

Summary
The mainstay of therapy for epilepsy is anti‐seizure drugs (ASDs, also referred to as anticonvulsants and anti‐epileptic medications). Through much of the past century, only a handful for ASDs were available for clinical use. However, with the creation of the U.S. National Institutes of Health/National Institute of Neurological Disorders and Stroke (NINDS)–sponsored Anticonvulsant Screening Program (ASP), coupled with the emergence of high‐throughput screening platforms and methodologies, and advances in our understanding of the fundamental neurobiology of epilepsy, ASD development has greatly accelerated over the past 25 years. More than 18 new ASDs have been approved for clinical use since the inception of the ASP. Despite this remarkable success and the emergence of drugs possessing more favorable pharmacokinetic profiles that act on novel molecular targets, there has been increasing recognition that the paradigms for drug discovery have not yielded significant improvements in therapeutic efficacy, and that disease modification (i.e., anti‐epileptogenesis), among other challenges, must be addressed. Thus, with the renewed framework and mission of improving the lives of people with epilepsy, the name of the ASP was changed to the Epilepsy Therapy Screening Program (ETSP). This review briefly summarizes the history of ASD development and outlines some of the challenges and opportunities for the next generation of drug therapies for the epilepsy field.
Keywords: Epilepsy, Anti‐convulsant, Anti‐epileptic drug, Anti‐seizure drug, Medication, History, Drug development
Key Points.
Until recently, the history of anti‐seizure drugs (ASDs) has been one of serendipitous discovery, and a ceiling effect with respect to therapeutic efficacy
The creation of the National Institute of Neurological Disorders and Stroke (NINDS)–sponsored Anticonvulsant Screening Program (ASP) spurred the development of most clinically used ASDs today
The reframing of the ASP into the current Epilepsy Therapy Screening Program (ETSP) marks a significant turning point in the strategies and methodologies used to identify novel medications
Precision‐medicine approaches for epilepsy may afford age‐, syndrome‐, and etiology‐specific treatments for patients, to improve seizure control, to promote disease modification, and to mitigate attendant comorbidities
The identification and validation of disease‐specific biomarkers will advance our ability to conduct highly meaningful disease prevention and disease‐modifying clinical studies in the patient population at risk for developing epilepsy
Epilepsy is one of the most common neurologic disorders and occurs throughout the age‐span from a multiplicity of causes and is found in approximately 1% of the general population.1 Anti‐seizure drugs (ASDs) represent the mainstay of treatment for epilepsy, but at least one‐third of affected individuals continue to experience spontaneous recurrent seizures despite often aggressive medical trials. This large medically intractable group does not only have unremitting seizure activity but is also at risk for negative health consequences—such as cognitive impairment and comorbid mental health problems—and has a heightened risk for sudden unexpected death.1 Furthermore, patients receiving chronic ASD therapy often experience concomitant transient or lasting side‐effects, and in rarer instances idiosyncratic reactions that can be life‐threatening such as the ASD hypersensitivity syndrome. Notwithstanding these limitations and given the fundamental importance of ASDs, it is important to appreciate the history and evolution of this class of medications, and to leverage the tremendous advances in basic‐translational neuroscience over the past quarter century to develop more efficacious and better tolerated drugs.
Early History of Epilepsy Treatment
Prior to the 19th century, treatment for people with epilepsy was based largely on spiritual and supernatural beliefs; that is, epilepsy had been referred to at times as the “Sacred Disease.”2 Unfortunately, this societal view was presented from a wholly negative perspective, not one favoring a special place for afflicted individuals but long‐standing persecution and discrimination, the latter which continues to a considerable extent even today, particularly in developing countries throughout the world. The very early recognition by Hippocrates, the Greek physician and philosopher (circa 460–370 B.C.), that epilepsy was a disorder of the brain was dismissed for centuries.3 It was not until the late 1700s to mid‐1800s that the Hippocratic view of epilepsy began to take root. Even at that time, however, treatment consisted primarily of botanical/herbal remedies and various chemical and organic concoctions that were at best empiric and wholly lacking with respect to scientific scrutiny.3
In this climate of free‐for‐all interventions and claims emerged a critical serendipitous observation in 1857 by Sir Charles Locock, an obstetrician to Queen Victoria. In the mid‐1800s, it was recognized that various inorganic bromide salts resulted in sedative effects, and in the case of potassium bromide, caused impotence. Locock surmised that catamenial epilepsy (referred to at that time as “hysterical” epilepsy) might be amenable to potassium bromide treatment and found that this treatment effectively arrested epileptic seizures in 14 of 15 women.4 However, the larger adoption of bromides would not likely have been possible without independent evidence provided by Samuel Wilks (1824–1911), a prominent British physician, who was contemporaneous with Locock.4 Nevertheless, potassium bromide represented the first drug therapy for epilepsy (and is still used largely in dogs with epilepsy and very rarely in humans). This medication, unfortunately, possesses a small therapeutic index, and can induce a variety of severe skin reactions and central nervous system effects, including lethargy, cachexia, delirium/psychosis, paresis, and even exacerbation of seizure activity. Hence, its use has been extremely rare in clinical practice for decades.
Potassium bromide was the de facto treatment for epilepsy, but there was not a better drug until phenobarbital became available in 1912. Of interest, the discovery of phenobarbital as an effective ASD came serendipitously as well.5 At the dawn of the 20th century, German chemists working at Bayer noted that a lipophilic barbituric acid derivative—that is, barbital or 5,5‐diethylbarbituric acid—produced sedative and hypnotic effects in dogs. This group then synthesized a related compound, phenobarbital (5‐ethyl‐5‐phenylbarbituric acid) in 1911, and Bayer marketed this under the tradename Luminal for the treatment of human insomnia.5 It was not until Alfred Hauptmann used phenobarbital for his patients with epilepsy that this medication was recognized as a better alternative to potassium bromide, in terms of both efficacy and tolerability. To this day, phenobarbital remains one of the main treatments of patients, especially infants, with epilepsy, and young and old patients alike in developing countries.
The examples of potassium bromide and phenobarbital represent pure serendipity on the part of clinicians who made an intuitive link between sedative properties of drugs and the need to suppress epileptic seizures that were viewed as abnormally exaggerated behaviors.6 There was no systematic or scientific approach for drug discovery at the time, and no validated animal models of seizures/epilepsy that could be utilized to test the antiseizure efficacy of investigational compounds. The extreme dearth of ASDs necessitated the development of animal models to capitalize on the growing numbers of chemical agents being produced by the pharmaceutical industry.
Dawn of the Pre‐Modern Era
In the late 1800s, investigators began to elicit seizure activity in animals (i.e., dogs) using electrical stimulation or chemoconvulsants derived from naturally occurring chemicals (such as pentylenetetrazol, or PTZ, later shown to be a γ‐aminobutyric acid A [GABAA] receptor antagonist).7 Despite these encouraging developments, there was no commonly accepted algorithm for drug screening, and it was not feasible to screen for many hundreds (if not thousands) of compounds given the cost and labor‐intensive nature of seizure induction in these early models. Furthermore, the experimental protocols employed did not lead to consistent results.
However, this changed in the early 1930s when Merritt and Putnam adapted a simple and reliable electroshock seizure model and readout (i.e., the electroshock threshold test) in cats.8 Using this new tool, they were able to screen hundreds of compounds, and once again, in a somewhat serendipitous manner, discovered phenytoin (owned by Parke‐Davis), which appeared more effective and less sedating than either potassium bromide or phenobarbital.9 The clinical efficacy of phenytoin was shown by Merritt and Putnam in 1938, and this drug remained the primary treatment for various forms of epilepsy for decades to come. A phosphate‐ester prodrug of phenytoin (i.e., fosphenytoin), developed and initially marketed in the 1990s, remains today as one of the first‐line treatments for status epilepticus and seizure exacerbation.
The seminal discovery of phenytoin using a phenotypic readout in a highly efficient model validated the maximal electroshock seizure (MES) model and added credence to the systematic approach for ASD drug screening that was to become the mainstay of epilepsy experimental therapeutics for decades to come. The discovery of phenytoin was later followed by the identification of trimethadione in 1944 by Everett and Richards10 using another model, the PTZ seizure model in mice. Uniquely, trimethadione was able to block absence seizures in humans, unlike phenytoin, which was ineffective in the PTZ model. Trimethadione and the PTZ assay paved the way for the later discovery of succinimides (e.g., methsuximide and ethosuximide). The combination of the electroshock model used by Merritt and Putnam8, 9 and the PTZ model by Everett and Richards,10 laid the foundation for later efforts (and adaptations of these early animal models) for more expanded drug‐screening approaches, notably the U.S. National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS)–sponsored Anticonvulsant Screening Program (ASP) in the 1970s.11
Anticonvulsant Screening Program
The genesis of the NINDS ASP in 1975 was the culmination of the passionate efforts of several key individuals, who realized the need to develop new drugs for epilepsy using simple and effective screening approaches that would predict efficacy in humans.11 Ewart Swinyard, Dixon Woodbury, and colleagues at the University of Utah systematically utilized comparative assays in rodents to demonstrate that the MES and PTZ tests were useful for identifying various derivatives of the hydantoins, diones, barbiturates, and succinimides. However, only ethosuximide emerged from these efforts as a broadly utilized medication upon clinical approval in 1960.
Between the early 1960s and the mid‐1970s, only 2 major drugs for epilepsy were developed—valproate and carbamazepine—but these were developed initially in Europe and later brought to the United States. And although benzodiazepines were first discovered at Hoffman LaRoche by Leo Sternbach in New Jersey (U.S.A.), the first clinically available benzodiazepine, chlordiazepoxide (Librium), was launched in the United Kingdom in 1960, and this was followed by diazepam (Valium) in 1963.12
Despite the increased availability of newer ASDs, it was clear to many, including those at NINDS, that the drug discovery process was painstakingly slow and that there was a great need and opportunity to enhance throughput and uncover novel, more effective medications. The key individuals who helped create the NINDS ASP were Richard Masland, director of the NINDS at the NIH in the 1960s, J. Kiffin Penry who catalyzed efforts at the intramural epilepsy program, Harvey Kupferberg who ran the NINDS analytical laboratory, and NINDS Epilepsy Advisory Committee members Ewart Swinyard and Dixon Woodbury.11
The unique partnership between the government (i.e., NINDS), academia, and the pharmaceutical industry was established at the University of Utah (the awardee of the NINDS extramural request for proposal, or RFP), and remains to this day. Later principals of the ASP included Harold Wolf and H. Steve White, who directed the steady growth of the program, particularly with respect to the broader adoption of animal models and importantly, inclusion of models that represent pharmacoresistant epilepsy and that recapitulate key aspects of epileptogenesis.11, 13 To date, this preeminent drug‐development program has screened over 32,000 novel compounds, and most of these agents have come from the pharmaceutical industry. And of the 18 latest ASDs approved for clinical use in the United States, the ASP has been critical to the development of half of these, including ground‐breaking medications such as felbamate, topiramate, rufinamide, lacosamide, and retigabine (Fig. 1).11
Figure 1.
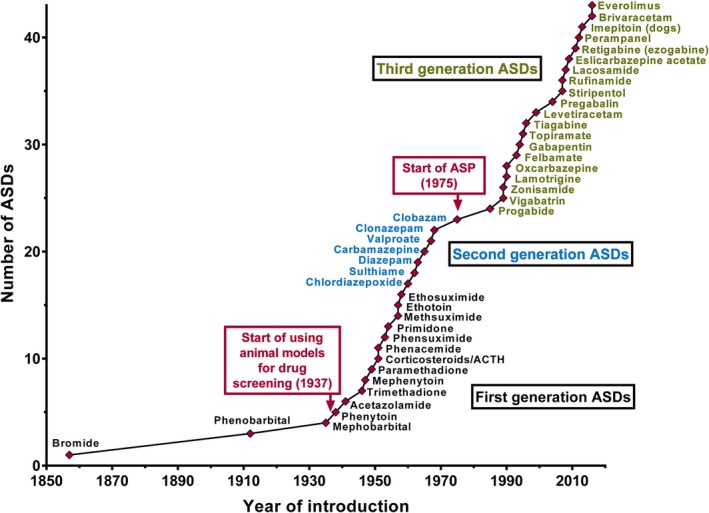
Introduction of ASDs to the worldwide market from 1853 to 2016. Although licensing has varied from country to country, here the year of first licensing or the first mention of clinical use in a country of Europe, the United States, or Japan is depicted. The earliest systematic drug screening in an animal model (by Merritt and Putnam in the 1930s) as well as the start the NINDS Anticonvulsant Screening Project (ASP) in 1975 are also noted. Reprinted intact and with permission from Löscher, Neurochem Res 2017.
The Post‐Modern Era
Despite the remarkable growth of new ASDs over the past 25 years, largely due to the NINDS ASP, there was a growing sense that the decades‐old paradigm for drug discovery had not led to substantive improvements in therapeutic efficacy, particularly for the medically intractable population with unremitting seizure activity.13 And this has remained true despite the recognition that many newer ASDs are believed to exert their clinical activity by interacting with highly novel molecular targets.14 This sobering reality is tempered, however, by the fact that the newer‐generation ASDs are often better tolerated and possess more favorable pharmacokinetic profiles than the older medications. Moreover, assuming that the results from well‐designed randomized double‐blinded clinical trials translate to the general epilepsy population, it is clear that the newer ASDs have not only lessened the seizure burden in patients with uncontrolled epilepsy but have also served patients whose seizures are considered well controlled. Ultimately, the goal is to uncover therapies that provide complete seizure freedom without producing adverse effects and drug–drug interactions. Nevertheless, in the absence of this “idealized therapy,” we should not underestimate the importance that even incremental advances in therapy can provide for patients and their caregivers.
Moving beyond the symptomatic treatment of epilepsy, it is important to note that both clinical and basic translational research has revealed new knowledge about the processes of seizure genesis and epileptogenesis.15, 16 With this increased knowledge, greater attention has been directed toward the question of whether ASDs can be designed to modify the disease processes and prevent, mitigate, or cure any form of epilepsy. Thus far, however, it appears that none of the clinically available ASDs can affect the epileptogenic process in humans.15, 16
Within this shifting backdrop, the ASP underwent a critical assessment from an external review panel composed of leading scientists from academic medical centers and the pharmaceutical industry. In 2015, the ASP became the Epilepsy Therapy Screening Program (ETSP), a name that better reflects the broad nature of emerging and recently validated treatment approaches for epilepsy, and not one that is solely restricted to ASDs uncovered using both in vitro and in vivo models that have historically reflected normal, not epileptic, brain.17
With respect to ASD development in the new ETSP, there is a multistep process beginning with an identification phase utilizing the MES test and the 6‐Hz test. Other screening tests are employed to broaden the net, including a kindling model (Fig. 2).17 Along the way, preclinical toxicity is assessed using a variety of rodent behavioral assays. The next step is the differentiation phase that employs models that mirror certain aspects of human temporal lobe epilepsy, pharmacoresistant epilepsy, epilepsy arising from induced status epilepticus, primary genetic forms of epilepsy, and even neuroinflammation‐related epilepsy (secondary to virus‐induced encephalitis).17 In addition, the potential of novel compounds to influence epileptogenesis is studied in several chronic models of epilepsy.
Figure 2.
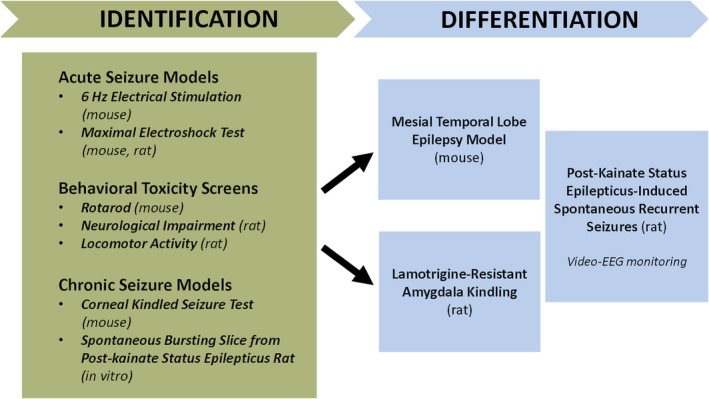
Workflow for identification and differentiation of agents screened to address pharmacoresistance in the Epilepsy Therapy Screening Program (ETSP). Reprinted intact with permission from Kehne et al., Neurochem Res 2017.
Of course, given the growing reality that epilepsy is not a singular disease, but more accurately reflects heterogeneous “epilepsies,” there is no ideal animal model that mirrors all the key features of even one human epileptic condition.16, 18 The core strength of the current staged and multipronged approach toward ASD development lies in the use of multiple animal models, including the kindling model, and the inherent flexibility to capture promising drugs without “missing” potentially impactful agents,17 such as what nearly happened with levetiracetam when it was evaluated by the ASP in the 1990s.19 Levetiracetam was found to be inactive in both the MES and subcutaneous PTZ tests, the work‐horse screening models of the ASP at the time. Hence, the agent failed the initial screen; however, levetiracetam was highly effective in blocking the fully expressed kindled seizure of the kindled rodent and subsequently emerged as one of the most widely prescribed ASDs in modern history.19
The Future of ASD Discovery
Clearly, the last generation has witnessed the beneficial fruits of the steady and visionary work of many individuals and the implementation of unique programs/platforms in epilepsy experimental therapeutics. The current ETSP is only one partner in the evolving and complex landscape of ASD discovery.17 Creative funding avenues and collaborative research initiatives, coupled with rapidly growing knowledge about epilepsy, are coming together to both challenge and direct future efforts. These advances include among others increased knowledge of the genetic underpinnings of various forms of epilepsy, appreciation for the biologic complexities inherent in brain network function, the environmental influences affecting epigenetic regulation of epilepsy and epileptogenesis, and a greater understanding of the features unique to special populations of patients with epilepsy (i.e., infants and children, the elderly, and gender differences). In the current era of Precision Medicine, it is the hope that patients will benefit from age‐, syndrome‐, and etiology‐specific treatments, not only to mitigate or eliminate spontaneous recurrent seizures, but also to address the attendant comorbidities mentioned earlier.20
Other model systems are also being employed, such as epileptic zebrafish, since these are very amenable to genetic manipulation and higher‐throughput screening.21 And the traditional focus on cellular membrane–bound ion channels and transporters, or synaptic neurotransmitter systems, is being supplemented by a brain metabolic framework as biochemical pathways are being increasingly recognized as potent modulators of the epileptic state.22 For example, one recent approach has been the clinical validation of 2‐deoxyglucose, which induces glycolytic restriction.23
With a greater appreciation for the factors that underlie the development of epilepsy and the attenuation of seizure activity comes the opportunity for rethinking our approach to clinical evaluation of an investigational therapy; that is, with precision medicine, we will be able to enrich the trial population in such a way that new drugs will be evaluated in smaller patient cohorts that are perhaps more representative of target patient populations.20 It is notable that our ability to advance a truly novel therapy for the prevention of epilepsy in susceptible patients will clearly benefit from the many ongoing efforts to develop biomarkers that track with disease onset and progression. The ability to conduct targeted trials with a validated biomarker will allow for the evaluation of more mechanistically directed therapies as they emerge from preclinical studies and will likely reshape our approach to clinical prevention and disease modification trials.
In the end, for the epilepsy field to truly enhance the lives of people with epilepsy and to optimize efficacy and maximize tolerability, a dynamic, expanded and integrated approach toward drug screening—one that keeps pace with the ever‐accelerating research discoveries—is necessary. All partners and stakeholders in this effort are critically important, whether they are from government agencies and policymakers, from private industry, academic medical centers and healthcare professionals, community and private organizations, and importantly, the patients and their families. For novel ASD discovery, the workflows must incorporate the innovation represented by these and more. For transformative knowledge transfer, the challenge will be to organize, store, retrieve, and analyze the bewildering amount and array of data that will come from these and other sectors of society, for the benefit of discovering more effective and safer medications for patients with epilepsy.
Summary
For much of recorded history, effective medical treatments for epilepsy have been sparse. Within the last century, but especially over the past 50 years, there has been a virtual explosion in the development of ASDs. Although newer ASDs have yet to demonstrate increased efficacy over traditional medications, the pharmacokinetic and safety profiles of drugs approved for use in the past 25 years have improved measurably. Future ASDs, whether they represent novel modifications aimed at traditional molecular targets or whether their origin is from a radically different understanding of the neurobiology of epilepsy, will require expanded collaborative and integrated efforts of many stakeholders—even beyond what was originally conceptualized in the creation of the NINDS ASP. Although the field of epilepsy therapeutics has yet to achieve the vision espoused by the recently reformulated ETSP, the future nevertheless remains promising for people with epilepsy.
Conflict of Interest
Dr. Rho has served as a paid consultant for Accera Pharma, Xenon Pharmaceuticals, Danone Nutricia, and Ajinomoto U.S.A. Dr. Rho has received royalties from UpToDate for a chapter about the mechanisms of seizures and epilepsy. In the last 24 months, Dr. White has served as a paid consultant to Xenon Pharmaceuticals, Bial, Greenwich Biosciences, and Biogen. He has also served as a Research Advisor to Citizens United for Research in Epilepsy (CURE) and has received research funding from a collaborative subaward from the University of Utah Prime Contract with the ETSP and the Department of Defense. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
Dr. Rho's research has been supported by the Canadian Institutes of Health Research. Dr. White's research has been supported by grants and contracts from the National Institute of Neurological Disorders and Stroke and the Department of Defense.
Biography
Dr. Jong M. Rho is professor of pediatrics, clinical neurosciences, physiology & pharmacology at the Cumming School of Medicine, University of Calgary.

References
- 1. Devinsky O, Vezzani A, O'Brien TJ, et al. Epilepsy. Nat Rev Dis Primers 2018;4:18024. [DOI] [PubMed] [Google Scholar]
- 2. Eadie MJ, Bladin PF. A disease once sacred: a history of the medical understanding of epilepsy. Montrouge, France: John Libbey Eurotext; 2001. [Google Scholar]
- 3. Temkin O. The falling sickness: a history of epilepsy from the Greeks to the beginnings of modern neurology. 2nd Ed Baltimore, MD: The Johns Hopkins University Press; 1994. [Google Scholar]
- 4. Eadie MJ. Sir Charles Locock and potassium bromide. J R Coll Physicians Edinb 2012;42:274–279. [DOI] [PubMed] [Google Scholar]
- 5. Yasiry Z, Shorvon SD. How phenobarbital revolutionized epilepsy therapy: the story of phenobarbital therapy in epilepsy in the last 100 years. Epilepsia 2012;53(Suppl 8):26–39. [DOI] [PubMed] [Google Scholar]
- 6. Porter RJ, Rogawski MA. New antiepileptic drugs: from serendipity to rational discovery. Epilepsia 1992;33(Suppl 1):S1–S6. [DOI] [PubMed] [Google Scholar]
- 7. Löscher W. Animal models of seizures and epilepsy: past, present and future role for the discovery of antiseizure drugs. Neurochem Res 2017;42:1873–1888. [DOI] [PubMed] [Google Scholar]
- 8. Putnam TJ, Merritt HH. Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 1937;85:525–526. [DOI] [PubMed] [Google Scholar]
- 9. Merritt HH, Putnam TJ. Sodium diphenyl hydantoinate in the treatment of convulsive disorders. JAMA 1984;251:1062–1067. [DOI] [PubMed] [Google Scholar]
- 10. Richards RK, Everett GM. Tridione: a new anticonvulsant drug. J Lab Clin Med 1946;31:1330–1336. [PubMed] [Google Scholar]
- 11. Porter RJ, Kupferberg HJ. The anticonvulsant screening program of the national institute of neurological disorders and stroke, NIH: history and contributions to clinical care in the twentieth century and beyond. Neurochem Res 2017;42:1889–1893. [DOI] [PubMed] [Google Scholar]
- 12. Wick JY. The history of benzodiazepines. Consult Pharm 2013;28:538–548. [DOI] [PubMed] [Google Scholar]
- 13. French JA, White HS, Klitgaard H, et al. Development of new treatment approaches for epilepsy: unmet needs and opportunities. Epilepsia 2013;54(Suppl 4):3–12. [DOI] [PubMed] [Google Scholar]
- 14. Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med 2016;6:a022780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pitkänen A, Lukasiuk K, Dudek FE, et al. Epileptogenesis. Cold Spring Harb Perspect Med 2015;5:a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barker‐Haliski ML, Friedman D, French JA, et al. Disease modification in epilepsy: from animal models to clinical applications. Drugs 2015;75:749–767. [DOI] [PubMed] [Google Scholar]
- 17. Kehne JH, Klein BD, Raeissi S, et al. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res 2017;42:1894–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bialer M, White HS. Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov 2010;9:68–82. [DOI] [PubMed] [Google Scholar]
- 19. Klitgaard H, Verdru P. Levetiracetam: the first SV2A ligand for the treatment of epilepsy. Expert Opin Drug Discov 2007;2:1537–1545. [DOI] [PubMed] [Google Scholar]
- 20. EpiPM Consortium . A roadmap for precision medicine in the epilepsies. Lancet Neurol 2015;14:1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Griffin A, Krasniak C, Baraban SC. Advancing epilepsy treatment through personalized genetic zebrafish models. Prog Brain Res 2016;226:195–207. [DOI] [PubMed] [Google Scholar]
- 22. Kobow K, Auvin S, Jensen F, et al. Finding a better drug for epilepsy: antiepileptogenesis targets. Epilepsia 2012;53:1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bialer M, Johannessen SI, Levy RH, et al. Progress report on new antiepileptic drugs: a summary of the Thirteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIII). Epilepsia 2017;58:181–221. [DOI] [PubMed] [Google Scholar]