, Adams-Campbell LL, Woods N, Ockene J; Women’s Health Initiative Cohort Study . Recreational physical activity and the risk of breast cancer in postmenopausal women: the Women’s Health Initiative Cohort Study. JAMA 290: 1331–1336, 2003. doi: 10.1001/jama.290.10.1331. [DOI] [PubMed] [Google Scholar]
- 41.Mijwel S, Cardinale DA, Norrbom J, Chapman M, Ivarsson N, Wengström Y, Sundberg CJ, Rundqvist H. Exercise training during chemotherapy preserves skeletal muscle fiber area, capillarization, and mitochondrial content in patients with breast cancer. FASEB J 32: 5495–5505, 2018. doi: 10.1096/fj.201700968R. [DOI] [PubMed] [Google Scholar]
- 42.Miller MS, Callahan DM, Tourville TW, Slauterbeck JR, Kaplan A, Fiske BR, Savage PD, Ades PA, Beynnon BD, Toth MJ. Moderate-intensity resistance exercise alters skeletal muscle molecular and cellular structure and function in inactive older adults with knee osteoarthritis. J Appl Physiol (1985) 122: 775–787, 2017. doi: 10.1152/japplphysiol.00830.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min K, Kwon O-S, Smuder AJ, Wiggs MP, Sollanek KJ, Christou DD, Yoo J-K, Hwang M-H, Szeto HH, Kavazis AN, Powers SK. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J Physiol 593: 2017–2036, 2015. doi: 10.1113/jphysiol.2014.286518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyatake S, Bilan PJ, Pillon NJ, Klip A. Contracting C2C12 myotubes release CCL2 in an NF-κB-dependent manner to induce monocyte chemoattraction. Am J Physiol Endocrinol Metab 310: E160–E170, 2016. doi: 10.1152/ajpendo.00325.2015. [DOI] [PubMed] [Google Scholar]
- 45.Montgomery MK, Osborne B, Brown SHJ, Small L, Mitchell TW, Cooney GJ, Turner N. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J Lipid Res 54: 3322–3333, 2013. doi: 10.1194/jlr.M040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrella JK, Kim JS, Cross JM, Kosek DJ, Bamman MM. Efficacy of myonuclear addition may explain differential myofiber growth among resistance-trained young and older men and women. Am J Physiol Endocrinol Metab 291: E937–E946, 2006. doi: 10.1152/ajpendo.00190.2006. [DOI] [PubMed] [Google Scholar]
- 47.Piscitelli SC, Rodvold KA, Rushing DA, Tewksbury DA. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clin Pharmacol Ther 53: 555–561, 1993. doi: 10.1038/clpt.1993.69. [DOI] [PubMed] [Google Scholar]
- 48.Powers SK, Morton AB, Ahn B, Smuder AJ. Redox control of skeletal muscle atrophy. Free Radic Biol Med 98: 208–217, 2016. doi: 10.1016/j.freeradbiomed.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poynton RA, Hampton MB. Peroxiredoxins as biomarkers of oxidative stress. Biochim Biophys Acta 1840: 906–912, 2014. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Rengo JL, Callahan DM, Savage PD, Ades PA, Toth MJ. Skeletal muscle ultrastructure and function in statin-tolerant individuals. Muscle Nerve 53: 242–251, 2016. doi: 10.1002/mus.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 29: 1774–1785, 2010. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103: 16260–16265, 2006. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schakman O, Kalista S, Barbé C, Loumaye A, Thissen JP. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol 45: 2163–2172, 2013. doi: 10.1016/j.biocel.2013.05.036. [DOI] [PubMed] [Google Scholar]
- 54.Simonini A, Long CS, Dudley GA, Yue P, McElhinny J, Massie BM. Heart failure in rats causes changes in skeletal muscle morphology and gene expression that are not explained by reduced activity. Circ Res 79: 128–136, 1996. doi: 10.1161/01.RES.79.1.128. [DOI] [PubMed] [Google Scholar]
- 55.Tarnopolsky MA, Pearce E, Smith K, Lach B. Suction-modified Bergström muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve 43: 716–725, 2011. doi: 10.1002/mus.21945. [DOI] [PubMed] [Google Scholar]
- 56.Tegeder I, Bräutigam L, Seegel M, Al-Dam A, Turowski B, Geisslinger G, Kovács AF. Cisplatin tumor concentrations after intra-arterial cisplatin infusion or embolization in patients with oral cancer. Clin Pharmacol Ther 73: 417–426, 2003. doi: 10.1016/S0009-9236(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 57.Toth MJ, Gottlieb SS, Fisher ML, Poehlman ET. Skeletal muscle atrophy and peak oxygen consumption in heart failure. Am J Cardiol 79: 1267–1269, 1997. doi: 10.1016/S0002-9149(97)00098-2. [DOI] [PubMed] [Google Scholar]
- 58.Toth MJ, Miller MS, Callahan DM, Sweeny AP, Nunez I, Grunberg SM, Der-Torossian H, Couch ME, Dittus K. Molecular mechanisms underlying skeletal muscle weakness in human cancer: reduced myosin-actin cross-bridge formation and kinetics. J Appl Physiol (1985) 114: 858–868, 2013. doi: 10.1152/japplphysiol.01474.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toth MJ, Ward K, van der Velden J, Miller MS, Vanburen P, Lewinter MM, Ades PA. Chronic heart failure reduces Akt phosphorylation in human skeletal muscle: relationship to muscle size and function. J Appl Physiol (1985) 110: 892–900, 2011. doi: 10.1152/japplphysiol.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villaseñor A, Ballard-Barbash R, Baumgartner K, Baumgartner R, Bernstein L, McTiernan A, Neuhouser ML. Prevalence and prognostic effect of sarcopenia in breast cancer survivors: the HEAL Study. J Cancer Surviv 6: 398–406, 2012. doi: 10.1007/s11764-012-0234-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, Trivedi T, Charkhzarrin S, Khatiwada P, Wronska A, Haynes A, Benassi MS, Witzmann FA, Zhen G, Wang X, Cao X, Roodman GD, Marks AR, Guise TA. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat Med 21: 1262–1271, 2015. doi: 10.1038/nm.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE, Carson JA. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2: 14, 2012. doi: 10.1186/2044-5040-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–E1052, 2013. doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wojtala A, Bonora M, Malinska D, Pinton P, Duszynski J, Wieckowski MR. Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol 542: 243–262, 2014. doi: 10.1016/B978-0-12-416618-9.00013-3. [DOI] [PubMed] [Google Scholar]
- 65.Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167: 661–672, 2004. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med 6: 229ra243, 2014. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]