

Abstract
Although hyperhomocysteinemia (HHcy) occurs because of the deficiency in cystathionine-β-synthase (CBS) causing skeletal muscle dysfunction, it is still unclear whether this effect is mediated through oxidative stress, endoplasmic reticulum (ER) stress, or both. Nevertheless, there is no treatment option available to improve HHcy-mediated muscle injury. Hydrogen sulfide (H2S) is an antioxidant compound, and patients with CBS mutation do not produce H2S. In this study, we hypothesized that H2S mitigates HHcy-induced redox imbalance/ER stress during skeletal muscle atrophy via JNK phosphorylation. We used CBS+/− mice to study HHcy-mediated muscle atrophy, and treated them with sodium hydrogen sulfide (NaHS; an H2S donor). Proteins and mRNAs were examined by Western blots and quantitative PCR. Proinflammatory cytokines were also measured. Muscle mass and strength were studied via fatigue susceptibility test. Our data revealed that HHcy was detrimental to skeletal mass, particularly gastrocnemius and quadriceps muscle weight. We noticed that oxidative stress was reversed by NaHS in homocysteine (Hcy)-treated C2C12 cells. Interestingly, ER stress markers (GRP78, ATF6, pIRE1α, and pJNK) were elevated in vivo and in vitro, and NaHS mitigated these effects. Additionally, we observed that JNK phosphorylation was upregulated in C2C12 after Hcy treatment, but NaHS could not reduce this effect. Furthermore, inflammatory cytokines IL-6 and TNF-α were higher in plasma from CBS as compared with wild-type mice. FOXO1-mediated Atrogin-1 and MuRF-1 upregulation were attenuated by NaHS. Functional studies revealed that NaHS administration improved muscle fatigability in CBS+/− mice. In conclusion, our work provides evidence that NaHS is beneficial in mitigating HHcy-mediated skeletal injury incited by oxidative/ER stress responses.
Keywords: cellular stress, cystathionine-β-synthase, inflammation, muscle atrophy, reactive oxygen species
INTRODUCTION
Homocysteine (Hcy) is a sulfur-containing non-proteinogenic amino acid that is generated during methionine metabolism via the methionine cycle (57). In healthy subjects, synthesis and elimination of Hcy are balanced; however, if Hcy metabolism is disturbed, then its plasma levels are elevated, leading to hyperhomocysteinemia (HHcy) (13, 53, 64, 65, 73). Children born with HHcy due to cystathionine-β-synthase (CBS) deficiency die shortly after birth, but children heterozygous for CBS mutation (CBS+/−) can survive (22, 65, 66). How HHcy triggers such pathological effects in skeletal muscle are not fully understood.
Previous studies revealed that Hcy contains an -SH group like thiols (RSH), which can undergo oxidation to form a disulfide (RSSR) even at physiological pH in the presence of metal catalysts and molecular oxygen [O2·] (18). Further, Hcy can also produce hydrogen peroxide (H2O2, a pro-oxidant molecule) during metal-catalyzed oxidation step and peroxynitrite (ONOO−, a powerful oxidant) in the presence of nitric oxide (NO) and superoxide anion (O2·−) (29). Although these phenomena have been studied in multiple tissue types, whether HHcy exerts its detrimental effects on muscle through these mechanisms is not yet elucidated. Oxidative stress has been implicated in many diseases associated with protein misfolding (36, 39, 62). Likewise, studies have also reported that HHcy could induce endoplasmic reticulum (ER) stress in hepatocytes as well as in vascular endothelial and aortic smooth muscle cells, but the cellular pathways that are involved in these stress-related conditions are not adequately studied (69, 76). It is well known that after translation the protein folding occurs inside the ER, but during stress conditions misfolded proteins can accumulate inside ER lumen, inducing the unfolded protein response (UPR) (42). In mammals, there are three branches of UPR: inositol-requiring enzyme-1 (IRE1), PRKR-like ER kinase (PERK), and activating transcription factor-6 (ATF6) (23, 37, 50). During severe ER stress conditions, activated IRE1α recruits TNF receptor-associated factor-2 (TRAF2) and apoptosis signal-regulating kinase-1 (ASK1), which further activate c-Jun N-terminal kinase (JNK) (6, 26, 52). Activation of JNK phosphorylates c-Jun at Ser63 and 73 residues in NH2-terminal (7, 10). JNK along with c-Jun makes up the activator protein-1 (AP-1) transcription factor, which regulates the expression of several proinflammatory genes (16, 21). JNK also regulates maturation and activity of T cells in addition to the synthesis of proinflammatory cytokines, such as interleukin-2 (IL-2), IL-6, and TNF-α (14, 40, 72). Whether HHcy can compromise muscle survival via activation of JNK is not currently known.
Chronic systemic inflammation is an important driver for muscle wasting, which can be dysregulated by HHcy conditions (28). Proinflammatory cytokines, especially TNF-α and IL-6, regulate this process via the forkhead box protein O (FOXO) pathway by activating the ubiquitin-proteasome system (55). These cytokines work synergistically, promoting muscle atrophy due to the cross-talk between inflammatory cells and organs, resulting in reduced protein synthesis and increased protein degradation, and ultimately leading to muscle loss and functional impairment. Indeed, several E3 ubiquitin ligases, such as muscle RING-finger protein-1 (MuRF-1), muscle atrophy F-box (MAFBx), Nedd4.1, TRAF6, and MUSA1, have been identified that mediate degradation of both thick and thin filaments during skeletal muscle atrophy (1, 2). Therefore, identification of the precise molecular mechanism(s) as to how these E3 ubiquitin ligases are regulated during HHcy is essential to devise future preventive strategies.
Hydrogen sulfide (H2S) is increasingly being recognized as an important signaling molecule in the cardiovascular and nervous systems via its ability to neutralize a variety of reactive oxygen species (ROS) (70, 71, 75) and reduction of the disulfide bonds (9, 59). Cystathionine γ-lyase and CBS can irreversibly remove Hcy by converting it into H2S, since patients with CBS lack H2S, making them vulnerable to oxidative stress damage (59). Hence, the purpose of our study was to understand the effect(s) of HHcy-mediated oxidative and ER stress responses in muscle and the beneficial effects of an H2S donor [sodium hydrogen sulfide (NaHS)], employing both in vitro (C2C12 cells) and in vivo model (CBS+/−) systems on stress responses and muscle biology. Our results indicate that H2S could be developed as a potential therapeutic target in various forms of musculopathies wherein HHcy is linked with metabolic dysfunction.
MATERIALS AND METHODS
Animal maintenance and diet protocol.
Male wild-type (WT; C57BL/6J) and CBS+/− (B6.129P2-Cbstm1Unc/J 002853) mice were purchased from Jackson Laboratory (Bar Harbor, ME) (68). All animals were ∼8–12 wk old and were maintained in 12:12 h light-dark cycle with regular mouse chow diet in the animal facility of the University of Louisville. All animal protocols and care were carried out according to the guidelines of National Institutes of Health (NIH Pub. No. 86–23, revised 1985) and were approved by the Institutional Animal Care and Use Committee of the University of Louisville. Animals were divided into four experimental groups: 1) WT C57BJ/L6 mice (WT); 2) CBS+/− heterozygous mice fed with methionine (CBS+Met); 3) NaHS-supplemented wild-type mice (WT+NaHS); and 4) NaHS-supplemented CBS+/−+Met (CBS+Met+NaHS). Mice were treated with NaHS for 8 wk (30 μM·kg−1·day−1 ip) and fed with a methionine-enriched and low folate, vitamin B6, and vitamin B12 diet (TD 97345, Harlan Teklad, Madison, WI) as described previously (17, 19, 20, 58), whereas the WT mice were given 0.9% normal saline (vehicle control) and fed with normal chow (Purina, Farmer’s Exchange, Framingham, MA).
Body weight and physical activity monitoring and tissue collection.
We measured body weights at 0 wk, 4 wk and after 8-wk intervals during NaHS treatment. Further, the animals were routinely inspected for any discomfort, body posture, skin integrity (injury), and fur appearance to monitor their physical activity. At the end of the experiment, animals were euthanized by using 2× tribromoethanol, and both blood and muscle samples were collected for further analysis.
Genotyping analysis of the heterozygous CBS+/− mouse.
After purchase, mice were cross bred, yielding around 10% CBS−/−, 60% CBS+/−, and 25% CBS+/+. For genotyping, tail samples were collected, and genotypic analysis was performed using PCR by targeted disruption of the CBS gene at loci, as shown in Fig. 1A. The PCR products were run on 1.2% agarose gel (prepared in TAE buffer, pH 8.4) in the presence of ethidium bromide, and the images were recorded in a gel documentation system (25). CBS+/−heterozygote gene-positive mice produced two bands (450 and 308 bp), whereas CBS+/+ mice represented only one band (308 bp).
Fig. 1.

Genotype-phenotype correlation and effect of HHcy on muscle mass loss. A: genotyping of CBS+/− and WT (C57BL/6) mice. B: morphological difference of skeletal muscle in hind limb between CBS+Met and WT mice. C: different muscle weights in hindlimb: gastrocnemius (Gastroc), quadriceps (Quad), tibialis anterior (TA), extensor digitorium longus (EDL), and soleus. D: tibia length measurements. E: body weight measurements. F: total homocysteine (tHcy) measurements. Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; mice number (n) = 4–6. Statistical difference *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; HHcy, hyperhomocysteinemia; Met, methionine; ns, not significant; WT, wild type.
Reagents and antibodies.
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from American Type Culture Collection (Manassas, VA), and trypsin EDTA was from VWR (Radnor, PA). ECL reagent and polyvinylidene difluoride (PVDF) membrane were from Bio-Rad (Hercules, CA). Dihydroethidium (DHE) was purchased from Thermo Fisher Scientific (Waltham, MA). All other reagents and chemicals were ordered from Sigma–Aldrich or available highest grade.
The antibodies for GRP78 (cat. no. sc-13968), IRE1α (cat. no. sc-20790), ATF6 (cat. no. sc-22799), X-box binding protein (XBP1; cat. no. sc-7160), rabbit anti-mouse (cat. no. sc-358914), mouse anti-rabbit (cat. no. sc-2357), and mouse anti-goat (cat. no. sc-2354) were ordered from Santa Cruz Biotechnology (Dallas, TX). The antibody for GAPDH (cat. no. MAB-374) was ordered from EMD Millipore (Burlington, MA). The remainder of the antibodies, for p-IRE1α (S724; cat. no. ab48187), p-JNK (T183/Y185; cat. no. ab4821), t-JNK (cat. no. ab85139), p-cJun (S73; cat. no. ab30620), t-cJun (cat. no. ab32137), p-FOXO1A (S256; cat. no. ab131339), t-FOXO1A (cat. no. ab70382), Atrogin-1 (cat. no. ab92281), myosin heavy chain-I (MHC-I; cat. no. ab11083), MuRF-1 (cat. no. ab172479), and Laminin (cat. no. ab11575), were ordered from Abcam (Cambridge, MA) and used for Western blot and immunohistochemistical (IHC) analysis as per the manufacturer’s protocol.
Cell culture and treatments.
C2C12 cells (immortalized mouse myoblast cell line, ATCC) were cultured in Corning T-75 flasks in ATCC-formulated DMEM (cat. no. 30-2002) supplemented with 10% FBS and 0.1% of penicillin and streptomycin at 37°C with 5% CO2. C2C12 cells were grown to 80% confluence and were plated for 4 different experimental groups: Group 1: CT (PBS as vehicle control); Group 2: Hcy (500 µM); Group 3: Hcy+NaHS (250 µM); and Group 4: NaHS. The Hcy and NaHS concentrations for the individual treatments were chosen as described previously (60, 65). A stock solution of Hcy and NaHS was prepared by directly dissolving Hcy and NaHS in basal DMEM medium (serum-free media). Following 24 h of treatment as mentioned earlier, cells were processed for quantitative (q)PCR, Western blotting, DHE staining, and other biochemical analysis.
Total RNA extraction.
Total RNA was extracted from muscle samples and cells using a Trizol method as described previously (48). Total RNA quality was determined by NanoDrop ND-1000, and RNA with high purity (260/280~2.00 and 260/230~2.00) was used for qPCR.
Reverse transcription and real-time real-time quantitative PCR.
Reverse transcription was performed according to manufacturer’s protocol using a high-capacity cDNA reverse transcription kit from Applied Biosystems (Foster City, CA) for the primer sequences listed in Table 1. For RT-qPCR, a SYBR Green-based kit was used to measure the relative expression of each mRNA-specific primer. Briefly, a three-step cycling protocol was performed using 20 ng of cDNA template in a 20-μl reaction volume under the following conditions: denaturation at 95°C for 15 min followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 70°C for 34 s in which fluorescence was acquired and detected by Roche LightCycler 96 Real-Time PCR System (Roche Diagnostics). Following RT-qPCR, analysis of melt curve was performed to validate the specific generation of the expected PCR product. GAPDH was used as an endogenous control (Quanta Biosciences).
Table 1.
Primers used for RT-qPCR analysis
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GRP78 | 5′-ATTGGTGGCCGTTAAGAATG-3′ | 5′-CAGTGTTGTCTCGGCCAGTA-3′ |
| ERN1 (IRE1) | 5′-CCCAAATGTGATCCGCTACT-3′ | 5′-TTGAGAGAATGCAGGTGTGC-3′ |
| ATF6 | 5′-GGCCAGACTGTTTTGCTCTC-3′ | 5′-CCCATACTTCTGGTGGCACT-3′ |
| XBP1 | 5′-TGAATGGCCCTTAGCATTTC-3′ | 5′-CACAGAACAGGACGCTGTGT-3′ |
Western blotting.
All protein expressions of both tissues and cells were assessed by Western blotting as described previously (4). Briefly, all protein lysates were made in RIPA buffer (containing 5 mM ethylenediamine-tetraacetic acid), which was supplemented with PMSF (1 mM), Na-orthovanadate (1 mM), and a protease inhibitor cocktail (10 μl/ml of lysis buffer) following centrifugation. The protein samples were estimated by Bradford assay. Equal amounts of protein (50 μg) were resolved on SDS-PAGE (8%, 10%, 12%) and transferred onto a PVDF membrane. The blots were visualized using ECL Luminata Forte (Millipore, Temecula, CA) in a Bio-Rad ChemiDoc system. The band intensity was normalized to GAPDH for all of the proteins and quantified using Bio-Rad Image Lab Software.
Total homocysteine measurement.
Total tHcy levels were measured from plasma of experimental mice using homocysteine assay kit (Crystal Chem) as per manufacturer’s instructions.
Intracellular ROS imaging by confocal scanning microscopy.
The cell-permeable fluorescent dye dihydroethidium (DHE) was used to detect intracellular ROS (15). C2C12 cells were incubated in DHE (10 μM/l) for 20 min in a humidified chamber at room temperature in the dark. At the end of the incubation, cells were washed with PBS, and fluorescence images were scanned using a laser confocal microscope (Olympus FluoView1000, Pittsburgh, PA), as this is routinely performed in our laboratory (15). Fluorescent intensity was quantified using ImageJ software (NIH, Bethesda, MD; https://imagej.nih.gov/ij/).
Assessment of lipid peroxidation.
Malondialdehyde (MDA), a metabolite of lipid peroxidation and an indicator of oxidative stress, was measured by the method previously described by Okhawa et al. (see Ref. 47). Briefly, after treatment, the cells were washed with PBS, followed by sonication (5 Amps/5 min) and centrifugation at 13,000 revolutions/min for 10 min. To the supernatant 100 μl of 8.1% SDS, 20% acetic acid, and 0.8% thiobarbituric acid were added. The samples were incubated at 95°C for 60 mins, and then 100 μl of deionized H2O and 100 μl of 1-butanol were added. After spinning at 4,000 revolutions/min for 10 min, the top layer was collected in a 96-well plate and read at a wavelength of 532 nm. We plotted the OD values of our unknown samples with the known standard to determine the intracellular malondialdehyde levels.
Assessment of hydrogen peroxide and total ROS.
H2O2 was measured from cells by Amplex red assay kit (Invitrogen, cat no. A-22188) according to the manufacturer’s protocol. Whereas we used 2′,7′-dichlorofluorescein diacetate (2′,7′-DCFDA) cellular ROS detection assay kit for detection of total ROS (Abcam, cat no. ab113851). After treatment, cells were washed two times with PBS and incubated with 10 µM DCFDA probe for 15 min in serum-free media. Final fluorescence intensity was measured in a microplate reader (Ex, Em = 485, 535 nm).
Measurement of GSH/GSSG ratio.
Assessment of GSH versus GSSG ratio was carried out in all four treatment groups using GSH/GSSG ratio detection kit (ab138881, Abcam) according to the manufacturer’s protocol.
Assessment of proinflammatory cytokines.
To measure all proinflammatory cytokines (IL1α, IL2, IL4, IL6, IL12A, IFN-γ, and TNF-α), we used multi-analyte enzyme-linked immunosorbent assay kit (Qiagen, Germantown, MD) following the manufacturer’s protocol.
Immunohistochemistry.
For IHC, we used cryo-tissue sections of the gastrocnemius muscle (7 µm) that were labeled for immunofluorescence following the standard protocol. Briefly, the tissue sections were fixed in 4% paraformaldehyde and permeabilized with 0.25% Triton X-100 in PBS. Then the sections were incubated overnight in primary antibodies anti-MuRF-1, anti-laminin, and anti-MHC-I at 4°C, and then secondary antibodies labeled with either Alexa Fluor-488 or Alexa Fluor-594 (Invitrogen) appropriate to the primary antibody species were applied. Sections were coverslipped with ProLong Gold Antifade Mountant. Stained images were visualized and analyzed for fluorescence intensity under an EVOS FL Auto Imaging System (Thermo Fisher Scientific) using an appropriate filter. To study the cross-sectional area in the gastrocnemius muscle from each group, we analyzed 30–40 fields per mouse.
Muscle fatigability tests.
The muscle fatigability test was developed from recommendations listed in the Resource Book for the Design of Animal Exercise Protocols by the American Physiological Society with minor modifications. First, one mouse at a time was allowed to swim for 10 min on 4 different days for acclimatization to the environment. We used a swimming tub with water temperature usually between 32°C and 36°C. The depth of water was maintained at a minimum of 30 cm so that mice could not touch the bottom, and 10–15 cm distance was left from the top to prevent animals from climbing or jumping out. On the final day, each mouse was placed in the water to swim to check their maximum swimming capacity. To monitor their live motion, we used Clever Sys (Reston, VA) systems in a method previously discussed (63). If the mice discontinued swimming for 2 s, they were gently nudged to promote their movement, and if they were drowning in the water, then they were immediately taken out from the water as per protocol.
To determine muscle grip strength of our experimental mice, we used a rotarod instrument (San Diego Instruments, San Diego, CA) and grip strength test meter (Bioseb) as in previously described methods with minor modifications (12, 33). Briefly, rotarod performance was done by placing mice on the rotarod and allowing them to run at a constant low speed (12 revolutions/min) for 5 min on 4 different days (acclimatization steps). Following acclimatization, on the final day mice were placed in the rotarod (12 revolutions/min) and allowed to run until they fall off the apparatus (time was recorded). For grip strength, both forelimbs and hindlimbs were measured by using a grip strength test meter. Each mouse was held by the base of the tail and placed in front of the grasping grid. Once the mouse grasped the grid, it was slowly pulled back until the pulling force overcame the mouse’s grip strength. This process was repeated 5 times for each mouse with a 15 min gap between each repeat, and all measurements of grip strength were recorded.
Statistics.
All values are expressed as mean ± SE. The interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred, whereas unpaired t-test was used for comparison between two groups. The threshold for significance was set at P < 0.05, and a minimum of three biological replicates was used for each experiment. For all in vivo experiments, number of mice (n) = 4–5 in each group, and for all exercise capacity tests n = 11 mice were used in each group. Differences between inflammatory cytokines between experimental groups were tested using unpaired Student’s t-test. For all statistical calculation, GraphPad Prism (version 7, GraphPad Software) was used.
RESULTS
HHcy causes skeletal muscle atrophy in CBS+Met mice.
In this study, we noticed CBS+Met mice had significantly low body weight, most likely because of excessive muscle wasting in comparison to WT mice (Fig. 1, B–E). Although we did not observe any changes in tibial length between groups of experimental mice (Fig. 1D), gastrocnemius and quadriceps muscle weights were significantly reduced in CBS compared with WT mice (Fig. 1C). Also, we did not notice any difference in weight for tibialis anterior, extensor digitorium longus, and soleus muscle between CBS and WT mice (Fig. 1C). After administration of NaHS for 8 wk, we noticed an improvement in overall body weight and muscle mass, as shown in Fig. 1, C and E. We also noticed that tHcy levels in plasma were significantly increased in CBS+Met than WT mice, and that was similar in NaHS-treated CBS+Met mice group (Fig. 1F).
NaHS treatment improves muscle fatigability in CBS+Met mice.
To measure HHcy effect on muscle fatigability, we performed a swimming capacity test for all experimental groups. We noticed that CBS+Met mice moved less distance and spent less time during swimming, whereas NaHS supplementation improved their capacities significantly (Fig. 2, A and B). Similar findings were also observed for the muscle grip strength test as shown by less latency to fall from the rotarod and grip strength-to-body weight ratio in CBS+Met as compared with WT mice. After 8 wk of NaHS administration, these effects were substantially improved in CBS+Met mice (Fig. 2, C and D).
Fig. 2.

NaHS treatment improves muscle fatigability in CBS+Met mice. A: measurement of muscle fatigability was done by swimming test. Left: total distance moved. Right: total time spent during swimming performance test. B: images captured during live recording of swim test (top) and representative animal tracings after motion performance in swimming test for each group (bottom). C: results of grip strength using rotarod performance test (left) and grip strength test (right). D: images captured in rotarod (top) and in Bioseb grip strength meter (bottom) during grip strength test. Interaction between multiple groups was determined by one-way, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; mice number (n) = 11 mice. *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; Met, methionine; NaHS, sodium hydrogen sulfide; WT, wild type.
Hcy induces oxidative stress in C2C12 cells.
DHE-staining results showed that Hcy significantly induced ROS in C2C12 as compared with control cells (PBS treated), whereas NaHS treatment potentially mitigated the effects of Hcy (Fig. 3A). To detect superoxide-, peroxide- and peroxynitrite-mediated oxidative chemistry, we used a DCFDA fluorescent probe. A similar finding was also observed in the levels of total ROS in Hcy-treated cells compared with vehicle controls as measured by a fluorometric method (Fig. 3B). Hcy treatment showed significant induction of H2O2 production in C2C12, and it was reduced by NaHS (Fig. 3C). Similarly, malondialdehyde levels (a marker of lipid peroxidation) was significantly increased upon Hcy treatment in C2C12, and NaHS reversed this effect (Fig. 3D). Furthermore, we noticed a significant reduction in GSH/GSSG ratios in Hcy-treated cells as compared with controls, whereas NaHS treatment was found to attenuate this effect (Fig. 3E).
Fig. 3.

Elevated levels of oxidative stress parameters after treatment with Hcy in C2C12 cells (immortalized mouse myoblast cell line). A: Hcy treatment (1 mM) for 24 h resulted in the generation of cellular oxidative stress in C2C12 cells as detected by DHE staining. Images captured using confocal microscope are at left and their quantification is at right (under ×60, bar, 20 μm). B: Hcy treatment induces superoxide-, peroxide-, and peroxynitrite-mediated oxidative chemistry detected by CM-H2DCFDA (chloromethyl derivative-2′,7′-dichlorodihydrofluorescein diacetate) probe. C: Hcy exposure induces H2O2 production measured by Amplex red assay. D: Hcy treatment induces lipid peroxidation measured by malondialdehyde (MDA) assay. E: Hcy treatment reduces GSH vs. GSSG ratio. Interaction between multiple groups was determined by one-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; n = 3–4 biological replicates. *P < 0.05 vs. CT and #P < 0.05 vs. Hcy. CT, control; DHE, dihydroethidium; Hcy, homocysteine; RFU, relative fluorescence units.
Hcy induces ER stress response in skeletal muscle.
To examine whether HHcy induces ER stress via redox imbalance mechanism, we performed Western blot analyses of samples from in vitro and in vivo models. We found that Hcy significantly induced ER stress markers, such as GRP78, ATF6, and p-IRE1α, and that this was mitigated by NaHS in C2C12, as shown in Fig. 4, A and B. However, we did not notice any significant changes in XBP1 levels after Hcy treatment (Fig. 4, A and B). In addition, the ratios of p-IRE1α/tIRE1α were found to be significantly altered in C2C12 after Hcy treatment compared with controls, whereas NaHS supplementation attenuated this effect (Fig. 4C). In qPCR analysis, we noticed mRNA levels of GRP78, ATF6, IRE1α, and XBP1 were also increased significantly by post Hcy treatment of C2C12 compared with vehicle controls, and these effects were also similarly mitigated by NaHS (Fig. 4D). In addition, our in vivo model revealed similar types of changes in Western blot and qPCR data from the gastrocnemius muscle (Fig. 4, E–H).
Fig. 4.

A–D: high Hcy mediates ER stress response in muscle cells in vitro. A: Western blot analysis showing Hcy treatment induces ER stress markers, such as GRP78, ATF6, p-IRE1α (S724), t-IREα, and XBP1, whereas NaHS treatment mitigates this effect. B: densitometric measurement of ER stress markers from the Western blot images in A. C: quantification of p-IRE1α/t-IREα ratio from the Western blot images in A. D: Hcy induced expression mRNA levels of GRP78, ATF6, IRE1α, and XBP1 in C2C12 cells (log transformed data). Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are mean ± SE; n = 3–4 biological replicates. *P < 0.05 vs. CT and #P < 0.05 vs. Hcy. E–H: high Hcy mediates ER stress response in muscle in vivo. E: Western blot analysis showing induced expression of ER stress markers, such as GRP78, ATF6, p-IRE1α (S724), and t-IREα in CBS+Met mice compared with WT mice. NaHS treatment mitigates this effect. F: densitometric quantification of the Western blots of ER stress markers in E. G: densitometric analysis of p-IRE1α/IREα ratio from the Western blots in E. H: quantitative PCR analysis showing that mRNA levels of GRP78, ATF6, IRE1α, and XBP1 (log-transformed data) were increased in skeletal muscle of CBS+Met mice compared with WT mice. Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; mice number (n) = 4–5. *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; CT, control; ER, endoplasmic reticulum; Hcy, homocysteine; Met, methionine; WT, wild type.
Hcy enhanced JNK phosphorylation, Atrogin-1, and MuRF-1 expression in skeletal muscle.
To confirm whether high oxidative and ER stress responses can induce JNK phosphorylation during HHcy, we employed Western blotting for p-JNK (Fig. 5A). We found that Hcy significantly induced phosphorylation of JNK in comparison with controls; however, we did not find a concomitant reduction in JNK phosphorylation via NaHS treatment, as shown in Fig. 5, A and B. To study whether JNK phosphorylation is mediated via ER stress mechanism(s), we also treated cells with tunicamycin (a known positive inducer). Results revealed that JNK phosphorylation was inhibited by SP600125 (JNK inhibitor) and induced by tunicamycin (Fig. 5, A and B). Additionally, we observed that phosphorylation of c-Jun was elevated in C2C12 post Hcy treatment compared with vehicle controls (Fig. 5, A and B). Expression of Atrogin-1 and MuRF-1 were higher in Hcy-treated C2C12 in comparison to vehicle controls, and this effect was attenuated by NaHS treatment (Fig. 5, C and D). Similar findings were observed in muscle collected from experimental mice (Fig. 5, E–H). Moreover, the findings revealed upregulation of FOXO1A phosphorylation in CBS mice compared with WT, and this was attenuated via NaHS administration (Fig. 5, E–H). In addition, IHC experiments confirmed higher MuRF-1 and reduced myosin heavy chain type-I (MHC-I) expression in skeletal muscle of CBS+Met as compared with WT mice, and this effect was alleviated by NaHS treatment (Fig. 6, A–C). The Western blot results also confirmed a similar association as seen in IHC staining (Fig. 6, D and E).
Fig. 5.

A–D: high Hcy induces JNK phosphorylation and induces atrophic markers in cells in vitro. A: Western blot data showing Hcy-mediated JNK phosphorylation and induced protein expression of p-cJun (S73) in C2C12 cells, where SP600125 was used as a JNK inhibitor and tunicamycin was used as positive control. B: densitometric measurements of p-JNK/t-JNK and p-cJun/t-cJun ratios from the Western blot images shown in A. C: Western blot data showing expression of Atrogin-1 and MuRF-1 were elevated in C2C12 cells post Hcy treatment compared with control. D: densitometric measurement of Atrogin-1 and MuRF-1 expression from the Western blot images shown in C. Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data is mean ± SE, where n = 3–4 biological replicates and *P < 0.05 vs. CT and #P < 0.05 vs. Hcy. E–H: high Hcy induces JNK phosphorylation and induces atrophic markers in vivo. E: Western blot data showing Hcy-mediated JNK phosphorylation and induced protein expression of p-cJun (S73) in muscle. F: Western blot data showing expression of Atrogin-1 and MuRF-1 were elevated in the muscle of CBS+Met mice compared with WT mice. G: densitometric measurements of p-JNK/t-JNK, p-cJun/t-cJun, and p-FOXO1/t-FOXO1 ratios from the Western blot images shown in E. H: densitometric measurement of Atrogin-1 and MuRF-1 expression from the Western blot images shown in F. Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; mice number (n) = 4–5. *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; CT, control; Hcy, homocysteine; Met, methionine; WT, wild type.
Fig. 6.

High Hcy mediates elevated expression of MuRF-1 and degradation of its target protein MHC-I in skeletal muscle of CBS mice compared with WT mice. A: IHC analysis showing representative images of elevated expression of MuRF-1 and reduced expression of MHC-I in tissue cross section of the GA muscle in CBS+Met mice compared with WT mice. Arrows indicate the respective expression levels of MuRF-1 (low) and MHC-I (high) in the fiber content of GA muscle (bar, 200 μm). B: quantification of MHC-I expression from the IHC images shown in A. C: quantification of MuRF-1 expression from the IHC images shown in A. D: Western blot data showing protein expression of MHC-I and MuRF-1 in the muscle of CBS+Met mice compared with WT mice. E: densitometric measurement of MHC-I and MuRF-1 from the Western blot images shown in D. Interaction between multiple groups was determined by one-way or two-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE; mice number (n) = 4–5. *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; GA, gastrocnemius; Hcy, homocysteine; IHC, immunohistochemistry; Met, methionine; WT, wild type.
HHcy induced proinflammatory milieu in CBS+Met mice.
To identify weather HHcy-mediated oxidative and ER stress responses could modulate proinflammatory cytokines via the JNK/c-Jun axis, we measured levels of these cytokines in plasma samples collected from experimental mice. Results showed that IL1α, IL2, IL4, IL6, IL12A, IFN-γ, and TNF-α, were elevated in CBS+Met in comparison with WT mice (Fig. 7A). IL6 and TNF-α levels were found to be significantly induced in plasma of CBS+Met as compared with the WT mice. However, we did not notice any improvement by NaHS administration (data are not shown).
Fig. 7.

A–C: HHcy-induced proinflammatory milieu in CBS+Met mice and its effect in morphological parameters of skeletal muscle in vivo. A: levels of inflammatory cytokines, such as IL-1α, IL-2, IL-4, IL-6, IL-12A, IFN-γ, and TNF-α, were elevated in the plasma of diet-induced hyperhomocysteinemic mice in comparison to WT mice. B: laminin staining showing cross-sectional area measurements are low in gastrocnemius (GA) muscle of CBS+Met mice compared with WT mice (bar, 200 μm). To study the cross-sectional area in GA muscle from each group, we analyzed 30–40 fields/mouse. Only the representative image from each group of mice is shown here. C: quantification of cross-sectional area from the laminin staining images shown in B. Interaction between multiple groups was determined by one-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Unpaired t-test was used for comparison between two groups. Data are means ± SE; mice number (n) = 4–5. *P < 0.05 vs. WT. D–G: HHcy-induced morphological changes in skeletal muscle in vivo. D: hematoxylin and eosin (H&E) staining in cross-sectional area of gastrocnemius muscle showing morphological differences in muscle fibers between CBS+Met and WT mice. Images were taken using EVOS FL Auto Cell Imaging System; mice number (n) = 4–5. Bar, 400 μm. Representative areas from H&E staining images are zoomed in to show more clear view. E: quantification of regenerated muscle fibers as quantified by centrally located nuclei from H&E staining images. F: Masson’s Trichrome staining in cross-sectional area of gastrocnemius muscle showing blue color stain (white arrowheads) as collagen deposition in the muscle of CBS+Met mice compared with WT mice. Images were taken using EVOS FL Auto Cell Imaging System; mice number (n) = 4–5. Bar, 400 μm. G: quantification of collagen deposition as quantified by blue color stain from Masson’s Trichrome staining images. The interaction between multiple groups was determined by one-way ANOVA, including a Tukey’s post hoc analysis when significant interaction occurred. Data are means ± SE and mice number (n) = 4–5. *P < 0.05 vs. WT and #P < 0.05 vs. CBS+Met. CBS, cystathionine-β-synthase; HHcy, hyperhomocysteinemia; Met, methionine; WT, wild type.
HHcy induced morphological changes in skeletal muscle in vivo.
To understand the effect of HHcy-mediated oxidative and ER stress responses on cross-sectional areas of skeletal muscle fibers, we did laminin staining. Results of the staining showed a significant reduction of cross-sectional areas in gastrocnemius muscle isolated from CBS+Met in comparison to WT mice (Fig. 7, B and C). Additionally, we did notice an improvement after 8 wk NaHS treatment (Fig. 7, B and C). Hematoxylin-eosin and Trichrome staining revealed a higher level of fibrosis and collagen deposition in muscle derived from CBS+Met as compared with WT mice. Interestingly, treatment with NaHS was found to alleviate this effect in the CBS+Met group of mice (Fig. 7, D–G).
DISCUSSION
Previously, we reported that HHcy was detrimental to muscle force generation and was responsible for muscle fatigability in CBS+/− mice (65). However, molecular mechanisms underlying the detrimental effects of HHcy on muscles were not precisely studied. To our knowledge, this is the first study elaborating the mechanistic roles of homocysteine during oxidative and ER stress responses, which can potentiate the skeletal muscle atrophy via JNK phosphorylation. Data obtained in the present study from both physiological and biochemical investigations suggest that increase in tHcy levels leads to severe muscle atrophy via oxidative and ER stress-dependent mechanisms and that NaHS treatment could successfully mitigate these harmful effects.
Findings from our laboratory and others have demonstrated the effects of HHcy on oxidative stress in cardiac microvascular endothelial cells (61), vascular smooth muscle cells (75), and liver tissue (34). The results from the present study add to the growing body of evidence that HHcy can in fact induce pathological changes in muscles via inducing ROS moieties, given that both ROS generation and lipid peroxidation are equally elevated by Hcy in C2C12 cells and that NaHS treatment was found to improve these conditions. Also, these results suggest that HHcy as a result of CBS heterozygosity is equally detrimental to skeletal muscle, which may result from higher ROS generation and a concomitant reduction in glutathione and H2S levels.
Although oxidative stress has been implicated in many diseases encompassing protein misfolding and disruption of protein folding pathways thereof (36, 39, 45, 62), to our knowledge this is the first study showing that HHcy could also induce severe ER stress response via induction of GRP78, ATF6, and IRE1α phosphorylation in skeletal muscle (Fig. 4). This finding also corroborates the previous observation showing that HHcy mediates higher ER stress responses in endothelial cells (24, 27, 44). A study by Malhotra et al. (31) indicated that antioxidants reduced ER stress, leading to improved protein secretion. Similarly, we also noticed that NaHS treatment has a beneficial effect toward mitigating UPR response in skeletal muscle (Fig. 4, E–H).
The JNK pathway is known to control cellular response to harmful extracellular stimuli (41, 49, 74); whether Hcy-mediated oxidative and ER stress responses can also induce similar responses in skeletal muscle has not been studied previously. This study shows that HHcy-mediated oxidative and ER stress responses induce JNK phosphorylation and subsequently upregulate the secretion of proinflammatory cytokines (TNF-α, IL-1, and IL-6) in CBS+Met mice (Fig. 7A). Our results are also in agreement with inflammatory bowel disease conditions wherein JNK upregulation plays a vital role (49). Oudi and colleagues (43) reported a similar inflammatory response in acute coronary syndrome patients, where tHcy, HsCRP, IL-6, and TNF-α were significantly elevated. In addition to these findings, studies from other groups have also reported that HHcy causes cardiovascular disease via increasing IL-1ra and IL-6 levels (8, 11). Furthermore, it is possible that during inflammation, immune cells (CD8+ T lymphocytes) may cause myocyte degeneration (autoimmune responses), skeletal muscle weakness, fibrosis along collagen deposition, and atrophic changes in the muscle (3). Previous reports showed that FOXO1 is induced by inflammatory cytokines (46). However, in this study we noticed that NaHS could not mitigate inflammation, but it did mitigate phosphorylation of FOXO1 in the skeletal muscle of CBS+Met mice, suggesting that during HHcy condition phosphorylation of FOXO1 may not be mediated via inflammatory pathways. Previous results also suggested that the role of FOXO1 transcription factors in the regulation of E3-ubiquitin ligases like Atrogin-1 and MuRF-1 (35, 51, 54, 67). Similarly, our results showed higher expression of Atrogin-1 and MuRF-1, suggesting that FOXO1 plays an important role in this process. We also noticed that expression of MHC-I was decreased in skeletal muscle of CBS+Met mice (Fig. 6), which indicates that higher expression of MuRF-1 could trigger muscle atrophy via proteasomal degradation of its target protein, such as MHC-I (38). Indeed, we found that CBS+Met mice had severe muscle fatigue syndrome during swimming and grip strength tests (Fig. 2). We noticed that NaHS was able to improve muscle functions during muscle fatigability tests, most likely via reduction of oxidative and ER stress responses in affected skeletal muscle.
In our study, we saw a difference in the phosphorylation status of the JNK levels between in vivo and in vitro conditions. To the best of our knowledge, there could be more than one explanation for the observed difference, such as the acute homocysteine-mediated effect on the phosphorylation event taking place posttranscriptionally in the in vitro setting. Most likely we might have missed the phosphorylation time point for the JNK phosphorylation in the in vitro settings, since we only studied a single readout (24 h post HHcy), unlike the in vivo CBS model wherein we were able to observe the continuous or prolonged effect of the NaHS-mediated phospho-JNK status. Although JNK phosphorylation was not affected by NaHS, the mitigation of FOXO1 phosphorylation was prominent despite the fact we do not know the exact nature of putative mediators that might be at play and are involved in phosphorylation of FOXO1 during HHcy condition. Several models of atrophy previously showed that inhibition of PI3K/Akt signaling induces nuclear import of FOXO1 regulating the Atrogin-1 and MuRF-1 expression dynamics (5, 56). Future work should explore whether a reduction in FOXO1, Atrogin-1, and MuRF-1 is mediated via the Akt/PI3K axis or other similar mechanisms(s) (32). Since HHcy is known to cause hypermethylation of genes (30), however, we did not test the possibility of methylation status that may or may not be a factor in muscle atrophy and related muscular pathologies.
The findings from this study have been summarized in Fig. 8, highlighting the sequential events during HHcy and its effects on skeletal muscle atrophy. Further work is required that might shed light on whether H2S could be developed as a potential therapeutic target for treating skeletal muscle atrophy and related metabolic disorders.
Fig. 8.
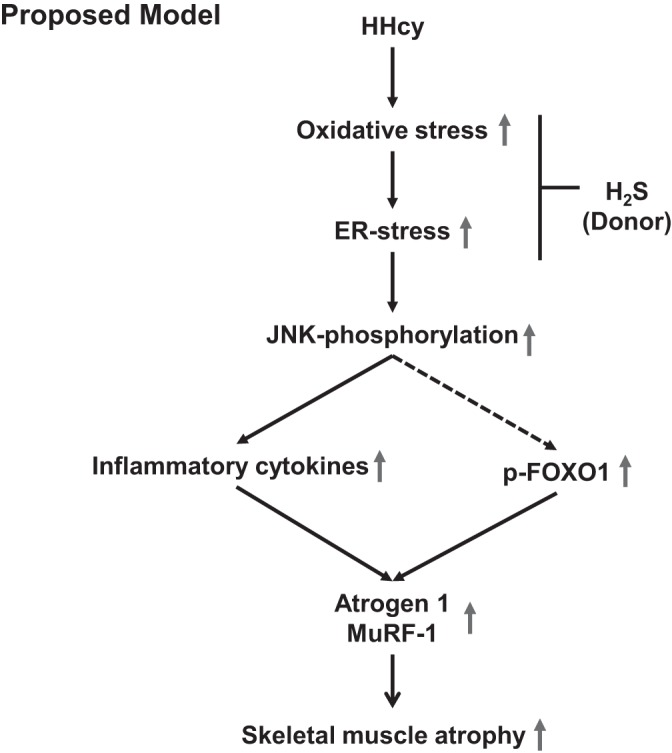
A schematic representation of a model showing how high homocysteine (HHcy) can induce muscle atrophy via oxidative and endoplasmic reticulum (ER) stress, alteration of proinflammatory milieu, and elevation of atrophic markers in skeletal muscles.
GRANTS
The work was supported by National Institutes of Health National Heart, Lung, and Blood Institute Grants HL-74815 and HL-107640 and National Institute of Neurological Disorders and Stroke Grant NS-084823.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M., J.B., and S.C.T. conceived and designed research; A.M., N.T.T., and N.M. performed experiments; A.M. and M.S. analyzed data; A.M., M.S., A.K.G., and N.T. interpreted results of experiments; A.M. and N.M. prepared figures; A.M., M.S., J.B., and A.K.G. drafted manuscript; A.M., M.S., A.K.G., and S.C.T. edited and revised manuscript; A.M., M.S., N.T., N.M., and S.C.T. approved final version of manuscript.
REFERENCES
- 1.Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab 307: E469–E484, 2014. doi: 10.1152/ajpendo.00204.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 6: 25–39, 2013. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brustolin S, Giugliani R, Felix TM. Genetics of homocysteine metabolism and associated disorders. Rev Bras Pesqui Med Biol 43: 1–7, 2010. doi: 10.1590/S0100-879X2009007500021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaturvedi P, Kalani A, Givvimani S, Kamat PK, Familtseva A, Tyagi SC. Differential regulation of DNA methylation versus histone acetylation in cardiomyocytes during HHcy in vitro and in vivo: an epigenetic mechanism. Physiol Genomics 46: 245–255, 2014. doi: 10.1152/physiolgenomics.00168.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clavel S, Siffroi-Fernandez S, Coldefy AS, Boulukos K, Pisani DF, Dérijard B. Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol 30: 470–480, 2010. doi: 10.1128/MCB.00666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem 276: 23681–23688, 2001. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 7.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76: 1025–1037, 1994. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 8.Gori AM, Corsi AM, Fedi S, Gazzini A, Sofi F, Bartali B, Bandinelli S, Gensini GF, Abbate R, Ferrucci L. A proinflammatory state is associated with hyperhomocysteinemia in the elderly. Am J Clin Nutr 82: 335–341, 2005. doi: 10.1093/ajcn/82.2.335. [DOI] [PubMed] [Google Scholar]
- 9.Greiner R, Pálinkás Z, Bäsell K, Becher D, Antelmann H, Nagy P, Dick TP. Polysulfides link H2S to protein thiol oxidation. Antioxid Redox Signal 19: 1749–1765, 2013. doi: 10.1089/ars.2012.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 7: 2135–2148, 1993. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 11.Holven KB, Aukrust P, Retterstol K, Hagve TA, Mørkrid L, Ose L, Nenseter MS. Increased levels of C-reactive protein and interleukin-6 in hyperhomocysteinemic subjects. Scand J Clin Lab Invest 66: 45–54, 2006. doi: 10.1080/00335510500429821. [DOI] [PubMed] [Google Scholar]
- 12.Hwee DT, Kennedy A, Ryans J, Russell AJ, Jia Z, Hinken AC, Morgans DJ, Malik FI, Jasper JR. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS One 9: e96921, 2014. doi: 10.1371/journal.pone.0096921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368, 2010. doi: 10.1074/jbc.M110.147439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishizuka T, Terada N, Gerwins P, Hamelmann E, Oshiba A, Fanger GR, Johnson GL, Gelfand EW. Mast cell tumor necrosis factor alpha production is regulated by MEK kinases. Proc Natl Acad Sci USA 94: 6358–6363, 1997. doi: 10.1073/pnas.94.12.6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.John AMSP, Kundu S, Pushpakumar S, Fordham M, Weber G, Mukhopadhyay M, Sen U. GYY4137, a hydrogen sulfide donor modulates miR194-dependent collagen realignment in diabetic kidney. Sci Rep 7: 10924, 2017. doi: 10.1038/s41598-017-11256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta 1773: 1341–1348, 2007. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalani A, Kamat PK, Givvimani S, Brown K, Metreveli N, Tyagi SC, Tyagi N. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. J Mol Neurosci 52: 202–215, 2014. doi: 10.1007/s12031-013-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalra BR, Ghose S, Sood NN. Homocystinuria with bilateral absolute glaucoma. Indian J Ophthalmol 33: 195–197, 1985. [PubMed] [Google Scholar]
- 19.Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, Tyagi N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 252: 302–319, 2013. doi: 10.1016/j.neuroscience.2013.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamath AF, Chauhan AK, Kisucka J, Dole VS, Loscalzo J, Handy DE, Wagner DD. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 107: 591–593, 2006. doi: 10.1182/blood-2005-06-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaminska B. Molecular characterization of inflammation-induced JNK/c-Jun signaling pathway in connection with tumorigenesis. Methods Mol Biol 512: 249–264, 2009. doi: 10.1007/978-1-60327-530-9_13. [DOI] [PubMed] [Google Scholar]
- 22.Kanwar YS, Manaligod JR, Wong PW. Morphologic studies in a patient with homocystinuria due to 5, 10-methylenetetrahydrofolate reductase deficiency. Pediatr Res 10: 598–609, 1976. doi: 10.1203/00006450-197606000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389–1398, 2002. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokame K, Kato H, Miyata T. Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. GRP78/BiP and novel genes. J Biol Chem 271: 29659–29665, 1996. doi: 10.1074/jbc.271.47.29659. [DOI] [PubMed] [Google Scholar]
- 25.Kumar M, Tyagi N, Moshal KS, Sen U, Kundu S, Mishra PK, Givvimani S, Tyagi SC. Homocysteine decreases blood flow to the brain due to vascular resistance in carotid artery. Neurochem Int 53: 214–219, 2008. doi: 10.1016/j.neuint.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437, 2003. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lentz SR, Sadler JE. Homocysteine inhibits von Willebrand factor processing and secretion by preventing transport from the endoplasmic reticulum. Blood 81: 683–689, 1993. [PubMed] [Google Scholar]
- 28.Londhe P, Guttridge DC. Inflammation induced loss of skeletal muscle. Bone 80: 131–142, 2015. doi: 10.1016/j.bone.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lubos E, Handy DE, Loscalzo J. Role of oxidative stress and nitric oxide in atherothrombosis. Front Biosci 13: 5323–5344, 2008. doi: 10.2741/3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majumder A, Behera J, Jeremic N, Tyagi SC. Hypermethylation: causes and consequences in skeletal muscle myopathy. J Cell Biochem 118: 2108–2117, 2017. doi: 10.1002/jcb.25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA 105: 18525–18530, 2008. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274, 2007. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuo N, Takao K, Nakanishi K, Yamasaki N, Tanda K, Miyakawa T. Behavioral profiles of three C57BL/6 substrains. Front Behav Neurosci 4: 29, 2010. doi: 10.3389/fnbeh.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matté C, Stefanello FM, Mackedanz V, Pederzolli CD, Lamers ML, Dutra-Filho CS, Dos Santos MF, Wyse AT. Homocysteine induces oxidative stress, inflammatory infiltration, fibrosis and reduces glycogen/glycoprotein content in liver of rats. Int J Dev Neurosci 27: 337–344, 2009. doi: 10.1016/j.ijdevneu.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 35.McLoughlin TJ, Smith SM, DeLong AD, Wang H, Unterman TG, Esser KA. FoxO1 induces apoptosis in skeletal myotubes in a DNA-binding-dependent manner. Am J Physiol Cell Physiol 297: C548–C555, 2009. doi: 10.1152/ajpcell.00502.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merad-Boudia M, Nicole A, Santiard-Baron D, Saillé C, Ceballos-Picot I. Mitochondrial impairment as an early event in the process of apoptosis induced by glutathione depletion in neuronal cells: relevance to Parkinson’s disease. Biochem Pharmacol 56: 645–655, 1998. doi: 10.1016/S0006-2952(97)00647-3. [DOI] [PubMed] [Google Scholar]
- 37.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101: 451–454, 2000. doi: 10.1016/S0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 38.Moriscot AS, Baptista IL, Bogomolovas J, Witt C, Hirner S, Granzier H, Labeit S. MuRF1 is a muscle fiber-type II associated factor and together with MuRF2 regulates type-II fiber trophicity and maintenance. J Struct Biol 170: 344–353, 2010. doi: 10.1016/j.jsb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura T, Lipton SA. Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell Mol Life Sci 64: 1609–1620, 2007. doi: 10.1007/s00018-007-6525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishina H, Bachmann M, Oliveira-dos-Santos AJ, Kozieradzki I, Fischer KD, Odermatt B, Wakeham A, Shahinian A, Takimoto H, Bernstein A, Mak TW, Woodgett JR, Ohashi PS, Penninger JM. Impaired CD28-mediated interleukin 2 production and proliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen-activated protein kinase kinase 4 (MKK4)-deficient T lymphocytes. J Exp Med 186: 941–953, 1997. doi: 10.1084/jem.186.6.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26: 9220–9231, 2006. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490: 71–92, 2011. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oudi ME, Aouni Z, Mazigh C, Khochkar R, Gazoueni E, Haouela H, Machghoul S. Homocysteine and markers of inflammation in acute coronary syndrome. Exp Clin Cardiol 15: e25–e28, 2010. [PMC free article] [PubMed] [Google Scholar]
- 44.Outinen PA, Sood SK, Liaw PC, Sarge KD, Maeda N, Hirsh J, Ribau J, Podor TJ, Weitz JI, Austin RC. Characterization of the stress-inducing effects of homocysteine. Biochem J 332: 213–221, 1998. doi: 10.1042/bj3320213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plaisance V, Brajkovic S, Tenenbaum M, Favre D, Ezanno H, Bonnefond A, Bonner C, Gmyr V, Kerr-Conte J, Gauthier BR, Widmann C, Waeber G, Pattou F, Froguel P, Abderrahmani A. Endoplasmic reticulum stress links oxidative stress to impaired pancreatic beta-cell function caused by human oxidized LDL. PLoS One 11: e0163046, 2016. doi: 10.1371/journal.pone.0163046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp Diabetes Res 2012: 939751, 2012. doi: 10.1155/2012/939751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puntel RL, Roos DH, Grotto D, Garcia SC, Nogueira CW, Rocha JB. Antioxidant properties of Krebs cycle intermediates against malonate pro-oxidant activity in vitro: a comparative study using the colorimetric method and HPLC analysis to determine malondialdehyde in rat brain homogenates. Life Sci 81: 51–62, 2007. doi: 10.1016/j.lfs.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 48.Rio DC, Ares M Jr, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc 2010: pdb.prot5439, 2010. doi: 10.1101/pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 49.Roy PK, Rashid F, Bragg J, Ibdah JA, Hepatology DG, Medicine UMS, Columbia, Missouri, States U. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol 14: 200–202, 2008. doi: 10.3748/wjg.14.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol 14: 20–28, 2004. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103: 16260–16265, 2006. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833: 3460–3470, 2013. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schalinske KL, Smazal AL. Homocysteine imbalance: a pathological metabolic marker. Adv Nutr 3: 755–762, 2012. doi: 10.3945/an.112.002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J 22: 3836–3845, 2008. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sente T, Van Berendoncks AM, Fransen E, Vrints CJ, Hoymans VY. Tumor necrosis factor-α impairs adiponectin signalling, mitochondrial biogenesis, and myogenesis in primary human myotubes cultures. Am J Physiol Heart Circ Physiol 310: H1164–H1175, 2016. doi: 10.1152/ajpheart.00831.2015. [DOI] [PubMed] [Google Scholar]
- 56.Shimizu H, Langenbacher AD, Huang J, Wang K, Otto G, Geisler R, Wang Y, Chen JN. The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. eLife 6: e27955, 2017. doi: 10.7554/eLife.27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 34: 17–32, 2011. doi: 10.1007/s10545-009-9006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab 33: 708–715, 2013. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6: 917–935, 2007. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 60.Tyagi N, Moshal KS, Sen U, Vacek TP, Kumar M, Hughes WM Jr, Kundu S, Tyagi SC. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid Redox Signal 11: 25–33, 2009. doi: 10.1089/ars.2008.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol 289: H2649–H2656, 2005. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- 62.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513–517, 2006. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 63.Veeranki S, Givvimani S, Pushpakumar S, Tyagi SC. Hyperhomocysteinemia attenuates angiogenesis through reduction of HIF-1α and PGC-1α levels in muscle fibers during hindlimb ischemia. Am J Physiol Heart Circ Physiol 306: H1116–H1127, 2014. doi: 10.1152/ajpheart.00003.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veeranki S, Tyagi SC. Mechanisms of hyperhomocysteinemia induced skeletal muscle myopathy after ischemia in the CBS−/+ mouse model. Int J Mol Sci 16: 1252–1265, 2015. doi: 10.3390/ijms16011252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Veeranki S, Winchester LJ, Tyagi SC. Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta 1852: 732–741, 2015. doi: 10.1016/j.bbadis.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voskoboeva E, Semyachkina A, Yablonskaya M, Nikolaeva E. Homocystinuria due to cystathionine beta-synthase (CBS) deficiency in Russia: molecular and clinical characterization. Mol Genet Metab Rep 14: 47–54, 2018. doi: 10.1016/j.ymgmr.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab 295: E785–E797, 2008. doi: 10.1152/ajpendo.00646.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA 92: 1585–1589, 1995. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 107: 1263–1273, 2001. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem 90: 765–768, 2004. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 71.Whiteman M, Cheung NS, Zhu YZ, Chu SH, Siau JL, Wong BS, Armstrong JS, Moore PK. Hydrogen sulphide: a novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem Biophys Res Commun 326: 794–798, 2005. doi: 10.1016/j.bbrc.2004.11.110. [DOI] [PubMed] [Google Scholar]
- 72.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med (Berl) 74: 589–607, 1996. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 73.Winchester L, Veeranki S, Givvimani S, Tyagi SC. Exercise mitigates the adverse effects of hyperhomocysteinemia on macrophages, MMP-9, skeletal muscle, and white adipocytes. Can J Physiol Pharmacol 92: 575–582, 2014. doi: 10.1139/cjpp-2014-0059. [DOI] [PubMed] [Google Scholar]
- 74.Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med 51: 1365–1375, 2011. doi: 10.1016/j.freeradbiomed.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 75.Yan SK, Chang T, Wang H, Wu L, Wang R, Meng QH. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cells. Biochem Biophys Res Commun 351: 485–491, 2006. doi: 10.1016/j.bbrc.2006.10.058. [DOI] [PubMed] [Google Scholar]
- 76.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455–462, 2008. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]