

Abstract
The composition of the gut microbiome is altered in obesity and type 2 diabetes; however, it is not known whether these alterations are mediated by dietary factors or related to declines in metabolic health. To address this, cecal contents were collected from age-matched, chow-fed male University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats before the onset of diabetes (prediabetic PD; n = 15), 2 wk recently diabetic (RD; n = 10), 3 mo (D3M; n = 11), and 6 mo (D6M; n = 8) postonset of diabetes. Bacterial species and functional gene counts were assessed by shotgun metagenomic sequencing of bacterial DNA in cecal contents, while metabolites were identified by gas chromatography-quadrupole time-off-flight-mass spectrometry. Metagenomic analysis showed a shift from Firmicutes species in early stages of diabetes (PD + RD) toward an enrichment of Bacteroidetes species in later stages of diabetes (D3M + D6M). In total, 45 bacterial species discriminated early and late stages of diabetes with 25 of these belonging to either Bacteroides or Prevotella genera. Furthermore, 61 bacterial gene clusters discriminated early and later stages of diabetes with elevations of enzymes related to stress response (e.g., glutathione and glutaredoxin) and amino acid, carbohydrate, and bacterial cell wall metabolism. Twenty-five cecal metabolites discriminated early vs. late stages of diabetes, with the largest differences observed in abundances of dehydroabietic acid and phosphate. Alterations in the gut microbiota and cecal metabolome track diabetes progression in UCD-T2DM rats when controlling for diet, age, and housing environment. Results suggest that diabetes-specific host signals impact the ecology and end product metabolites of the gut microbiome when diet is held constant.
Keywords: diabetes, metabolomics, metagenomics, microbiota
INTRODUCTION
Case control studies in humans and in animals models have repeatedly shown differing compositions of gut microbial populations between obese or insulin-resistant groups compared with metabolically healthy controls (3, 26, 31, 36, 38, 39, 52, 61). However, differentiating between the direct effects of diet and dietary components vs. changes in host metabolic health (e.g., glucose homeostasis, insulin resistance, or overt diabetes) on microbial ecology has not been systematically investigated. Studies in rodent models of diabetes, including ob/ob mice and Zucker diabetic fatty rats, have identified differences in the gut microbiota between mutant and wild-type animals fed similar diets (24, 25, 50, 61). Although these models control for diet type, the insulin resistance and diabetes phenotype in these animals are driven through defects in leptin production or leptin signaling, which are extremely rare in the pathogenesis of obesity and type 2 diabetes mellitus (T2DM) in humans. A recent study by Carmody et al. (15) reported that diet-related alterations of the gut microbiota overwhelmed genetic effects in several outbred mice strains, including several (e.g., ob/ob and NOD2−/−) used to investigate microbial mechanisms affecting insulin resistance and type 2 diabetes. While the importance of dietary components on the composition, function, and metabolic end products of the gut microbiota should not be minimized, diet-independent alterations of the gut microbiota have been previously reported. For example, mice lacking Toll-like receptor 5 (TLR5) have a disparate microbiome and are more susceptible to impairments in glucose homeostasis compared with their wild-type counterpart under similar feeding regimens (16, 63).
Fecal microbiota transplantation studies in which obesogenic phenotypes have been transmitted into germ-free recipients have confirmed that the gut microbiota is an additional modulating factor in the development of obesity, insulin resistance, and T2DM (2, 56, 61). A key feature of this dysbiosis is the disruption of gastrointestinal epithelial barrier function and increased gut permeability (17, 28, 46). Indeed, several investigators have demonstrated that high-fat feeding promotes metabolic endotoxemia, which is characterized by the presence of systemic low-grade inflammation induced by very low circulating concentrations of lipopolysaccharides and other bacterial cell wall components that traverse the gastrointestinal tract (11–13). While the underlying cause of increased gut permeability is not currently known, it is becoming increasingly clear that bacterial metabolic end products are a key regulator of this process in addition to host metabolism. Short-chain fatty acids and indoles (bacterial catabolic products of tryptophan) have both been shown to be protective of intestinal barrier integrity, whereas hydrogen sulfide and p-cresol have been associated with increased gut permeability (4). Recently, Byndloss et al. (8) proposed a model of host-microbe cross talk where commensal obligate anaerobic bacteria are maintained by colonic surface hypoxia via butyrate activation of epithelial peroxisome proliferator-activated receptor-γ and colonic regulatory T cells. Disruption of this homeostasis resulted in decreased epithelial hypoxia, expansion of facultative anaerobes, and increased epithelial inflammation. Although Byndloss and Baümler (7) were investigating known pathogen expansion (Escherichia and Salmonella spp.), their model provides a clear mechanistic foundation for host-microbe synergistic cross talk and its influence on host health outcomes (10).
To better understand the relationship between host metabolic health and microbial ecology, we utilized the Univeristy of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) Rat model to investigate the compositional and genetic changes in the gut microbiome, as well as the cecal metabolome, at different periods of diabetes progression. We have previously utilized this model to investigate how the diabetes phenotype impacts the systemic metabolome (54). The UCD-T2DM rat is an optimal model to investigate diabetes-related alterations in the gut microbiome for several reasons: 1) male and female rats spontaneously develop polygenic adult-onset obesity and insulin resistance under standard chow conditions while fully maintaining leptin signaling (18); therefore, a diabetic phenotype that closely models the progression of T2DM in humans transpires without dietary manipulation; 2) using an inbred rat strain minimizes much of the potential variability in the gut microbiome due to genetic differences; 3) all animals have been bred and maintained in the same vivarium at UCD for >12 yr, potentially reducing environmental differences known to affect the gut microbiome; and 4) rats within disparate stages of T2DM can be studied within a similar age range. Thus removing several potential modulators of the gut microbiome (e.g., diet, strain, sex, age, and environment) allows us to identify specific taxonomic, genetic, and intracecal metabolic alterations associated with the decline in metabolic control associated with diabetes progression. We hypothesized that the deterioration of the host’s metabolic homeostasis would directly contribute to a significant alteration of the gut microbiome and associated outcomes such as altered bacterial metabolism. To our knowledge, this is the first study to determine these parameters in the periods spanning prediabetes, early diabetes, and later stage established diabetes.
MATERIAL AND METHODS
Animals.
All animal studies were approved by the UCD Institutional Animal Care and Use Committee. In-depth details of the UCD-T2DM rat lineage and breeding strategy have been previously published (18). Briefly, the UCD-T2DM rat was created by crossing obese Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) prone to adult-onset obesity and insulin resistance with Zucker diabetic fatty lean rats (ZDF-lean; Charles River Laboratories) homozygous wild-type (+/+) at the leptin receptor locus. The resulting phenotype of the UCD-T2DM rat includes a polygenic origin of obesity, an inherited β-cell defect, and retention of functional leptin signaling (18). Lean Sprague-Dawley (LSD) rats (Harlan Laboratories, Indianapolis, IN) were also included in the studies as healthy nondiabetic control animals.
Experimental design.
The UCD-T2DM and LSD colonies have been maintained at the animal facility in the Department of Nutrition at UCD for >12 yr. All animals were singly housed within the same room in polycarbonate cages on a 14:10-h light-dark schedule. Cages had Carefresh bedding, and the light cycle, temperature, and humidity were consistent among all animals studied. All rats had ad libitum access to standard chow (2018 Telkad Global; Harlan Laboratories). Onset of diabetes in the UCD-T2DM rat was defined as nonfasted blood glucose concentration >200 mg/dl over 2 consecutive weeks. Nonfasting blood glucose concentrations were assessed with a glucose meter (LifeScan One-Touch Ultra, Milpitas, CA) from a drop of blood collected from the tail using a lancet (between ca. 1300 and 1600, tested weekly). Male UCD-T2DM rats were selected for this study if ~180 days old and were prediabetic with blood sugar < 200 mg/dL (PD; n = 15), were ~180 days old and met the criteria for diabetes with “recent diabetes” (RD; ~2 wk postdiabetes onset; n = 10), or were ~180 days old and 3 mo postonset of diabetes (D3M; n = 11). An additional set of D3M rats was aged an extra 90 days to assess long-term effects of uncontrolled diabetes (D6M; n = 8); therefore, D6M rats were not age-matched to all other animals. Male LSD rats (n = 12) were aged ~180 days before being selected for this study. Median age of rats at euthanization were as follows: LSD: 184 days; PD: 182 days; RD: 183 days; D3M: 184 days; and D6M: 266.5 days. We also used an “Early” and “Late” diabetes classifier, which consisted of combining the PD and RD groups together as the Early group and combining D3M and D6M together as the Late group. These classifiers were based on our previous report that showed no differences in the plasma metabolome between PD and RD groups and no differences between D3M and D6M groups (54). Rat specimens were collected in years 2014 (n: LSD = 5; PD = 6; D3M = 6; and D6M = 5) and 2016 (n: LSD = 7; PD = 9; RD = 10; D3M = 5; and D6M = 2).
Cecal samples.
After a ~13-h fast, rats used were given a 200 mg/kg ip dose of pentobarbital sodium to render them unconscious, and then exsanguinated via cardiac puncture between ca. 0800 and 1100. Cecal tissue was immediately removed after exsanguination and blotted dry. A hole was punched into the cecum tissue, and a cecal content sample was collected into a sterile 1.5-ml microcentrifuge tube by squeezing the contents into the tube. Cecal contents were immediately placed in a −80°C freezer until analysis.
Metagenomic sequencing.
Bacterial DNA was isolated from cecal contents (0.1–0.3 g) using the DNeasy PowerSoil HTP 96 Kit (Qiagen, Germantown, MD). The isolation followed the instructions provided by the manufacturer. Amplification of the V4 variable region of the 16S rRNA gene (50 ng) using 515F/806R forward and reverse primers was conducted by polymerase chain reaction (33). Pooled amplicons were then paired-end sequenced (2 × 250 bp) with ~30% PhiX DNA using an Illumina Miseq (5). The Quantitative Insight into Microbial Ecology (QIIME) pipeline was utilized for operational taxonomic unit (OTU) clustering and taxonomic identification of amplicon sequences (14). A similarity threshold of 97% was used to cluster amplicon sequences followed by taxonomic annotation of OTUs using the Greengenes 16S rRNA database with a confidence threshold of 80%. Microbial sequencing data provided as sequence counts. There was a single sample from the D6M group (K2501) that sequenced poorly, resulting in very few sequencing reads for that sample. After the defective sample was removed, the median sample depth was 45,662 reads. In total, 50,533 OTUs (2,496,494 total reads) were identified, which was highly skewed toward low abundant OTUs. Therefore, we filtered low abundant OTUs if >6% of all samples had ≤5 sequence counts. We used a low prevalence criteria to ensure the lowest sized group (D6M) had at least 50% coverage of OTUs before filtering OTUs. This resulted in 1,752 OTUs (2,195,413 sequence reads) that were used for statistical analysis.
DNA extractions from 16S rRNA analysis were used for metagenomic shotgun sequencing as described above. Genomic DNA (0.5 ng) was utilized for generation of sequencing libraries using Nextera XT reagents including dual indexes following manufacturer’s protocols. Libraries were quantitated using Qubit dsDNA reagents and pooled and sequenced using a NextSeq 500 high output reagents (150 bp paired reads). Resulting reads were analyzed using MEGAN Community Edition software (27), which performs taxonomic binning by assigning reads to nodes in the National Center for Biotechnology Information (NCBI) taxonomy. The DIAMOND (6) program is used for the ultrafast alignment of reads against NCBI-nr, and Meganizer performs both taxonomic and functional analysis. Clustering of genes with similar functional roles was performed in MEGAN using the SEED classification system (49). Briefly, SEED clusters genes into a rooted tree, where nodes represent “subsystems” of allied roles that make up a metabolic pathway, a complex, or a class of proteins (45). Gene annotations mapped to eukaryotes were removed from subsequent analyses. Sample number K2501 was found to have insufficient sequencing depth and was removed from the analysis. The median sample depth for metagenomic sequencing was 2.3 Gbp. Species and functional gene clusters were removed if <6% of samples had <190.5 sequence reads (0.5 × the median of the minimum nonzero counts of each gene cluster). As described above, the low prevalence cutoff was to ensure adequate coverage within the lowest sized group.
All sequencing data are publicly available at the NCBI Sequence Read Archive at https://www.ncbi.nlm.nih.gov/sra (Accession No. SRP140861).
Metabolomics.
Frozen cecal contents (~35 mg) were analyzed by the West Coast Metabolomics Center at UCD for metabolomics assessment of metabolites associated with primary metabolism using gas chromatography-time-off-flight-mass spectrometry (GC-TOF-MS). Details of this assay have been previous published (21). Briefly, frozen cecal contents were pulverized by bead beating and then extracted with degassed acetonitrile:isopropanol:water (3:3:2; vol/vol/vol) at −20°C. Samples were centrifuged, decanted, and then dried. A set of 13 fatty acid methyl esters was added as an internal control followed by derivatization with methoxyamine hydrochloride in pyridine (10 μl) and 90 μl N-methyl-N-(trimethylsilyl)-trifluoroacetamide. Samples were then assessed by GC-TOF-MS. Raw spectra data were processed by ChromaTOF for spectral deconvolution and peak detection. Apex masses were imported to the West Coast Metabolomics Center BinBase database for peak annotation by matching mass spectrum information and retention index against spectra from the Fiehn laboratory mass spectral library (1,200 in-house standards) and the NIST05 commercial library. Metabolites were reported if present in >25% of all samples, and true peak detection must occur in at least 50% of a given condition (58). Peaks that were not structurally matched to known metabolite databases were considered as “unknown” metabolites and assigned with a BinBase numerical identifier. All metabolites were normalized by the sum of known metabolite intensities and reported as quantifier ion peak heights.
Statistical analysis.
All statistical analyses were conducted in the R Statistical Language (v 3.4.1). Group differences in α-diversity measurements were assessed with Kruskal-Wallis tests with post hoc analysis by Dunn’s test. β-diversity was assessed with Bray-Curtis dissimilarities and visualized with principal coordinate analysis (PCoA) and tested with permutational multivariate ANOVA with 500 permutations on log-shifted data. Group differences in individual taxa were determined with negative binomial Wald test from the DESeq2 Bioconductor package (40). Differentially abundant taxa were considered significant at false discovery rate <0.1. Sequencing data were not rarified (42). All other tests were considered significant at P < 0.05 unless otherwise noted. Partial least squares-discriminant analysis (PLS-DA) was used to identify discriminant features in metagenomic taxonomy (species level) and functional genetic (SEED Subsystem Level 2) and metabolomics data using the pls package (44). Data used in PLS-DA models were split sample-wise (stratified by experimental groups) into training (2/3 of data) and test (1/3 or data) sets. Training data were used for PLS-DA model development and feature selection. Test data were only utilized to determine PLS-DA model prediction accuracy. Before modeling, the metagenomic sequencing data had zeros imputed with a Bayesian-multiplicative treatment (41) and then normalized by centered log transformations. Metabolomics data were normalized by unit variance transformations. Feature selection was implemented by bootstrapping variable importance in projection (VIP). A cutoff of VIP >1 has previously been noted as an acceptable threshold to identify important features discriminating PLS-DA classifiers (64); however, others have suggested that this criterion is likely not fixed (43). Thus we calculated the adjusted bootstrap percentile confidence intervals (1,000 bootstrap replicates) for each VIP calculation and then identified discriminant features as features with a lower bound bootstrapped confidence interval (VIPbootCI) >1. Principal component analysis was used reduce the dimensionality of metabolomics data. Correlations were assessed with Spearman’s rank order correlations.
RESULTS
PD and RD UCD-T2DM rat groups had significantly greater total body weight compared with age-matched LSD rats; however, total body weights of D3M and D6M groups were significantly lower compared with PD and RD groups (Fig. 1). In-depth metabolic characterization of the UCD-T2DM rat model has been previously described (18), and we have described similar weight declines in D3M and D6M rat groups attributed to the catabolic state associated with uncontrolled insulinopenic disease (54).
Fig. 1.
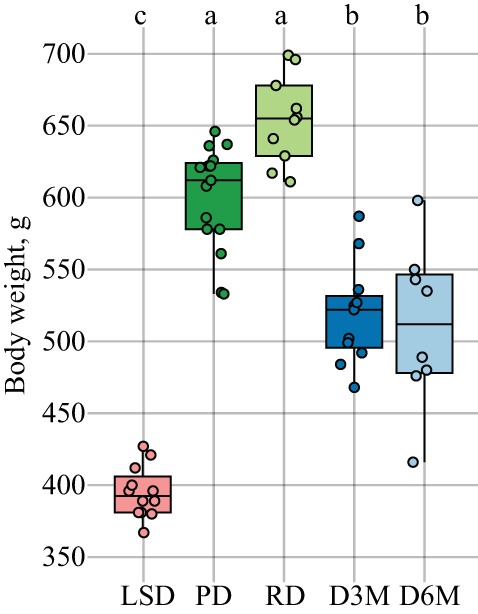
Total body weights (g) of adult male lean Sprague-Dawley (LSD) and University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats. Rats include LSD (n = 12), UCD-T2DM rats before onset of diabetes (PD; n = 15) or 2 wk (RD; n = 10), 3 mo (D3M; n = 11), and 6 mo (D6M; n = 7) postonset of diabetes. Rats within LSD, PD, RD, and D3M groups were age matched (~180 days old), while rats within D6M group are technically D3M rats aged an additional 90 days. Points in plots are individual rats and are jittered for clarity. Statistical difference assessed by Kruskal-Wallis test followed by Dunn’s test. Boxes without a common letter differ. All boxplots were constructed using default settings in the R graphics::boxplot() function.
Composition of cecal microbiome is altered in later stages of diabetes progression relative to early stages.
We initially utilized 16S rRNA amplicon sequencing data to identify whether there were large compositional differences in the cecal microbiota during the progression of diabetes. PCoA visualization of Bray-Curtis Dissimilarities of OTUs found distinct separation of LSD rats from UCD-T2DM rats along PC1 with no visible discrimination within the UCD-T2DM rat groups in the two-dimensional plot (Fig. 2A). This discrimination was found to be statistically significant by permutational multivariate ANOVA (P < 0.05). It is important to note that the LSD group is genetically distinct from the UCD-T2DM rat model and numerous studies have shown significant variation in the gut microbiome among rodent strains (15, 20, 34, 51). Therefore, we excluded LSD rats and reassessed the PCoA with only UCD-T2DM rats. Dropping the LSD rats revealed that RD and PD groups clustered together, but separate from D3M and D6M groups, which clustered together (Fig. 2B). The discrimination of UCD-T2DM in early stages of diabetes from those in later stages of diabetes was found along PC1 of the PCoA (Fig. 2B). Next, we overlaid the year each sample was collected on the same PCoA shown in Fig. 2B and found that the variance explaining year of collection was also explained along PC1 (Fig. 2C). This suggests that there was a batch effect by collection year and this effect overlaps with the variance associated with early and late stages of diabetes. As described in the methods, RD rats (indicated with “x” in Fig. 2C) were only assessed in the 2016 cohort, which is likely driving this observation. Thus techniques commonly used to correct for batch effects (29) were not effective at minimizing the variance associated with year of collection in β-diversity analyses.
Fig. 2.

Operational taxonomic unit level discrimination of adult male lean Sprague-Dawley (LSD) and University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats by 16S rRNA amplicon sequencing. Principal coordinate analysis (PCoA) samples plot of Bray-Curtis dissimilarity measurements (A) of LSD rats (n = 12, pink lines) and UCD-T2DM rats groups: before onset of diabetes (PD; n = 15; dark green lines) or 2 wk (RD; n = 10; light green lines), 3 mo (D3M; n = 11; dark blue lines), and 6 mo (D6M; n = 7; light blue lines) postonset of diabetes; UCD-T2DM rats (B) overlaid by group: PD (dark green), RD (light green), D3M (dark blue), and D6M (light blue); or UCD-T2DM (C) rats overlaid by collection year: 2014 (orange lines) and 2016 (purple lines). Symbols at the end of colored lines denote study group: PD (o), RD (x), D3M (∆), and D6M (+). Colored lines in PCoA plots extend from group medoids to the PCoA scores that represent an individual rat. PCoA plots in B and C are the same PCoA but overlaid with different class assignments.
Taxonomic distribution in shotgun metagenomic sequencing confirms 16S rRNA sequencing data at phylum level.
The positive findings in the 16S rRNA amplicon sequencing data suggest that later stage established diabetes contributes to compositional differences in the cecal microbiome in the UCD-T2DM rat; however, 16S rRNA amplicon sequencing data do not provide species level coverage nor provide information related to the genetic capacity of the bacterial ecosystem. Therefore, we further assessed the cecal microbiome with shotgun metagenomics to give us species level depth in the gut microbiome.
The phylum distribution of shotgun metagenomic reads showed similar patterns when compared with the 16S rRNA sequencing reads, i.e., Firmicutes, Bacteroidetes, and Proteobacteria made up the majority of reads while Verrucomicrobia was more proliferative in LSD rats and showed similar trends in several UCD-T2DM rats (Fig. 3A). There were several additional lower abundant phyla identified in the shotgun metagenomic data not identified in the 16S rRNA data; however, these phyla consisted of 1–2% of total reads. Similar to the 16S rRNA amplicon sequencing data, PCoA of metagenomic species counts showed a significant (P < 0.05) separation of early and late stages of UCD-T2DM rats along the first PC (Fig. 3B). Variance associated with year of collection was also apparent along PC1 of the PCoA (P < 0.05), suggesting a significant batch effect across different platforms (Fig. 3C).
Fig. 3.

Comparison of 16S rRNA amplicon sequencing and shotgun metagenomic taxonomy data from cecal contents of lean Sprague-Dawley (LSD) and University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats. A: stacked bar charts of the phylum level percent relative abundances among LSD and UCD-T2DM rats. Individual rats are represented column-wise between both plots. Phyla detected by technologies are represented by a single color in both graphs. Only phylum found with a mean %relative abundance >0.05% were included in shotgun metagenome color legend. B: principal coordinate analysis (PCoA) of Bray Curtis dissimilarities of species level taxonomy data from shotgun metagenome assessment. Samples plot overlaid with UCD-T2DM rats groups: before onset of diabetes (PD; n = 15; dark green lines) or 2 wk (RD; n = 12; light green lines), 3 mo (D3M; n = 11; dark blue lines), and 6 mo (D6M; n = 8; light blue lines) postonset of diabetes. C:UCD-T2DM rats overlaid by collection year: 2014 (orange lines), 2016 (purple lines). Symbols at the end of colored lines denote study group: PD (o), RD (x), D3M (∆), and D6M (+). Colored lines in PCoA plots extend from group medoids to the PCoA scores that represent an individual rat. Ellipses are the SE of the point scores along the 2 components. PCoA plots in B and C are the same PCoA but overlaid with different class assignments.
Cecal microbiome and metabolome are altered by early and late stages of diabetes in the UCD-T2DM rats, independent of diet.
We then utilized PLS-DA to predict whether bacteria species in the cecal content can predict stages of diabetes progression in the UCD-T2DM rat. First, we developed PLS-DA models predicting all four UCD-T2DM groups and found that this model correctly predicted <31% of group classifiers in the test data (Table 1), suggesting that bacterial species could not fully distinguish the deterioration of metabolic health when four groups were considered together. We further used PLS-DA to assess whether the bacterial genetic data could predict all four UCD-T2DM groups, and although the overall prediction accuracy of test samples was greater than the taxonomy PLS-DA model, it only managed to correctly classify <62% of held out samples (Table 1). As the bacterial genetic data from the shotgun metagenomic sequencing cannot determine the presence of DNA translation or actual metabolic activities of the microbe populations, we coupled the sequencing data with untargeted metabolomics to predict enzymatic function in the cecal environment. Again, using PLS-DA for all four classification groups, we found similar results to the metagenomic genetic PLS-DA model with <62% classification accuracy of test data (Table 1). Together, these results suggest that the cecal microbiome and metabolome cannot capture acute changes in the progression of diabetes in the UCD-T2DM rat when all four groups are included in the analysis.
Table 1.
Partial least squares-discriminant analysis model performance for 4-class modela
| Training Data |
Test Data |
||||
|---|---|---|---|---|---|
| Factor Level | Latent Variableb | Cross validation Accuracyc | Overall Accuracyd | Sensitivitye | Specificitye |
| Metagenomics species taxonomyf | 1 | 54.5% | 30.8% | ||
| PD | 0.60 | 0.38 | |||
| RD | 0.00 | 1.0 | |||
| D3M | 0.33 | 0.60 | |||
| D6M | 0.00 | 1.0 | |||
| Metagenomics functional gene clustersf | 4 | 57.4% | 61.5% | ||
| PD | 0.80 | 0.75 | |||
| RD | 0.67 | 1.0 | |||
| D3M | 0.33 | 0.80 | |||
| D6M | 0.50 | 0.91 | |||
| Metabolomics: primary metabolismf | 9 | 66.1% | 61.5% | ||
| PD | 0.60 | 0.80 | |||
| RD | 1.0 | 1.0 | |||
| D3M | 0.33 | 0.80 | |||
| D6M | 0.5 | 0.82 | |||
Four-class partial least squares-discriminant analysis (PLS-DA) model includes classifiers for age-matched, male University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats before the onset of diabetes (PD; n = 15), 2 wk postonset of diabetes (RD; n = 10), 3 mo postonset of diabetes (D3M; n = 11), and 6 mo postonset of diabetes (D6M; n = 7). Data were split into training and test sets (67%:33%). Training and test splits were stratified by classifiers (i.e., distribution of within groups were equal in test and training sets). Models were preprocessed and fit with training data only. Test data were used only for class prediction.
Latent variable associated with highest overall 6-fold cross validation prediction accuracy.
Prediction accuracy from 6-fold cross validation.
Prediction classification accuracy of test data (n = 13) using the number of latent variables with highest classification accuracy from cross validation results in training set.
Sensitivity and specificity calculated for each group by comparing each group against all remaining levels in test set, i.e., one level vs. all remaining samples.
Count data from metagenomics analysis were normalized by centered log transformations before modeling. Metabolomics quantifier ion peak height data were log transformed and scaled to unit variance before modeling.
Based on our observation that microbiota patterns appeared to discriminate early and late diabetes (Figs. 2B and 3B), and our previous finding that the plasma metabolome in this model distinguishes early and late stages of disease (54), we next modeled the microbiome and metabolomics data using Early and Late classifiers: e.g., combinations of PD and RD groups (Early) and D3M and D6M groups (Late). This resulted in at least 85% overall prediction accuracy in test samples across all analytical platforms (Table 2). We also developed PLS-DA models predicting the year of collection as unsupervised methods showed evidence that collection year contributed to a significant portion of the overall variance in the microbial data (Fig. 3C). Overall prediction accuracy of these models ranged between 69 and 77% in held out samples, which were lower than the two-class PLS-DA models predicting Early/Late classifiers (Table 2). Overall, the results strongly suggest a batch effect in samples collected in 2014 and 2016.
Table 2.
Partial least squares-discriminant analysis model performance for 2-class modela
| Training Data |
Test Data |
||||
|---|---|---|---|---|---|
| Model | Latent variableb | Cross validation accuracyc | Overall accuracyd | Sensitivitye | Specificitye |
| Metagenomics species taxonomyf | |||||
| Early/Late | 2 | 87.2% | 84.6% | 0.88 | 0.80 |
| Collection | 2 | 89.7% | 76.9% | 0.57 | 1.0 |
| Metagenomics functional gene clustersf | |||||
| Early/Late | 2 | 76.9% | 84.6% | 0.88 | 0.80 |
| Collection | 6 | 89.2% | 69.2% | 0.57 | 0.83 |
| Metabolomics: primary metabolismf | |||||
| Early/Late | 4 | 83.6% | 84.6% | 0.88 | 0.80 |
| Collection | 9 | 80.8% | 76.9% | 0.57 | 1.0 |
Two-class partial least squares-discriminant analysis (PLS-DA) model includes classifiers for age-matched, male University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats in early stages of diabetes (n = 25; combining rats sampled before diabetes onset and 2 wk postonset of diabetes) and later stages of diabetes (n = 18; combining rats sampled 3 mo and 6 mo postonset of diabetes). Training and test splits were stratified by classifiers (i.e., distribution of within groups were equal in test and training sets). Models were preprocessed and fit with training data only. Test data were used only for class prediction.
Latent variable associated with highest overall 6-fold cross validation prediction accuracy.
Average prediction classification accuracy of out-of-bag samples from 6-fold cross validation.
Average prediction classification accuracy of test data (n = 13) using the number of latent variables with highest classification accuracy from cross validation results in training set.
Sensitivity and specificity calculated for each group by comparing each group against all remaining levels in test set, i.e., 1 level vs. all remaining samples.
Count data from metagenomics analysis were normalized by centered log transformations before modeling. Metabolomics quantifier ion peak height data were log transformed and scaled to unit variance before modeling.
Prevotella and Bacteroides spp. are enriched in later stages of diabetes in the UCD-T2DM rat.
We next used a conservative filtering approach to identify features that contributed to the discrimination of Early and Late stages of diabetes in the UCD-T2DM rat. First, we identified bacterial species with a VIPbootCI >1 in PLS-DA models predicting Early/Late and collection year classifiers, and then, we filtered bacterial species that met this criterion in both models and then selected those that still remained as discriminators of the Early/Late PLS-DA model (Supplementary Table 1; Supplemental Material for this article is available online at the Journal website). A total of 45 bacterial species met these filtering criteria, and all belonged to Bacteroidetes and Firmicutes phyla (Fig. 4). All selected Firmicutes species were lower in later stages of diabetes (except Clostridium sp. CAG:964), and all Bacteroidetes species were greater in later stages of diabetes with the exception of Prevotella scopos (Fig. 4). Bacteroides and Prevotella spp. accounted for > 55% of all selected bacterial species, with P. sp. MA2016, P. bergensis, P. maculosa, P. sp. CAG:592, P. sp. BP1-148, P. albensis, P. sp. Kh1p2, P. sp. KHD1, B. plebeius, B. nordii, and B. timonensis having eight times greater abundances in late stages of diabetes relative to early stages.
Fig. 4.
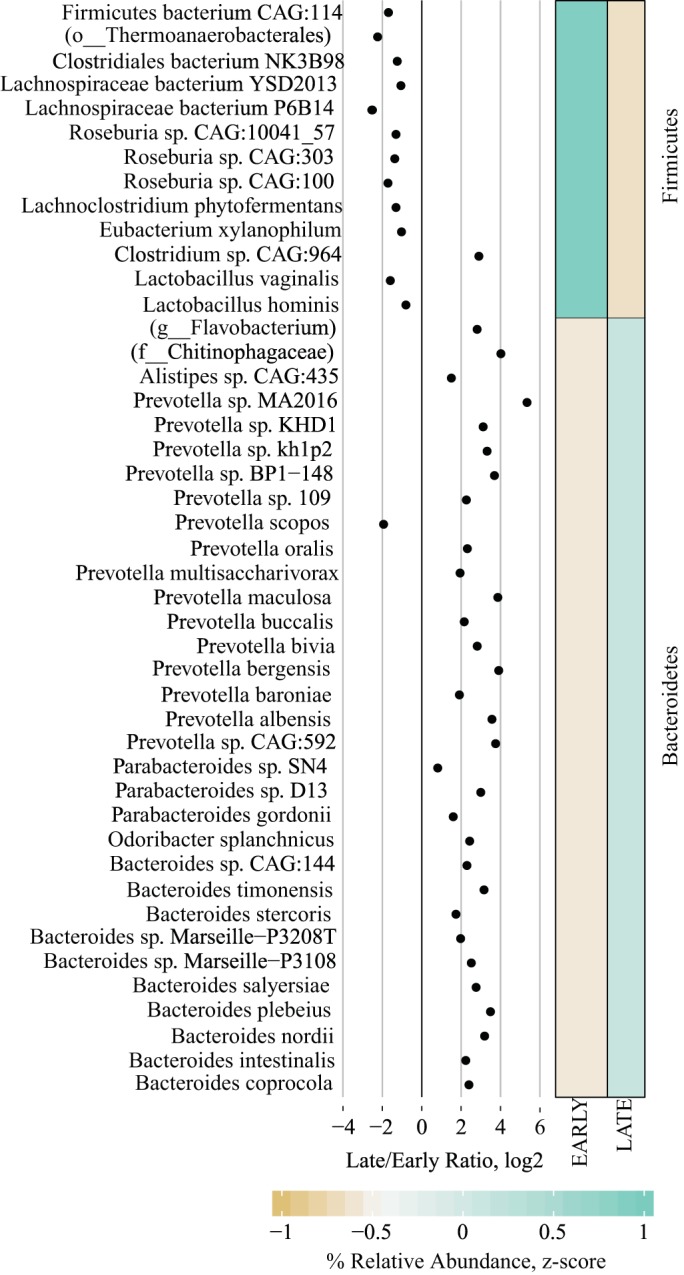
Differences in Early and Late stages of diabetes progression in cecal contents from University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats using taxonomic species count data from shotgun metagenomic sequencing. Each point in the dot plot represents the log2-fold ratio between Early and Late stages of diabetes progression in UCD-T2DM rat. Heatmaps represent the median values of scaled relative abundances. Discriminant features determined as follows: Separate partial least squares-discriminant analysis (PLS-DA) models were fit for Early and Late stages of diabetes and year of collection classifiers. Lower bounds of adjusted bootstrap percentile confidence intervals of variable importance in projection calculations (VIPbootCI) were assessed both models. Featured variables were selected if VIPbootCI >1 in Early vs. Late PLS-DA and VIPbootCI <1 in collection year model. UCD-T2DM rats included rats before onset of diabetes (PD; n = 15) or 2 wk (RD; n = 10), 3 mo (D3M; n = 11), and 6 mo (D6M; n = 7) postonset of diabetes. Rats in PD and RD groups were classified as “Early” and D3M and D6M were classified as “Late.” Classifiers for year of collection were Y14 (n = 17) and Y16 (n = 26).
Bacterial gene clusters associated with amino acid, carbohydrates, vitamin, cell wall metabolism, and stress response were enriched in later stages of diabetes in the UCD-T2DM rat.
We used the same filtering approach as described above for PLS-DA models fit with bacterial genetic data (Supplemental Table 2) and identified 61 gene clusters that discriminated Early and Late classifiers, with the majority of these elevated in later stages of diabetes relative to earlier stages (Fig. 5). Enriched SEED Level 1 subpathways included gene clusters associated with amino acid, carbohydrate, cofactor/vitamin, cell wall metabolism, and stress response (Fig. 5). Other than a single carbohydrate-based gene cluster (Citrate Metabolism KE2), all gene clusters within these groups were elevated in later stages of diabetes.
Fig. 5.
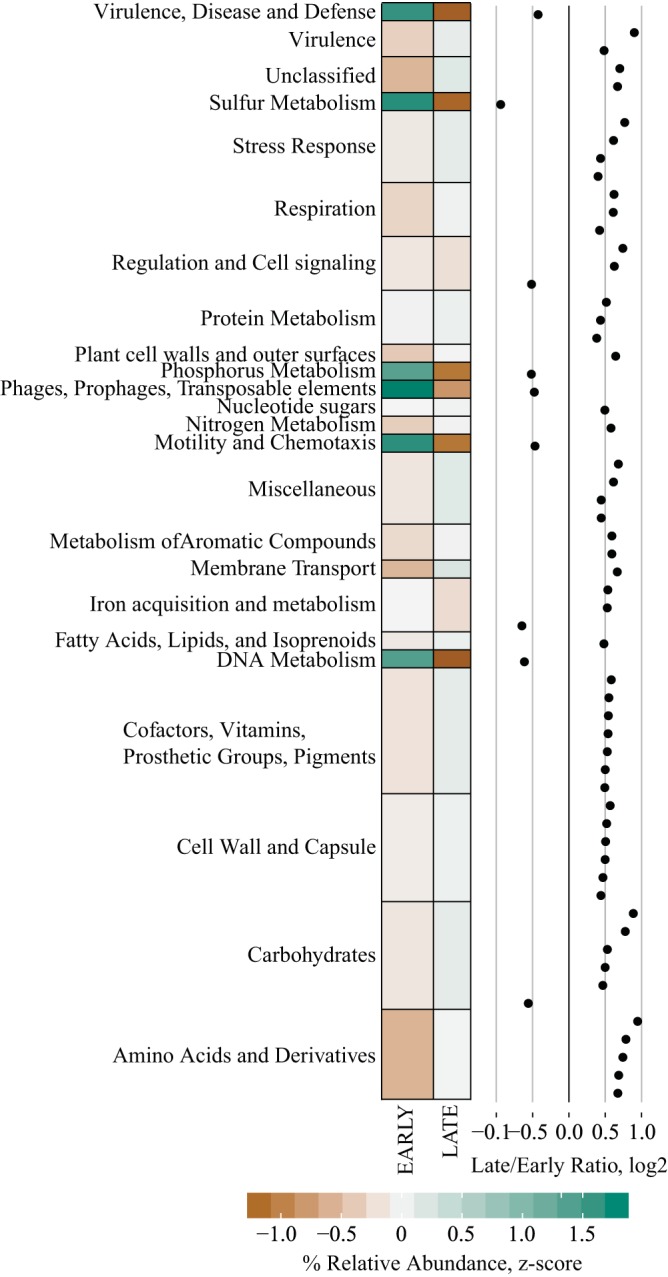
Differences in Early and Late stages of diabetes progression in cecal contents from University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats using SEED level 2 subsystem data from shotgun metagenomic sequencing. Each point in the dot plot represents the log2-fold ratio between Early and Late stages of diabetes progression in UCD-T2DM rat. Heatmaps represent the median values of summed relative abundances of all SEED level 2 subsystems within their respective level 1 subsystem. Discriminant features determined as follows: Separate partial least squares-discriminant analysis (PLS-DA) models were fit for Early and Late stages of diabetes and year of collection classifiers. Lower bounds of adjusted bootstrap percentile confidence intervals of variable importance in projection calculations (VIPbootCI) were assessed both models. Featured variables were selected if VIPbootCI >1 in Early vs. Late PLS-DA and VIPbootCI <1 in collection year model. UCD-T2DM rats included rats before onset of diabetes (PD; n = 15) or 2 wk (RD; n = 10), 3 mo (D3M; n = 11), and 6 mo (D6M; n = 7) postonset of diabetes. Rats in PD and RD groups were classified as “Early” and D3M and D6M were classified as “Late.” Classifiers for year of collection were Y14 (n = 17) and Y16 (n = 26).
Phosphate, vitamin E, and lipid metabolism indexes are elevated in cecal contents at later stages of diabetes and correlate to enrichment of select Bacteroidetes species in UCD-T2DM rats.
Again, using the same filtering approach described above for metabolomics data (Supplemental Table 3), 25 cecal content metabolites discriminated UCD-T2DM rats classified by Early and Late stages of diabetes. Cecal content abundances of dehydroabietic acid, cholesterol, arachidonic acid, hexuronic acid, lysine, N-acetylornithine, butyrolactam, and several unknown metabolites were reduced in later stages of diabetes relative to early stages (Fig. 6A), while phosphate, inositol-4-monophosphate, γ-tocopherol, arachidic acid, and stearic acid were greater in later stages of diabetes compared with early stages (Fig. 6A).
Fig. 6.

Metabolomics differences in Early and Late stages of diabetes progression in cecal contents from and University of California, Davis Type 2 Diabetes Mellitus (UCD-T2DM) rats. A: discriminant metabolites from untargeted metabolomics assessment of primary metabolism (gas chromatography-quadrupole time-off-flight-mass spectrometry). Each point represents the log2-fold ratio of a selected metabolite between Early and Late stages of diabetes progression in UCD-T2DM rat. Heatmaps represent the medians of scaled quantifier peak ion heights. Discriminant metabolites determined as follows: separate partial least squares-discriminant analysis (PLS-DA) models were fit for Early and Late stages of diabetes and year of collection classifiers. Lower bounds of adjusted bootstrap percentile confidence intervals of variable importance in projection calculations (VIPbootCI) were assessed both models. Featured variables were selected if VIPbootCI >1 in Early vs. Late PLS-DA and VIPbootCI <1 in collection year model. UCD-T2DM rats included rats before onset of diabetes (PD; n = 15) or 2 wk (RD; n = 10), 3 mo (D3M; n = 11), and 6 mo (D6M; n = 7) postonset of diabetes. Rats in PD and RD groups were classified as “Early” and D3M and D6M were classified as “Late.” Classifiers for year of collection were Y14 (n = 17) and Y16 (n = 26). B: scores plot (i.e., sample plot) of unsupervised principal component analysis of discriminant metabolites selected in A. Colored lines in principal coordinate analysis (PCoA) plots extend from group medoids to the PCoA scores that represent an individual rat. Ellipses are the SE of the point scores along principal component analysis of the 2 components. Metabolite loadings are overlaid on top of scores plot. Boxplot on top of graph indicate groups differences of scores along principal component 1 (PC1). *Statistical differences between groups, as assessed by Kruskal-Wallis test. Outliers in boxplots are displayed as a black dot extending from whisker(s). C and D: radial plots of significant correlations (Spearman’s) among PC1 from B and featured bacterial species (top) and functional gene cluster (bottom). Radial circles indicate Spearman’s rank correlation coefficient (rho) level. Orange lines extending from middle are positive rho coefficients and blue lines are negative rho coefficients.
Unsupervised principal component analysis was utilized to reduce the discriminant metabolomics data into a single variable that captured variance associated with discrimination of early and late stages of diabetes (44.1% explained variance along PC1; Fig. 6B). This component was then correlated among diabetes-altered bacterial species and gene clusters. PC1 negatively correlated with species within the Bacteroidetes phylum and positively correlated to species in the Firmicutes phylum, suggesting the shift to Bacteroidetes species was concurrent with elevations in phosphate, inositol-3-phosphate, γ-tocopherol, arachidic acid, and stearic acid (Fig. 6C). PC1 also had primarily negative correlations with gene subsystems, with the strongest correlations among bacterial osmoregulation, terminal cytochrome oxidases, cell envelope metabolism, phosphotransferases, urease subunits, and urea decomposition, and CBSS-452863.6-peg.1046 (Fig. 6D).
DISCUSSION
To our knowledge, the results herein are the first to comprehensively describe the cecal microbiome and metabolome during the progression of a type 2 diabetes phenotype. We found robust differences between UCD-T2DM rat groups that can accurately discriminate later stages of diabetes compared with early stages of the disease, across all analysis platforms. The results indicate that severe loss of metabolic control, i.e., dysregulation associated with long-term uncontrolled diabetes, alters the taxonomic and genetic composition of the cecal microbiota and cecal metabolome, independent of diet, housing environment, sex, and age in the UCD-T2DM rat. Although there is a large body of literature describing microbial differences between healthy rodent models and obese-diabetic/insulin resistant models (1, 26, 39, 47, 61, 65), very little is known about transition from the symbiotic relationship between the host and gut microbes to potential dysbiosis that accompanies deterioration of metabolic homeostasis in T2DM. It should be emphasized again that the UCD-T2DM rats in this study consumed the same diet at all stages of diabetes, unlike diet-induced obesity models of obesity (22, 26, 47, 60). Therefore, alterations in the cecal microbiome and metabolome are likely influenced by host-mediated factors in this model. While these factors remain unknown, gastrointestinal complications of T2DM that could influence microbial populations include alterations in gut motility, digestion, and absorption (35) and chronic activation of the innate immune system (13). Notably, our results suggest an upregulation of microbial genes associated with bacterial cell wall metabolism in later stages of diabetes, including capsular polysaccharides and lipopolysaccharides, which have previously been linked to obesity and insulin resistance (11). Further studies are required to determine if these factors play a role in linking the T2DM phenotype to changes in the gut microbiome.
Several species belonging to the Prevotella and Bacteroides genera dominated the Bacteroidetes shift in later stages of diabetes in the UCD-T2DM rat. Species from both genera (Prevotella copri and Bacteroides vulgatus) have been associated with T2DM phenotypes in humans (38, 52), but these particular species were not discriminant of diabetes progression in our rat model. Prevotella and Bacteroides spp. are saccharolytic (32) and would likely flourish in a progressive hyperphagic response to a carbohydrate rich chow diet observed in UCD-T2DM rats (18); this may explain species-specific differences in the bacterial shifts associated with the diabetes phenotype. Several functional gene clusters associated with oxidative stress (e.g., glutaredoxin, glutathione, and periplasmic stress) and osmoregulation were increased in late diabetes, hinting at a challenging intestinal environment coincident with diabetes. Similar to our findings, bacterial upregulation of genes associated with glutathione has been identified with diabetes in humans (55). Functional gene clusters associated with capsular polysaccharides and lipopolysaccharides were also greater in UCD-T2DM rats from later stages of diabetes progression, which could suggest that these microbes exacerbate intestinal inflammation and host immune response. It has been shown that lipopolysaccharide derived from Bacteroides and Prevotella spp. can stimulate several cytokines in vitro, but the responses are variable between species and are lower compared with responses of other known pathogens like Escherichia coli (62). Altogether, while speculative, the evidence here alludes to a situation where worsening metabolic health promotes the colonization of lower intestine bacteria that exacerbate host inflammatory and immune responses in the gut.
As noted above, the UCD-T2DM rat is hyperphagic compared with control strains and increase their food consumption as they progress through diabetes (18). By 3 mo postonset of diabetes, the UCD-T2DM rat has lost half of its total body weight despite a 30–40% increase in energy consumption (18, 19). This loss of energy has mainly been attributed to glucosuria and polyuria but could also be due to malabsorption caused by gastrointestinal complications associated with T2DM (35). In the latter case, the large intestine would be presented with a high volume of undigested material, which would likely radically alter the composition of the gut microbiome independent of alterations in host metabolism. In addition to the direct effects of excess nutrient availability, severe polyuria would alter fluid absorption and retention in the large intestine, further disrupting the environment of the lumen (e.g., alterations in luminal pH and osmolarity). Interestingly, bacterial genes associated with osmoregulation were altered with diabetes progression in UCD-T2DM rats and were also significantly correlated to changes in the cecal content metabolome, suggesting that changes in intestinal fluid dynamics are occurring in this model. Still, investigations of hyperphagia and its effect on the gut microbiome under wasting conditions have not been assessed and future studies will be required to differentiate the host-mediated effects of later stage established diabetes from malabsorption issues related to hyperphagia.
To better understand how shifts in microbial populations lead to changes in bacterial metabolism and molecular signaling, we utilized untargeted metabolomics to identify a wide range of cecal metabolites during diabetes progression. Several metabolites were discriminant of diabetes progression and correlated to bacterial gene clusters; however, only phosphate and inositol-3-phosphate were directly related to differentially abundant gene clusters related to phosphate metabolism (phosphotransferase, phosphoenolpyruvate phosphomutase, and DNA phosphorothioation). Although the present study design cannot prove mechanistic links, it is known that alterations in available phosphate or phosphate substrates can have profound effects on bacterial energy and carbon metabolism in addition to facilitating environmental niches for opportunistic taxa (59). While other metabolites may not directly relate to functional pathways identified herein, they do provide new insight into the changing intestinal environment as diabetes progresses. Dehydroabietic acid, a diterpene found primarily in plants, was lower in UCD-T2DM rats from later stages of diabetes. This metabolite has previously been reported to reduce epididymal white adipose tissue mRNA levels and circulating concentrations of monocyte chemoattractant protein-1 and tumor necrosis factor-α when administered experimentally to obese diabetic KK-Ay mice (30). However, we did not detect this compound in plasma using the same study paradigm in the UCD-T2DM rat (54), which suggests that it is either poorly absorbed or further metabolized by the host. Derivatives of dehydroabietic acid have antibacterial and antifungal properties (57), which could help control gut colonization by pathogenic bacteria in early stages of diabetes. γ-Tocopherol (vitamin E) was also increased in later stages of diabetes, suggesting a potential difference in redox balance in the cecal environment. Several bacterial gene clusters associated with vitamin K were also enriched in later stages of diabetes, suggesting that microbial and/or host fat-soluble vitamin metabolism may be altered in the gut during the progression of diabetes. The Fiehn chemical standard library does not currently include phylloquinone or menaquinone; thus we were unable to confirm whether these metabolites are reflective of alterations in bacterial gene expression. Still, bacterial metabolism of fat soluble vitamins during diabetes is not known and may be significant to host vitamin status during the progression of the disease.
Our untargeted metabolomics approach (GC-QTOF-MS) identifies a wide range of metabolites associated with cellular metabolism that includes but was not limited to mammalian and bacterial sources. Thus, we detected a limited set of xeno-metabolites (metabolites derived or modified from nonhost origin) including polyamines (spermidine, putrescine), tryptophan catabolic products (indoles), phenols [e.g., 3-(4-hydroxyphenyl)-propionic acid), and certain bile acids (e.g., deoxycholic acid). Many of these specific metabolites have been shown to modify the luminal environment (e.g., pH and osmolarity) and/or regulate intestinal epithelial energy metabolism, proliferation, and barrier integrity (4). Furthermore, these xeno-metabolites may possess biological activity in peripheral tissues, which may have positive or deleterious effects at these sites depending on a myriad of factors in the organism (48). We did not observe major differences in these more well characterized xeno-metabolites as a function of early and late stages of diabetes in the UCD-T2DM rats even though we would hypothesize that later stages of diabetes is characterized by altered colonic epithelial metabolism and decreased barrier function. Our inability to identify a diabetes-specific effect in more well characterized xeno-metabolites may be due to our conservative statistical methods combined with a potentially low precision of these metabolites in an untargeted metabolomics approach and/or absence of potentially discriminant metabolites in the database used for metabolite identification. It should be emphasized that untargeted metabolomics is limited in its reliability of quantification due its inherent lack of metabolite standards, which minimizes its comparability between studies (9). Thus results from this study should be considered as discovery-based findings (hypothesis generating) and needs to be verified in future studies.
We utilized the UCD-T2DM model to keep all external inputs fixed and consistent. Furthermore, we used only used age-matched male animals in a colony that has been in the same facility and vivarium for >12 yr. Thus, 2 yr after our initial collection, we opportunistically added an additional time to study (RD) and increased the total sample size in other groups. Several studies suggest that environment is the key driver of the gut microbiota in rodent colonies. For example, stable gut microbiome communities were observed in transgenerational C57BL/6J mice regardless of ob genotype (39) and between mothers and their transplanted offspring from different genetic backgrounds (23). Thus we expected a relatively stable microbiome in the UCD-T2DM colony; however, we found a significant batch effect between the two collection periods. Given the unbalanced study design between collection years, we were unable to minimize the variance associated with the time of collection with commonly used statistical approaches (37). Instead, we utilized a very conservative filtering approach to determine specific bacterial and metabolite features that discriminate diabetes progression despite the variability by year. Our results suggest that the gut microbiome is not stable across UCD-T2DM rat generations, and this highlights that future studies in rodent models should take sampling time into account when assessing the microbiome.
Our understanding of the complex relationship between the gut microbiome and host metabolic health and regulation is still evolving. Results from this study suggest that T2DM-related alterations in the microbiome are not solely driven by diet and the environment of the host but are likely an interaction of internal and external factors that provide niches for opportunistic bacteria that associate with metabolic dysregulation in the host. Our descriptive assessment of the bacterial composition and metagenome in the UCD-T2DM rat model aligns with results from human studies, suggesting an oxidative environment that promotes Prevotella and Bacteroides spp. that have the potential to cause dysbiosis and inflammation in the host. Alterations in the cecal metabolome correlate to changes in bacterial gene clusters and could also influence intestinal redox status and contribute to bacterial colonization. Identifying and understanding diabetes-associated host signals (e.g., metabolites, hormones, inflammation and immune regulators) or intestinal alterations (e.g., immune cell infiltration, mucin thickness, tissue morphology, motility) that shape the gut microbial ecology present an exciting new frontier and challenge.
GRANTS
This research was supported by intramural U.S. Department of Agriculture-Agricultural Research Service Projects 5306-51530019-00 and 6026-51000-010-05S and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant R01-DK-078328-01 (to S. H. Adams). Additional funding support from the University of Arkansas for Medical Sciences College of Medicine Research Council and the Sturgis Foundation for Diabetes Research and National Institute of General Medical Sciences Grants P20-GM-121293 and P20-GM-125503. P. J. Havel’s laboratory also received funding during the project period from National Heart, Lung, and Blood Institute Grants R01-HL-091333, R01-HL-107256, and R01-HL-121324 and NIDDK Grants U24-DK-092993, RC1-DK-087307, and R01-DK-095060 and a Multicampus Award from the University of California, Office of the President (Award 142691).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.D.P., P.J.H., and S.H.A. conceived and designed research; B.D.P., J.L.G., K.L.S., K.E.M., U.D.W., K.S., and P.J.H. performed experiments; B.D.P., I.N., and S.V.C. analyzed data; B.D.P. and S.H.A. interpreted results of experiments; B.D.P. prepared figures; B.D.P. drafted manuscript; B.D.P., P.J.H., and S.H.A. edited and revised manuscript; B.D.P., J.L.G., K.L.S., I.N., K.E.M., S.V.C., U.D.W., K.S., P.J.H., and S.H.A. approved final version of manuscript.
REFERENCES
- 1.Arora T, Seyfried F, Docherty NG, Tremaroli V, le Roux CW, Perkins R, Bäckhed F. Diabetes-associated microbiota in fa/fa rats is modified by Roux-en-Y gastric bypass. ISME J 11: 2035–2046, 2017. doi: 10.1038/ismej.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723, 2004. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhute SS, Suryavanshi MV, Joshi SM, Yajnik CS, Shouche YS, Ghaskadbi SS. Gut microbial diversity assessment of indian type-2-diabetics reveals alterations in eubacteria, archaea, and eukaryotes. Front Microbiol 8: 214, 2017. doi: 10.3389/fmicb.2017.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blachier F, Beaumont M, Andriamihaja M, Davila AM, Lan A, Grauso M, Armand L, Benamouzig R, Tomé D. Changes in the luminal environment of the colonic epithelial cells and physiopathological consequences. Am J Pathol 187: 476–486, 2017. doi: 10.1016/j.ajpath.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10: 57–59, 2013. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59–60, 2015. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 7.Byndloss MX, Bäumler AJ. The germ-organ theory of non-communicable diseases. Nat Rev Microbiol 16: 103–110, 2018. doi: 10.1038/nrmicro.2017.158. [DOI] [PubMed] [Google Scholar]
- 8.Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, Litvak Y, Lopez CA, Xu G, Napoli E, Giulivi C, Tsolis RM, Revzin A, Lebrilla CB, Bäumler AJ. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357: 570–575, 2017. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cajka T, Fiehn O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal Chem 88: 524–545, 2016. doi: 10.1021/acs.analchem.5b04491. [DOI] [PubMed] [Google Scholar]
- 10.Cani PD. Gut cell metabolism shapes the microbiome. Science 357: 548–549, 2017. doi: 10.1126/science.aao2202. [DOI] [PubMed] [Google Scholar]
- 11.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti J-F, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 12.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–1481, 2008. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 13.Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 3: 279–288, 2012. doi: 10.4161/gmic.19625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336, 2010. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmody RN, Gerber GK, Luevano JM Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17: 72–84, 2015. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chassaing B, Ley RE, Gewirtz AT. Intestinal epithelial cell toll-like receptor 5 regulates the intestinal microbiota to prevent low-grade inflammation and metabolic syndrome in mice. Gastroenterology 147: 1363–1377.e17, 2014. doi: 10.1053/j.gastro.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox AJ, Zhang P, Bowden DW, Devereaux B, Davoren PM, Cripps AW, West NP. Increased intestinal permeability as a risk factor for type 2 diabetes. Diabetes Metab 43: 163–166, 2017. doi: 10.1016/j.diabet.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Cummings BP, Digitale EK, Stanhope KL, Graham JL, Baskin DG, Reed BJ, Sweet IR, Griffen SC, Havel PJ. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am J Physiol Regul Integr Comp Physiol 295: R1782–R1793, 2008. doi: 10.1152/ajpregu.90635.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummings BP, Stanhope KL, Graham JL, Baskin DG, Griffen SC, Nilsson C, Sams A, Knudsen LB, Raun K, Havel PJ. Chronic administration of the glucagon-like peptide-1 analog, liraglutide, delays the onset of diabetes and lowers triglycerides in UCD-T2DM rats. Diabetes 59: 2653–2661, 2010. doi: 10.2337/db09-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ericsson AC, Davis JW, Spollen W, Bivens N, Givan S, Hagan CE, McIntosh M, Franklin CL. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS One 10: e0116704, 2015. doi: 10.1371/journal.pone.0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiehn O, Kind T. Metabolite profiling in blood plasma. Methods Mol Biol 358: 3–17, 2007. doi: 10.1007/978-1-59745-244-1_1. [DOI] [PubMed] [Google Scholar]
- 22.Fleissner CK, Huebel N, Abd El-Bary MM, Loh G, Klaus S, Blaut M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br J Nutr 104: 919–929, 2010. doi: 10.1017/S0007114510001303. [DOI] [PubMed] [Google Scholar]
- 23.Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS One 5: e8584, 2010. doi: 10.1371/journal.pone.0008584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu C, Yang Y, Xiang H, Li S, Liang L, Sui H, Zhan L, Lu X. Deciphering bacterial community changes in zucker diabetic fatty rats based on 16S rRNA gene sequences analysis. Oncotarget 7: 48941–48952, 2016. doi: 10.18632/oncotarget.10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakkak R, Korourian S, Foley SL, Erickson BD. Assessment of gut microbiota populations in lean and obese Zucker rats. PLoS One 12: e0181451, 2017. doi: 10.1371/journal.pone.0181451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen Y-Y, Knight R, Ahima RS, Bushman F, Wu GD. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137: 1716–1724.e1, 2009. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh HJ, Tappu R. MEGAN Community Edition–interactive exploration and analysis of large-scale microbiome sequencing data. PLOS Comput Biol 12: e1004957, 2016. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, Mohan V, Balasubramanyam M. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem 388: 203–210, 2014. doi: 10.1007/s11010-013-1911-4. [DOI] [PubMed] [Google Scholar]
- 29.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127, 2007. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 30.Kang M-S, Hirai S, Goto T, Kuroyanagi K, Kim YI, Ohyama K, Uemura T, Lee JY, Sakamoto T, Ezaki Y, Yu R, Takahashi N, Kawada T. Dehydroabietic acid, a diterpene, improves diabetes and hyperlipidemia in obese diabetic KK-Ay mice. Biofactors 35: 442–448, 2009. doi: 10.1002/biof.58. [DOI] [PubMed] [Google Scholar]
- 31.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498: 99–103, 2013. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 32.Könönen E, Wade WG, Citron DM. Bacteroides, porphyromonas, prevotella, fusobacterium, and other anaerobic gram-negative rods. In: Manual of Clinical Microbiology (11th ed.). Washington, DC: American Society of Microbiology, 2015, p. 967–993. doi: 10.1128/9781555817381.ch54. [DOI] [Google Scholar]
- 33.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79: 5112–5120, 2013. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreznar JH, Keller MP, Traeger LL, Rabaglia ME, Schueler KL, Stapleton DS, Zhao W, Vivas EI, Yandell BS, Broman AT, Hagenbuch B, Attie AD, Rey FE. Host genotype and gut microbiome modulate insulin secretion and diet-induced metabolic phenotypes. Cell Reports 18: 1739–1750, 2017. doi: 10.1016/j.celrep.2017.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan B, Babu S, Walker J, Walker AB, Pappachan JM. Gastrointestinal complications of diabetes mellitus. World J Diabetes 4: 51–63, 2013. doi: 10.4239/wjd.v4.i3.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5: e9085, 2010. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28: 882–883, 2012. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leite AZ, Rodrigues NC, Gonzaga MI, Paiolo JCC, de Souza CA, Stefanutto NA, Omori WP, Pinheiro DG, Brisotti JL, Matheucci Junior E, Mariano VS, de Oliveira GL. Detection of increased plasma interleukin-6 levels and prevalence of Prevotella copri and Bacteroides vulgatus in the feces of type 2 diabetes patients. Front Immunol 8: 1107, 2017. doi: 10.3389/fimmu.2017.01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102: 11070–11075, 2005. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martín-Fernández JA, Hron K, Templ M, Filzmoser P, Palarea-Albaladejo J. Bayesian-multiplicative treatment of count zeros in compositional data sets. Stat Model 15: 134–158, 2014. doi: 10.1177/1471082X14535524. [DOI] [Google Scholar]
- 42.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLOS Comput Biol 10: e1003531, 2014. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehmood T, Liland KH, Snipen L, Saebo S. A review of variable selection methods in partial least squares regression. Chemom Intel Lab 118: 62–69, 2012. doi: 10.1016/j.chemolab.2012.07.010. [DOI] [Google Scholar]
- 44.Mevik BH, Wehrens R. The pls package: Principal component and partial least squares regression in R. J Stat Softw 18: 1–23, 2007. doi: 10.18637/jss.v018.i02. [DOI] [Google Scholar]
- 45.Mitra S, Rupek P, Richter DC, Urich T, Gilbert JA, Meyer F, Wilke A, Huson DH. Functional analysis of metagenomes and metatranscriptomes using SEED and KEGG. BMC Bioinformatics 12, Suppl 1: S21, 2011. doi: 10.1186/1471-2105-12-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mokkala K, Pellonperä O, Röytiö H, Pussinen P, Rönnemaa T, Laitinen K. Increased intestinal permeability, measured by serum zonulin, is associated with metabolic risk markers in overweight pregnant women. Metabolism 69: 43–50, 2017. doi: 10.1016/j.metabol.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 47.Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, Clarke SF, O’Toole PW, Quigley EM, Stanton C, Ross PR, O’Doherty RM, Shanahan F. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59: 1635–1642, 2010. doi: 10.1136/gut.2010.215665. [DOI] [PubMed] [Google Scholar]
- 48.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science 336: 1262–1267, 2012. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 49.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42, D1: D206–D214, 2014. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park MY, Kim SJ, Ko EK, Ahn SH, Seo H, Sung MK. Gut microbiota-associated bile acid deconjugation accelerates hepatic steatosis in ob/ob mice. J Appl Microbiol 121: 800–810, 2016. doi: 10.1111/jam.13158. [DOI] [PubMed] [Google Scholar]
- 51.Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, Mehrabian M, Ursell LK, He A, Castellani LW, Zinker B, Kirby M, Drake TA, Drevon CA, Knight R, Gargalovic P, Kirchgessner T, Eskin E, Lusis AJ. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17: 141–152, 2013. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BAH, Forslund K, Hildebrand F, Prifti E, Falony G, Le Chatelier E, Levenez F, Doré J, Mattila I, Plichta DR, Pöhö P, Hellgren LI, Arumugam M, Sunagawa S, Vieira-Silva S, Jørgensen T, Holm JB, Trošt K, Kristiansen K, Brix S, Raes J, Wang J, Hansen T, Bork P, Brunak S, Oresic M, Ehrlich SD, Pedersen O; MetaHIT Consortium . Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535: 376–381, 2016. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- 53.Peregrín-Alvarez JM, Sanford C, Parkinson J. The conservation and evolutionary modularity of metabolism. Genome Biol 10: R63, 2009. doi: 10.1186/gb-2009-10-6-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piccolo BD, Graham JL, Stanhope KL, Fiehn O, Havel PJ, Adams SH. Plasma amino acid and metabolite signatures tracking diabetes progression in the UCD-T2DM rat model. Am J Physiol Endocrinol Metab 310: E958–E969, 2016. doi: 10.1152/ajpendo.00052.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto J-M, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490: 55–60, 2012. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 56.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214, 2013. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Savluchinske-Feio S, Curto MJ, Gigante B, Roseiro JC. Antimicrobial activity of resin acid derivatives. Appl Microbiol Biotechnol 72: 430–436, 2006. doi: 10.1007/s00253-006-0517-0. [DOI] [PubMed] [Google Scholar]
- 58.Scholz M, Fiehn O. SetupX–a public study design database for metabolomic projects. Pac Symp Biocomput 169–180, 2007. [PubMed] [Google Scholar]
- 59.Shimizu K. Metabolic regulation of a bacterial cell system with emphasis on escherichia coli metabolism. ISRN Biochem 2013: 645983, 2013. doi: 10.1155/2013/645983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223, 2008. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031, 2006. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 62.Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hämäläinen A-M, Peet A, Tillmann V, Uibo R, Mokurov S, Dorshakova N, Ilonen J, Virtanen SM, Szabo SJ, Porter JA, Lähdesmäki H, Huttenhower C, Gevers D, Cullen TW, Knip M, Xavier RJ; DIABIMMUNE Study Group . Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165: 842–853, 2016. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328: 228–231, 2010. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wold S, Sjostrom M, Eriksson L. PLS-regression: a basic tool of chemometrics. Chemom Intell Lab 58: 109–130, 2001. doi: 10.1016/S0169-7439(01)00155-1. [DOI] [Google Scholar]
- 65.Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, Zhang X, Zhao L. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci Rep 5: 14405, 2015. doi: 10.1038/srep14405. [DOI] [PMC free article] [PubMed] [Google Scholar]