

Summary
West syndrome, an age‐specific epileptic encephalopathy, manifests with infantile spasms (IS) and impaired neurodevelopmental outcomes and epilepsy. The multiple‐hit rat model of IS is a chronic model of IS due to structural etiology, in which spasms respond partially to vigabatrin analogs. Using this model, we investigated whether IS due to structural etiology may have deficits in parvalbumin (PRV) and somatostatin (SST) immunoreactive (‐ir) interneurons, and calretinin‐ir (CR‐ir) neurons of the primary somatosensory cortex of postnatal day (PN) 20–24 rats, using specific immunohistochemical assays. PN3 Sprague‐Dawley male rats underwent the multiple‐hit induction protocol, were monitored until PN20–24, and were transcardially perfused to collect brains for histology. Age‐matched sham and naive control male rats were also used. Coronal brain cryosections were stained with anti‐PRV, anti‐CR, and anti‐SST antibodies, and regions of interest (ROIs) from the primary somatosensory cortices were selected to determine PRV‐, CR‐, and SST‐ir cell counts and cortical ROI volumes, with blinding to experimental group. Statistical analyses were done using a linear mixed model accounting for repeated measures. We found PRV‐ir interneuronal selective reduction, sparing of the CR‐ir and SST‐ir neurons, and bilateral cortical atrophy. Our findings provide evidence for acquired PRV‐selective interneuronopathy, possibly underlying the pathogenesis of IS, neurodevelopmental deficits, and epilepsy, and potentially contributing to the partial response to vigabatrin analogs in this model.
Keywords: GABA, Parvalbumin, Interneurons, Calretinin, Somatostatin, Vigabatrin
Key Points.
The multiple‐hit rat model is a chronic model of infantile spasms due to structural lesion
In the multiple‐hit model, parvalbumin interneurons are selectively reduced in the somatosensory cortex of juvenile rats
The numbers of calretinin neurons and somatostatin interneurons are not altered in the cortex of multiple‐hit model
The interneuronal deficit may contribute to the pathogenesis of epilepsy and neurodevelopmental deficits of the multiple‐hit model
If the interneuronal deficit is confirmed also in younger ages, it might underlie the partial responsiveness to vigabatrin in this model
Infantile spasms (IS) are age‐specific seizures that occur in a severe epileptic encephalopathy of infancy, West syndrome. West syndrome typically begins during the first year of life, although later onset has been reported; it manifests with IS, multifocally epileptic, high amplitude and disorganized interictal background (hypsarrhythmia), and often dismal neurodevelopmental outcomes.1 A wide spectrum of etiologies, including structural‐metabolic, genetic, infectious/inflammatory, and metabolic, have been associated with West syndrome, yet in a third of the cases the etiology still remains unknown. IS due to structural lesions occur in ~60% of infants with IS, with a worse prognosis than those of unknown etiology, both with respect to neurodevelopmental outcomes and response to treatments.2
The available treatments include hormonal therapies such as the adrenocorticotropic hormone (ACTH) and glucocorticoid steroids, γ‐aminobutyric acid (GABA) aminotransferase inhibitors like vigabatrin, the ketogenic diet, and vitamin B6.2, 3 However, treatments are not always effective, adverse effects may limit their use, and even if effective on spasms, they may not prevent the persistence of epilepsy and neurodevelopmental disorders in the long‐term. Vigabatrin, an inhibitor of GABA aminotransferase, is one of the very few available medications, but is not always effective and can have adverse effects.2, 4 In prospective studies of the efficacy of vigabatrin in infants with IS, vigabatrin stopped spasms in 11–54% of the infants.5, 6, 7 In infants with tuberous sclerosis complex (TSC), vigabatrin is more effective in stopping spasms than in IS of non‐TSC etiology.2, 3, 8, 9, 10
Several acute or chronic models of IS or epileptic spasms have been developed to model various etiologies or pathologies or seizure phenotype reviewed in Refs 11 and 12. We have developed the multiple‐hit rat model, a nongenetic model of IS caused by a structural lesion that demonstrates a chronic phenotype.13 The multiple‐hit model is induced by right intracerebral injections of doxorubicin (DOX) and lipopolysaccharide (LPS) on postnatal day (PN)3, followed by intraperitoneal (i.p.) injections of p‐chlorophenylalanine (PCPA) on PN5 (herein called DLP rats).13 The induced lesion is at the right cortical and subcortical regions but may progress to the left periventricular regions with age.11, 13, 14, 15 Spasms with ictal electrodecremental responses on electroencephalography (EEG) appear after the infusions of DOX and LPS (PN4–13). DLP pups also manifest neurodevelopmental13, 15, 16 and sociability deficits, as well as other types of seizures including spontaneous motor seizures in adulthood.17 Spasms in the multiple‐hit model are not responsive to ACTH administration and are partially responsive to vigabatrin or its analog, CPP‐115,13, 15 which was proposed to have a lower risk for retinal toxicity than vigabatrin.18 The DLP model has been used extensively to screen for new therapies for IS, with several additional drugs emerging as promising candidates, including carisbamate,19 as well as rapamycin, an inhibitor of the mechanistic target of rapamycin.16
The poor responsiveness of DLP spasms to ACTH and vigabatrin is consistent with the lower responsiveness of IS due to structural lesion to treatment.2 The underlying pathology that could explain the partial responsiveness to vigabatrin and its analogs has not been clarified. Inhibition of GABA aminotransferase is expected to inhibit GABA metabolism, thereby increasing GABA availability. We therefore speculated that one of the reasons for the partial responsiveness to vigabatrin and its analog could be an underlying deficit or dysfunction of GABAergic interneurons.
The term interneuronopathy20 now refers to disorders associated with defective development, migration, or function of interneurons, many of which have been implicated in early life epilepsies and neurodevelopmental disorders, including West syndrome.21 Interneuronopathies can, however, be a feature of autism spectrum disorders, as well as neuropsychiatric diseases like schizophrenia, in which epilepsy is absent.21 Cortical GABAergic interneurons are inhibitory cells comprising only 10–25% of cortical neurons. However, their contribution to the neuronal circuitry and network activity of the central nervous system (CNS) is crucial.21 Parvalbumin (PRV) interneurons constitute almost half of the GABAergic cortical interneuronal population, followed by somatostatin (SST) interneurons and calretinin (CR) cells.22, 23, 24 Disrupted or dysfunctional GABAergic interneuronal migration can affect the proper balance of excitation and inhibition in the mammalian CNS, leading to neurodevelopmental disorders.25, 26, 27, 28, 29, 30, 31, 32
In this study, we utilized the multiple‐hit rat model of IS due to structural lesions, to determine whether interneuronopathy could also be a feature in a model of IS due to acquired structural etiologies. We provide proof‐of‐concept evidence for a selective reduction in the number of PRV GABAergic interneurons in the sensory (S1HL) cortex of PN20–24 DLP male rats, both contralateral (left: LCCX) and ipsilateral to the induced lesion at the right cortex (RCCX), while neocortical CR neurons or SST interneurons were spared.
Methods
Animals, multiple‐hit model induction
All of the experimental protocols used were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee, and all procedures and experiments were in accordance with the guidelines of the American Association for the Accreditation of Laboratory Animal Care, National Institutes of Health Guide for the Care and Use of Laboratory Animals and Animal Research: Reporting of In Vivo Experiments guidelines. Sprague‐Dawley rats (Taconic Farms, Germantown, NY, U.S.A.) were housed in litters of 10 male pups with their dam, on a 14 h light/10 h dark cycle. Laboratory rodent diet 5,001 (Labdiet, St. Louis, MO, U.S.A.) and water were provided ad libitum to the dam. The rats assigned to the multiple‐hit group were subjected to the induction protocol described in reports of our earlier studies to induce spasms.13, 14, 15, 16, 19, 33 Briefly, PN3 male rats were stereotactically injected with DOX (5 μg/2.5 μl saline, right intracerebroventricularly) and LPS (3 μg/1.5 μl saline, right intraparietal) under isoflurane anesthesia (Henry Schein, Melville, NY, U.S.A.).13, 14, 16, 19, 33 On PN5, rats received 200 mg/kg PCPA in saline, i.p. Controls underwent the same housing conditions but were not exposed to surgery or handling for monitoring. Sham‐operated rats received vehicle injections instead of LPS, DOX, or PCPA, and were bred and handled similarly to their DLP littermates. Video‐monitoring was performed on a daily basis between 10:00 and 12:00 and 14:00 and 16:00, from PN3 to PN19 to confirm the presence of spasms in the DLP group and their absence in the sham rats, as described previously.13, 14, 15, 16, 33 Chemicals were purchased from Sigma‐Aldrich (St. Louis, MO, U.S.A.).
Immunohistochemistry
Between PN20 and 24, rats were transcardially perfused in ice‐cold phosphate‐buffered saline (PBS), after euthanasia doses of pentobarbital, followed by 10% buffered formalin (Sigma‐Aldrich). Cryoprotection, storage, cryosectioning in a microtome (Microm 505 E Cryostat, Belair Instrument company, Inc., Springfield, NJ, U.S.A.), and immunochemical staining of free‐floating sections were done as described in established protocols34 with the following modifications. For the anti‐PRV and anti‐CR immunohistochemical assays, free‐floating sections were incubated initially in 1% H2O2 in PBS (pH = 7.4) for 30 minutes and either 10% normal horse (NHS; for mouse anti‐PRV) or goat (NGS; for anti‐CR) serum, respectively, diluted in PBS with 0.1% (ml/100 ml) Triton X‐100 and 0.1% (g/100 ml) bovine serum albumin (BSA, Sigma‐Aldrich) for 1 hour. Subsequently, incubation with either mouse anti‐PRV (1:3,000; Sigma‐Aldrich), or rabbit anti‐CR (1:3,000; Sigma‐Aldrich) antibodies, diluted in PBS with 0.1% Triton, 0.1% BSA, and 1.5% NHS (for anti‐PRV) or 3% NGS (for anti‐CR) for 2 days at 4°C on a shaker was done. Incubation in biotinylated secondary antisera (horse anti‐mouse [for anti‐PRV] or goat anti‐rabbit [for anti‐CR] immunoglobulin [IgG; 1:200, Vector Laboratories]) at room temperature was done for 90 min. Rinses with PBS were done between steps. Subsequent steps and substrate reaction were done according to manufacturer's protocol, using the Ultra‐Sensitive ABC Peroxidase Standard Staining Kit (ThermoFisher Scientific, Waltham, MA, U.S.A.) and Vector NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories, Burlingame, CA, U.S.A.). Sections were mounted on slides, dehydrated, and coverslipped. For the anti‐SST immunohistochemistry, the process was similar, except that Tris‐buffered saline (1× TBS; Tris HCl 7.88 g/L, NaCl 8.76 g/L) was used instead of PBS, and sections were incubated with the primary rabbit anti‐SST‐14 polyclonal antibody (1:5,000; Peninsula Laboratories International, Inc, San Carlos CA; Catalogue No. T‐4103) diluted in 2% NGS and 0.3% Triton in TBS for 72 hours at 4°C on a shaker.35 The numbers of rats included in our study are shown in the Figures 1, 2, and 3.
Figure 1.
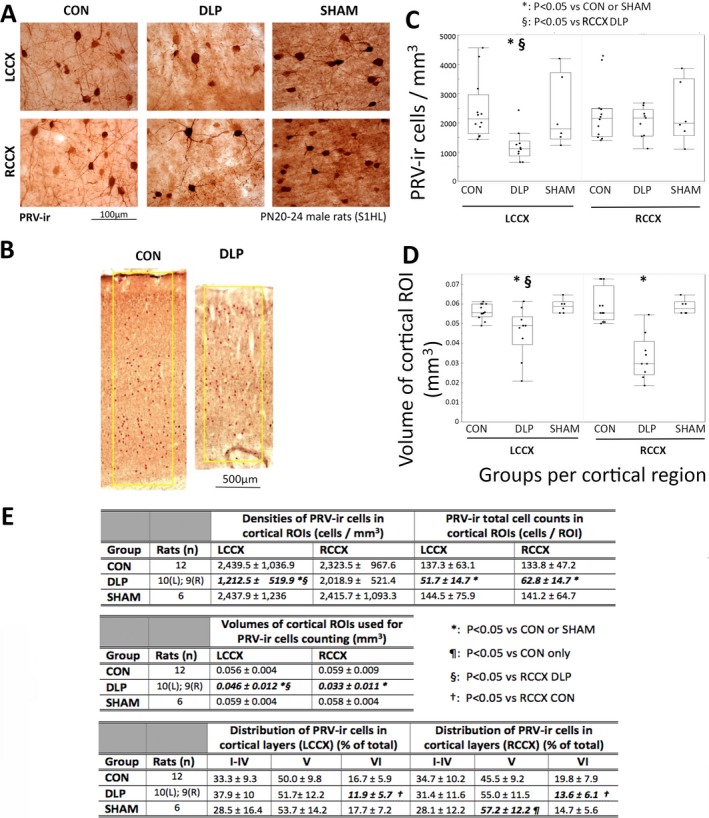
Cortical (S1HL) PRV‐ir interneurons are preferentially reduced in layer VI of PN20–24 DLP male rats. A, Examples of PRV‐ir interneurons in layer V of S1HL cortex of PN20–24 control (CON), DLP, and SHAM male rats. PRV‐ir GABAergic interneuronal densities are reduced in the cortex of PN20–24 DLP male rats compared to the other groups. B, The yellow boxes indicate the ROIs from which PRV‐ir interneurons were counted and include all cortical layers (0.7 mm width, 40 μm thick). Cortical thickness is reduced in DLP rats compared to CON. C, PRV‐ir GABAergic interneuronal densities are reduced in the LCCX of PN20–24 DLP male rats, compared to CON and SHAM (*: p < 0.05). PRV‐ir interneuronal densities are also decreased in the LCCX compared with the RCCX of PN20–24 DLP male rats (§: p < 0.05). D, DLP male pups have reduced cortical volumes bilaterally, but more prominently at the RCCX. E, Tables with the means and standard deviations (SDs) of the PRV‐ir cell densities, total cell counts, volumes of the cortical ROIs, as well as distribution of PRV‐ir neurons across the layers I–IV, V, and VI, presented as % of total PRV‐ir cell counts. There is significant reduction in PRV‐ir cell densities at the LCCX, bilateral reduction in total PRV‐ir cells/ROI, and bilateral, but right more than left, reduction in cortical volume of the selected ROIs in DLP rats compared to both CON and SHAM. PRV‐ir cells were preferentially reduced from the layer VI of the S1HL bilaterally in the DLP rats compared to CON and SHAM, but were preserved at the layers I–V. Graphs in panels C‐D show outlier box plots with interquartile differences. Significances are indicated with bold italic fonts. (*): p < 0.05 versus CON and SHAM. (¶): p < 0.05 versus CON only. (§): p < 0.05 versus RCCX of DLP rats. (†): p < 0.05 versus RCCX CON. CON: control; GABA: γ‐aminobutyric acid; LCCX, left cerebral cortex; PN, postnatal; PRV‐ir, parvalbumin‐immunoreactive; RCCX, right cerebral cortex; ROI, region of interest; S1HL, somatosensory cortex hindlimb region.
Figure 2.
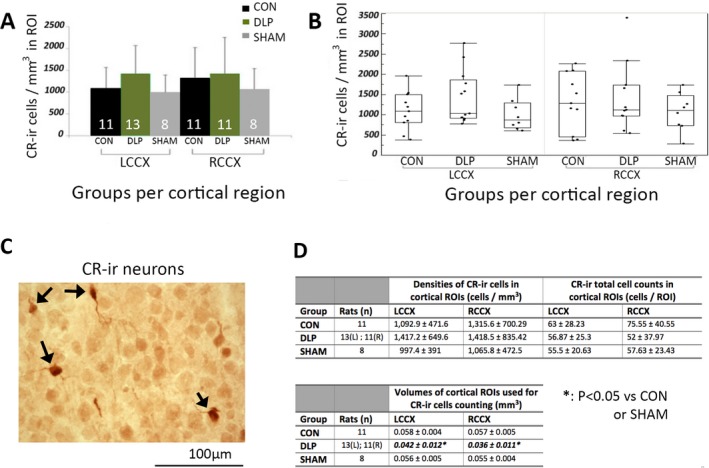
Cortical CR‐ir neuronal numbers in the S1HL ROIs are not altered in PN20–24 male DLP pups. A, There are no differences in the cell densities of CR‐ir neurons in the LCCX or RCCX of PN20–24 DLP male rats compared to CON or SHAM. Numbers of rats (n) are shown in the bars. B, Outlier box plots of cortical CR‐ir densities (cells/mm3) in the LCCX and RCCX of each group. C, Representative 400× magnified photograph of cortical CR‐ir stained neurons in a PN20 male rat (stained with red‐brown; black arrows). The bar scale indicates 100 μm. D, Tables with the means and standard deviations (SDs) of the CR‐ir cell densities, total cell counts, and volumes of the cortical ROIs. There is no difference in the cell densities or total cell counts of the CR‐ir cells in the cortical ROIs. There is significant reduction in cortical volumes of the selected ROIs in DLP rats compared to both CON and SHAM. Significances are indicated with bold italic fonts. (*): p < 0.05 versus CON or SHAM groups. CON: control; CR‐ir: calretinin‐immunoreactive; LCCX, left cerebral cortex; RCCX, right cerebral cortex; ROI, region of interest.
Figure 3.
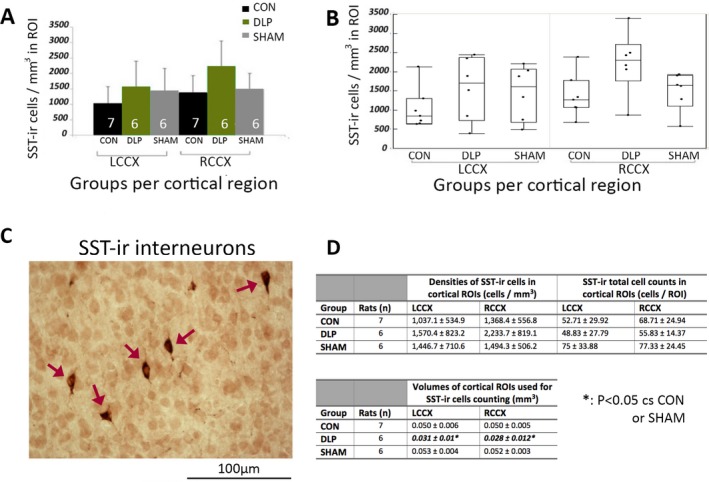
Cortical SST‐ir neuronal numbers in the S1HL ROIs are not altered in PN20–24 male DLP pups. A, There were no differences in the cell densities of SST‐ir neurons in the LCCX or RCCX of PN20–24 DLP male rats compared to controls or shams. Numbers of rats (n) are shown in the bars. B, Outlier box plots of cortical SST‐ir densities (cells/mm3) in the LCCX and RCCX of each group. C, Representative 400× magnified photograph of SST‐ir stained neurons (stained with red‐brown; red arrows) in the cortical ROI of a PN23 male rat. The bar scale indicates 100 μm. (D) Tables with the means and standard deviations (SD) of the SST‐ir cell densities, total cell counts, and ROI volumes in the cortical ROIs. There is no difference in the cell densities or total cell counts of the SST‐ir cells in the cortical ROIs. There is significant reduction in cortical volumes of the selected ROIs in DLP rats compared to both CON and SHAM. Significances are indicated with bold italic fonts. (*): p < 0.05 versus CON or SHAM groups. CON, control; LCCX, left cerebral cortex; RCCX, right cerebral cortex; ROI, region of interest; SST‐ir, somatostatin‐immunoreactive.
Cell counting
We selected one coronal section per rat brain for counting PRV‐, CR‐, or SST‐immunoreactive (‐ir) neurons, which corresponded to Atlas Figures 21–23.36 When the RCCX of DLP rats (one in the PRV group, 2 in the CR group) were severely injured and precluded staining and cell counting, the numbers of rats (and therefore sections) for RCCX and LCCX were different, as presented in the figure tables. Stained sections were imaged in a Nikon ECLIPSE E1000M microscope, captured at 40× magnification, and photographs were imported into the IMAGE J software (Wayne Rasband, NIH special volunteer), and were merged using the Adobe Photoshop application (version 8.0; Adobe) to reconstruct the whole thickness of the cortex. The total cell counts per region of interest (ROI), cell densities of (cells/mm3), and cortical ROI volumes (mm3) were determined at specific ROIs that included all cortical layers from the left and right primary somatosensory cortex hindlimb region (S1HL), from the cortical surface until the corpus callosum. The ROIs had similar width (0.7 mm) and section thickness (40 μm). Volumes of cortical ROIs were calculated by the formula: volume = cortical thickness × width of ROI × section thickness. To determine the numbers of PRV‐ir neurons across the cortical layers of the selected ROI, we defined the borders of layer V, outlining the region in the middle of the section where large size pyramidal neurons were located. Layers I‐IV encompassed the superficial regions, whereas layer VI the more deeper ROI regions, in reference to layer V. Morphology of PRV‐ir interneurons was scored by inspection of the PRV‐stained sections for presence of PRV‐ir cells with small somata (logged as Yes/No) or for short or no processes stemming out of PRV‐ir cells (logged as Yes/No). The descriptors small somata and short processes were given relative to the morphology of cortical PRV‐ir neurons from the same ROI of a PN24 male control brain. Counting of the cells and cellular morphology assessment was done by an investigator (AMK) blinded to experimental group.
Statistics
A linear mixed model considering repeated measures was used for statistics of the interneuron counts, whereas post hoc comparisons were done with a t‐test (JMP 10.0.0 version was used (SAS Institute Inc, Cary, NC, U.S.A.). Nonparametric analyses were done with chi‐square tests. All studies were done in a blinded fashion. Differences were considered statistically significant at p < 0.05.
Results
PRV‐ir interneurons are selectively reduced at the S1HL cortex of PN20–24 male DLP rats
PN20–24 DLP male rats had significantly fewer PRV‐ir interneuronal densities at the S1HL cortex compared to controls and sham‐treated rats at the LCCX (p < 0.05), whereas no differences were seen in the RCCX of DLP male rats compared to the rest of the groups (Fig. 1). The values of the PRV‐ir cell densities and numbers of rats per group are included in the table of Figure 1E. Comparisons were done with linear mixed model considering repeated measures [F(1,24.12)region = 6.6, p = 0.017); F(2,25)group = 2.7, p = 0.086); F(2,24.13)group*region = 13.4, p = 0.0001].
To determine whether the lack of differences in the PRV‐ir cell densities at ipsilateral to the lesion cortex (RCCX) could be due to a wider nonspecific cell loss secondary to the induced lesion, we compared the cortical thickness at the ROIs where cell counts were done (Fig. 1B). A significant bilateral cortical volume reduction was seen in DLP pups compared to each of the other groups (sham and control). Furthermore, RCCX volumes were lower in the DLP rats compared to their LCCX volumes [F(2,25.2)group = 18.4, p < 0.0001; F(1,24.5)region = 5.9, p = 0.02; F(2,24.5)group*region = 13.7, p < 0.0001]. Similarly, the total PRV‐ir cell counts in the cortical ROIs were significantly lower in DLP rats than in sham or controls [F(2,25)group = 9.2, p = 0.001]. Significant reductions in PRV‐ir interneuronal counts were found in both RCCX (62.8 ± 14.7 cells/ROI) and LCCX (51.7 ± 14.7 cells/ROI) of DLP rats compared to the other groups (LCCX: controls 137.3 ± 63.1, sham 144.5 ± 75.9 cells/ROI; RCCX: controls 133.8 ± 47.2, sham 141.2 ± 64.7 cells/ROI). The more prominent reduction of PRV‐ir cell densities at the LCCX versus the RCCX of DLP pups may therefore reflect the greater cortical atrophy that this group of DLP rats had at the RCCX versus the LCCX.
To determine whether the PRV‐ir cell loss was diffuse or layer specific, we studied the distribution of PRV‐ir cells across the layer V, outer layers (I–IV), and an inner layer (VI) of cortical S1HL ROIs (Fig. 1E). Fewer PRV‐ir cells were present at layer VI of DLP rats bilaterally (F(2,26.01)group = 3.96, p = 0.0315). No significant group differences were seen for the other layers [layer I‐IV: F(2,25.28)group = 1.08, p = 0.36); layer V: F(2,25.75)group = 2.02, p = 0.15)].
Morphologically, most PRV‐ir neurons in DLP rats had small somata (RCCX/LCCX: 89%/80% of sections), whereas lower percentages of small size PRV‐ir neurons were observed in controls (RCCX/LCCX: 50%/42% of sections) or shams (RCCX/LCCX: 33.3%/33.3% of sections), although this did not reach significance (chi‐square Pearson: RCCX = 5.37, p = 0.07; LCCX = 4.48, p = 0.1). PRV‐ir neurons with short or no processes were seen in 78–80% (RCCX and LCCX) of sections from DLP rats, compared with 42–50% of control sections (LCCX and RCCX respectively) and 33.3% of sections from shams (not statistically significant).
To further confirm whether the PRV‐ir cell loss is cell‐type specific, we studied the cell densities of the CR‐ir and SST‐ir interneurons.
Cortical CR‐ir and SST‐ir cell densities at S1HL are not affected in PN20–24 male DLP pups
No statistical differences were observed in the cell densities of CR‐ir neuronal populations at the cortical ROIs (S1HL) of PN20–24 male DLP rats compared to controls and the shams (F(2,27.79)group = 1.24, p = 0.3; F(2,29.03)group*region = 0.46, p = 0.64) (Fig. 2). Cortical volumes of ROIs used in CR‐ir cell counts showed reduced cortical volumes in the DLP group compared to controls or shams (F(2,27.66)group = 21.4, p < 0.0001; F(2,28.01)region = 9.94, P = 0.0038; F(2,28.03)group*region = 5.86, p = 0.0075). Despite the cortical volume differences, the total CR‐ir cell counts in the cortical ROIs were not significantly different in DLP versus control or sham rats [F(2, 27.59)group = 0.84, p = 0.44].
There were also no significant differences in SST‐ir interneuronal densities in the S1HL region of PN23 male DLP rats compared to the other groups (F(2,14.46)group = 2.32, p = 0.13; F(2,16.57)group*region = 16.57, p = 0.37). Cortical volumes were reduced bilaterally in the DLP rats compared to controls or shams (F(2,14.9)group = 19.61, p < 0.0001; F(2,16.91)region = 0.97, p = 0.3374 (Fig. 3). Despite the cortical volume differences, the total SST‐ir cell counts in the cortical ROIs were not significantly different in DLP versus control or sham rats [F(2, 14.63)group = 1.64, p = 0.23].
In summary, our results show a selective reduction in PRV‐ir cell population bilaterally in the primary somatosensory cortex of PN20–24 DLP male pups, with relative sparing of CR‐ir and SST‐ir cells, despite the bilateral cortical atrophy seen at this age.
Discussion
We report that in the multiple‐hit rat model of IS due to chemically induced structural lesion there is a cell‐type selective reduction in the number of PRV‐ir interneurons at the primary somatosensory cortex (S1HL) bilaterally. This deficit was due mainly to reduction in PRV‐ir interneurons from the deeper layers (VI) of the cortex. The PRV interneuronal deficit reported here was documented after spasms (PN4–13), other types of early life seizures (PN9 and on), learning deficits (PN16–19), and sociability deficits (PN12–20) manifested13, 15, 16 and could therefore contribute to these cognitive deficits and chronic epileptic phenotype, as proposed in other models.21 The densities and total numbers of CR‐ir and SST‐ir neurons were not different across PN20–24 DLP, control, or sham groups, supporting that the observed interneuronopathy is PRV‐specific. In our study, quantitative measures of cortical ROI volumes also demonstrated bilateral cortical atrophy in PN20–24 male DLP rats. We have previously published the evolution of injury in the DLP model, during the first 3 weeks of life, which is initially mostly right hemispheric but may progress with age to extend to periventricular left hemispheric regions (left hippocampus and/or cortex).13, 14, 15
It is yet unclear whether the PRV‐ir cell deficit precedes or follows the expression of spasms in the DLP model. PRV‐ir interneurons are first seen in the rat primary somatosensory cortex around PN8–9 and reach the adult expression patterns around PN21.37 The reduced PRV‐ir cell counts could represent migration deficit or cell loss or alternatively reduction in PRV‐ir expression. Currently, however, there is no specific marker to identify cells destined to selectively develop into PRV‐ir interneurons, prior to the time when PRV‐ir is evident, to help resolve this question. In our study, the PRV‐ir deficit was more pronounced in the deeper layers of the cortex (layer VI), suggesting that either the migration, targeting and residence in specific cortical layers, or survival of these interneurons may be preferentially disrupted in this model. Comparison of the interneuronal morphology suggested that DLP rats have smaller cortical PRV interneurons with shorter or no processes compared to controls or shams. Although our findings did not reach statistical significance, these trends may warrant more systematic quantitative morphometric analyses of the somata and arborization of interneurons, to confirm whether there is also disrupted connectivity and integration of the remaining PRV interneurons in the local circuits.
Layer VI interneurons mainly regulate neighboring neurons, but more distal connections with other cortical layers (e.g., IV) have also been reported.38 PRV‐ir interneurons specifically were reported as inhibiting local layer VI neurons39 and are therefore involved in the regulation of corticothalamic circuits.38 Dysregulation of these corticothalamic circuits may contribute to altered brain excitability and disruption of normal sleep regulation or of somatosensory and multisensory information integration processes.40 It would therefore be of interest to determine in future studies whether early treatments that may improve the cognitive and neurodevelopmental deficits and epilepsy16 might prevent the cortical PRV interneuronal loss in PN20–24 DLP rats.
Interneuronopathies are implicated in the pathogenesis of numerous neurologic conditions and epilepsies, in which deficits in the migration, differentiation, survival, or function of interneurons have been discussed as pathogenic or contributory factors (reviewed in Ref 21). Loss of interneurons has been described in acquired models of temporal lobe epilepsy,41, 42 in human patients or animal models of IS due to ARX (aristaless homeobox X‐linked) mutation.20, 43, 44, 45, 46 However, interneuronopathy deficits have not yet been described in animal models of IS due to nongenetic etiologies.
ARX mutations impair GABAergic interneuronal migration.20 The knockin mouse model Arx (GCG) 10+7 has decreased cortical calbindin (CB)‐ir interneurons, but no PRV‐ir neuronal loss, and demonstrates spontaneous seizures and spasms and cognitive and behavioral deficits.44, 47 Similar CB‐selective interneuronal deficits, with sparing of the CR‐ir and SST‐ir neurons, have been reported in 2 other models of polyalanine (PA1 or PA2) expansion.46 In the conditional knockout (cKO) Arx mouse model, deficits in all interneuronal subtypes and a shift to more ventral locations were reported; these deficits followed a cell type‐, sex‐, and region‐specific manner.45 On PN14, PRV‐ir neurons were increased in the cortex and reduced in the hippocampus in male Arx cKO, whereas in adults, nonsignificant trends for reduction of PRV‐ir interneurons for both regions were reported.45 None of these Arx models have been screened for the efficacy of vigabatrin on spasms. These deficits are distinct from those in the multiple‐hit, but also vary among the existing Arx models, suggesting that etiology and possibly phenotype may underlie these differences. It is unclear, yet possible, that this variability may contribute to the final phenotype and might determine the efficacy of treatments. For example, 17β‐estradiol given daily between PN3 and 10, but not at later developmental periods, to Arx (GCG) 10+7 mice prevents spasms and epilepsy expression and restores interneuronal deficits.47 However, in the DLP model, a similar treatment with 17β‐estradiol (PN3–10) had no effect on spasms, suggesting different etiopathogenic mechanisms.33
The interneuronal deficits in humans and animal models of IS may provide a reason for vigabatrin as a possible rational treatment, aimed at enhancing the already deficient GABA availability, but may also provide a basis for the relative refractoriness to the drug, the action of which depends on the presence of sufficient numbers of GABA‐producing cells at locations that would be important for the control of spasms. Vigabatrin and its analogs, for example, CPP–115, limit GABA metabolism by inhibiting GABA aminotransferase, a mitochondrial enzyme that degrades GABA to succinic semialdehyde. In the rat somatosensory cortex, GABA aminotransferase is expressed at low levels in the first 2 weeks of life and increases around PN16–18 and onward.48 GABA aminotransferase is expressed in both neuronal and glial cells; however, it has a predilection for GABAergic interneurons, including PRV‐ir.48, 49 These studies, along with the developmental patterns of migration and differentiation of GABAergic interneurons in the neocortex, suggest that responsiveness to GABA aminotransferase inhibitors could be lower in very young rats and increases around the third week of life. It is yet unclear how the developmental trajectory of GABA aminotransferase expression in rodents may compare to humans, to extrapolate these findings to the clinical use of vigabatrin and its analogs. Our studies further add that in the DLP model, there is a selective deficit of cortical PRV‐ir interneurons, out of proportion to the induced injury, which may further compromise the efficacy of these drugs by removing GABA‐producing cells.13, 15 Specifically, vigabatrin had only a transient and partial effect on spasms,13 whereas the vigabatrin analog CPP‐155, given between PN4 and 12 and after the onset of spasms, reduced spasms between PN5 and 7, but had a more rapid (evident within 2 hours), although still partial, effect when given on PN7.15 Although our current study was targeted on PN20–24 rats, when the interneuronal populations in the neocortex have reached adult levels, further studies tracking the trajectory of these model‐specific deficits at earlier ages would be interesting, when selective markers become available.
It is yet unclear whether the interneuronal deficit is caused by the induction method or is a consequence of the epileptic phenotype. The intracerebroventricular administration of DOX, a DNA‐intercalating drug that may halt cell replication, could potentially interfere with both the ongoing interneuronal progenitors’ replication and migration at the induction age (PN3) and contribute to the observed interneuronal deficit. The delayed appearance of PRV‐ir cells in the neocortex compared to CR‐ir neurons (present on PN0, increase between PN5 and 10 and decline to adult levels by PN15)50, 51 or SST‐ir interneurons (present at PN0 and drastically increase between PN3 and 21)52 may potentially render PRV interneuronal progenitors more vulnerable to the induction protocol. Systemic DOX administration in PN17 mice also causes brain volume reductions, as assessed by magnetic resonance imaging (MRI) studies, attributed to proinflammatory effects.53 Histopathologic findings in DLP rodent brains have shown that DOX, a cytotoxic chemotherapy drug, and LPS, a Toll‐like receptor 4 agonist, can cause prominent inflammation in the peri‐infusional cortical area.16 Whether these inflammatory changes contribute to the bilateral reduction in PRV interneurons and cortical atrophy, for example, via leakage of cytokines in the cerebroventricular space, remains to be investigated. Maternal neonatal separation has also been reported to reduce the numbers of PRV‐ir interneurons in the hippocampus54 or prefrontal cortex in a sex‐specific manner (reduced in PN25–27 female rats but not in male rats).55 However, it is unlikely that maternal separation contributed to our findings, since PRV‐ir cortical interneuronal densities were not reduced in the SHAM rats.
In summary, we showed that in the multiple‐hit rat model of IS due to structural lesions there is an acquired PRV‐preferential interneuronopathy, different from the interneuronal deficits described in genetic models, like the Arx. The distinct PRV‐ir interneuronal deficit may underlie the partial responsiveness to vigabatrin analogs in our model and the different model responses to 17β‐estradiol mediated disease modification and restoration of the Arx‐related interneuronopathy. Our findings emphasize the diversity of pathologies in various models of IS and the need to develop tools to identify them in vivo so that vulnerable populations are more likely to benefit from individualized pathology‐specific targeted treatment approaches.
Disclosure of conflicts of interest
The authors have no conflicts of interest with regard to this manuscript. SLM discloses that he is serving as Associate Editor of Neurobiology of Disease and is on the editorial boards of Brain and Development, Pediatric Neurology, and Physiological Research. He receives from Elsevier an annual compensation for his work as Associate Editor of Neurobiology of Disease and royalties from 2 books that he coedited. He received a consultant fee from UCB for participation in a Data Safety Monitoring Board. He has also received honorarium for participation in an advisory board meeting of Mallinckrodt and UCB, but there are no conflicts of interest with regard to the contents of this manuscript. ASG discloses that she is co‐Editor‐in‐Chief of Epilepsia Open and has received royalties for publications from Elsevier for book publications. She has also received honorarium for participation in an advisory board meeting of Mallinckrodt, but there is no conflict of interest with regard to the contents of this manuscript. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
Solomon L. Moshé is the Charles Frost Chair in Neurosurgery and Neurology and partially funded by grants from NIH U54 NS100064 (EpiBioS4Rx), and NS43209, US Department of Defense (W81XWH‐13‐1‐0180 and W81XWH‐18‐1‐0612), the Heffer Family and the Segal Family Foundations, and the Abbe Goldstein/Joshua Lurie and Laurie Marsh/Dan Levitz families. Aristea S. Galanopoulou acknowledges grant support from NINDS RO1 NS091170, NINDS U54 NS100064 (EpiBioS4Rx), the US Department of Defense (W81XWH‐13‐1‐0180 and W81XWH‐18‐1‐0612), the CURE Infantile Spasms Initiative, and research funding from the Heffer Family and the Segal Family Foundations and the Abbe Goldstein/Joshua Lurie and Laurie Marsh/Dan Levitz families. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Biography
Dr. Anna‐Maria Katsarou is a postdoctoral research fellow of neurology at Albert Einstein College of Medicine.

Contributor Information
Anna‐Maria Katsarou, Email: anna.katsarou@einstein.yu.edu.
Aristea S. Galanopoulou, Email: aristea.galanopoulou@einstein.yu.edu
References
- 1. Galanopoulou AS, Moshe SL. Neonatal and infantile epilepsy: acquired and genetic models. Cold Spring Harb Perspect Med 2015;6:a022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pellock JM, Hrachovy RA, Shinnar S, et al. Infantile spasms: a U.S. consensus report. Epilepsia 2010;51:2175–2189. [DOI] [PubMed] [Google Scholar]
- 3. Go CY, Mackay MT, Weiss SK, et al. Evidence‐based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2012;78:1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riikonen R. Long‐term outcome in children with infantile spasms treated with vigabatrin: a cohort of 180 patients. Epilepsia 2015;56:807–809. [DOI] [PubMed] [Google Scholar]
- 5. Knupp KG, Leister E, Coryell J, et al. Response to second treatment after initial failed treatment in a multicenter prospective infantile spasms cohort. Epilepsia 2016;57:1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riikonen R. Recent advances in the pharmacotherapy of infantile spasms. CNS Drugs 2014;28:279–290. [DOI] [PubMed] [Google Scholar]
- 7. Faulkner MA, Tolman JA. Safety and efficacy of vigabatrin for the treatment of infantile spasms. J Cent Nerv Syst Dis 2011;3:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database Syst Rev 2008;6:CD001770. [DOI] [PubMed] [Google Scholar]
- 9. Muzykewicz DA, Lyczkowski DA, Memon N, et al. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 2009;50:1118–1126. [DOI] [PubMed] [Google Scholar]
- 10. Vigevano F, Cilio MR. Vigabatrin versus ACTH as first‐line treatment for infantile spasms: a randomized, prospective study. Epilepsia 1997;38:1270–1274. [DOI] [PubMed] [Google Scholar]
- 11. Galanopoulou AS, Moshe SL. Pathogenesis and new candidate treatments for infantile spasms and early life epileptic encephalopathies: a view from preclinical studies. Neurobiol Dis 2015;79:135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galanopoulou AS, Moshé SL. Infantile spasms In Pitkänen A, Buckmaster PS, Galanopoulou AS, et al. (Eds) Models of seizures and epilepsy. Elsevier Inc, Academic Press: New York; 2017:977–993. [Google Scholar]
- 13. Scantlebury MH, Galanopoulou AS, Chudomelova L, et al. A model of symptomatic infantile spasms syndrome. Neurobiol Dis 2010;37:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jequier Gygax M, Klein BD, White HS, et al. Efficacy and tolerability of the galanin analog NAX 5055 in the multiple‐hit rat model of symptomatic infantile spasms. Epilepsy Res 2014;108:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Briggs SW, Mowrey W, Hall CB, et al. CPP‐115, a vigabatrin analogue, decreases spasms in the multiple‐hit rat model of infantile spasms. Epilepsia 2014;55:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raffo E, Coppola A, Ono T, et al. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis 2011;43:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Akman O, Briggs SW, Galanopoulou AS. Long‐term follow up of the multiple‐hit model of symptomatic infantile spasms. Epilepsy Curr 2012;13:147. [Google Scholar]
- 18. Pan Y, Gerasimov MR, Kvist T, et al. (1S, 3S)‐3‐amino‐4‐difluoromethylenyl‐1‐cyclopentanoic acid (CPP‐115), a potent gamma‐aminobutyric acid aminotransferase inactivator for the treatment of cocaine addiction. J Med Chem 2012;55:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ono T, Moshe SL, Galanopoulou AS. Carisbamate acutely suppresses spasms in a rat model of symptomatic infantile spasms. Epilepsia 2011;52:1678–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kato M, Dobyns WB. X‐linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol 2005;20:392–397. [DOI] [PubMed] [Google Scholar]
- 21. Katsarou AM, Moshé SL, Galanopoulou AS. Interneuronopathies and their role in early life epilepsies and neurodevelopmental disorders. Epilepsia Open 2017;2:284–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu Q, Cobos I, De La Cruz E, et al. Origins of cortical interneuron subtypes. J Neurosci 2004;24:2612–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wichterle H, Turnbull DH, Nery S, et al. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 2001;128:3759–3771. [DOI] [PubMed] [Google Scholar]
- 24. Butt SJ, Fuccillo M, Nery S, et al. The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron 2005;48:591–604. [DOI] [PubMed] [Google Scholar]
- 25. Tanaka DH, Oiwa R, Sasaki E, et al. Changes in cortical interneuron migration contribute to the evolution of the neocortex. Proc Natl Acad Sci USA 2011;108:8015–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levitt P. Disruption of interneuron development. Epilepsia 2005;46(Suppl. 7):22–28. [DOI] [PubMed] [Google Scholar]
- 27. Galanopoulou AS. GABA(A) receptors in normal development and seizures: friends or foes? Curr Neuropharmacol 2008;6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galanopoulou AS. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res 2008;80:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Giorgi FS, Galanopoulou AS, Moshe SL. Sex dimorphism in seizure‐controlling networks. Neurobiol Dis 2014;72(Pt B):144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ben‐Ari Y. Excitatory actions of Gaba during development: the nature of the nurture. Nat Rev Neurosci 2002;3:728–739. [DOI] [PubMed] [Google Scholar]
- 31. Kato M. Genotype‐phenotype correlation in neuronal migration disorders and cortical dysplasias. Front Neurosci 2015;9:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Danglot L, Triller A, Marty S. The development of hippocampal interneurons in rodents. Hippocampus 2006;16:1032–1060. [DOI] [PubMed] [Google Scholar]
- 33. Galanopoulou AS, Mowrey WB, Liu W, et al. Preclinical screening for treatments for infantile spasms in the multiple hit rat model of infantile spasms: an update. Neurochem Res 2017;42:1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galanopoulou AS. Sex‐ and cell‐type‐specific patterns of GABAA receptor and estradiol‐mediated signaling in the immature rat substantia nigra. Eur J Neurosci 2006;23:2423–2430. [DOI] [PubMed] [Google Scholar]
- 35. Besser S, Sicker M, Marx G, et al. A transgenic mouse line expressing the red fluorescent protein tdTomato in GABAergic neurons. PLoS ONE 2015;10:e0129934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paxinos G, Watson C. The rat brain, in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- 37. Alcantara S, Ferrer I, Soriano E. Postnatal development of parvalbumin and calbindin D28K immunoreactivities in the cerebral cortex of the rat. Anat Embryol (Berl) 1993;188:63–73. [DOI] [PubMed] [Google Scholar]
- 38. Briggs F. Organizing principles of cortical layer 6. Front Neural Circuits 2010;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arzt M, Sakmann B, Meyer HS. Anatomical correlates of local, translaminar, and transcolumnar inhibition by layer 6 GABAergic interneurons in somatosensory cortex. Cereb Cortex 2018;28:2763–2774. [DOI] [PubMed] [Google Scholar]
- 40. Henschke JU, Noesselt T, Scheich H, et al. Possible anatomical pathways for short‐latency multisensory integration processes in primary sensory cortices. Brain Struct Funct 2015;220:955–977. [DOI] [PubMed] [Google Scholar]
- 41. Dudek FE, Shao LR. Loss of GABAergic interneurons in seizure‐induced epileptogenesis. Epilepsy Curr 2003;3:159–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci 2006;26:4613–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marsh E, Fulp C, Gomez E, et al. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 2009;132:1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Price MG, Yoo JW, Burgess DL, et al. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10 + 7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci 2009;29:8752–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marsh ED, Nasrallah MP, Walsh C, et al. Developmental interneuron subtype deficits after targeted loss of Arx. BMC Neurosci 2016;17:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee K, Ireland K, Bleeze M, et al. ARX polyalanine expansion mutations lead to migration impediment in the rostral cortex coupled with a developmental deficit of calbindin‐positive cortical GABAergic interneurons. Neuroscience 2017;357:220–231. [DOI] [PubMed] [Google Scholar]
- 47. Olivetti PR, Maheshwari A, Noebels JL. Neonatal estradiol stimulation prevents epilepsy in Arx model of X‐linked infantile spasms syndrome. Sci Transl Med 2014;6:220ra212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kristt DA, Waldman JV. Late postnatal changes in rat somatosensory cortex. Temporal and spatial relationships of GABA‐T and AChE histochemical reactivity. Anat Embryol (Berl) 1986;174:115–122. [DOI] [PubMed] [Google Scholar]
- 49. Yung KK, Kwok KH, Gao ZG, et al. Expression of GABA transaminase immunoreactivity in interneurons of the rat neostriatum. Neurochem Int 1998;33:567–572. [DOI] [PubMed] [Google Scholar]
- 50. Schierle GS, Gander JC, D'Orlando C, et al. Calretinin‐immunoreactivity during postnatal development of the rat isocortex: a qualitative and quantitative study. Cereb Cortex 1997;7:130–142. [DOI] [PubMed] [Google Scholar]
- 51. Fonseca M, del Rio JA, Martinez A, et al. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol 1995;361:177–192. [DOI] [PubMed] [Google Scholar]
- 52. Bendotti C, Hohmann C, Forloni G, et al. Developmental expression of somatostatin in mouse brain. II. In situ hybridization. Brain Res Dev Brain Res 1990;53:26–39. [DOI] [PubMed] [Google Scholar]
- 53. Spencer Noakes TL, Przybycien TS, Forwell A, et al. Brain development and heart function after systemic single‐agent chemotherapy in a mouse model of childhood leukemia treatment. Clin Cancer Res (in press 2018). 10.1158/1078-0432. [DOI] [PubMed] [Google Scholar]
- 54. Katahira T, Miyazaki N, Motoyama J. Immediate effects of maternal separation on the development of interneurons derived from medial ganglionic eminence in the neonatal mouse hippocampus. Dev Growth Differ 2018;60:278–290. [DOI] [PubMed] [Google Scholar]
- 55. Holland FH, Ganguly P, Potter DN, et al. Early life stress disrupts social behavior and prefrontal cortex parvalbumin interneurons at an earlier time‐point in females than in males. Neurosci Lett 2014;566:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]