

Summary
Rational prescribing should be based on the assessment of high‐quality evidence about the benefits and risks of available treatment options. Because clinical trials, particularly randomized controlled trials (RCTs), provide the best source of evidence, their design and results need to be carefully scrutinized. The majority of RCTs of antiepileptic drugs (AEDs) have been designed to address regulatory requirements, and generally they involve restrictive eligibility criteria, rigid dosing schemes, short duration of follow‐up, and comparison with placebo rather than standard treatments. Although these studies have high internal validity, they are conducted in a setting that is distant from routine clinical practice and therefore their usefulness in guiding treatment decisions is limited. Information more directly applicable to clinical practice can be derived from a relatively small number of comparative effectiveness monotherapy RCTs, although the design of some of these studies was probably biased in favor of the sponsor's product. Alarmingly, there is a paucity of well‐designed trials in epilepsy syndromes other than focal epilepsies, and no RCTs at all in most of the less common epileptic syndromes of infancy and childhood. In the light of these shortcomings, there is scope for re‐assessing regulatory requirements to facilitate generation of data more directly applicable to the routine clinical setting. Likewise, research‐funding organizations should be sensitized about the lack of adequate evidence to guide therapeutic practice in epilepsy, and the need to promote high‐quality comparative effectiveness trials. Future prospective pragmatic trials may benefit from the increasingly widespread availability of electronic health records.
Keywords: Clinical trials, Seizures, Epilepsy, Antiepileptic drugs, Comparative effectiveness, Drug therapy
Key Points.
Clinical trials, particularly randomized controlled trials (RCTs), provide the best source of evidence for clinical decision‐making
Most RCTs of antiepileptic drugs (AEDs) have been designed to fulfil regulatory requirements, and their usefulness in guiding clinical practice is limited
Information more readily applicable to routine practice can be derived from a few comparative effectiveness monotherapy trials, including pragmatic trials
There a paucity of RCTs in generalized epilepsies, and no RCTs in most of the less common epileptic syndromes of infancy and childhood
Funding organizations should be sensitized to the need to promote high‐quality comparative effectiveness RCTs, particularly in areas where evidence is lacking
Physicians managing people with epilepsy need to make treatment decisions based on the best available evidence. Although the value of observational studies and personal clinical experience cannot be discounted, unquestionably the highest quality of evidence comes from well‐designed clinical trials and, particularly, randomized controlled trials (RCTs).1 Although a large number of clinical trials of antiepileptic drugs (AEDs) have been completed over the last several decades, their usefulness in guiding treatment decisions is less than desirable. In fact, a 2013 systematic review conducted by an International League Against Epilepsy (ILAE) Subcommission concluded that ”many RCTs and especially those involving new AEDs are methodologically flawed and cannot answer important clinical questions” and “there continues to be an alarming lack of well designed, properly conducted epilepsy RCTs for patients with generalized seizures/epilepsies and in children in general.”2 Clearly, there is a need for improvement in the methodology and scope of future epilepsy trials.
Many parties have a stake in how clinical trials are designed and conducted. These include people with epilepsy and their families above all, but also physicians and other healthcare providers, industry involved in the development of pharmaceuticals and devices, regulatory agencies, professional and scientific societies, and organizations involved in funding clinical care and clinical research. These stakeholders may differ in their goals, obligations, and perspectives, a fact that needs to be taken into consideration to understand the implications of study designs and associated limitations.
The present article appraises critical issues that affect the design of AED trials and their relevance to the practical management of people with epilepsy and discusses ways by which trials could be improved in the future. It is hoped that the article will be of interest not only to physicians involved in the care of people with epilepsy, but also to individuals engaged in lay epilepsy organizations, and to scientists involved in drug development and regulation.
Regulatory Trials
During the first phases of drug development, the primary purpose for conducting clinical trials is to obtain a marketing license, and therefore such trials are designed primarily to fulfill regulatory requirements.3 Further regulatory trials may be conducted in the postmarketing phase, most notably to extend indications for additional seizure types, different populations (for example, pediatric age groups), or other treatment modalities (for example, monotherapy use). Typically, regulatory studies comply with high‐quality scientific standards and sound methodologic procedures as codified by Good Clinical Practice (GCP) regulations.4, 5 As outlined below, however, these trials are conducted under conditions that deviate markedly from those applicable to routine clinical practice (Table 1). This could restrict severely the generalizability of the data, and may limit considerably the possibility of using the results as a guide to rational prescribing in routine clinical settings.
Table 1.
Advantages and limitations of regulatory trials, conventional pragmatic comparative effectiveness trials, and comparative effectiveness trials using electronic health record (EHR) data
| Regulatory trials | Nonregulatory comparative effectiveness trials | Comparative effectiveness trials using EHR data |
|---|---|---|
|
Advantages
High internal validity (very high scientific standards) Comparison with placebo typically included (for adjunctive‐therapy trials) Rigorous assessment of dose‐response relationships |
Advantages
High external validity (procedures mimic routine clinical care) High internal validity (if well designed and well conducted) Modest interference with routine clinical management Endpoints usually of direct clinical relevance Compatible with high scientific standards (randomization, double‐blind design, quality assurance, and monitoring) Nature of the trial motivates patients and investigators to take part |
Advantages
High external validity (results derive from routine clinical care) Minimal intrusiveness into routine clinical management Efficient use of existing infrastructure and personnel Easier enrollment, minimal selection bias,feasibility of large sample size Simple data collection and source verification Faster implementation Feasibility of long‐term follow‐up Lower costs |
|
Limitations
Low external validity (highly selected patient groups, rigid treatment protocols) Question addressed in the trial often differs from question asked by physicians in their daily practice Duration of treatment often limited by protocol constraints Endpoints often of questionable clinical relevance Ethical concerns with prolonged exposure to placebo or fixed‐dose treatments Enrollment can be difficult if drugs being tested are already available in the market |
Limitations
Generally only applicable to investigation of licensed drugs Double‐blinding may be difficult to apply when multiple treatments are compared Implementation of rigorous monitoring procedures may be limited by suboptimal funding |
Limitations
Lower internal validity (less certainty about the validity of baseline and outcome data, and higher susceptibility to confounding) Only applicable to settings with adequate health informatics systems For multicenter studies, compatibility of EHR systems can be an issue Only suitable to assess licensed drugs Open label (and associated bias). Blinded adjudication committees may be needed to reduce bias Monitoring and validation still needed Choice of endpoints limited to data collected in routine clinical care |
Adjunctive‐therapy trials
Because it is generally considered unethical to expose epilepsy patients to prolonged monotherapy with an agent whose clinical efficacy has not yet been established, traditionally investigational AEDs are tested initially as adjunctive therapy in patients whose seizures have not been controlled by existing treatments.5, 6 In most cases, these trials provide the only data available to physicians at the time a new AED is introduced to the market. Therefore, the implications of these trials for optimal use of the drug need to be scrutinized carefully. By doing so, it is clear that there is room for improvement in several areas of clinical drug development, particularly to enable generation of information that could guide better the use of the drug in routine clinical practice.3
Initially, investigational AEDs are often tested in open‐label uncontrolled exploratory trials, the primary purpose of which is to identify dose‐limiting adverse effects, to refine knowledge of pharmacokinetic properties, and to assess potential interactions with concomitantly taken AEDs. Although these studies can provide preliminary signals of an antiseizure effect, demonstration of efficacy is ultimately dependent on the conduction of RCTs. The standard design involves randomization of patients to double‐blind treatment with a range of doses of the investigational agent and placebo, administered in addition to preexisting AEDs (Fig. 1). Typically, the protocol includes a prospective baseline, a dose‐titration period of variable length, and a 12‐week maintenance period.5 Demonstration of efficacy relies on evidence that, compared with baseline, percent decrease in seizure frequency (or proportion of patients exhibiting at least a 50% reduction in seizure frequency) is greater in the active treatment group(s) than in the placebo group. This design has been utilized successfully to demonstrate efficacy and obtain marketing approval for over a dozen second‐generation AEDs,5, 7 even though its sensitivity in differentiating active from inactive treatments may have weakened over the years in relation to a rising placebo response7 and the geographical setting in which trials are conducted.8, 9, 10 In addition, ethical concerns have been raised following demonstration that patients randomized to placebo in these trials have a 6‐fold increase in risk of sudden unexpected death in epilepsy (SUDEP) compared with patients exposed to effective treatments.11 This could be interpreted as violating the ethical rule governing the feasibility of placebo use, that is, the requirement that “delaying or withholding the established effective intervention will result in no more than a minor increase above minimal risk to the participant.”12
Figure 1.
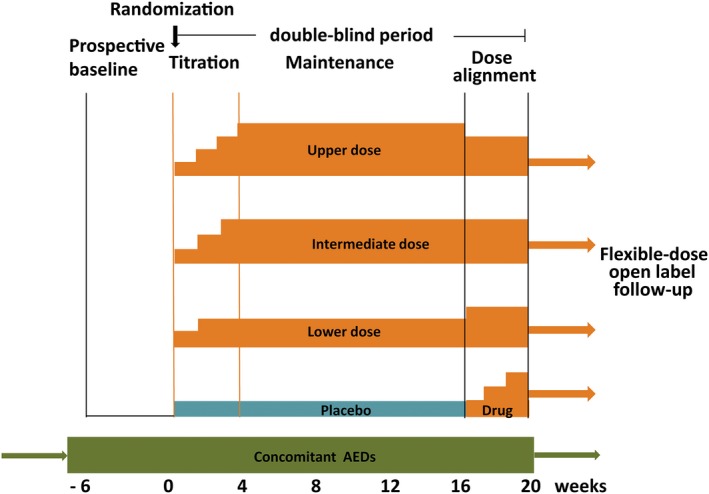
Representative double‐blind trial design for assessing the efficacy and tolerability of investigational new antiepileptic drugs (AEDs) given as adjunctive therapy. After an initial 4‐ to 8‐week prospective evaluation to establish a baseline, patients are randomized to 3 parallel‐dose groups or placebo. Treatment generally includes a titration period of variable length and modalities, and a 12‐week maintenance period. Efficacy is evaluated by comparing changes in seizure frequency or responder rate (versus baseline) between each dose group and the placebo group. The treatment period used for efficacy assessment typically includes the titration and maintenance period combined (FDA‐preferred analysis) or the maintenance period alone (EMA‐preferred analysis). In the trial design illustrated in the figure, the maintenance phase is followed by a dose‐alignment phase during which all patients are blindly converted to a common dose in order to preserve the double‐blind. When dose alignment is completed, open‐label flexible‐dose treatment can be continued long‐term as clinically indicated. The dose alignment phase also allows patients initially exposed to placebo to receive a trial treatment with the investigational drug.
Obtaining a marketing license requires demonstration of efficacy and safety. For AEDs, this requirement is generally met by providing evidence that the product of interest is superior to placebo in reducing seizure frequency without causing undue toxicity.5, 6 The question addressed by regulatory trials, however, is different from the question faced by physicians when prescribing treatments in daily practice.3 Not only do physicians need to be assured that a new treatment is better than placebo, but they also want to know how that treatment compares in terms of effectiveness and safety with other available agents. Due to the many trial‐specific variables that influence clinical outcomes, this information cannot be derived from comparison of data across trials but needs to be obtained through head‐to‐head comparisons.6 Regrettably, however, comparative effectiveness trials are generally not available at the time a new product is released into the market. To make matters worse, such trials may not be conducted for many years postmarketing and, for some AEDs, may never be performed.
There are other limitations associated with currently used adjunctive‐therapy designs. To minimize the influence of confounders, these trials typically incorporate strict eligibility criteria and may exclude patients outside a predefined age range, those with a low (or high) seizure frequency, those with certain comorbidities or conditions, and those taking certain comedications. As a result, populations included in regulatory adjunctive‐therapy trials differ considerably from those managed in everyday clinical practice. Moreover, unlike standard practice, regulatory study protocols allow little or no flexibility in dosing schemes, permit little or no individualization of dosage, and they generally forbid changes in concomitant AED therapy during the duration of the trial. Overall, these features may affect the generalizability of results because they do not ascertain the effectiveness of the drug under conditions of optimal use. An additional concern arises from the fact that efficacy endpoints used in regulatory adjunctive‐therapy trials (50% responder rate, or median percent change in seizure frequency) have modest clinical relevance, because substantial improvement in quality of life can only be expected when sustained seizure freedom is achieved.13, 14
Double‐blind adjunctive‐therapy trials are typically followed by an open‐label flexible‐dose extension phase. This phase addresses the ethical requirement of permitting continuation of treatment for those participants who had shown clinical benefit and offers the opportunity of a trial on active medication for those who were randomized initially to placebo. Open‐label extension can provide valuable information about potentially delayed adverse effects. However, the lack of a control group and the influence of confounders, including possible changes in dosage or type of concomitant AEDs, make it difficult to draw conclusions about long‐term maintenance of antiseizure effects.
Can the adjunctive‐therapy design be improved?
Based on the considerations above, there is scope for considering whether currently used regulatory adjunctive‐therapy trials could be improved to address ethical concerns and provide data of greater relevance to clinical practice.
Can duration of placebo exposure be minimized?
As discussed above, prolonged exposure of patients with uncontrolled seizures to placebo raises ethical concerns because of the associated increase in mortality risk.11 In addition, inclusion of placebo in these trials discourages patients from participating, thereby hampering recruitment and introducing selection bias, which may impact negatively on the generalizability of the results.5 One way to address these concerns while maintaining a placebo control is to require individuals to exit the trial once a small, predefined number of seizures have occurred. By doing so, patients whose seizures do not improve during treatment are protected from remaining on an ineffective treatment, and the efficacy of the AED being tested can be demonstrated by a longer time to exit compared with placebo. To date, time‐to‐event designs in epilepsy have been applied mostly to monotherapy trials,6 but their feasibility for adjunctive‐therapy trials has also been demonstrated. In a recent post hoc analysis of data from 3 adjunctive‐therapy trials, perampanel doses of 4–12 mg/day were found to be associated across all trials with a statistically significant prolongation of time to prerandomization monthly seizure count, generally by more than 1 week, compared with placebo, consistent with results obtained using the original primary endpoints assessed over the full duration of the trial.15 Although time‐to‐event trials may be perceived by physicians as difficult to interpret in terms of clinical significance, their results can easily be translated into conventional outcome measures. For example, in a trial using time to prerandomization monthly seizure count as primary endpoint, a median time to event of 3 months in the active treatment arm would mean that 50% of patients on active treatment continued in the study for 3 months, or that 50% of patients had their seizure frequency reduced by at least two‐thirds.15 A drawback of a markedly shortened placebo exposure period is that no placebo control group is available to assess longer‐term safety data. The latter shortcoming, however, could be partly addressed by introducing an active control as an additional arm (see below). In addition to time‐to‐event and active control designs, alternative approaches to minimize placebo exposure have been proposed, including placebo add‐on to background therapy with adjustment, adaptive designs, platform trials with a pooled placebo control group, pharmacokinetic/pharmacodynamic modeling, and shorter trials. Advantages and limitations of these options are discussed in a recent publication.16
Should an active control be included?
Adjunctive‐therapy active control trials have been performed rarely, mainly because they are not required by current regulatory guidelines and their sensitivity in differentiating between active treatments is considered to be low, unless a new treatment with outstanding greater efficacy emerges someday. If 2 active treatments are not found to differ and placebo is not included as a comparator, the argument could be made that in the specific setting in which the trial was conducted, both treatments might have been equally ineffective.6, 17 The latter possibility is far from remote because, historically, adjunctive‐therapy RCTs have not always differentiated established active treatments from placebo, at least in certain settings.9, 10, 18 Recently, concerns with assay sensitivity in active control trial designs were rekindled by the results of a double‐blind flexible‐dose RCT in which no difference in seizure outcomes was found between pregabalin and gabapentin, contrary to the expectation from earlier trials that pregabalin would show superior efficacy.19
Despite these limitations, there is an increasing interest in including established active controls in adjunctive‐therapy trials of new AEDs. This approach would address physicians’ requests for comparative effectiveness data as a guide to rational prescribing and would provide industry with comparative data, which are increasingly required by national health services or insurers to make decisions on reimbursement prices of newly approved medicines. The issue of assay sensitivity in these trials could be addressed by including a placebo arm in addition to an active control, an approach that has been found to be feasible.8 Active control adjunctive‐therapy trials including a placebo arm could also use a time‐to‐event design, thereby minimizing ethical concerns related to prolonged exposure to placebo. To avoid selection bias, RCTs including an established active treatment would have to exclude patients already on that treatment, or previously exposed to it.17 This could severely limit enrollment rate, and in some settings these trials may not be feasible or would take a long time to complete.
Should trial designs be adjusted to facilitate generalizability?
There are many ways by which generalizability of trial results could be improved. First, efforts could be made to broaden eligibility criteria for phase III trials, to allow inclusion of patients who are more representative of everyday practice. Second, although fixed‐dose designs still need to be conducted to establish dose–response relationships, there is scope also for conducting flexible‐dose studies under conditions mimicking to the extent possible routine clinical use. The feasibility of such trials, and their sensitivity in demonstrating the added value of individualized dosing, has been demonstrated.20
A special issue related to generalizability is the possibility of extrapolating clinical trial results to different populations. In recent years, there has been increasing attention to the need to obtain as early as possible information on the benefits and risks of newly developed AEDs at the extremes of age. Regulatory agencies realize that, under certain conditions, efficacy and safety data obtained in adults can be extrapolated to children with the same disease.21, 22, 23 A convincing case has been made that, at least for patients with focal epilepsies, efficacy data can be extrapolated from adults to children down to the age of 2 years, provided careful pharmacokinetic and tolerability studies are conducted beforehand to permit appropriate dose adjustments in relation to age.24 It might be argued that efficacy data could be extrapolated from adults to children also for generalized tonic–clonic seizures associated with idiopathic (genetic) generalized epilepsies, even though for this seizure type there is little historical evidence from previous trials to support the contention that AED responsiveness for this seizure type in children younger than 12 years of age is comparable to that for older children or adults. Obviously, for epilepsies of infancy and childhood, such as West syndrome, Dravet syndrome, Lennox‐Gastaut syndrome, or childhood absence epilepsy, no extrapolation is possible and specific efficacy studies need be conducted in the pediatric populations of interest. In the case of trials conducted in pediatric epilepsies, however, consideration should be given to whether the results could be extrapolated from children to adults with the same syndrome, provided seizure manifestations (and, presumably, pathophysiology) did not change from childhood to adulthood. As for the elderly, an effort should be made to conduct specific studies in patients of older age, or at least to enroll a sizeable number of patients over 65 in trials conducted in adult epilepsies.25 If pharmacokinetic differences between elderly and nonelderly adults are prominent, patients above a certain age may need to be assigned to different dosages to ensure comparable levels of exposure.
When considering the applicability of clinical trial results to routine clinical practice, a major issue relates to the feasibility of extrapolating results of adjunctive‐therapy RCTs to the monotherapy setting. As discussed below, this issue has become especially critical following a deliberation by the U.S. Food and Drug Administration (FDA) that, under certain conditions, a monotherapy license can be granted based on an analysis of efficacy and safety data obtained in add‐on trials.26
Monotherapy trials
For the past several decades, adjunctive‐therapy trials of new AEDs permitted only regulatory approval for add‐on use. Therefore, specific monotherapy trials were required to obtain a monotherapy license. Monotherapy designs to fulfill this requirement have differed widely between the 2 sides of the Atlantic.
The European paradigm: noninferiority trials in newly diagnosed patients
The guidelines of the European Medicines Agency (EMA) state that “dose‐response relationships from add‐on studies in refractory patients may not be applicable to use in monotherapy” not only because of the possible confounding influence of pharmacodynamic and pharmacokinetic interactions, but also because most newly diagnosed patients “have milder, more responsive forms of epilepsy.” 4 The EMA guidelines emphasize the importance of conducting studies in patients with newly diagnosed epilepsy and recommend the use of randomized, double‐blind active controlled designs “aiming to demonstrate at least a similar benefit/risk balance of the test product as compared to an acknowledged standard product at its optimal dose.”4, 27 The primary efficacy endpoint recommended by the EMA guidelines is the proportion of patients achieving seizure freedom for at least 6 months (excluding the dose escalation period), although a minimum follow‐up of 1 year is recommended to assess longer‐term safety and maintenance of efficacy. A number of noninferiority RCTs conducted according to EMA guidelines have been completed in recent years, and they led to approval of a European monotherapy license for levetiracetam,28 zonisamide,29 lacosamide,30 and eslicarbazepine acetate31 for the treatment of focal seizures in adults.
In terms of generalizability and relevance to clinical practice, studies fulfilling the EMA requirements are valuable because they are conducted in populations for which monotherapy is most commonly applied, permit individualization of dose, use clinically meaningful endpoints and, most importantly, provide comparative effectiveness data using as reference an established comparator at its optimal dose.6 Some of these trials, however, have restrictive eligibility criteria, which may render the population not fully representative of the spectrum of patients with newly diagnosed epilepsy. When assessing these studies, consideration should also be given to any measures set in place for diagnostic validation prior to enrollment. An important conceptual concern with these trials relates to uncertainty about assay sensitivity, although in at least one case, application of this design did allow to clearly differentiate an effective from an ineffective (or less effective) treatment.32 From the industry's perspective, these trials are expensive to conduct, mainly because patients need to be followed for 12 months, or much longer depending on the time required to optimize dose. The industry's desire to streamline time and costs has led over the years to limit the duration of these trials, thereby preventing adequate follow‐up of those patients who did not respond to the initial dose and required subsequent dose adjustments. In fact, a possible explanation for pregabalin being found to be inferior to lamotrigine in one of these trials is that the initial maintenance dose of pregabalin (150 mg/day) was poorly effective, and the overall duration of the trial did not permit assessment of comparative effectiveness at higher doses.32
The EMA guidelines are currently undergoing revision, and it is possible that some of the issues outlined above, including the agency's position on the value of noninferiority trials, may change in the future.
The U.S. conundrum: from conversion to monotherapy to extrapolation from adjunctive‐therapy trials
The FDA considers noninferiority active control trials inadequate to provide evidence of efficacy for AEDs, due to concerns with assay sensitivity. Instead, seeking a monotherapy license from the FDA has traditionally required demonstration of superiority over a comparator.6 Because placebo cannot be ethically justified as sole therapy in active epilepsy, early trials conducted to address FDA requirements involved comparison with a suboptimal (low‐dose) active control. Most of these trials used the conversion to monotherapy design, whereby AED‐treated patients with uncontrolled seizures are randomized to receive a high dose of the investigational agent or a low‐dose active control, often referred to as “pseudo‐placebo.” The underlying AEDs are then removed gradually, and patients who meet predefined criteria for seizure deterioration are required to exit the study.6 Proof of efficacy in these trials is obtained by demonstrating that patients randomized to the high‐dose investigational agent exit at a lower rate and are more likely to achieve conversion to monotherapy compared with patients randomized to suboptimal treatment.
Over the years, the conversion to monotherapy trial design has come under increasing criticism on ethical grounds due to lack of equipoise, and the risks associated with worsened seizure control (particularly high in the group allocated to suboptimal treatment) following removal of underlying AEDs.6 The observation that exit rates in groups randomized to suboptimal treatment were similar across conversion‐to‐monotherapy trials led the FDA to accept in 2010 a new paradigm whereby subsequent trials could use historical suboptimally treated controls as comparator.33, 34 This approach has been used to obtain a U.S. monotherapy license for several AEDs.35, 36, 37, 38 However, the removal of a suboptimally treated control arm did not eliminate ethical concerns associated with exposing patients to withdrawal of concomitant AEDs. In addition, demonstration of an increase in placebo response over time7 led to questioning the validity of using control data from trials conducted many years earlier.34
A turning point in the U.S. regulatory approach occurred in the last few years. The argument was raised that, provided appropriate assessments are done to exclude (or to control for) the influence of potential drug–drug interactions, there is no sound reason to believe that an AED found to be efficacious and safe when prescribed as add‐on therapy could lose its activity when used in monotherapy.39 In 2017, this proposition was accepted by the FDA, at least for focal seizures, with perampanel and brivaracetam being the first AEDs to be granted a monotherapy license in the United States based on analysis of data from adjunctive‐therapy trials.40, 41
Based on available evidence, it may be possible to establish with reasonable confidence whether the efficacy of an AED is affected by the presence or absence of concomitant medications. If pharmacokinetic drug interactions occur, dose adjustments can be made to ensure comparable levels of exposure between the monotherapy and the polytherapy setting. However, there are limitations in extrapolating findings from adjunctive‐therapy studies, or from conversion to monotherapy trials, to the clinical settings where monotherapy is typically used. Although the range of doses or serum AED concentrations found to be efficacious in adjunctive‐therapy trials in patients with pharmacoresistant epilepsy may overlap with those required in monotherapy to control seizures in newly diagnosed patients,39 optimal dosing schedules may differ between these populations. Indeed, there is evidence that patients with newly diagnosed epilepsy often achieve sustained seizure freedom at doses that are in the lower range of those associated with clinical benefit in patients with refractory epilepsy.6, 42 Moreover, adverse effects associated with any given serum AED concentration may differ between monotherapy and polytherapy, due to the influence of pharmacodynamic drug interactions.43 Therefore, from the viewpoint of the practicing physicians, it would be desirable to have access to data from monotherapy trials in newly diagnosed patients, and most importantly, trials in which newly developed AEDs are compared with established agents.
Nonregulatory Trials
Nonregulatory trials are designed primarily to address goals other than obtaining a regulatory license or fulfilling commitments required by regulators. Nonregulatory trials can be conducted by different stakeholders and may serve different purposes and differ widely in quality, methodology, and value in informing rational prescribing. Two categories of nonregulatory trials of particular relevance because of their influence on clinical practice are discussed below.
Comparative effectiveness trials
Randomized comparative effectiveness trials are designed to compare the effects of 2 or more currently used treatments on clinical outcomes in order to guide therapeutic decision making.44 If well designed, these trials provide highly valuable information and have a significant impact on clinical decision‐making. Despite their common goal, comparative effectiveness trials may differ greatly in methodology, and in the extent to which they reproduce conditions that apply to routine clinical practice (Table 1).
Some comparative effectiveness trials utilize rigorous scientific methodology, such as a double‐blind design, and require investigators to follow precise rules in applying and evaluating the treatment being compared. Examples are the trials supported by the U.S. Department of Veteran Affairs to compare the effectiveness of several AEDs in patients with previously untreated or undertreated focal epilepsy.45, 46, 47 These trials had a double‐blind randomized design and used double‐dummies to mask the identity of treatments. Physicians were instructed to aim at a predefined target dose or serum drug concentration, with further dose adjustments being permitted according to individual response. Clinically significant differences in outcomes could be demonstrated among various AEDs, and results have been influential in guiding physicians in their daily practice. Although these trials provide highly valuable information, their generalizability to routine care is likely to be limited to some extent. In one of the trials, for example, 765 of the 1358 patients (56%) screened for possible enrolment failed to meet inclusion criteria, including 131 patients who declined to participate,47 and the protocol‐defined procedures may have altered usual clinical management.
An alternative approach in evaluating comparative effectiveness is represented by pragmatic randomized trials. These trials are designed to reproduce more closely clinical practice, primarily by permitting physicians to apply treatments as done in routine practice. The Multicentre Study of Early Epilepsy and Single Seizures trial, which compared outcomes following immediate or deferred treatment for early epilepsy or single seizures, is an example of a typical pragmatic trial.48 In this trial, choice of AED, choice of doses, and duration of treatment were dependent on the clinician's usual practice, and the trial simply determined which of the 2 management policies (immediate treatment or deferred treatment) had the best outcomes in a setting that mimicked routine care. A pragmatic approach was also applied in the Standard versus New Antiepileptic Drugs studies,49, 50 even though these studies had shortcomings in trial design that may have adversely affected generalizability.51 Because clinical management in pragmatic trials resembles routine care, these trials tend to be easily accepted by physicians and patients alike, thereby facilitating enrolment of large sample sizes.48, 49, 50 However, for open‐label studies, lack of masking can introduce bias in the assessment of outcomes.
In the last few years, there has been an increased interest in conducting comparative effectiveness trials, which are even more closely integrated into clinical care. The feasibility of these trials is facilitated by the availability of networks utilizing electronic health records (EHRs), which permit collection of outcome data as part of routine care.44 Point‐of‐care pragmatic trials using individual or cluster randomization can take advantage of the infrastructure of existing networks and use EHR as a source for most data (Table 1).52 Similar approaches can be applied to registry‐based trials.53 To ensure optimal exploitation of these opportunities, however, a number of challenges need to be addressed. One relates to concerns about accuracy of diagnostic and outcome data in EHRs and large registries. For multicenter studies, the feasibility of harmonizing EHR systems across networks of participating centers also needs to be addressed. In Europe, for example, the Epicare epilepsy network, which is part of the European Reference Networks (ERNs) for rare and complex diseases, is already engaged in developing an EHR infrastructure that can be used for registries and clinical trials.54
A broader challenge to the implementation of non‐regulatory trials relates to the need to address the bureaucratic hurdles that hamper the conduction of non–industry‐sponsored trials. Over the last decades, there has been a staggering increase in regulations that impact all aspects of clinical trial implementation, including regulatory approvals; documents to be supplied to ethics committees; modalities of written informed consent; and requirements for data collection, data management systems, quality assurance, and site‐based monitoring. In many settings, not only has strict adherence to GCP guidelines become an absolute requirement, but there has also been a tendency to overinterpret GCP regulations.52, 53 This has resulted in increased trial complexity, delays in trial initiation, longer trial duration, and exploding implementation costs, which severely limit the possibility of conducting academic trials.55 The need to streamline procedures to improve efficiency and generalizability, particularly for low‐risk pragmatic trials, has been emphasized repeatedly.44, 52, 53
Clinical trials as a tool for drug promotion in the market place
At times, clinical trials supported directly or indirectly by the pharmaceutical industry are designed and used as a marketing tool to promote the sponsor's products. These trials are often referred to as “seeding trials,: that is, trials of little or no scientific value that are intended to familiarize clinicians with the use of recently introduced products, increase the utilization of the same products through enrolment of patients in a “study,” generate misleading information about the value of specific drugs and, at times, promote off‐label use for nonapproved indications.56 The overwhelming majority of clinical trials conducted postmarketing consists of uncontrolled trials, and these trials can represent an excellent tool for drug promotion because they typically overestimate the clinical benefit of any treatment.1, 57, 58
A more insidious way to generate data useful for drug promotion consists of conducting RCTs that incorporate bias favoring the sponsor's product.1, 2, 59, 60 These bias can affect any aspect of a trial design, from the choice of eligibility criteria to the interpretation of the results (Table 2). In fact, many comparative‐effectiveness monotherapy trials performed in the last 2 decades that reported a better tolerability of recently introduced AEDs compared with older agents were affected by subtle bias, which probably led to overestimate the value of the sponsor's product.1, 2, 61
Table 2.
Potential sources of bias in randomized controlled trials that may favor outcomes associated with the sponsor's product
| Source of bias | Example |
|---|---|
| Inclusion criteria | Inclusion of a pooled population of patients with focal seizures and generalized tonic–clonic seizures may bias efficacy outcomes in favor of AEDs that have broad spectrum antiseizure activity |
| Exclusion criteria | Exclusion criteria may preselect a population less likely to experience adverse effects with one of the products being compared |
| Choice of the comparator | The comparator may not be the most appropriate for the study population (for example, phenytoin in a study comparing first‐line treatments for children with focal seizures) |
| Choice of sample size | An underpowered trial is unlikely to reveal a potentially inferior efficacy of the sponsor's product |
| Mode of use of comparator | Comparator may be suboptimally used (for example, immediate‐release carbamazepine given twice daily) |
| Dosage of comparator | Underdosing or overdosing of the comparator may lead to underestimation of its efficacy, or to overestimation of its adverse effects |
| Duration of treatment | Tolerability of an investigational AED causing weight gain during prolonged treatment can be overestimated by keeping trial duration short |
| Choice of endpoints | Using effectiveness (retention in the trial) as primary endpoint may not highlight a potentially inferior antiseizure activity of one of the treatments, if patients with uncontrolled seizures are not required to exit before completion of trial duration |
| Presentation of results | Proportion of patients who completed the full duration of the trial and were responders may not be reported |
| Interpretation of results | Advantages in outcomes associated with the sponsor's treatment may be overemphasized, or bias and limitations may not be discussed |
For more information and specific examples, refer to Perucca and Wiebe.1
From Clinical Trials to Clinical Practice
Despite the limitations discussed in the previous sections of this article, clinical trials provide invaluable information about risks and benefits of individual treatments. Their usefulness in guiding treatment decisions, however, is variable, being dependent on design specificities that affect the quality of the data (internal validity) and their generalizability to routine clinical care (external validity). There is often a tug‐of‐war between internal and external validity, which explains why at times medications that produced highly promising results in regulatory trials turn out to be of disappointing value in routine practice, and vice versa. Indeed, a number of approaches have been proposed to bridge the so called efficacy‐effectiveness gap, where, in this context, efficacy describes how a drug performs under conditions of clinical trials, whereas effectiveness describes how it performs under conditions of everyday clinical practice.62
To make optimal use of trial data in clinical practice, it is essential for trial results to be interpreted correctly. Many pitfalls that can lead to misinterpretation of the data, and physicians need to scrutinize carefully methodology and results without necessarily accepting the authors’ conclusions, as discussed in detail in a recent publication. 1 Apparently minor methodologic details, such as choice of dosing schemes and pharmaceutical formulation,1, 63 can have a profound impact on clinical outcomes, and introduce bias in the assessment of comparative effectiveness. In addition, attention needs to be paid to the presentation of results. For example, responder rates may differ markedly depending on whether seizure outcomes are calculated over the entire treatment period or over the maintenance period, particularly when efficacy assessments based on the maintenance period exclude from the denominator those patients who tolerated the treatment poorly and exited during dose titration.64 Furthermore, not all physicians seem to be aware that as a rule responder rates are calculated by using the last‐observation‐carried‐forward (LOCF) analysis, that is, by including in the efficacy analysis those patients who exited the trial prematurely.1 For example, if in a 20‐week trial a patient exited after 2 weeks because of adverse effects and did not experience any seizure during those 2 weeks, that patient will be considered in the final analysis as having achieved seizure freedom. It could be argued that the measure of efficacy most meaningful to the practicing physician is the proportion of patients who had their seizures improved and were able to complete the trial. Yet, this information is rarely reported.65 In a systematic review of randomized placebo‐controlled adjunctive‐therapy trials, only 3 of the 63 trials conducted in adults with focal epilepsy reported the proportion of patients who completed the trial successfully, that is, who had a >50% reduction seizure in seizure frequency and were able to complete the trial.7
Finally, it should be remarked that there are still many areas where evidence from RCTs is lacking, particularly with respect to most rare pediatric epilepsy syndromes. There is also a paucity of trials that address important aspects such as disease prevention and disease modification, etiology‐based treatments, the comparative value of potentially synergistic AED combinations, biomarker‐guided treatments, and treatments targeting common comorbidities. There is also a need for improved communication between preclinical and clinical scientists in improving the translational value of research.66 Finally, even in areas where RCTs exist, there are still important deficiencies in the quality of the information available. As eloquently stated in a recent metanalysis of pediatric RCTs in epilepsy, “the quality of studies should be improved through the use of comparative designs, relevant outcomes, appropriate follow‐up length, and more reliable inclusion criteria.”67
Conclusions
Physicians need to make treatment decisions based on evidence, and well‐designed clinical trials represent the best source of evidence. Regrettably, as discussed in this article, there many areas related to epilepsy treatment where RCTs have not been conducted. Moreover, most RCTs that have been conducted in patients with seizure disorders have been designed to address regulatory requirements, and their usefulness as a guide to rational prescribing is limited. Any adjustment in regulatory requirements to improve generalizability of clinical trial data to routine clinical practice would be welcome. Research‐funding organizations should be sensitized about the lack of adequate evidence to guide therapeutic practice in epilepsy, and the need to promote high‐quality comparative effectiveness trials, including pragmatic trials.
Acknowledgments and disclosures
The author confirms that he has read the Journal's position on issues involved in ethical publication and affirms that this report is consistent with those guidelines. This work was not supported by any funding source. The author received research funds from the European Union, the Italian Medicines Agency, the Italian Ministry of Health, and the Italian Ministry for Education, University and Research. He also received speaker's or consultancy fees from Eisai, GW Pharma, LivaNova, Medichem, Mylan, Sandoz, Sanofi, Sun Pharma, Takeda, and UCB Pharma.
Biography
Dr. Emilio Perucca is Professor in the Department of Internal Medicine and Therapeutics at the University of Pavia.

References
- 1. Perucca E, Wiebe S. Not all that glitters is gold: a guide to critical interpretation of drug trials in epilepsy. Epilepsia 2016;1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glauser T, Ben‐Menachem E, Bourgeois B, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2013;54:551–563. [DOI] [PubMed] [Google Scholar]
- 3. Walker MC, Sander JW. Difficulties in extrapolating from clinical trial data to clinical practice: the case of antiepileptic drugs. Neurology 1997;49:333–337. [DOI] [PubMed] [Google Scholar]
- 4. European Medicines Agency . Guideline on clinical investigation of medicinal products in the treatment of epileptic disorders. London: European Medicines Agency; 2010. (CPM/EWP/566/98/rev 2) [Google Scholar]
- 5. Perucca E. What clinical trial designs have been used to test antiepileptic drugs and do we need to change them? Epileptic Disord 2012;14:124–131. [DOI] [PubMed] [Google Scholar]
- 6. Perucca E. Designing clinical trials to assess antiepileptic drugs as monotherapy: difficulties and solutions. CNS Drugs 2008;22:917–938. [DOI] [PubMed] [Google Scholar]
- 7. Rheims S, Perucca E, Cucherat M, et al. Factors determining response to antiepileptic drugs in randomized controlled trials. A systematic review and meta‐analysis. Epilepsia 2011;52:219–233. [DOI] [PubMed] [Google Scholar]
- 8. Baulac M, Leon T, O'Brien TJ, et al. A comparison of pregabalin, lamotrigine, and placebo as adjunctive therapy in patients with refractory partial‐onset seizures. Epilepsy Res 2010;91:10–19. [DOI] [PubMed] [Google Scholar]
- 9. French JA, Krauss GL, Biton V, et al. Adjunctive perampanel for refractory partial‐onset seizures: randomized phase III study 304. Neurology 2012;79:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. French JA, Baroldi P, Brittain ST, et al. Efficacy and safety of extended‐release oxcarbazepine (Oxtellar XR™) as adjunctive therapy in patients with refractory partial‐onset seizures: a randomized controlled trial. Acta Neurol Scand 2014;129:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta‐analysis of placebo‐controlled randomised trials. Lancet Neurol 2011;10:961–968. [DOI] [PubMed] [Google Scholar]
- 12. Council for International Organizations of Medical Sciences (CIOMS) . International ethical guidelines for health‐related research involving humans. Geneva: Council for International Organizations of Medical Sciences (CIOMS); 2016. https://cioms.ch/wp-content/uploads/2017/01/WEBCIOMS-EthicalGuidelines.pdf. Accessed 14 November 2017 [Google Scholar]
- 13. Birbeck GL, Hays RD, Cui X, et al. Seizure reduction and quality of life improvements in people with epilepsy. Epilepsia 2002;43:535–538. [DOI] [PubMed] [Google Scholar]
- 14. Luoni C, Bisulli F, Canevini MP, et al. Determinants of health‐related quality of life in pharmacoresistant epilepsy: results from a large multicenter study of consecutively enrolled patients using validated quantitative assessments. Epilepsia 2011;52:2181–2191. [DOI] [PubMed] [Google Scholar]
- 15. French JA, Gil‐Nagel A, Malerba S, et al. Time to prerandomization monthly seizure count in perampanel trials: A novel epilepsy endpoint. Neurology 2015;84:2014–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fureman BE, Friedman D, Baulac M, et al. Reducing placebo exposure in trials: considerations from the Research Roundtable in Epilepsy. Neurology 2017;89:1507–1515. [DOI] [PubMed] [Google Scholar]
- 17. Leber PD. Hazards of inference: the active control investigation. Epilepsia 1989;30(Suppl 1):S57–S63. [DOI] [PubMed] [Google Scholar]
- 18. Xiao Z, Li JM, Wang XF, et al. Efficacy and safety of levetiracetam (3,000 mg/Day) as an adjunctive therapy in Chinese patients with refractory partial seizures. Eur Neurol 2009;61:233–239. [DOI] [PubMed] [Google Scholar]
- 19. French J, Glue P, Friedman D, et al. Adjunctive pregabalin vs gabapentin for focal seizures: Interpretation of comparative outcomes. Neurology 2016;87:1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elger CE, Brodie MJ, Anhut H, et al. Pregabalin add‐on treatment in patients with partial seizures: a novel evaluation of flexible‐dose and fixed‐dose treatment in a double‐blind, placebo‐controlled study. Epilepsia 2005;46:1926–1936. [DOI] [PubMed] [Google Scholar]
- 21. Dunne J, Rodriguez WJ, Murphy MD, et al. Extrapolation of adult data and other data in pediatric drug‐development programs. Pediatrics 2011;128:e1242–e1249. [DOI] [PubMed] [Google Scholar]
- 22. European Medicines Agency . Human Medicines Development and Evaluation. Concept paper on extrapolation of efficacy and safety in medicine development, 2012. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129285.pdf. Accessed December 30, 2017
- 23. European Medicines Agency . Human Medicines Development and Evaluation. Reflection paper on the use of extrapolation in the development of medicines for paediatrics, 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/04/WC500204187.pdf. Accessed December 30, 2017
- 24. Pellock JM, Arzimanoglou A, D'Cruz O, et al. Extrapolating evidence of antiepileptic drug efficacy in adults to children ≥2 years of age with focal seizures: the case for disease similarity. Epilepsia 2017;58(10):1686–1696. [DOI] [PubMed] [Google Scholar]
- 25. Perucca E, Richens A. Trials in the elderly. Epilepsy Res 2001;45:149–151. [DOI] [PubMed] [Google Scholar]
- 26. FDA Communication . Reference ID: 3985169, 2016. Eisai News Release, 2017. http://www.eisai.com/news/news201737.html. Accessed January 2, 2017
- 27. European Medicines Agency . Note for guidance on clinical investigation of medicinal products in the treatment of epileptic disorders. London: European Medicines Agency; 2000. (CPMP/EWP/566/98/re v1) [Google Scholar]
- 28. Brodie MJ, Perucca E, Ryvlin P, et al. Comparison of levetiracetam and controlled‐release carbamazepine in newly diagnosed epilepsy. Neurology 2007;68:402–408. [DOI] [PubMed] [Google Scholar]
- 29. Baulac M, Patten A, Giorgi L. Long‐term safety and efficacy of zonisamide versus carbamazepine monotherapy for treatment of partial seizures in adults with newly diagnosed epilepsy: results of a phase III, randomized, double‐blind study. Epilepsia 2014;55:1534–1543. [DOI] [PubMed] [Google Scholar]
- 30. Baulac M, Rosenow F, Toledo M, et al. Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled‐release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double‐blind, non‐inferiority trial. Lancet Neurol 2017;16:43–54. [DOI] [PubMed] [Google Scholar]
- 31. Trinka E, Ben‐Menachem E, Kowacs PA, et al. Efficacy and safety of eslicarbazepine acetate versus controlled release carbamazepine monotherapy in newly diagnosed epilepsy: a phase III double‐blind, randomized, parallel‐group, multicenter study. Epilepsia 2018;59:479–491. [DOI] [PubMed] [Google Scholar]
- 32. Kwan P, Brodie MJ, Kälviäinen R, et al. Efficacy and safety of pregabalin versus lamotrigine in patients with newly diagnosed partial seizures: a phase 3, double‐blind, randomised, parallel‐group trial. Lancet Neurol 2011;10:881–890. [DOI] [PubMed] [Google Scholar]
- 33. French JA, Wang S, Warnock B, et al. Historical control monotherapy design in the treatment of epilepsy. Epilepsia 2010;51:1936–1943. [DOI] [PubMed] [Google Scholar]
- 34. Perucca E. When clinical trials make history: demonstrating efficacy of new antiepileptic drugs as monotherapy. Epilepsia 2010;51:1933–1935. [DOI] [PubMed] [Google Scholar]
- 35. Chung S, Ceja H, Gawłowicz J, et al. Levetiracetam extended release conversion to monotherapy for the treatment of patients with partial‐onset seizures: a double‐blind, randomised, multicentre, historical control study. Epilepsy Res 2012;101:92–102. [DOI] [PubMed] [Google Scholar]
- 36. French JA, Temkin NR, Shneker BF, et al. Lamotrigine XR conversion to monotherapy: first study using a historical control group. Neurotherapeutics 2012;9:176–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wechsler RT, Li G, French J, et al. Conversion to lacosamide monotherapy in the treatment of focal epilepsy: results from a historical‐controlled, multicenter, double‐blind study. Epilepsia 2014;55:1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sperling MR, Harvey J, Grinnell T, et al. Efficacy and safety of conversion to monotherapy with eslicarbazepine acetate in adults with uncontrolled partial‐onset seizures: a randomized historical‐control phase III study based in North America. Epilepsia 2015;56:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mintzer S, French JA, Perucca E, et al. Is a separate monotherapy indication warranted for antiepileptic drugs? Lancet Neurol 2015;14:1229–1240. [DOI] [PubMed] [Google Scholar]
- 40. Eisai Inc., press release, Woodcliff Lake, NJ, 2017; http://eisai.mediaroom.com/2017-07-26-FDA-Approves-Eisais-FYCOMPA-R-perampanel-for-Use-as-Monotherapy-for-the-Treatment-of-Partial-Onset-Seizures (accessed 14 November 2017)
- 41. UCB Pharma, press release, Brussels, 2017. https://www.ucb.com/stories-media/Press-Releases/article/New-indication-for-BRIVIACT-brivaracetam-UCB-s-newest-antiepileptic-drug-approved-by-FDA-as-monotherapy-treatment-of-partial-onset-seizures-in-adults (accessed 14 November 2017)
- 42. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia 2001;42:1255–1260. [DOI] [PubMed] [Google Scholar]
- 43. Perucca E. The pharmacology of new antiepileptic drugs: does a novel mechanism of action really matter? CNS Drugs 2011;25:907–912. [DOI] [PubMed] [Google Scholar]
- 44. Fiore LD, Lavori PW. Integrating randomized comparative effectiveness research with patient care. N Engl J Med 2016;374:2152–2158. [DOI] [PubMed] [Google Scholar]
- 45. Mattson RH, Cramer JA, Collins JF, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic‐clonic seizures. N Engl J Med 1985;313:145–151. [DOI] [PubMed] [Google Scholar]
- 46. Mattson RH, Cramer JA, Collins JF, et al. A comparison of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic‐clonic seizures in adults. N Engl J Med 1992;327:765–771. [DOI] [PubMed] [Google Scholar]
- 47. Rowan AJ, Ramsay RE, Collins JF, et al. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005;64:1868–1873. [DOI] [PubMed] [Google Scholar]
- 48. Marson A, Jacoby A, Johnson A, et al. Immediate versus deferred antiepileptic drug treatment for early epilepsy and single seizures: a randomised controlled trial. Lancet 2005;365:2007–2013. [DOI] [PubMed] [Google Scholar]
- 49. Marson AG, Al‐Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007;369:1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marson AG, Al‐Kharusi AM, Alwaidh M, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet 2007;369:1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perucca E, Alexandre V Jr, Tomson T. Old versus new antiepileptic drugs: the SANAD study. Lancet 2007;370:313. [DOI] [PubMed] [Google Scholar]
- 52. Zannad F, Pfeffer MA, Bhatt DL, et al. Streamlining cardiovascular clinical trials to improve efficiency and generalisability. Heart 2017;103:1156–1162. [DOI] [PubMed] [Google Scholar]
- 53. Mentz RJ, Hernandez AF, Berdan LG, et al. Good clinical practice guidance and pragmatic clinical trials: balancing the best of both worlds. Circulation 2016;133:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Epicare – A ERN for epilepsy. https://www.epilepsyallianceeurope.org/programmes/epicare/(accessed 18 December 2017)
- 55. Morice AH. The death of academic clinical trials. Lancet 2003;361:1568. [DOI] [PubMed] [Google Scholar]
- 56. Kessler DA, Rose JL, Temple RJ, et al. Therapeutic‐class wars–drug promotion in a competitive marketplace. N Engl J Med 1994;331:1350–1353. [DOI] [PubMed] [Google Scholar]
- 57. Spilker B, Segreti A. Validation of the phenomenon of regression of seizure frequency in epilepsy. Epilepsia 1984;25:443–449. [DOI] [PubMed] [Google Scholar]
- 58. Barnett AG, van der Pols JC, Dobson AJ. Regression to the mean: what it is and how to deal with it. Int J Epidemiol 2005;34:215–220. [DOI] [PubMed] [Google Scholar]
- 59. Lexchin J, Bero LA, Djulbegovic B, et al. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326:1167–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Every‐Palmer S, Howick J. How evidence‐based medicine is failing due to biased trials and selective publication. J Eval Clin Pract 2014;20:908–914. [DOI] [PubMed] [Google Scholar]
- 61. Perucca E, Tomson T. Monotherapy trials with the new antiepileptic drugs: study designs, practical relevance and ethical implications. Epilepsy Res 1999;33:247–262. [DOI] [PubMed] [Google Scholar]
- 62. Eichler HG, Abadie E, Breckenridge A, et al. Bridging the efficacy‐effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat Rev Drug Discov 2011;10:495–506. [DOI] [PubMed] [Google Scholar]
- 63. Perucca E. Extended‐release formulations of antiepileptic drugs: rationale and comparative value. Epilepsy Curr 2009;9:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. French JA, Abou‐Khalil BW, Leroy RF, et al. Randomized, double‐blind, placebo‐controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology 2011;76:1555–1563. [DOI] [PubMed] [Google Scholar]
- 65. Gazzola DM, Balcer LJ, French JA. Seizure‐free outcome in randomized add‐on trials of the new antiepileptic drugs. Epilepsia 2007;48:1303–1307. [DOI] [PubMed] [Google Scholar]
- 66. Barker‐Haliski M, Friedman D, White HS, et al. How clinical development can, and should, inform translational science. Neuron 2014;84:582–593. [DOI] [PubMed] [Google Scholar]
- 67. Rosati A, Ilvento L, Lucenteforte E, et al. Comparative efficacy of antiepileptic drugs in children and adolescents: a network meta‐analysis. Epilepsia 2017;59:297–314. [DOI] [PubMed] [Google Scholar]