

Abstract
Intrauterine growth restriction (IUGR) is linked to increased risk for chronic disease. Placental ischemia and insufficiency in the mother are implicated in predisposing IUGR offspring to metabolic dysfunction, including hypertension, insulin resistance, abnormalities in glucose homeostasis, and nonalcoholic fatty liver disease (NAFLD). It is unclear whether these metabolic disturbances contribute to the developmental origins of exaggerated cardiovascular-renal disease (CVRD) risk accompanying IUGR. IUGR impacts the pancreas, adipose tissue, and liver, which are hypothesized to program for hepatic insulin resistance and subsequent NAFLD. NAFLD is projected to become the major cause of chronic liver disease and contributor to uncontrolled type 2 diabetes mellitus, which is a leading cause of chronic kidney disease. While NAFLD is increased in experimental models of IUGR, lacking is a full comprehension of the mechanisms responsible for programming of NAFLD and whether this potentiates susceptibility to liver injury. The use of well-established and clinically relevant rodent models, which mimic the clinical characteristics of IUGR, metabolic disturbances, and increased blood pressure in the offspring, will permit investigation into mechanisms linking adverse influences during early life and later chronic health. The purpose of this review is to propose mechanisms, including those proinflammatory in nature, whereby IUGR exacerbates the pathogenesis of NAFLD and how these adverse programmed outcomes contribute to exaggerated CVRD risk. Understanding the etiology of the developmental origins of chronic disease will allow investigators to uncover treatment strategies to intervene in the mother and her offspring to halt the increasing prevalence of metabolic dysfunction and CVRD.
Keywords: hypertension, inflammation, insulin resistance, placental ischemia, steatosis
INTRODUCTION
Environmental perturbations in early life predispose offspring to chronic disease. This is the basis for the “developmental origins of health and disease” theory set forth by Dr. David Barker and colleagues. The importance of understanding the developmental programming of chronic disease is becoming increasingly relevant due to the rising exposure rates of offspring to poor intrauterine environments, such as maternal smoking and drug use, maternal under- or overnutrition, obesity, maternal diabetes, environmental toxins, or hypertensive disorders of pregnancy (25, 57, 107, 216). Hypertensive disorders of pregnancy, including preeclampsia (PE), affect ~9% of pregnancies in the United States (retrieved February 12, 2018: https://www.cdc.gov/reproductivehealth/maternalinfanthealth/pregnancy-complications-data.htm). Hypertensive disorders of pregnancy are associated with IUGR (216). IUGR programs offspring for obesity, proinflammatory status (157), insulin resistance, and type 2 diabetes mellitus (T2DM) (51).
In addition to metabolic abnormalities, intrauterine growth restriction (IUGR) is associated with developmental programming of cardiovascular-renal disease (CVRD). CVRD is the leading cause of death worldwide, with the main modifiable risk factor being hypertension (232). Blood pressure in IUGR adolescents is already increased by 6 yr of age (186) implicating a higher risk of hypertension in IUGR offspring as they age. Obesity and resulting metabolic diseases, like T2DM, are thought to contribute to 70% of essential hypertensive cases (85). While evidence demonstrates that T2DM exacerbates hypertensive renal disease in experimental animals (213), it is unknown whether developmental programming of metabolic disease risk factors contributes to exaggerated risk for CVRD in IUGR offspring.
It is well established that nonalcoholic fatty liver disease (NAFLD) and T2DM coexist (38). Yet, how these two morbidities are linked is not fully understood. Several studies implicate a role for insulin resistance in the etiology of steatosis (fatty liver) (97, 176), whereas there is no consensus that NAFLD initiates T2DM (16, 76, 106). Complexity in the timing of presentation of these comorbidities is evidenced in adolescents and young adults, whereby some individuals with T2DM have no evidence of NAFLD at the time of diagnosis of T2DM while others have both T2DM and NAFLD (154). Abnormal birth weight is a suggested contributor to the development of advanced NAFLD in children (153), but it is unclear if a history of complications from IUGR predicts or exaggerates the risk for metabolic disease. Although not all patients with NAFLD and T2DM were IUGR, experimental studies suggest that IUGR programs earlier insulin resistance with additional studies indicating an increased risk for NAFLD in IUGR offspring. It has not yet been examined whether early insulin resistance drives accelerated development of NAFLD in IUGR. It is established that accumulation of hepatic lipids fosters hepatic insulin resistance (165, 218). Furthermore, progression of NAFLD and liver injury may worsen insulin resistance making diabetes more difficult to treat. Liver-specific knockout of the insulin receptor (IR) results in insulin resistance, glucose intolerance, and reduced insulin-mediated suppression of hepatic glucose production suggesting the importance of protecting against hepatic insulin resistance (142). Combined hepatic insulin resistance, NAFLD, and diabetes may contribute to the greater risk for cardiovascular disease (81). Currently, it has not been substantiated whether IUGR hastens the progression of hepatic insulin resistance to NAFLD and liver injury and whether the latter worsens T2DM and diabetes-induced CVRD in these at-risk offspring. Therefore, the purpose of this review is to propose that developmental programming for increased susceptibility to metabolic diseases, like insulin resistance, contributes to the incidence of IUGR-induced chronic disease.
DEVELOPMENTAL PROGRAMMING OF CHRONIC DISEASE: IUGR AND EPIDEMIOLOGY
Barker et al. (23, 24) proposed that developmental programming of chronic disease is dependent on reduced fetal growth independent of gestational age. The definition of impaired fetal growth varies. Low birth weight (LBW) is defined as birth weight <10th percentile (2,500 g) regardless of gestational week. LBW is often used as a surrogate marker for IUGR. IUGR, when associated with an increased brain-to-liver weight ratio, is indicative of asymmetric IUGR and predicts an enhanced risk for future pathologies in the offspring. However, being born LBW is not always associated with inappropriate gestational age at birth. Small-for-gestational age (SGA) is defined as birth weight <10th percentile for gestational week; those associated with IUGR are termed “SGA associated with IUGR.” Alarmingly, the prevalence of IUGR is increasing in the United States (132).
Although the origins of IUGR are multifactorial, a main cause within the Western world is due to placental insufficiency and ischemia (84). Placental ischemia is an initiating event in PE (75). PE is defined as new-onset hypertension in the presence of neural, vascular, uterine, and/or renal abnormalities in the mother occurring after the 20th week of gestation. PE is a contributor to both maternal and fetal morbidity immediately during pregnancy and long term. PE is one of the major risk factors for IUGR in conjunction with unemployment, smoking, drug abuse, and maternal age >34 yr (109). PE, which includes preterm PE with delivery <34 wk, contributes to a respective 2.7 and 4.3 times increase in the odds of having an IUGR baby at <10 and <5% of fetal growth, as calculated using gestational age at delivery and absolute birth weight (192). There is no current treatment for IUGR beyond close monitoring of the pregnancy and early delivery (22), which highlights the necessity for preclinical research to identify novel prophylactic strategies. Preclinical research using experimental animal models is required to develop cost-effective therapeutic strategies to treat pregnancy-related disorders, such as PE, to attenuate the incidence of IUGR. Furthermore, it is critical to study IUGR offspring to identify mechanistic targets to prevent long-lasting adversities on chronic health, which include the developmental programming of NAFLD and T2DM.
In this review, we will highlight the epidemiology of NAFLD and the evidence suggesting that it magnifies metabolic dysfunction. Next, we will provide an overview of evidence from experimental models that suggest the development of accelerated insulin resistance in IUGR offspring, which is likely mediated via programming of pancreatic β-cell loss and dysfunction of insulin-signaling pathways in adipose tissue and liver leading to steatosis. Earlier onset of insulin resistance may promote more progressive steatosis, NAFLD, and liver injury in patients with a history of IUGR. We will detail mechanisms of liver injury, as discovery of approaches to reverse liver injury may also allow for improvement in clinically uncontrolled metabolic diseases, such as T2DM (52). Lastly, we will explore the hypothesis that metabolic disease mediates the known exaggerated risk for hypertensive CVRD in IUGR offspring with a focus on a role for proinflammatory pathways.
DIAGNOSING NAFLD
NAFLD is the most common form of chronic liver disease (184) and is divided into nonalcoholic fatty liver (NAFL) and its advanced form of nonalcoholic steatohepatitis (NASH). Upon biopsy and histological inspection, NAFL is defined as isolated hepatic steatosis with or without mild lobular inflammation, while advancement to NASH includes features of inflammation and hepatocyte ballooning (39). The appearance of fibrosis is indicative of ongoing NASH (2). Although patients are diagnosed with NAFLD by noninvasive factors, presently, and despite its invasive nature, the gold standard for diagnosing NAFLD is by histological staging of liver biopsies (11). This diagnosis is defined by the NAFLD activity score (NAS) and includes the presence of steatosis (0–3), inflammation (0–2), hepatocellular ballooning (0–2), and fibrosis (0–4) (12). NAS scores ≥5 are diagnosed as NASH (110). In a follow-up study of a median 6.6 yr, 37% of NAFL patients and 43% of NASH patients had progressive fibrosis (139). The fibrotic process of NAFLD may be worsened by T2DM, and NAFLD is proposed to render control of diabetes more challenging (38). Therefore, it is important understand the role that NAFLD plays in the etiology of T2DM and whether approaches to target and attenuate this liver disease allow for better control of metabolic disease.
NAFLD IS A MAJOR RISK FACTOR FOR UNCONTROLLED METABOLIC DISEASE
Health care burden of NAFLD.
It is observed that 10–20% of patients with NAFL progress to NASH (139, 205), and ~20% of patients with NASH advance to cirrhosis (173). Between 1999 and 2013, the death rate from chronic liver disease and cirrhosis rose by 22% in America (45). In 2013, chronic liver disease and cirrhosis accounted for 101,000 discharges as the first-listed diagnosis and contributed to 36,427 deaths in America (79). According to the National Institute of Diabetes and Digestive and Kidney Diseases, NASH is the third leading cause of cirrhosis after hepatitis C and alcoholic liver disease in patients awaiting liver transplantation (Cirrhosis; retrieved Sept. 1, 2017: https://www.niddk.nih.gov/health-information/liver-disease/cirrhosis). Forty-six percent of patients with NASH develop decompensated cirrhosis within 10 yr and require transplantation (72, 179). Currently, the only steadfast treatment for liver failure is transplantation (135, 136). NASH has increased as a cause for liver transplantation from 1.2 to 9.7% (128) and is projected to become the primary reason for liver transplantation (3, 220, 236).
NAFLD exaggerates metabolic dysfunction in T2DM.
Studies suggest that diagnosing NAFLD may identify those individuals at greater risk for metabolic dysfunction in T2DM (143, 228). Epidemiological studies suggest that NAFLD and T2DM coexist (56, 66). In a Chinese cohort, NAFLD was implicated in advancing T2DM (123). The prominent histological feature in patients with NAFLD that is associated with long-term outcomes, including advancement of T2DM, is liver fibrosis (15). NAFLD-induced liver fibrosis and injury may exaggerate insulin resistance. Indeed, in a Korean population, ultrasonography, fibrotic scoring (49, 196), and the fatty liver index (calculated using body mass index, waist circumference, triglycerides, and GGT levels) revealed that diabetes is greater with increasing NAFLD and liver fibrosis scores (227). The World Health Organization reports that T2DM or high blood glucose is associated with 3.8 million deaths in 2012 (WHO Diabetes Fact Sheet;. retrieved October 7, 2017: http://www.who.int/en/news-room/fact-sheets/detail/diabetes). T2DM is projected to be the seventh leading cause of worldwide death by 2030 (137) underscoring the importance of understanding the mechanisms that encourage its pathology, which includes determining the impact of developmental programming of metabolic disease risk factors, like NAFLD.
Do IUGR and developmental programming contribute to the increasing incidence of NAFLD?
In America, between 80 and 90% of obese adults have some level of NAFLD, resulting in ~95 million Americans with some stage of the disease. The incidence of NAFLD is increasing in all races in the United States and throughout the world, including in adolescents (215). The incidence has risen in obese children to 40–70% (58). The rising prevalence of NAFLD may be partly explained by fetal programming of NAFL and NASH by the in utero environment. Children with LBW and high birth weights are at increased odds for this disease compared with their normal birth weight counterparts (153). Moreover, experimental studies highlight that early adversities predisposed offspring for chronic metabolic disease are suggested in male rats born from high-fat diet (HFD)-fed dams that develop exaggerated HFD-induced obesity, increased liver weight and circulating leptin levels, significantly reduced adiponectin levels, an insulin-sensitizing adipokine, and insulin resistance (185, 202). Similar diet protocols in male mice offspring from dams on a HFD have exaggerated HFD- or methionine choline-deficient diet-induced NASH, as detected by profibrotic gene expression (150, 214). To aid in understanding the mechanisms mediating fetal programming of chronic disease in adult offspring, including NAFLD, researchers have utilized the knowledge that placental ischemia and insufficiency are potent stimuli for IUGR. Experimental animal models of placental ischemia and insufficiency have been developed to study the impact of IUGR on programming of chronic disease.
EXPERIMENTAL MODELS OF IUGR
Models of placental ischemia and insufficiency used to identify placental factors mediating IUGR.
Induction of placental ischemia and insufficiency contributes to the pathogenesis of hypertension in PE and the development of IUGR. The model of surgical-induced reduced uterine perfusion pressure (RUPP) promotes hypertension in the dam (Fig. 1A) and reduces placental mass (Fig. 1B) and fetal weight (Fig. 1C) by gestational day 19 in once normotensive pregnant rats. To produce the RUPP model, a silver clip is placed on the abdominal aorta, directly above the iliac bifurcation and on both branches of the ovarian arteries at day 14 of gestation. At delivery, the RUPP dams give birth to male and female pups with reduced body weight indicative of IUGR (Fig. 1D). Along with the RUPP rat, additional experimental models are proving useful to support the causal role of placental insufficiency on inducing IUGR and that these IUGR offspring are programmed for increased odds of chronic disease as they age.
Fig. 1.

Reduced uterine perfusion pressure (RUPP) produces maternal hypertension (A), reduced placental weight (B), and reduced fetal weight (C) by gestational day (GD) 19. RUPP reduces birth weight in both male and female offspring (D). *P < 0.05 vs. normal pregnant; **P < 0.05 vs. corresponding control offspring of same sex from normal pregnant dams. [Panel A adapted from Alexander et al. (6) with permission from Wolters Kluwer Health Inc. Copyright © 2001 American Heart Association Inc.; panels B and C adapted from Spradley et al. (191) with permission from Wolters Kluwer Health, Inc. Copyright © 2016 American Heart Association, Inc.; panel D adapted from Intapad et al. (95) with permission from Wolters Kluwer Health, Inc. Copyright © 2015 American Heart Association, Inc.]
Antiangiogenic placental factors induce IUGR.
It is reported that factors released from the ischemic placenta induce IUGR. Circulating and placental levels of the antiangiogenic factor soluble Fms-like tyrosine kinase (sFlt-1), which binds and quenches bioavailable vascular endothelial growth factor and placental growth factor that are important for maintenance of vascular health, are increased in RUPP dams (191). In pregnant Sprague-Dawley rats, chronic excess levels of sFlt-1 elicit hypertension and IUGR (37). Furthermore, adenovirus-driven chronic increases in sFlt-1 during pregnancy in mice result in smaller offspring at weaning and a significant increase in blood pressure and vascular dysfunction in adult male but not female IUGR offspring (41) (130). When chronic sFlt-1 excess is combined with maternal HFD, these offspring have a further increase in mean arterial pressure and circulating inflammatory markers relative to IUGR counterparts retained on regular fat chow (42). Furthermore, the chronic sFlt-1 excess model is associated with mechanisms that support the pathogenesis of NAFLD. Offspring of chronic sFlt-1 dams have increased fetal hepatic peroxisome proliferator-activated receptor (PPAR)-α levels (194). In contrast, hepatic mRNA PPAR-α levels are reduced in male and female IUGR rats at gestational day 20 in the nutrient-restricted rodent model of IUGR (238). It has not been examined whether these changes in expression remain as IUGR offspring age. Yet, it is important to define the timing of altered PPAR-α expression and signaling during development. PPAR-α is implicated to mediate early lipogenesis in the development of steatosis (229) whereas it is also reported to be protective against inflammation and fibrosis in progression to NASH (161). The role of hepatic PPAR-α in controlling the progression of NAFLD and liver injury has not been examined in adults from IUGR models of developmental insult. Moreover, it is unclear if antiangiogenic factors are involved in fetal programming of liver disease directly by signaling in the fetal liver or indirectly by encouraging placental insufficiency and IUGR.
Proinflammatory placental factors induce IUGR.
Inflammatory factors contribute to the etiology of IUGR. In the RUPP dams, tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and agonistic autoantibodies to the angiotensin II type 1 receptor (AT1-AA) are elevated and additional studies indicate each of these contribute to the development of maternal hypertension by late gestation in this model of PE (190). Adoptive transfer of CD4+ T cells or B cells from RUPP dams induces hypertension and increase sFlt-1 and TNF-α levels in once normotensive pregnant rats. Although sFlt-1 or TNF-α antagonism reduces blood pressure in RUPP dams, long-term fetal metabolic or CVRD outcomes of such treatments have only just started to be examined. Pharmacological-induced proliferation of endogenous anti-inflammatory regulatory T cells using a rat CD28 superagonist improves blood pressure by 50% in the RUPP hypertensive model reducing circulating levels of agonistic autoantibodies to the angiotensin II type 1 receptor and a slight improvement in fetal weights (90). This improvement in maternal blood pressure and fetal weights is also found following antibody-mediated depletion of natural killer cells in RUPP dams (60), although this did not increase fetal weights to those values observed in normal pregnant dams. The inability of these treatment strategies to fully restore fetal weight in RUPP dams is most likely due to the chronic constriction produced by the clipping procedure. Thus a shortcoming of the RUPP model is the inability to fully realize the potential protective impact of targeted in utero therapies against IUGR in the face of mechanical uterine vascular constriction. Genetic models are also available and support the targeting of proinflammatory mechanisms during pregnancy to attenuate IUGR. In pregnant rats that are transgenic for the human renin-angiotensin system, the CD28 superagonist significantly improves fetal weight and brain-to-liver weight ratio without reducing maternal blood pressure (169). In summary, these data collectively support that the placental ischemia-derived milieu of antiangiogenic and proinflammatory factors induce IUGR. As mentioned above, investigators have used IUGR offspring from models of placental ischemia and insufficiency to highlight the impact that IUGR has on developmental programming of CVRD and metabolic diseases. We propose that this predisposition for metabolic disease synergizes with the programming of cardiac and renal abnormalities in IUGR offspring to accelerate the progression of CVRD-related morbidity and mortality.
Developmental programming of cardiac and renal abnormalities in IUGR offspring.
Various preclinical models of IUGR are utilized to investigate the developmental programming of CVRD. These include models of surgical-induced placental ischemia and insufficiency, including induction of the RUPP procedure in rats (191) and mice (96), dams with bilateral uterine ligation (189), or maternal hypoxia (178); infusion of antiangiogenic or proinflammatory factors into dams (41, 68); and models of dietary-induced placental insufficiency, such as maternal protein restriction (74, 208), maternal HFD or high-sucrose diets (67, 182), or maternal administration of betamethasone (34). Importantly, it is demonstrated by utilizing each of these models that there are overarching similarities by which IUGR offspring are predisposed to CVRD, which supports the developmental programming of chronic disease. IUGR offspring from many of these models have altered cardiomyocyte biology, including early reductions in cardiomyocyte numbers reported in offspring from dams with bilateral uterine ligation (30), decreased cardiomyocyte proliferation by postnatal day 1 in offspring of dams with maternal hypoxia (171), decreased cardiac expression of genes involved in heart’s ability to produce energy from fatty acid oxidation in mice offspring exposed to maternal HFD by 27 wk of age (112), and increased contractile function of isolated left ventricles by 12 wk of age in offspring from nutrient-restricted dams (80). Moreover, data from these various models of developmental programming of CVRD in IUGR offspring show reductions in nephron endowment. For example, a reduced nephron complement is observed in IUGR offspring following maternal uterine ligation (71), maternal hypoxia (223), maternal low-protein diet, and maternal betamethasone administration (34). The reduced nephron number is also detected in human IUGR offspring (102). This nephron deficit is accompanied by persistent glomerular hyperfiltration to maintain renal excretory function at the cost of accelerated development of renal injury, loss of renal function, and hypertension (131). Indeed, glomerular sclerosis and increased blood pressure are present by 4 mo of age in offspring of rabbits exposed to maternal HFD and high-sucrose diets (221) and in IUGR rats from dams with altered nutritional status (35). Hypertension and vascular dysfunction, noted by reduced endothelium-dependent vasorelaxation, are evident by adulthood in offspring from RUPP dams (5, 162), bilateral uterine ligation (30), maternal excess of sFlt-1 (41, 130), and HFD (221). Coronary artery endothelial dysfunction is present in IUGR in offspring from ewes with placental disease produced by placental embolization following injection of microspheres into the fetal circulation (40). In summary, the utilization of different experimental models of IUGR provide credence to the developmental programming of CVRD in offspring (14).
Developmental programming of accelerated insulin resistance in IUGR offspring.
Experiments utilizing a number of IUGR models have implicated reduced maternal health in the developmental programming of metabolic dysfunction, including mechanisms related to insulin resistance, hepatic steatosis, and gluconeogenesis (122). Fetuses of obese mothers have insulin resistance when examined in utero (47). Moreover, in a series of elegant studies from the laboratory of Simmons et al. (28, 54, 175, 189, 193), they demonstrated that IUGR is associated with abnormal insulin biology. These studies indicated that bilateral ligation of the uterine arteries in the pregnant rat produces metabolic abnormalities in offspring including increased basal insulin section, hepatic glucose production, hyperinsulinemia, and hyperglycemia and attenuated glucose-stimulated insulin secretion. β-Cell dysfunction is detected in IUGR from models induced via maternal nutrient restriction (28, 54, 175, 189, 193) and bilateral uterine ligation (189, 193). Also, reduced insulin-mediated suppression of hepatocyte glucose production is detected in IUGR offspring from bilateral uterine artery-ligated dams (99, 170). These hepatic changes occur by 6 days of life and before the onset of hyperglycemia at 3–6 mo old (211). Prevention of reductions in β-cell vascularity and islet number and increased inflammation by treatment in early life with IL-4-neutralizing antibodies reduced T-helper 2 lymphocytes and macrophages and abolished fasting hyperglycemia, but assessments of hepatic insulin signaling were not reported. Fasting hyperglycemia is also detected in male and female IUGR offspring from RUPP rats (93, 94). Under standard chow conditions, by 12 mo of age, male IUGR offspring from RUPP dams have an increased area under the curve to glucose tolerance testing without a significant change in insulin secretion in response to a fasting blood glucose challenge (94). Interestingly, these male IUGR offspring had increased fasting glucose (Fig. 2), which is indicative of hepatic insulin resistance (167), and a tendency for increased liver fat content without dramatic changes in body fat composition compared with their normal birthweight controls. In females, although steatosis was not examined, IUGR offspring from RUPP have increased body fat, reduced oral glucose tolerance, and increased fasting blood glucose. In these IUGR females, there was unaltered insulin tolerance or circulating insulin levels but glucose simulation of insulin release is impaired in response to a glucose challenge in the fasted state (93). Molecular mechanisms whereby female IUGR offspring from RUPP have impaired glucose homeostasis may involve lower pancreatic expression of glucose transporter (GLUT)2. There is no difference in muscle GLUT4 or IRβ expression, but dorsal white adipose tissue has reduced GLUT4 and increased IRβ expression implicating dysfunctional insulin signaling in adipose tissue as a contributor to developmental programming of hyperglycemia. Thus several models of IUGR provide evidence for developmental programming of insulin resistance in males and females. Whether the mechanisms leading to this point are similar between both sexes or involve sex-specific mechanisms including a role for the sex hormones is understudied. Overall, these data implicate IUGR in the acceleration of insulin resistance in these at-risk offspring. However, it is not yet known if IUGR-mediated programming of insulin resistance fosters a heightened risk for development of NAFLD (177).
Fig. 2.
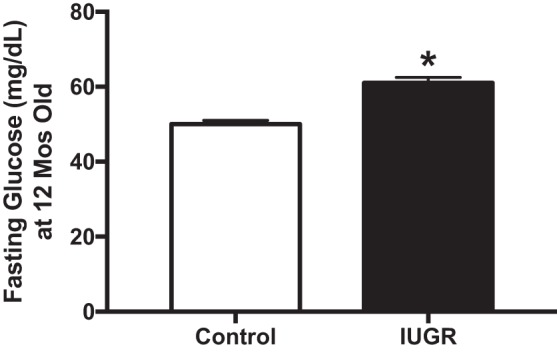
By 12 mo of age, male intrauterine growth restriction (IUGR) offspring from reduced uterine perfusion pressure (RUPP) pregnancies have elevated fasting glucose levels on normal chow diet. *P < 0.05 vs. control offspring from normal pregnant rats. [Adapted from Intapad et al. © 2017 (94), licensed under Creative Commons Attribution CC-BY 4.0.]
PROGRAMMING OF INSULIN RESISTANCE IN IUGR OFFSPRING AND ITS IMPACT ON PROMOTING NAFLD
In humans, IUGR is associated with the development of insulin resistance and T2DM (217). As stated above, studies indicate that the predisposition for T2DM in IUGR offspring is dependent on reduced pancreatic β-cell size (32). However, experimental evidence suggests that additional mechanisms outside of the pancreas contribute to developmental programming of insulin resistance in IUGR offspring. In a study by Um et al. (207), mouse dams with global S6 kinase (S6K1) deficiency resulted in reduced placental weight and IUGR offspring that exhibit reduced β-cell number and size and lower whole body insulin content but increased insulin sensitivity. This latter finding is due to the ability of this enzyme to reduce insulin signaling. To determine if the reduced placental weight potentially mediates β-cell dysfunction in these knockout offspring, placental growth was restored in the knockout offspring using a technique to generate blastocyst-stage embryos, which were produced by aggregation of tetraploid wild-type embryos with diploid S6K1 knockout embryonic stem cells. These were implanted into uteri of pseudopregnant CD1 outbred female recipient mice. At embryonic day 16.5, the fetuses and placentas were examined. These fetuses were genotyped and determined to be SGK1 knockouts, but they had increased placental weight and rescued fetal growth. However, there was still lower fetal insulin content in the S6K1 knockout mice even though there was restoration of placental weight. This implicates S6K1 as being critical for proper insulin production regardless of placental disease and IUGR. Conversely, reexpression of S6K1 specifically in β-cells of S6K1 global knockout embryos restored circulating insulin levels but did not protect against IUGR in these offspring. This pancreatic reexpression of S6K1 in the S6K1 knockout offspring did not completely restore the insulin tolerance test to levels seen in control wild-type normal birthweight offspring. Thus dysfunction of pancreatic S6K1 during fetal development is capable of reducing β-cell growth and inducing the long-term development of insulin resistance regardless of IUGR. However, another important take-home message from the studies by Um et al. is that manipulation of pancreatic S6K1 did not completely restore insulin tolerance in IUGR offspring, but the contribution to changes in extra-pancreatic tissue S6K1 expression was not studied. Others have shown that skeletal muscle and liver are sites for increased S6K1 signaling in the development of insulin resistance in HFD-induced obesity (188), but it is unknown whether changes in S6K1 contribute to the insulin resistance found in adult IUGR offspring from dams exposed to hypoxia (177) or other models of IUGR. Interestingly, S6K1 expression is reduced in myotubules isolated from skeletal muscle of neonatal pigs with reduced birth weight (50) suggestive of early compensatory mechanisms to preserve insulin sensitivity in the various IUGR models mentioned above with reduced insulin production. However, the temporal role of S6K1 as a regulator of insulin signaling, including its role in adipose tissue or liver, in IUGR offspring requires further investigation. In humans, expression of S6K1 in visceral adipose tissue is upregulated in obesity and associated with insulin resistance and inflammation (46), but the impact of IUGR of these signaling pathways is unknown. Collectively, these studies advocate for further study of mechanisms whereby IUGR provokes insulin resistance to identify potential therapeutic targets to intervene in developmental programming of metabolic disease.
Insulin resistance in adipose tissue.
Several studies suggest that proper insulin signaling in adipose tissue promotes glucose homeostasis. While overstimulation of insulin signaling in adipose tissue promotes obesity and hyperglycemia (31), insulin resistance in adipocytes attenuates glucose uptake and contributes to hyperglycemia (198). Adipose tissue inactivation of GLUT4 results in blunted insulin-stimulated glucose uptake, glucose intolerance, and hyperinsulinemia along with attenuated insulin-mediated prevention of β2-adrengergic receptor-induced lipolysis in small and large adipocytes (1). As mentioned above, female IUGR offspring from RUPP dams have adipose tissue inactivation of GLUT4 resulting in reduced insulin-stimulated glucose uptake in muscle, although GLUT4 expression remains normal in this tissue, and reduced insulin-mediated suppression of hepatic glucose production. Intriguingly, ewes with spontaneous IUGR have increased fetal hepatic gluconeogenic capacity marked by increased expression of proliferator-activated receptor-γ coactivator-1α and phos-cAMP-response element-binding protein/total cAMP-response element-binding protein ratio in their livers (203) (Fig. 3). With regards to NAFLD, studies suggest that compensatory mechanisms allow buffering of insulin-mediated lipolysis and subsequent ectopic accumulation of hepatic lipids, but it is unknown if these systems fail leading to increased risk for NAFLD and metabolic dysfunction in the face of IUGR. For example, adipocytes from obese humans and mice have increased expression of Snail1, which is a Snail transcription factor family member shown to inhibit adipocyte differentiation and expression of lipogenic and gluconeogenic genes. Insulin stimulates production of Snail1. Mice having adipocyte deletion of Snail1 have increased adipose tissue lipolysis and liver fat under normal or HFD conditions (195), but this is not yet studied in developmental programming of insulin resistance in IUGR offspring.
Fig. 3.
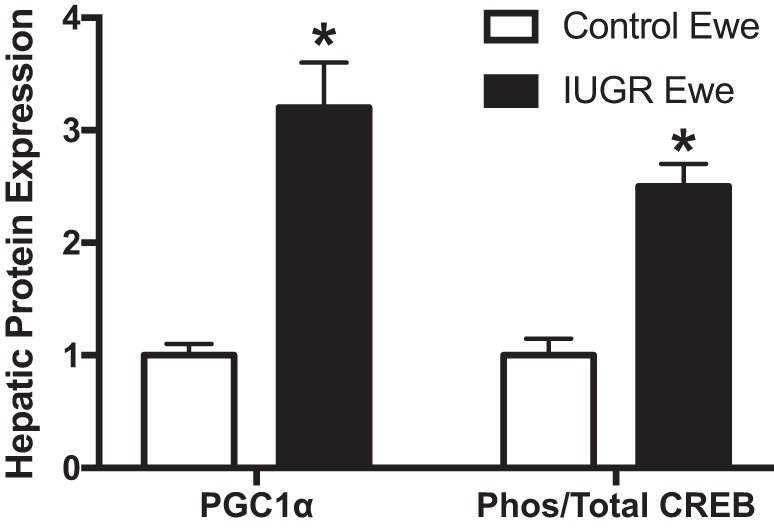
Hepatic protein expression of gluconeogenic factors in intrauterine growth restriction (IUGR) ewes at approximately gestation day (GD) 134, which were generated by exposing pregnant ewes to elevated ambient room temperature from approximately GD 38–120. Control fetuses were from pregnant ewes exposed to normal ambient temperatures. *P < 0.05 vs. control. [Adapted from Thorn et al. (203) with permission from Oxford University Press. Copyright © 2009 The Endocrine Society.]
Pathological insulin signaling in immune cells might also lead to downstream adipose tissue dysfunction and allow for the progression of NAFLD. Increased insulin signaling in inflammatory cells is implicated in development of adipose tissue dysfunction. In myeloid cells, IR deficiency attenuates HFD-induced macrophage invasion into white adipose tissue and circulating TNF-α (138). In these mice with IR deficiency in their myeloid cells, the euglycemic- hyperinsulinemic clamp studies reveal that they are protected against obesity-induced insulin resistance by decreased basal hepatic glucose production and increased insulin-stimulated glucose disposal in skeletal muscle. These studies suggest that increased insulin signaling in inflammatory cells during obesity leads to hepatic insulin resistance. This may act in concert with the finding that, in mice, hyperinsulinemia leads to reduced hepatic insulin receptor substrate (IRS)-2 expression while continuing to increase production of sterol regulatory element-binding protein-1c (SREBP-1c), a transcription factor that elicits fatty acid synthesis (187). In IUGR female rats from food-restricted dams, there are slight increases in SREBP-1c mRNA levels by gestational day 20 that persist as they age (238). These data allude to increases in insulin signaling in immune cells and adipose tissue, but insulin resistance in liver, as pathogenic in the development of NAFLD. It remains to be determined whether inflammatory mechanisms mediate these NAFLD and metabolic responses in IUGR in both males and females, especially in the face of diet-induced NAFLD.
Increased inflammation in adipose tissue is thought to maintain ectopic lipid accumulation in NAFLD and propagation of hepatic insulin resistance and T2DM (101, 180). This is based on evidence that hepatic lipid overload is mediated by increased hepatic de novo lipogenesis from glucose diverted from lipid-overloaded, insulin-resistant skeletal muscle. This excess glucose reduces hepatic insulin sensitivity and glycogen storage and allows for lipogenesis and increased release of VLDL cholesterol. The resulting hyperglycemia and hyperlipidemia subsequently leads to adipose tissue inflammation, which directs additional fat to the liver culminating in hepatic gluconeogenesis and maintained hyperglycemia (209). In mesenteric white adipose tissue, a visceral adipose tissue depot, inflammation and IL-6 signaling promote lipolysis and contribute to obesity-associated hepatic steatosis and insulin resistance (222). In humans, omental fat IL-6 mRNA levels are positively correlated with hepatic steatosis and insulin resistance and the greater visceral adipose-to-subcutaneous adipose tissue ratio in these individuals. Several studies suggest that visceral adipose tissue produces greater proinflammatory cytokines, like IL-6, than subcutaneous fat (144). Infiltration of macrophages in visceral adipose tissue in obese adolescents is associated with reduced ability of subcutaneous adipose tissue to expand and store lipids along with hypertriglyceridemia, hypoadiponectinemia, and systemic insulin resistance (113). Collectively, these studies suggest that shunting of excess lipids to visceral adipose depots or ectopically to organs, like the liver, sets off a cascade of events that promote inflammation and the onset of metabolic dysfunction. A complete understanding of how alterations in adipose tissue expansion and inflammation encourage the progression of NAFLD during the developmental programming of hyperglycemia and diabetes is warranted.
Adipocyte dysfunction and inflammation.
Adipocyte expansion allows for proper lipid storage in times of energy surplus (147). However, a chronic increase in adipocyte size is an independent risk factor for NAFLD (166). In obesity, adipocyte hypertrophy is associated with adipocyte apoptosis (9) and the onset of inflammation (36) and steatosis (10). In humans, increased adipose tissue expression of proinflammatory cytokines is associated with the severity of NAFLD (103). In mice, inhibition of adipocyte apoptosis by knocking out Bid, a proapoptotic member of the Bcl-2 family, protects against macrophage infiltration into adipose tissue and the development of hepatic steatosis (8). Studies support that this subsequently leads to the development of hepatic insulin resistance. One potential mechanism mediating this response is increased adipose tissue SREBP-1a function. SREBP-1a, sterol regulatory element-binding protein-1a, is a transcription factor that increases expression of enzymes in adipose and liver involved in fatty acid and cholesterol synthesis (13). SREBP-1a overexpression in adipose tissue in mice results in adipocyte hypertrophy, increased fatty acid secretion, increased circulating free fatty acids, and steatosis (86). This is associated with increased white adipose tissue levels of fatty acid synthase (FAS), glycerol-3-phosphate acyltransferase, and acetyl-coA carboxylase, which are enzymes linked to biosynthesis of fatty acid and triacylglycerol. Increases in these enzymes are detectable in adipose tissue isolated from models of IUGR. In an IUGR rat model produced by maternal food restriction, male IUGR offspring at 6 mo of age have greater fatty acid de novo synthesis in both subcutaneous and visceral adipose tissues with increases in acetyl-coA carboxylase-α and fatty acid synthase in these tissues (231) (Fig. 4). In pigs with IUGR, which are defined as newborns with a body weight of 2 SD below the average birth weight of all piglets from the same litter, HFD feeding from postnatal days 28–178 results in greater body fat mass, circulating triglycerides, and insulin along with total hepatic and semitendinosus muscle fat (230). Hepatic IL-6 mRNA levels are greater in IUGR pigs when fed HFD (124). In a guinea pig model of IUGR induced surgically by uterine artery ablation, there are increased lipid synthesis-related genes in visceral epididymal white adipose tissue (181). Thus there is valid rationale to examine the pathways of adipose tissue expansion, hypertrophy, and inflammation in the pathogenesis of metabolic dysfunction and NAFLD in IUGR offspring.
Fig. 4.

Subcutaneous (SC) adipose tissue (A and B) and visceral adipose tissue (C and D) expression of lipogenic proteins acetyl-coenzyme A carboxylase (ACCα) and fatty acid synthase (FASN) in 6-mo-old intrauterine growth restriction (IUGR) male offspring produced by maternal protein restriction. *P < 0.05 vs. control offspring. [Adapted from Yee et al. (231) with permission from John Wiley and Sons. Copyright © American Oil Chemists' Society 2016 (AOCS).]
Adipokines in the pathogenesis of NAFLD.
Adipose tissue also produces adipokines that have a role in the pathogenesis of NAFLD. Preventing adipose tissue hypertrophy in global knockout of caspase-2 mice prevents HFD-induced abdominal adiposity, T2DM, dyslipidemia, and steatosis (133). Furthermore, the epididymal white adipose tissue in the caspase-2 knockout mice is more proliferative with smaller adipocytes; they have increased mitochondrial function suggestive of ability to burn energy surplus; and they are resistant to adipocyte hypertrophy and death. These changes correspond with alterations in circulating adipokine levels noted by lower leptin and greater adiponectin levels mirrored by their expression in the fat tissue. In humans, in both lean and overweight individuals, the levels of leptin increase with the severity of NAFLD (87), and there are increased circulating leptin receptor levels in the morbidly obese with insulin resistance and NAFLD (140), suggestive of reduced leptin signaling. It is established that obesity is a state of leptin resistance, and there is hepatic leptin resistance in NAFLD (226). This promotes early fat accumulation, as suggested in mice with selective deletion of leptin receptors in hepatocytes where they have a slight but significant increase in liver triglyceride and cholesterol content (89). This occurs despite no change in body weight and while being maintained on a standard chow diet. Reversal studies in humans with lipodystrophy found that leptin treatment for ~6 mo results in increased serum leptin from 1.4 ± 0.3 to 25.7 ± 9.0 ng/ml and reduced steatosis (100). However, in the setting of a proinflammatory environment, as mimicked by low-dose endotoxin, leptin exaggerates the progression to NASH during HFD (91). Therefore, the role of leptin in the pathogenesis of progressive NASH is complex with even less understood about its effects in liver pathology in IUGR offspring.
With regards to adiponectin, its levels are reduced in association with increasing adipocyte size (83). Adiponectin is important in maintaining hepatic insulin sensitivity (21). Adiponectin levels are lower in mothers with IUGR (114). With regards to fetal levels, the available data are variable ranging from no change to reduced levels (210), as measured in blood from doubly clamped umbilical cords. By 7 yr of age, adiponectin levels are increased in lean SGA offspring and lower in overweight SGA offspring compared with lean appropriate-for-gestational age children all born at term (129). This was associated with increased homeostatic model assessment of insulin resistance (HOMA-IR) only in the overweight SGA group suggesting compensatory effects of adiponectin in lean SGA to protect against metabolic disease, which is lost in the face a “second hit,” such as obesity. These studies provide credence for investigation into the role of adiponectin in controlling insulin resistance and the proinflammatory mechanisms in progressive NAFLD in IUGR offspring. This is especially true in light of findings that plasma levels of adiponectin are reduced in NASH resulting in a loss of adiponectin’s protective effects against hepatic inflammation, insulin resistance, and fibrosis (65, 158). Adiponectin administration into obese ob/ob mice, which had reduced adiponectin in their adipose tissue, ameliorated hepatomegaly, steatosis, glucose and insulin tolerance tests, ALT levels, and hepatic TNF-α mRNA expression (224). However, the importance of altered adiponectin signaling in mediating proinflammatory mechanisms and increased risk for NAFLD in IUGR remains unknown.
PROINFLAMMATORY MECHANISMS MEDIATING NAFLD-INDUCED LIVER INJURY: ARE THESE MECHANISMS MORE PRONOUNCED IN IUGR TO ACCELERATE ONSET OF CHRONIC LIVER DISEASE?
Immune cells and cytokines.
A study by Campanati et al. (43) demonstrated that 24 wk of treatment with etanercept, a soluble TNF-α receptor antagonist, reduces the AST/ALT ratio, circulating levels of the inflammatory marker C-reactive protein, and fasting insulin levels in patients with established NAFLD implicating a role for proinflammatory mediators in the etiology of NAFLD-induced liver injury. The role of TNF-α as a mediator of NAFLD risk is supported by experimental studies, which demonstrate that knockout of the TNF-receptor 1 in mice protects against diet-induced steatosis and liver injury (63). In vitro studies indicate that hepatocyte TNF receptor-associated factor 3 stimulates HFD-induced liver steatosis and insulin resistance (212). In vivo studies in HFD-fed mice demonstrate that TNF-α promotes lipid accumulation in the liver (159). Furthermore, an increase in free fatty acids stimulates TNF-α production in hepatocytes (63), collectively implicating a vicious cycle in the etiology of NAFLD. However, there is a paucity of data linking these proinflammatory mechanisms to NAFLD and liver injury in IUGR.
Although observational studies in humans demonstrate an association between IUGR and NAFLD (7, 61), these reports have not examined whether proinflammatory pathways mediate this increased risk for NAFLD in IUGR offspring. The benefit of using experimental animals is the ability to test the hypothesis that proinflammatory mechanisms mediate an accelerated risk for NAFLD in IUGR offspring. This has not yet been tested directly, but studies provide rationale to the validity of this hypothesis. Adult male and female IUGR offspring from food-restricted dams vs. control dams present with greater NAFLD under HFD as adults (238). Regarding examination of inflammatory factors, surgical-induced utero-placental insufficiency programs an increase in serum and adipose tissue levels of TNF-α by postnatal day 21 in male IUGR (172). Others show that poor maternal nutrition programs greater liver inflammation, oxidative stress, and fibrosis (200). However, it has not been determined if intervening in proinflammatory mechanisms blocks the increased risk for NAFLD and liver injury in IUGR offspring.
Perry et al. (163) showed that blocking actions of the proinflammatory cytokine IL-6 by utilizing infusion of neutralizing antibodies attenuated HFD-induced hepatic insulin resistance and T2DM in rats. These investigators propose that one of the primary sites for action of this mechanism is macrophage-mediated inflammation driving lipolysis and release of acetyl CoA from white adipose tissue. The acetyl CoA targets the liver where it attenuates insulin-induced suppression of hepatic glucose production. In another study, use of oral celecoxib, an inhibitor of the proinflammatory enzyme cyclooxygenase (COX)-2, lessened HFD high-sucrose diet-induced NASH along with being beneficial for insulin tolerance, HOMA-IR, liver expression of the proinflammatory transcription factor, NF-κB, and ALT and AST levels (204). COX-2 may be localized to infiltrating immune cells in the progression of this disease. In vivo studies suggest that homing of immune cells to the liver in the progression of NASH whereby bone marrow-derived macrophages in hyperlipidemic LDLr−/− mice are important in the progression of NASH (29). A study using a model of chronic liver injury generated by oral carbon tetrachloride (CCl4) demonstrated that local hepatic Kupffer cells, a resident macrophage cell type in the liver, and infiltrating macrophages express COX-2 during the development of liver injury (219). In contrast, COX-2 expression in hepatocytes protects against models of NASH and liver injury, including CCl4 treatment (149); this protective effect may be dependent on COX-2 production of prostaglandin E2. In summary, the relative role of hepatocyte vs. immune cell COX-2 (88) should be examined in the development of NAFLD. Furthermore, these mechanisms and interventions should be utilized to probe the hypothesis that proinflammatory cytokines and immune cells mediate the accelerated risk for obesity-induced NAFLD in IUGR offspring.
Kupffer cells, an innate immune cell to the liver, mediate hepatic monocyte recruitment via production TNF-α and monocyte chemoattractant protein-1 in the pathogenesis of NASH (206). Kupffer cells also have the ability to produce the CXC chemokine receptor 3 (148), which studies suggest is a major regulator in promoting lipid accumulation, immune cell infiltration, and tissue levels of TNF-α and NF-κB (239). The CXC chemokine receptor 3 signaling mechanism also contributes to NASH via impaired cell stress sensing pathways, including endoplasmic reticulum (ER) stress and autophagy (239). Overall, ER stress-induced autophagy is thought to provide protection against tissue injury (48, 237). Collectively, it remains unexplored whether ER stress or autophagy directly contributes at any step to the accelerated progression of obesity-induced inflammation, insulin resistance, or NAFLD and liver injury in IUGR.
ER stress and inflammation.
Folding of transmembrane and secretory proteins occurs in the ER before mediating their biological functions (234). In instances of heavy protein workload, accumulation of unfolded proteins in the lumen of the ER can trigger ER stress. If proteins are not folded in their allotted time, they are targeted for ER-associated degradation by several chaperone proteins including the immunoglobulin heavy chain binding protein/glucose-regulated protein 78 (Bip/Grp78). Restoration of cellular homeostasis and adaptation is supported by the unfolded protein response with Grp78-induced activation of ER transmembrane receptors PKR-like ER kinase (PERK), activating transcription factor (ATF)6, and inositol-requiring enzyme 1 and downstream signaling intermediates eukaryotic initiation factors-2α/ATF4/stanniocalcin 2, cleaved ATF6, and X-box binding protein-1 (XBP-1), respectively (225). Under irremediable ER stress, PERK activates ATF4 that increases expression of the CCAAT-enhancer-binding homologous protein (CHOP) transcription factor and CHOP-mediated inflammation and apoptosis (120, 155). Prolonged ER stress promotes apoptosis and profibrotic mechanisms (44).
It is hypothesized that ER stress-mediated apoptosis is an important upstream mechanism mediating immune cell infiltration in the liver (4). A main initiator of this process is lipotoxicity (174). Preventing hepatic triglyceride and cholesterol accumulation in a preclinical model of NASH is associated with lower levels of the ER stress marker CHOP, an ER stress protein that induces apoptosis and cytokine production in macrophages (155), and cytokine levels of IL-1β in liver tissue along with the development of NASH (26). However, this study did not examine immune cell infiltration or inflammasome activation. The inflammasome is a multiprotein system expressed in myeloid cells that promotes inflammation and apoptosis in target tissues (77). Interestingly, ER stress induces IL-1β expression that activates the inflammasome and resultant hepatocyte death (116). Inflammasome activation is thought to also involve mitochondria to propagate the cell-death response (116). Chronic enrichment of ER-mitochondria contacts leads to hepatic mitochondrial dysfunction in obesity (19). Using a controlled-release mitochondrial protonophore to pharmacologically induced mitochondrial uncoupling, which is hypothesized to protect cells from conditions that favor reactive oxygen species production (146), is beneficial in preventing the development of HFD-induced NAFLD in rats along with reducing hypertriglyceridemia, insulin resistance, hepatic steatosis, diabetes, and NASH in methionine/choline-deficient rats (164). Increased physical and molecular interactions between the mitochondria and ER elicit apoptosis. Collectively, these studies suggest that signaling between the ER and mitochondria play a major role in the core mitochondrial apoptosis pathway (82). This background information is necessary for the discussion below detailing mechanisms of NAFLD-mediated progression of liver injury.
ER stress in NAFLD and liver injury.
Studies in human liver biopsies implicate altered ER stress signaling in the development of NAFLD. In patients with NASH, there are significantly greater levels of the ER stress markers of spliced XBP-1s and STC2 with a trend for increases in c-Jun NH2-terminal kinase (JNK), ATF4, and CHOP (115). In a separate study, intervening with a chemical chaperone, tauroursodeoxycholic acid (TUDCA), to inhibit aberrant ER stress increases liver insulin sensitivity in obese men and women (105). In an experimental model of obesity, insulin resistance, and NASH, foz/foz mice on 6-wk HFD have activation of JNK, ATF4, and CHOP, whereas XBP-1 levels are not altered (119). Inhibition of ER stress reduces steatosis but does not improve glucose intolerance or hepatic inflammation and apoptosis in these mice. This lack of an effect on these injurious mechanisms may be due to the fact that other ER stress mediators, like XBP-1, are not altered in this specific model. To probe the specific ER stress factors that may promote the development of steatosis and liver injury, ER stress factor-deficient models have been developed. Although XBP-1-deficient mice have exaggerated fructose diet-induced ER stress noted by increased factors, like JNK, they have increased hepatic insulin sensitivity and reduced hepatic lipids (104). Furthermore, it was found that XBP-1 deficiency specifically in hepatocytes attenuates HFD and high-sucrose diet-induced steatosis, whereas these mice have exaggerated diet-induced mRNA levels for markers of inflammation, apoptosis, and injury associated with increased CHOP and phosphorylated-JNK and increased hepatic injury and fibrosis (125). Moreover, in mice with Xbp1 deficiency specifically in the liver, it was shown that this ER stress mediator is important for resolution of chronic ER stress-induced liver injury (156). These data suggest that XBP-1 is an important factor promoting steatosis but is protective against the progression of NAFLD-mediated liver injury. Not as much mechanistic information is known about the ER stress pathway in mediating the predisposition NAFLD or liver injury in IUGR offspring.
Information is beginning to surface regarding IUGR and its impact on ER stress proteins during the pathogenesis of NAFLD. In a mouse model of maternal caloric restriction, expression of the hepatic ER stress markers, like spliced XBP-1s, is increased in IUGR offspring on normal diet. These IUGR offspring have greater HFD-induced steatosis accompanied by lower hepatic SREBP1 levels and increased fatty acid synthase levels and liver macrophages (152). However, following the HFD regimen, XBP-1 expression was no longer different to normal birthweight counterparts. TUDCA reduced steatosis and circulating small dense LDL cholesterol more dramatically in the IUGR offspring and reduced XBP-1s only in this offspring group along with other ER stress mediators that were exaggerated in the HFD IUGR offspring. Furthermore, TUDCA reduced the greater ALT and AST levels in these IUGR offspring on HFD indicative of attenuated liver injury (Fig. 5). Similar trends were found for reduced nonfasting glucose, but this did not reach statistical significance. However, this study did not examine these responses in comparison to age-matched IUGR or control offspring maintained on normal diet, and it is unclear if the HFD-induced ER stress, metabolic disease, and liver injury were exaggerated in IUGR in comparison with control IUGR offspring on a control diet and over the HFD responses in normal birth weight counterparts. Furthermore, it has not yet been teased apart if different ER stress factors mediate the development of steatosis vs. liver injury in HFD IUGR offspring. Collectively, these data advocate for further study of the impact that ER stress has on metabolic disease and NAFLD.
Fig. 5.

Steatotic grade (A) and plasma small dense LDL (B) and circulating markers of liver injury ALT (C) and AST (D) in male intrauterine growth restriction (IUGR) offspring from calorically-restricted (CR) pregnant C57Bl/6 mice. All offspring were on high-fat diet (60% lipids) from 9 to 22 wk treated with vehicle or the endoplasmic reticulum stress inhibitor tauroursodeoxycholic acid (TUDCA; 0.5 g·kg−1·day−1). *P < 0.05 vs. vehicle-treated control rats from ad libitum dams; **P < 0.05 vs. TUDCA-treated control rats. #P < 0.05 vs. vehicle-treated controls. [Adapted from Muramatsu-Kato et al. (152), licensed under Creative Commons Attribution CC-BY 4.0.]
ER stress and autophagy in liver injury.
When discussing ER stress control of cell survival, autophagy is important to mention. Autophagy is an adaptive response activated during cell stress that delivers proapoptotic cytoplasmic components, like damaged cellular organelles or unused proteins, to the lysosome for degradation to encourage cell survival (121). Increases in splicing of the ER stress-associated protein XBP-1 and induction of ER stress is an early adaptive mechanism to protect against excessive cell stress by activation of autophagy (108). An inability to proceed through autophagy is thought to drive apoptosis in NAFLD (183) and marks the transition to cirrhosis (53). Studies using mice with hepatocyte-specific deletion of MAP kinase kinase transforming growth factor-β-activated kinase 1 (TAK1) revealed that this factor suppresses autophagy in response to HFD allowing for exaggeration of HFD-induced steatosis, inflammation, and fibrotic injury in the liver (92). In that same study, pharmacologic-mediated increases in autophagy by rapamycin-induced increases in mammalian target of rapamycin complex 1 (mTORC1) significantly prevented hepatic apoptosis, injury, and hepatocellular carcinoma (HCC) in the TAK1-deficient mice. Intriguingly, and with regards to IUGR, it was found that newborn IUGR piglets have reduced protein expression of factors linked to activation of autophagy, including mTOR, protein phosphatase 2A, and microtubule-associated protein 1A/1B-light chain 3 along with reduced autophagic vacuoles in their livers (126) (Fig. 6). At this juncture, it is unknown if this early indication of dysfunctional autophagy persists as IUGR offspring age to mediate programming of accelerated progression of NAFLD and liver injury.
Fig. 6.

Expression of proteins associated with activation of autophagy in liver tissue from newborn piglets with intrauterine growth restriction (IUGR). A: mammalian target of rapamycin. B: catalytic subunit of protein phosphatase 2A (PP2Ac). C: microtubule-associated protein 1A/1B-light chain 3 (LC3). D: autophagic vacuoles detected by electron microscopy. *P < 0.05 vs. control, normal-birthweight piglets. [Adapted from Long et al. (126) with permission from American Chemical Society. Copyright © 2016, American Chemical Society.]
NAFLD AS A NOVEL RISK FACTOR FOR CVRD
Does NAFLD exaggerate risk for CVRD?
NAFLD is linked to increased risk for coronary heart disease (69, 117, 141) independent of other metabolic factors, including obesity (20, 160). The incidence of atrial fibrillation is also higher in hospitalized patients with NAFLD and T2DM (199). It is likely that greater CVRD in these patients presenting with NAFLD and T2DM could be due to uncontrolled diabetes. Portillo-Sanchez et al. (168) report that patients with NAFLD and T2DM exhibit greater uncontrolled diabetes over those with T2DM alone. According to the American Heart Association (Cardiovascular Disease & Diabetes; retrieved September 2, 2017: http://www.heart.org/HEARTORG/Conditions/More/Diabetes/WhyDiabetesMatters/Cardiovascular-Disease-Diabetes_UCM_313865_Article.jsp#.WiWSK7aZPXS), adults with diabetes have quadrupled the risk of death from heart disease. In a 33-yr follow-up study, individuals with NASH had reduced survival mainly due to cardiovascular disease (59, 145). Longitudinal studies suggest that NAFLD and hepatic insulin resistance are associated with the onset of hypertension (33). Moreover, in a sample of patients from the UK, 80% of those individuals with biopsy-proven steatosis and progressive fibrosis were diabetic at follow-up compared with 25% that did not have progressive fibrotic disease (139). With the use of both ultrasound and histology, NAFLD was present in >50% of patients with T2DM and was higher in NASH (66). In genetically obese foz/foz mice, HFD-induced NASH and liver fibrosis were greater and corresponded with hyperinsulinemia, hyperglycemia, HOMA-IR, hypoadiponectinemia, and hepatic levels of TNF-α (62). It is not yet clear the relative role that NAFLD-related fibrosis has on advancement of metabolic disease and CVRD.
T2DM as a mediator of exaggerated CVRD risk in NAFLD.
Rodent studies indicate a synergistic action between high glucose levels and hypertension in promoting renal injury in male nonobese Goto-Kakizaki rats, a model of nonobese T2DM and hepatic insulin resistance (213). A positive relationship was detected between hypertension and the onset of NAFLD (64). Patients with baseline hypertension, hypertriglyceridemia, impaired fasting glucose, or T2DM have increased odds for steatosis (33). In spite of this finding, the relationship of hypertension, NAFLD, and liver injury is unknown. Although hydralazine-mediated lowering of blood pressure reduces immune cell counts in hypertensive rodents (98), the importance of proinflammatory mediators has not been examined in livers from hypertensive models of obesity and progressive NAFLD. In a series of studies performed by investigators at Kagoshima University, hepatic lipid levels (17) and circulating ALT and AST were increased in male spontaneously hypertensive rats compared with their normotensive Wistar-Kyoto counterparts on a normal diet of 10% fat (118). A hypertensive insult of chronic high-salt diet exaggerated their hypertension accompanied by increased liver weight/body weight and liver fibrosis (17) along with increased insulin, fasting blood glucose, and insulin resistance (111). Antihypertensive therapy in already hypertensive rats reduced these parameters accompanied by a reduction in IL-6 and increased anti-inflammatory and vasoprotective IL-10 levels in serum. It is unknown whether the liver is a source of the circulating cytokines or whether they contribute to alterations in hypertension-induced CVRD and damage. Future studies are required to fully examine the contributions of hypertension and inflammation on the development of NAFLD and ensuing metabolic dysfunction and CVRD (56, 127).
Inflammatory markers implicated in the pathogenesis of NAFLD and liver injury may also be responsible for the exaggerated CVRD in IUGR offspring.
The above highlights the complexity of the pathogenesis of programming for cardiometabolic disease and the need for research to explore the contribution of inflammatory pathways. Inflammatory markers that are implicated in the pathogenesis of NAFL and NASH may also be responsible for CVRD associated with IUGR. A redundancy in these inflammatory pathways may explain how a history of IUGR is related to the association between T2DM and NAFLD and exaggerated CVRD risk. The aforementioned anti-inflammatory treatment studies in IUGR pregnancies did not examine hepatic or CVRD outcomes in the offspring, including their known programming for decreased cardiomyocyte proliferation (171), increased cardiac fibrosis (14), or decreased renal nephron endowment (233). Moreover, hypertension is observed in the IUGR offspring of maternal nutrient restriction, hypoxia, and RUPP dams (5). In humans, there is an association between higher fetal-placental vascular resistance and systolic blood pressure in IUGR offspring (70), but the role of inflammation in mediating the increased CVRD risk in IUGR offspring has not been examined in humans or animal models.
PERSPECTIVES AND SIGNIFICANCE
NAFLD is an increasing public health concern (27). Evidence in humans implicate that a risk factor for accelerated progression of NAFLD is IUGR. The various experimental models of IUGR discussed in this review will allow for research to assist in a better understanding of the mechanisms mediating increased risk for NAFLD in IUGR offspring. Studies already being conducted in these models of IUGR suggest that developmental programming of insulin resistance is a determinative factor in predisposing for NAFLD is these at-risk offspring. Obesity-induced inflammation is an initial signal to drive the development of hepatic insulin resistance and steatosis, which ultimately results in progressive NAFLD and liver injury. This injury response is mediated by hepatic lipotoxicity-induced inflammation and alterations in ER stress and autophagy signaling. Further research is needed to determine if intervening in these pathways, which are illustrated in Fig. 7, will halt the developmental programming of NAFLD and liver injury in IUGR offspring. It is important to fully understand these mechanisms and to identify those populations at increased risk for NAFLD, such as those with a history of IUGR, because NAFLD is projected to become the major cause of chronic liver disease. We are not proposing that every infant with IUGR will develop NAFLD, but it is suggested that IUGR offspring are at greater risk for progression of this chronic liver disease when exposed to environmental insults that foster its pathogenesis, such as adverse diets and obesity.
Fig. 7.

Overall hypothetical scheme summarizing the integrative proposal that placental insufficiency and intrauterine growth restriction (IUGR) increases the risk for the development of metabolic dysfunction and exaggerated cardiovascular-renal disease (CVRD). Briefly, this is promoted by programming of immune cell activation (78, 197, 201) eliciting increased insulin resistance, hyperglycemia and hyperlipidemia, and nonalcoholic fatty liver disease (NAFLD). The events allow propagation of further hyperglycemia and hyperlipidemia to allow the progression of NAFLD, liver injury, which involves endoplasmic reticulum stress and reduced autophagy, and worsened type 2 diabetes mellitus (T2DM). The latter metabolic dysfunction combined with the hypertension found in IUGR may promote greater vascular dysfunction and drive exaggerated incidence of CVRD in these offspring. The dashed line indicates that it is important to tease out the contribution of inflammation to both the development of T2DM and NAFLD and the hypothesis that metabolic disease accelerates CVRD in IUGR. This overall response may be even further exaggerated by combined maternal obesity (*) or in the face of adverse diets (**) in these offspring. Dashed lines represent the role for additional integrative mechanisms that mediate IUGR-induced pancreatic and renal dysfunction.
Regarding treatment strategies, Bril and Cusi (38) suggest that finding approaches to manage NAFLD and liver injury may assist in the treatment of uncontrolled metabolic diseases, like T2DM. T2DM is a leading cause of CVRD. In this review, it was discussed that IUGR offspring are at greater risk for cardiovascular-renal abnormalities that predispose to CVRD morbidities, like hypertension. Although IUGR encourages multiple risk factors for CVRD, including metabolic diseases and hypertension, it is unknown if these chronic diseases have synergistic effects to mediated the exaggerated risk for CVRD in IUGR offspring. Understanding the etiology of the developmental origins of chronic disease will allow investigators to uncover treatment strategies to intervene in the mother and her offspring to halt CVRD. For instance, it is established that antidiabetic agents like metformin or inhibitors dipeptidyl peptidase-4 are beneficial in reducing advancing NAFLD and T2DM (18). Metformin during pregnancy may also provide benefit by reducing the incidence of preterm delivery and maternal hypertension (235), but the long-term effects of metformin treatment in pregnancies with IUGR on metabolic and CVRD outcomes in offspring are only now starting to be examined. Indicative of its protective effect in developmental programming of metabolic dysfunction is that metformin reduces central adiposity and insulin resistance in prepubertal children that were born SGA at full-term (55). Regarding the potential for other advances in the development of targeted strategies, downregulation of the transcription factor PPAR-γ contributes to hepatic lipid dysregulation and inflammation in IUGR (134). Furthermore, the Toll-like receptor-9 seems to be a potent factor in promoting immune cell infiltration in NASH (151) and compounds targeting this system are being developed for inflammatory diseases (73). Studies are needed in experimental models to determine if IUGR impacts the adverse pathways detailed in this review to allow for NAFLD-mediated exaggeration of metabolic dysfunction and subsequent hypertension-induced CVRD and damage.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 4R00-HL-130577-02.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.T.S., J.A.S., B.T.A., and C.A. conceived and designed research; F.T.S., J.A.S., B.T.A., and C.A. performed experiments; F.T.S., J.A.S., B.T.A., and C.A. analyzed data; F.T.S., J.A.S., B.T.A., and C.A. interpreted results of experiments; F.T.S., J.A.S., B.T.A., and C.A. prepared figures; F.T.S., J.A.S., B.T.A., and C.A. drafted manuscript; F.T.S., J.A.S., B.T.A., and C.A. edited and revised manuscript; F.T.S., J.A.S., B.T.A., and C.A. approved final version of manuscript.
REFERENCES
- 1.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409: 729–733, 2001. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 2.Abrams GA, Kunde SS, Lazenby AJ, Clements RH. Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology 40: 475–483, 2004. doi: 10.1002/hep.20323. [DOI] [PubMed] [Google Scholar]
- 3.Agopian VG, Kaldas FM, Hong JC, Whittaker M, Holt C, Rana A, Zarrinpar A, Petrowsky H, Farmer D, Yersiz H, Xia V, Hiatt JR, Busuttil RW. Liver transplantation for nonalcoholic steatohepatitis: the new epidemic. Ann Surg 256: 624–633, 2012. doi: 10.1097/SLA.0b013e31826b4b7e. [DOI] [PubMed] [Google Scholar]
- 4.Ahowesso C, Black PN, Saini N, Montefusco D, Chekal J, Malosh C, Lindsley CW, Stauffer SR, DiRusso CC. Chemical inhibition of fatty acid absorption and cellular uptake limits lipotoxic cell death. Biochem Pharmacol 98: 167–181, 2015. doi: 10.1016/j.bcp.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 41: 457–462, 2003. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 6.Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension 37: 1191–1195, 2001. doi: 10.1161/01.HYP.37.4.1191. [DOI] [PubMed] [Google Scholar]
- 7.Alisi A, Panera N, Agostoni C, Nobili V. Intrauterine growth retardation and nonalcoholic Fatty liver disease in children. Int J Endocrinol 2011: 269853, 2011. doi: 10.1155/2011/269853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol 5: 201–212, 2011. doi: 10.1586/egh.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, Schauer PR, Feldstein AE. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 285: 3428–3438, 2010. doi: 10.1074/jbc.M109.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen L, Ramalingam L, Menikdiwela K, Scoggin S, Shen CL, Tomison MD, Kaur G, Dufour JM, Chung E, Kalupahana NS, Moustaid-Moussa N. Effects of delta-tocotrienol on obesity-related adipocyte hypertrophy, inflammation and hepatic steatosis in high-fat-fed mice. J Nutr Biochem 48: 128–137, 2017. doi: 10.1016/j.jnutbio.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Alswat KA, Mumtaz K, and Jafri W. Liver biopsy for histological assessment: the case in favor. Saudi J Gastroenterol 16: 133–139, 2010. doi: 10.4103/1319-3767.61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aly FZ, Kleiner DE. Update on fatty liver disease and steatohepatitis. Adv Anat Pathol 18: 294–300, 2011. doi: 10.1097/PAP.0b013e318220f59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Sato R, Kimura S, Ishibashi S, Yamada N. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res 43: 1220–1235, 2002. [PubMed] [Google Scholar]
- 14.Amer MG, Mohamed NM, Shaalan AA. Gestational protein restriction: study of the probable effects on cardiac muscle structure and function in adult rats. Histol Histopathol 32: 1293–1303, 2017. doi: 10.14670/HH-11-883. [DOI] [PubMed] [Google Scholar]
- 15.Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, Saksena S, Burt AD, Bida JP, Lindor K, Sanderson SO, Lenzi M, Adams LA, Kench J, Therneau TM, Day CP. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 45: 846–854, 2007. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 16.Aparicio-Vergara M, Hommelberg PP, Schreurs M, Gruben N, Stienstra R, Shiri-Sverdlov R, Kloosterhuis NJ, de Bruin A, van de Sluis B, Koonen DP, Hofker MH. Tumor necrosis factor receptor 1 gain-of-function mutation aggravates nonalcoholic fatty liver disease but does not cause insulin resistance in a murine model. Hepatology 57: 566–576, 2013. doi: 10.1002/hep.26046. [DOI] [PubMed] [Google Scholar]
- 17.Arima S, Uto H, Ibusuki R, Kumamoto R, Tanoue S, Mawatari S, Oda K, Numata M, Fujita H, Oketani M, Ido A, Tsubouchi H. Hypertension exacerbates liver injury and hepatic fibrosis induced by a choline-deficient L-amino acid-defined diet in rats. Int J Mol Med 33: 68–76, 2014. doi: 10.3892/ijmm.2013.1544. [DOI] [PubMed] [Google Scholar]
- 18.Aroor AR, Habibi J, Ford DA, Nistala R, Lastra G, Manrique C, Dunham MM, Ford KD, Thyfault JP, Parks EJ, Sowers JR, Rector RS. Dipeptidyl peptidase-4 inhibition ameliorates Western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function. Diabetes 64: 1988–2001, 2015. doi: 10.2337/db14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20: 1427–1435, 2014. doi: 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assy N, Djibre A, Farah R, Grosovski M, Marmor A. Presence of coronary plaques in patients with nonalcoholic fatty liver disease. Radiology 254: 393–400, 2010. doi: 10.1148/radiol.09090769. [DOI] [PubMed] [Google Scholar]
- 21.Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K, Kobayashi M, Iwane A, Sasako T, Okazaki Y, Ohsugi M, Takamoto I, Yamashita S, Asahara H, Akira S, Kasuga M, Kadowaki T. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab 13: 401–412, 2011. doi: 10.1016/j.cmet.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Bamfo JE, Odibo AO. Diagnosis and management of fetal growth restriction. J Pregnancy 2011: 640715, 2011. doi: 10.1155/2011/640715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 23, Suppl: 588S–595S, 2004. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 24.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298: 564–567, 1989. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartsch E, Medcalf KE, Park AL, Ray JG; High Risk of Pre-eclampsia Identification Group . Clinical risk factors for pre-eclampsia determined in early pregnancy: systematic review and meta-analysis of large cohort studies. BMJ 353: i1753, 2016. doi: 10.1136/bmj.i1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bashiri A, Nesan D, Tavallaee G, Sue-Chue-Lam I, Chien K, Maguire GF, Naples M, Zhang J, Magomedova L, Adeli K, Cummins CL, Ng DS. Cellular cholesterol accumulation modulates high fat high sucrose (HFHS) diet-induced ER stress and hepatic inflammasome activation in the development of non-alcoholic steatohepatitis. Biochim Biophys Acta 1861: 594–605, 2016. doi: 10.1016/j.bbalip.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Berardis S, Sokal E. Pediatric non-alcoholic fatty liver disease: an increasing public health issue. Eur J Pediatr 173: 131–139, 2014. doi: 10.1007/s00431-013-2157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertin E, Gangnerau MN, Bellon G, Bailbé D, Arbelot De Vacqueur A, Portha B. Development of beta-cell mass in fetuses of rats deprived of protein and/or energy in last trimester of pregnancy. Am J Physiol Regul Integr Comp Physiol 283: R623–R630, 2002. doi: 10.1152/ajpregu.00037.2002. [DOI] [PubMed] [Google Scholar]
- 29.Bieghs V, Van Gorp PJ, Wouters K, Hendrikx T, Gijbels MJ, van Bilsen M, Bakker J, Binder CJ, Lütjohann D, Staels B, Hofker MH, Shiri-Sverdlov R. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS One 7: e30668, 2012. doi: 10.1371/journal.pone.0030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Black MJ, Siebel AL, Gezmish O, Moritz KM, Wlodek ME. Normal lactational environment restores cardiomyocyte number after uteroplacental insufficiency: implications for the preterm neonate. Am J Physiol Regul Integr Comp Physiol 302: R1101–R1110, 2012. doi: 10.1152/ajpregu.00030.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blüher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell 3: 25–38, 2002. doi: 10.1016/S1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- 32.Boehmer BH, Limesand SW, Rozance PJ. The impact of IUGR on pancreatic islet development and β-cell function. J Endocrinol 235: R63–R76, 2017. doi: 10.1530/JOE-17-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnet F, Gastaldelli A, Pihan-Le Bars F, Natali A, Roussel R, Petrie J, Tichet J, Marre M, Fromenty B, Balkau B; D.E.S.I.R., RISC Study Groups . Gamma-glutamyltransferase, fatty liver index and hepatic insulin resistance are associated with incident hypertension in two longitudinal studies. J Hypertens 35: 493–500, 2017. doi: 10.1097/HJH.0000000000001204. [DOI] [PubMed] [Google Scholar]
- 34.Boubred F, Daniel L, Buffat C, Tsimaratos M, Oliver C, Lelièvre-Pégorier M, Simeoni U. The magnitude of nephron number reduction mediates intrauterine growth-restriction-induced long term chronic renal disease in the rat. A comparative study in two experimental models. J Transl Med 14: 331, 2016. doi: 10.1186/s12967-016-1086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boubred F, Delamaire E, Buffat C, Daniel L, Boquien CY, Darmaun D, Simeoni U. High protein intake in neonatal period induces glomerular hypertrophy and sclerosis in adulthood in rats born with IUGR. Pediatr Res 79: 22–26, 2016. doi: 10.1038/pr.2015.176. [DOI] [PubMed] [Google Scholar]
- 36.Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia 59: 879–894, 2016. doi: 10.1007/s00125-016-3904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bridges JP, Gilbert JS, Colson D, Gilbert SA, Dukes MP, Ryan MJ, Granger JP. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am J Hypertens 22: 564–568, 2009. doi: 10.1038/ajh.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bril F, Cusi K. Management of nonalcoholic fatty liver disease in patients with type 2 diabetes: a call to action. Diabetes Care 40: 419–430, 2017. doi: 10.2337/dc16-1787. [DOI] [PubMed] [Google Scholar]
- 39.Brunt EM. Surgical assessment of significant steatosis in donor livers: the beginning of the end for frozen-section analysis? Liver Transpl 19: 360–361, 2013. doi: 10.1002/lt.23609. [DOI] [PubMed] [Google Scholar]
- 40.Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J Physiol 578: 871–881, 2007. doi: 10.1113/jphysiol.2006.121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byers BD, Betancourt A, Lu F, Hankins GD, Longo M, Saade GR, Bytautiene E. The effect of prepregnancy obesity and sFlt-1-induced preeclampsia-like syndrome on fetal programming of adult vascular function in a mouse model. Am J Obstet Gynecol 200: 432.e431–437, 2009. doi: 10.1016/j.ajog.2009.01.044. [DOI] [PubMed] [Google Scholar]
- 42.Bytautiene E, Tamayo E, Kechichian T, Drever N, Gamble P, Hankins GD, Saade GR. Prepregnancy obesity and sFlt1-induced preeclampsia in mice: developmental programming model of metabolic syndrome. Am J Obstet Gynecol 204: 398.e391–398, 2011. doi: 10.1016/j.ajog.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 43.Campanati A, Ganzetti G, Di Sario A, Damiani A, Sandroni L, Rosa L, Benedetti A, Offidani A. The effect of etanercept on hepatic fibrosis risk in patients with non-alcoholic fatty liver disease, metabolic syndrome, and psoriasis. J Gastroenterol 48: 839–846, 2013. doi: 10.1007/s00535-012-0678-9. [DOI] [PubMed] [Google Scholar]
- 44.Cao L, Quan XB, Zeng WJ, Yang XO, Wang MJ. Mechanism of hepatocyte apoptosis. J Cell Death 9: 19–29, 2016. doi: 10.4137/JCD.S39824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Case A, Deaton A. Rising morbidity and mortality in midlife among white non-Hispanic Americans in the 21st century. Proc Natl Acad Sci USA 112: 15078–15083, 2015. doi: 10.1073/pnas.1518393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Andrada P, Rotellar F, Valentí V, Moncada R, Martí P, Silva C, Salvador J, Frühbeck G. Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol 52: 257–266, 2015. doi: 10.1007/s00592-014-0632-9. [DOI] [PubMed] [Google Scholar]
- 47.Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 32: 1076–1080, 2009. doi: 10.2337/dc08-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandrika BB, Yang C, Ou Y, Feng X, Muhoza D, Holmes AF, Theus S, Deshmukh S, Haun RS, Kaushal GP. Endoplasmic reticulum stress-induced autophagy provides cytoprotection from chemical hypoxia and oxidant injury and ameliorates renal ischemia-reperfusion injury. PLoS One 10: e0140025, 2015. doi: 10.1371/journal.pone.0140025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang Y, Jung HS, Yun KE, Cho J, Cho YK, Ryu S. Cohort study of non-alcoholic fatty liver disease, NAFLD fibrosis score, and the risk of incident diabetes in a Korean population. Am J Gastroenterol 108: 1861–1868, 2013. doi: 10.1038/ajg.2013.349. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Zhu H, McCauley SR, Zhao L, Johnson SE, Rhoads RP, El-Kadi SW. Diminished satellite cell fusion and S6K1 expression in myotubes derived from skeletal muscle of low birth weight neonatal pigs. Physiol Rep 5: e13075, 2017. doi: 10.14814/phy2.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crume TL, Scherzinger A, Stamm E, McDuffie R, Bischoff KJ, Hamman RF, Dabelea D. The long-term impact of intrauterine growth restriction in a diverse U.S. cohort of children: the EPOCH study. Obesity (Silver Spring) 22: 608–615, 2014. doi: 10.1002/oby.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cusi K. Treatment of patients with type 2 diabetes and non-alcoholic fatty liver disease: current approaches and future directions. Diabetologia 59: 1112–1120, 2016. doi: 10.1007/s00125-016-3952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czaja MJ. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. Am J Physiol Cell Physiol 298: C973–C978, 2010. doi: 10.1152/ajpcell.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dahri S, Snoeck A, Reusens-Billen B, Remacle C, Hoet JJ. Islet function in offspring of mothers on low-protein diet during gestation. Diabetes 40, Suppl 2: 115–120, 1991. doi: 10.2337/diab.40.2.S115. [DOI] [PubMed] [Google Scholar]
- 55.Díaz M, Bassols J, López-Bermejo A, de Zegher F, Ibáñez L. Metformin treatment to reduce central adiposity after prenatal growth restraint: a placebo-controlled pilot study in prepubertal children. Pediatr Diabetes 16: 538–545, 2015. doi: 10.1111/pedi.12220. [DOI] [PubMed] [Google Scholar]
- 56.Ding X, Xu Y, Wang Y, Li X, Lu C, Su J, Ma Y, Chen Y, Yin Y, Zhang L, Wu Y, Jin Y, Zheng L, Xu S, Zhu X, Ma J, Yu L, Jiang J, Zhao N, Yan Q, Greenberg AS, Huang Q, Ren Q, Sun H, Gu M, Zhao L, Huang Y, Wu Y, Qian C, Peng Y. Nonalcoholic fatty liver disease and associated metabolic risks of hypertension in type 2 diabetes: a cross-sectional community-based study. Int J Endocrinol 2017: 5262560, 2017. doi: 10.1155/2017/5262560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ditchfield AM, Desforges M, Mills TA, Glazier JD, Wareing M, Mynett K, Sibley CP, Greenwood SL. Maternal obesity is associated with a reduction in placental taurine transporter activity. Int J Obes 39: 557–564, 2015. doi: 10.1038/ijo.2014.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM 103: 71–83, 2010. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61: 1547–1554, 2015. doi: 10.1002/hep.27368. [DOI] [PubMed] [Google Scholar]
- 60.Elfarra J, Amaral LM, McCalmon M, Scott JD, Cunningham MW Jr, Gnam A, Ibrahim T, LaMarca B, Cornelius DC. Natural killer cells mediate pathophysiology in response to reduced uterine perfusion pressure. Clin Sci (Lond) 131: 2753–2762, 2017. doi: 10.1042/CS20171118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faienza MF, Brunetti G, Ventura A, D’Aniello M, Pepe T, Giordano P, Monteduro M, Cavallo L. Nonalcoholic fatty liver disease in prepubertal children born small for gestational age: influence of rapid weight catch-up growth. Horm Res Paediatr 79: 103–109, 2013. doi: 10.1159/000347217. [DOI] [PubMed] [Google Scholar]
- 62.Farrell GC, Mridha AR, Yeh MM, Arsov T, Van Rooyen DM, Brooling J, Nguyen T, Heydet D, Delghingaro-Augusto V, Nolan CJ, Shackel NA, McLennan SV, Teoh NC, Larter CZ. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int 34: 1084–1093, 2014. doi: 10.1111/liv.12335. [DOI] [PubMed] [Google Scholar]
- 63.Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, Burgart LJ, Gores GJ. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40: 185–194, 2004. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 64.Ferreira VS, Pernambuco RB, Lopes EP, Morais CN, Rodrigues MC, Arruda MJ, Silva LM, Vilar L. Frequency and risk factors associated with non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. Arq Bras Endocrinol Metabol 54: 362–368, 2010. doi: 10.1590/S0004-27302010000400004. [DOI] [PubMed] [Google Scholar]
- 65.Finelli C, Tarantino G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J Gastroenterol 19: 802–812, 2013. doi: 10.3748/wjg.v19.i6.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Firneisz G. Non-alcoholic fatty liver disease and type 2 diabetes mellitus: the liver disease of our age? World J Gastroenterol 20: 9072–9089, 2014. doi: 10.3748/wjg.v20.i27.9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152: 2456–2464, 2011. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fung CM, White JR, Brown AS, Gong H, Weitkamp JH, Frey MR, McElroy SJ. Intrauterine growth restriction alters mouse intestinal architecture during development. PLoS One 11: e0146542, 2016. doi: 10.1371/journal.pone.0146542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 5: 1544–1560, 2013. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaillard R, Steegers EA, Tiemeier H, Hofman A, Jaddoe VW. Placental vascular dysfunction, fetal and childhood growth, and cardiovascular development: the generation R study. Circulation 128: 2202–2210, 2013. doi: 10.1161/CIRCULATIONAHA.113.003881. [DOI] [PubMed] [Google Scholar]
- 71.Gallo LA, Tran M, Cullen-McEwen LA, Denton KM, Jefferies AJ, Moritz KM, Wlodek ME. Transgenerational programming of fetal nephron deficits and sex-specific adult hypertension in rats. Reprod Fertil Dev 26: 1032–1043, 2014. doi: 10.1071/RD13133. [DOI] [PubMed] [Google Scholar]
- 72.Gan L, Chitturi S, Farrell GC. Mechanisms and implications of age-related changes in the liver: nonalcoholic fatty liver disease in the elderly. Curr Gerontol Geriatr Res 2011: 831536, 2011. doi: 10.1155/2011/831536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol 8: 508, 2017. doi: 10.3389/fphys.2017.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gonzalez PN, Gasperowicz M, Barbeito-Andrés J, Klenin N, Cross JC, Hallgrímsson B. Chronic protein restriction in mice impacts placental function and maternal body weight before fetal growth. PLoS One 11: e0152227, 2016. doi: 10.1371/journal.pone.0152227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, Bennett W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med 122: 383–392, 2006. [DOI] [PubMed] [Google Scholar]
- 76.Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta 1842: 2329–2343, 2014. doi: 10.1016/j.bbadis.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 77.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gyurko R, Siqueira CC, Caldon N, Gao L, Kantarci A, Van Dyke TE. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J Immunol 177: 7250–7256, 2006. doi: 10.4049/jimmunol.177.10.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hall MJ, Levant S, DeFrances CJ. Trends in inpatient hospital deaths: National Hospital Discharge Survey, 2000-2010. NCHS Data Brief 118: 1–8, 2013. [PubMed] [Google Scholar]
- 80.Harvey TJ, Murphy RM, Morrison JL, Posterino GS. Maternal nutrient restriction alters Ca2+ handling properties and contractile function of isolated left ventricle bundles in male but not female juvenile rats. PLoS One 10: e0138388, 2015. doi: 10.1371/journal.pone.0138388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hazlehurst JM, Woods C, Marjot T, Cobbold JF, Tomlinson JW. Non-alcoholic fatty liver disease and diabetes. Metabolism 65: 1096–1108, 2016. doi: 10.1016/j.metabol.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heath-Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2 protein family. Oncogene 27: 6419–6433, 2008. doi: 10.1038/onc.2008.309. [DOI] [PubMed] [Google Scholar]
- 83.Henninger AM, Eliasson B, Jenndahl LE, Hammarstedt A. Adipocyte hypertrophy, inflammation and fibrosis characterize subcutaneous adipose tissue of healthy, non-obese subjects predisposed to type 2 diabetes. PLoS One 9: e105262, 2014. doi: 10.1371/journal.pone.0105262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand 81: 112–114, 2002. doi: 10.1034/j.1600-0412.2002.810204.x. [DOI] [PubMed] [Google Scholar]
- 85.Henry SL, Barzel B, Wood-Bradley RJ, Burke SL, Head GA, Armitage JA. Developmental origins of obesity-related hypertension. Clin Exp Pharmacol Physiol 39: 799–806, 2012. doi: 10.1111/j.1440-1681.2011.05579.x. [DOI] [PubMed] [Google Scholar]
- 86.Horton JD, Shimomura I, Ikemoto S, Bashmakov Y, Hammer RE. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J Biol Chem 278: 36652–36660, 2003. doi: 10.1074/jbc.M306540200. [DOI] [PubMed] [Google Scholar]
- 87.Huang XD, Fan Y, Zhang H, Wang P, Yuan JP, Li MJ, Zhan XY. Serum leptin and soluble leptin receptor in non-alcoholic fatty liver disease. World J Gastroenterol 14: 2888–2893, 2008. doi: 10.3748/wjg.14.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hung JH, Su IJ, Lei HY, Wang HC, Lin WC, Chang WT, Huang W, Chang WC, Chang YS, Chen CC, Lai MD. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem 279: 46384–46392, 2004. doi: 10.1074/jbc.M403568200. [DOI] [PubMed] [Google Scholar]
- 89.Huynh FK, Levi J, Denroche HC, Gray SL, Voshol PJ, Neumann UH, Speck M, Chua SC, Covey SD, Kieffer TJ. Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 59: 3032–3040, 2010. doi: 10.2337/db10-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ibrahim T, Przybyl L, Harmon AC, Amaral LM, Faulkner JL, Cornelius DC, Cunningham MW, Hünig T, Herse F, Wallukat G, Dechend R, LaMarca B. Proliferation of endogenous regulatory T cells improve the pathophysiology associated with placental ischaemia of pregnancy. Am J Reprod Immunol 78: e12724, 2017. doi: 10.1111/aji.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, Kato S, Mawatari H, Shibata W, Kitani H, Ikejima K, Kirikoshi H, Nakajima N, Saito S, Maeyama S, Watanabe S, Wada K, Nakajima A. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab 16: 44–54, 2012. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 92.Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, Liang S, Pimienta M, Taniguchi K, Wu X, Asahina K, Lagakos W, Mackey MR, Akira S, Ellisman MH, Sears DD, Olefsky JM, Karin M, Brenner DA, Seki E. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest 124: 3566–3578, 2014. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Intapad S, Dasinger JH, Brown AD, Fahling JM, Esters J, Alexander BT. Glucose intolerance develops prior to increased adiposity and accelerated cessation of estrous cyclicity in female growth-restricted rats. Pediatr Res 79: 962–970, 2016. doi: 10.1038/pr.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Intapad S, Dasinger JH, Fahling JM, Backstrom MA, Alexander BT. Testosterone is protective against impaired glucose metabolism in male intrauterine growth-restricted offspring. PLoS One 12: e0187843, 2017. doi: 10.1371/journal.pone.0187843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Intapad S, Ojeda NB, Varney E, Royals TP, Alexander BT. Sex-specific effect of endothelin in the blood pressure response to acute angiotensin ii in growth-restricted rats. Hypertension 66: 1260–1266, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Intapad S, Warrington JP, Spradley FT, Palei AC, Drummond HA, Ryan MJ, Granger JP, Alexander BT. Reduced uterine perfusion pressure induces hypertension in the pregnant mouse. Am J Physiol Regul Integr Comp Physiol 307: R1353–R1357, 2014. doi: 10.1152/ajpregu.00268.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Irimia JM, Meyer CM, Segvich DM, Surendran S, DePaoli-Roach AA, Morral N, Roach PJ. Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J Biol Chem 292: 10455–10464, 2017. doi: 10.1074/jbc.M117.786525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, Konior A, Prejbisz A, Januszewicz A, Norlander AE, Chen W, Bonami RH, Marshall AF, Poffenberger G, Weyand CM, Madhur MS, Moore DJ, Harrison DG, Guzik TJ. Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 68: 123–132, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaeckle Santos LJ, Li C, Doulias PT, Ischiropoulos H, Worthen GS, Simmons RA. Neutralizing Th2 inflammation in neonatal islets prevents β-cell failure in adult IUGR rats. Diabetes 63: 1672–1684, 2014. doi: 10.2337/db13-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, Kleiner DE, Gorden P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology 41: 753–760, 2005. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- 101.Jelenik T, Kaul K, Séquaris G, Flögel U, Phielix E, Kotzka J, Knebel B, Fahlbusch P, Hörbelt T, Lehr S, Reinbeck AL, Müller-Wieland D, Esposito I, Shulman GI, Szendroedi J, Roden M. Mechanisms of insulin resistance in primary and secondary nonalcoholic fatty liver. Diabetes 66: 2241–2253, 2017. doi: 10.2337/db16-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones JE, Jurgens JA, Evans SA, Ennis RC, Villar VA, Jose PA. Mechanisms of fetal programming in hypertension. Int J Pediatr 2012: 584831, 2012. doi: 10.1155/2012/584831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jorge AS, Andrade JM, Paraiso AF, Jorge GC, Silveira CM, de Souza LR, Santos EP, Guimaraes AL, Santos SH, De-Paula AM. Body mass index and the visceral adipose tissue expression of IL-6 and TNF-alpha are associated with the morphological severity of non-alcoholic fatty liver disease in individuals with class III obesity. Obes Res Clin Pract 12: 1–8, 2018. doi: 10.1016/j.orcp.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 104.Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J Biol Chem 287: 2558–2567, 2012. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS, Klein S. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59: 1899–1905, 2010. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kato K, Takeshita Y, Misu H, Zen Y, Kaneko S, Takamura T. Liver steatosis is associated with insulin resistance in skeletal muscle rather than in the liver in Japanese patients with non-alcoholic fatty liver disease. J Diabetes Investig 6: 158–163, 2015. doi: 10.1111/jdi.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim YH, Lee HJ, Shin JE, Lee Y, Shin JC, Park TC, Park IY. The predictive value of the uterine artery pulsatility index during the early third trimester for the occurrence of adverse pregnancy outcomes depending on the maternal obesity. Obes Res Clin Pract 9: 374–381, 2015. doi: 10.1016/j.orcp.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 108.Kishino A, Hayashi K, Hidai C, Masuda T, Nomura Y, Oshima T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci Rep 7: 4442, 2017. doi: 10.1038/s41598-017-02960-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kleijer ME, Dekker GA, Heard AR. Risk factors for intrauterine growth restriction in a socio-economically disadvantaged region. J Matern Fetal Neonatal Med 18: 23–30, 2005. doi: 10.1080/14767050500127674. [DOI] [PubMed] [Google Scholar]
- 110.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321, 2005. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 111.Kozono M, Uto H, Ibusuki R, Arima S, Oda K, Taguchi H, Sasaki F, Nasu Y, Hashimoto S, Setoyama H, Kanmura S, Numata M, Tsubouchi H, Ido A. Antihypertensive therapy improves insulin resistance and serum levels of interleukin-6 and -10 in spontaneously hypertensive rats with steatohepatitis. Mol Med Rep 14: 5385–5394, 2016. doi: 10.3892/mmr.2016.5875. [DOI] [PubMed] [Google Scholar]
- 112.Kruse M, Fiallo A, Tao J, Susztak K, Amann K, Katz EB, Charron MJ. A high fat diet during pregnancy and lactation induces cardiac and renal abnormalities in GLUT4 +/− male mice. Kidney Blood Press Res 42: 468–482, 2017. doi: 10.1159/000479383. [DOI] [PubMed] [Google Scholar]
- 113.Kursawe R, Dixit VD, Scherer PE, Santoro N, Narayan D, Gordillo R, Giannini C, Lopez X, Pierpont B, Nouws J, Shulman GI, Caprio S. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes 65: 610–618, 2016. doi: 10.2337/db15-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kyriakakou M, Malamitsi-Puchner A, Militsi H, Boutsikou T, Margeli A, Hassiakos D, Kanaka-Gantenbein C, Papassotiriou I, Mastorakos G. Leptin and adiponectin concentrations in intrauterine growth restricted and appropriate for gestational age fetuses, neonates, and their mothers. Eur J Endocrinol 158: 343–348, 2008. doi: 10.1530/EJE-07-0692. [DOI] [PubMed] [Google Scholar]
- 115.Lake AD, Novak P, Hardwick RN, Flores-Keown B, Zhao F, Klimecki WT, Cherrington NJ. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci 137: 26–35, 2014. doi: 10.1093/toxsci/kft230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, Adam G, Lavallard VJ, Rovere C, Le Thuc O, Saint-Paul MC, Anty R, Schneck AS, Iannelli A, Gugenheim J, Tran A, Gual P, Bailly-Maitre B. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis 6: e1879, 2015. doi: 10.1038/cddis.2015.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee SH, Yun SJ, Kim DH, Jo HH, Park YS. Severity of nonalcoholic fatty liver disease on sonography and risk of coronary heart disease. J Clin Ultrasound 45: 391–399, 2017. doi: 10.1002/jcu.22472. [DOI] [PubMed] [Google Scholar]
- 118.Lee YJ, Jang YN, Han YM, Kim HM, Jeong JM, Seo HS. Fimasartan ameliorates nonalcoholic fatty liver disease through pparδ regulation in hyperlipidemic and hypertensive conditions. PPAR Res 2017: 8048720, 2017. doi: 10.1155/2017/8048720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Legry V, Van Rooyen DM, Lambert B, Sempoux C, Poekes L, Español-Suñer R, Molendi-Coste O, Horsmans Y, Farrell GC, Leclercq IA. Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin-resistant high-fat-diet-fed foz/foz mice. Clin Sci (Lond) 127: 507–518, 2014. doi: 10.1042/CS20140026. [DOI] [PubMed] [Google Scholar]
- 120.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16: 250–264, 2012. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li C, Shu ZJ, Lee S, Gupta MB, Jansson T, Nathanielsz PW, Kamat A. Effects of maternal nutrient restriction, intrauterine growth restriction, and glucocorticoid exposure on phosphoenolpyruvate carboxykinase-1 expression in fetal baboon hepatocytes in vitro. J Med Primatol 42: 211–219, 2013. doi: 10.1111/jmp.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li WD, Fu KF, Li GM, Lian YS, Ren AM, Chen YJ, Xia JR. Comparison of effects of obesity and non-alcoholic fatty liver disease on incidence of type 2 diabetes mellitus. World J Gastroenterol 21: 9607–9613, 2015. doi: 10.3748/wjg.v21.i32.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu J, He J, Yang Y, Yu J, Mao X, Yu B, Chen D. Effects of intrauterine growth retardation and postnatal high-fat diet on hepatic inflammatory response in pigs. Arch Anim Nutr 68: 111–125, 2014. doi: 10.1080/1745039X.2014.897532. [DOI] [PubMed] [Google Scholar]
- 125.Liu X, Henkel AS, LeCuyer BE, Schipma MJ, Anderson KA, Green RM. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am J Physiol Gastrointest Liver Physiol 309: G965–G974, 2015. doi: 10.1152/ajpgi.00132.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Long B, Yin C, Fan Q, Yan G, Wang Z, Li X, Chen C, Yang X, Liu L, Zheng Z, Shi M, Yan X. Global liver proteome analysis using iTRAQ reveals AMPK-mTOR-autophagy signaling is altered by intrauterine growth restriction in newborn piglets. J Proteome Res 15: 1262–1273, 2016. doi: 10.1021/acs.jproteome.6b00001. [DOI] [PubMed] [Google Scholar]
- 127.Long MT, Wang N, Larson MG, Mitchell GF, Palmisano J, Vasan RS, Hoffmann U, Speliotes EK, Vita JA, Benjamin EJ, Fox CS, Hamburg NM. Nonalcoholic fatty liver disease and vascular function: cross-sectional analysis in the Framingham heart study. Arterioscler Thromb Vasc Biol 35: 1284–1291, 2015. doi: 10.1161/ATVBAHA.114.305200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 10: 686–690, 2013. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 129.López-Bermejo A, Casano-Sancho P, Fernández-Real JM, Kihara S, Funahashi T, Rodríguez-Hierro F, Ricart W, Ibañez L. Both intrauterine growth restriction and postnatal growth influence childhood serum concentrations of adiponectin. Clin Endocrinol (Oxf) 61: 339–346, 2004. doi: 10.1111/j.1365-2265.2004.02102.x. [DOI] [PubMed] [Google Scholar]
- 130.Lu F, Bytautiene E, Tamayo E, Gamble P, Anderson GD, Hankins GD, Longo M, Saade GR. Gender-specific effect of overexpression of sFlt-1 in pregnant mice on fetal programming of blood pressure in the offspring later in life. Am J Obstet Gynecol 197: 418.e411–415, 2007. doi: 10.1016/j.ajog.2007.06.064. [DOI] [PubMed] [Google Scholar]
- 131.Luyckx VA, Brenner BM. Low birth weight, nephron number, and kidney disease. Kidney Int Suppl 68, Suppl: S68–S77, 2005. doi: 10.1111/j.1523-1755.2005.09712.x. [DOI] [PubMed] [Google Scholar]
- 132.MacDorman MF, Gregory EC. Fetal and perinatal mortality: United States, 2013. Natl Vital Stat Rep 64: 1–24, 2015. [PubMed] [Google Scholar]
- 133.Machado MV, Michelotti GA, Jewell ML, Pereira TA, Xie G, Premont RT, Diehl AM. Caspase-2 promotes obesity, the metabolic syndrome and nonalcoholic fatty liver disease. Cell Death Dis 7: e2096, 2016. doi: 10.1038/cddis.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Magee TR, Han G, Cherian B, Khorram O, Ross MG, Desai M. Down-regulation of transcription factor peroxisome proliferator-activated receptor in programmed hepatic lipid dysregulation and inflammation in intrauterine growth-restricted offspring. Am J Obstet Gynecol 199: 271.e271–275, 2008. doi: 10.1016/j.ajog.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Malhotra N, Beaton MD. Management of non-alcoholic fatty liver disease in 2015. World J Hepatol 7: 2962–2967, 2015. doi: 10.4254/wjh.v7.i30.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Manns MP. Liver cirrhosis, transplantation and organ shortage. Dtsch Arztebl Int 110: 83–84, 2013. doi: 10.3238/arztebl.2013.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3: e442, 2006. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Blüher M, Kolanus W, Kahn CR, Brüning JC. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet 6: e1000938, 2010. doi: 10.1371/journal.pgen.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 62: 1148–1155, 2015. doi: 10.1016/j.jhep.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 140.Medici V, Ali MR, Seo S, Aoki CA, Rossaro L, Kim K, Fuller WD, Vidovszky TJ, Smith W, Jiang JX, Maganti K, Havel PJ, Kamboj A, Ramsamooj R, Török NJ. Increased soluble leptin receptor levels in morbidly obese patients with insulin resistance and nonalcoholic fatty liver disease. Obesity (Silver Spring) 18: 2268–2273, 2010. doi: 10.1038/oby.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mehta R, Otgonsuren M, Younoszai Z, Allawi H, Raybuck B, Younossi Z. Circulating miRNA in patients with non-alcoholic fatty liver disease and coronary artery disease. BMJ Open Gastroenterol 3: e000096, 2016. doi: 10.1136/bmjgast-2016-000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6: 87–97, 2000. doi: 10.1016/S1097-2765(05)00015-8. [DOI] [PubMed] [Google Scholar]
- 143.Ming J, Xu S, Gao B, Liu G, Ji Y, Yang F, Jia Y, Fang Y, Ji Q. Non-alcoholic fatty liver disease predicts type 2 diabetes mellitus, but not prediabetes, in Xi’an, China: a five-year cohort study. Liver Int 35: 2401–2407, 2015. doi: 10.1111/liv.12851. [DOI] [PubMed] [Google Scholar]
- 144.Mirza MS. Obesity, visceral fat, and NAFLD: querying the role of adipokines in the progression of nonalcoholic fatty liver disease. ISRN Gastroenterol 2011: 592404, 2011. doi: 10.5402/2011/592404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Misra VL, Khashab M, Chalasani N. Nonalcoholic fatty liver disease and cardiovascular risk. Curr Gastroenterol Rep 11: 50–55, 2009. doi: 10.1007/s11894-009-0008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD. Mitochondrial uncoupling and lifespan. Mech Ageing Dev 131: 463–472, 2010. doi: 10.1016/j.mad.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Moreno-Indias I, Tinahones FJ. Impaired adipose tissue expandability and lipogenic capacities as ones of the main causes of metabolic disorders. J Diabetes Res 2015: 970375, 2015. doi: 10.1155/2015/970375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Morita A, Itoh Y, Toyama T, Fujii H, Nishioji K, Kirishima T, Makiyama A, Yamauchi N, Okanoue T. Activated Kupffer cells play an important role in intra-hepatic Th1-associated necro-inflammation in Concanavalin A-induced hepatic injury in mice. Hepatol Res 27: 143–150, 2003. doi: 10.1016/S1386-6346(03)00206-7. [DOI] [PubMed] [Google Scholar]
- 149.Motiño O, Agra N, Brea Contreras R, Domínguez-Moreno M, García-Monzón C, Vargas-Castrillón J, Carnovale CE, Boscá L, Casado M, Mayoral R, Valdecantos MP, Valverde AM, Francés DE, Martín-Sanz P. Cyclooxygenase-2 expression in hepatocytes attenuates non-alcoholic steatohepatitis and liver fibrosis in mice. Biochim Biophys Acta 1862: 1710–1723, 2016. doi: 10.1016/j.bbadis.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 150.Mouralidarane A, Soeda J, Sugden D, Bocianowska A, Carter R, Ray S, Saraswati R, Cordero P, Novelli M, Fusai G, Vinciguerra M, Poston L, Taylor PD, Oben JA. Maternal obesity programs offspring non-alcoholic fatty liver disease through disruption of 24-h rhythms in mice. Int J Obes 39: 1339–1348, 2015. doi: 10.1038/ijo.2015.85. [DOI] [PubMed] [Google Scholar]
- 151.Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, Ajamieh H, Adams L, Hamdorf JM, Teoh NC, Farrell GC. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci (Lond) 131: 2145–2159, 2017. doi: 10.1042/CS20160838. [DOI] [PubMed] [Google Scholar]
- 152.Muramatsu-Kato K, Itoh H, Kohmura-Kobayashi Y, Ferdous UJ, Tamura N, Yaguchi C, Uchida T, Suzuki K, Hashimoto K, Suganami T, Ogawa Y, Kanayama N. Undernourishment in utero primes hepatic steatosis in adult mice offspring on an obesogenic diet; involvement of endoplasmic reticulum stress. Sci Rep 5: 16867, 2015. doi: 10.1038/srep16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Newton KP, Feldman HS, Chambers CD, Wilson L, Behling C, Clark JM, Molleston JP, Chalasani N, Sanyal AJ, Fishbein MH, Lavine JE, Schwimmer JB; Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) . Low and high birth weights are risk factors for nonalcoholic fatty liver disease in children. J Pediatr 187: 141–146 e141, 2017. doi: 10.1016/j.jpeds.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Newton KP, Hou J, Crimmins NA, Lavine JE, Barlow SE, Xanthakos SA, Africa J, Behling C, Donithan M, Clark JM, Schwimmer JB; Nonalcoholic Steatohepatitis Clinical Research Network . Prevalence of prediabetes and type 2 diabetes in children with nonalcoholic fatty liver disease. JAMA Pediatr 170: e161971, 2016. doi: 10.1001/jamapediatrics.2016.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 151: 217–219, 2012. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 156.Olivares S, Henkel AS. Hepatic Xbp1 gene deletion promotes endoplasmic reticulum stress-induced liver injury and apoptosis. J Biol Chem 290: 30142–30151, 2015. doi: 10.1074/jbc.M115.676239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ortiz-Espejo M, Pérez-Navero JL, Olza-Meneses J, Muñoz-Villanueva MC, Aguilera-García CM, Gil-Campos M. Prepubertal children with a history of extra-uterine growth restriction exhibit low-grade inflammation. Br J Nutr 112: 338–346, 2014. doi: 10.1017/S0007114514000920. [DOI] [PubMed] [Google Scholar]
- 158.Pagano C, Soardo G, Esposito W, Fallo F, Basan L, Donnini D, Federspil G, Sechi LA, Vettor R. Plasma adiponectin is decreased in nonalcoholic fatty liver disease. Eur J Physiol 152: 113–118, 2005. . [DOI] [PubMed] [Google Scholar]
- 159.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140: 197–208, 2010. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Patil R, Sood GK. Non-alcoholic fatty liver disease and cardiovascular risk. World J Gastrointest Pathophysiol 8: 51–58, 2017. doi: 10.4291/wjgp.v8.i2.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 62: 720–733, 2015. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 162.Payne JA, Alexander BT, Khalil RA. Reduced endothelial vascular relaxation in growth-restricted offspring of pregnant rats with reduced uterine perfusion. Hypertension 42: 768–774, 2003. doi: 10.1161/01.HYP.0000084990.88147.0C. [DOI] [PubMed] [Google Scholar]
- 163.Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, Ruan HB, Yang X, Caprio S, Kaech SM, Sul HS, Birnbaum MJ, Davis RJ, Cline GW, Petersen KF, Shulman GI. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160: 745–758, 2015. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA, Shulman GI. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18: 740–748, 2013. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510: 84–91, 2014. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Petäjä EM, Sevastianova K, Hakkarainen A, Orho-Melander M, Lundbom N, Yki-Järvinen H. Adipocyte size is associated with NAFLD independent of obesity, fat distribution, and PNPLA3 genotype. Obesity (Silver Spring) 21: 1174–1179, 2013. doi: 10.1002/oby.20114. [DOI] [PubMed] [Google Scholar]
- 167.Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 13: 572–587, 2017. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Portillo-Sanchez P, Bril F, Maximos M, Lomonaco R, Biernacki D, Orsak B, Subbarayan S, Webb A, Hecht J, Cusi K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J Clin Endocrinol Metab 100: 2231–2238, 2015. doi: 10.1210/jc.2015-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Przybyl L, Ibrahim T, Haase N, Golic M, Rugor J, Luft FC, Bendix I, Serdar M, Wallukat G, Staff AC, Müller DN, Hünig T, Felderhoff-Müser U, Herse F, LaMarca B, Dechend R. Regulatory T cells ameliorate intrauterine growth retardation in a transgenic rat model for preeclampsia. Hypertension 65: 1298–1306, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Raab EL, Vuguin PM, Stoffers DA, Simmons RA. Neonatal exendin-4 treatment reduces oxidative stress and prevents hepatic insulin resistance in intrauterine growth-retarded rats. Am J Physiol Regul Integr Comp Physiol 297: R1785–R1794, 2009. doi: 10.1152/ajpregu.00519.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Reyes LM, Shah A, Quon A, Morton JS, Davidge ST. The role of the tumor necrosis factor (TNF)-related weak inducer of apoptosis (TWEAK) in offspring exposed to prenatal hypoxia. J Dev Orig Health Dis 2017 Dec 18 [Epub ahead of print]. doi: 10.1017/S2040174417001003. [DOI] [PubMed] [Google Scholar]
- 172.Riddle ES, Campbell MS, Lang BY, Bierer R, Wang Y, Bagley HN, Joss-Moore LA. Intrauterine growth restriction increases TNF α and activates the unfolded protein response in male rat pups. J Obes 2014: 829862, 2014. doi: 10.1155/2014/829862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 313: 2263–2273, 2015. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 174.Rius B, Duran-Güell M, Flores-Costa R, López-Vicario C, Lopategi A, Alcaraz-Quiles J, Casulleras M, Lozano JJ, Titos E, Clària J. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J 31: 5384–5398, 2017. doi: 10.1096/fj.201700394R. [DOI] [PubMed] [Google Scholar]
- 175.Rodríguez-Trejo A, Ortiz-López MG, Zambrano E, Granados-Silvestre ML, Méndez C, Blondeau B, Bréant B, Nathanielsz PW, Menjivar M. Developmental programming of neonatal pancreatic β-cells by a maternal low-protein diet in rats involves a switch from proliferation to differentiation. Am J Physiol Endocrinol Metab 302: E1431–E1439, 2012. doi: 10.1152/ajpendo.00619.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rotman Y, Neuschwander-Tetri BA. Liver fat accumulation as a barometer of insulin responsiveness again points to adipose tissue as the culprit. Hepatology 65: 1088–1090, 2017. doi: 10.1002/hep.29094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rueda-Clausen CF, Dolinsky VW, Morton JS, Proctor SD, Dyck JR, Davidge ST. Hypoxia-induced intrauterine growth restriction increases the susceptibility of rats to high-fat diet-induced metabolic syndrome. Diabetes 60: 507–516, 2011. doi: 10.2337/db10-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rueda-Clausen CF, Stanley JL, Thambiraj DF, Poudel R, Davidge ST, Baker PN. Effect of prenatal hypoxia in transgenic mouse models of preeclampsia and fetal growth restriction. Reprod Sci 21: 492–502, 2014. doi: 10.1177/1933719113503401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Samonakis DN, Koulentaki M, Coucoutsi C, Augoustaki A, Baritaki C, Digenakis E, Papiamonis N, Fragaki M, Matrella E, Tzardi M, Kouroumalis EA. Clinical outcomes of compensated and decompensated cirrhosis: a long term study. World J Hepatol 6: 504–512, 2014. doi: 10.4254/wjh.v6.i7.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126: 12–22, 2016. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sarr O, Thompson JA, Zhao L, Lee TY, Regnault TR. Low birth weight male guinea pig offspring display increased visceral adiposity in early adulthood. PLoS One 9: e98433, 2014. doi: 10.1371/journal.pone.0098433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sasson IE, Vitins AP, Mainigi MA, Moley KH, Simmons RA. Pre-gestational vs gestational exposure to maternal obesity differentially programs the offspring in mice. Diabetologia 58: 615–624, 2015. doi: 10.1007/s00125-014-3466-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol 11: 187–200, 2014. doi: 10.1038/nrgastro.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Setiawan VW, Stram DO, Porcel J, Lu SC, Le Marchand L, Noureddin M. Prevalence of chronic liver disease and cirrhosis by underlying cause in understudied ethnic groups: The multiethnic cohort. Hepatology 64: 1969–1977, 2016. doi: 10.1002/hep.28677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 294: R528–R538, 2008. doi: 10.1152/ajpregu.00316.2007. [DOI] [PubMed] [Google Scholar]
- 186.Shankaran S, Das A, Bauer CR, Bada H, Lester B, Wright L, Higgins R, Poole K. Fetal origin of childhood disease: intrauterine growth restriction in term infants and risk for hypertension at 6 years of age. Arch Pediatr Adolesc Med 160: 977–981, 2006. doi: 10.1001/archpedi.160.9.977. [DOI] [PubMed] [Google Scholar]
- 187.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 6: 77–86, 2000. doi: 10.1016/S1097-2765(05)00010-9. [DOI] [PubMed] [Google Scholar]
- 188.Shum M, Bellmann K, St-Pierre P, Marette A. Pharmacological inhibition of S6K1 increases glucose metabolism and Akt signalling in vitro and in diet-induced obese mice. Diabetologia 59: 592–603, 2016. doi: 10.1007/s00125-015-3839-6. [DOI] [PubMed] [Google Scholar]
- 189.Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50: 2279–2286, 2001. doi: 10.2337/diabetes.50.10.2279. [DOI] [PubMed] [Google Scholar]
- 190.Spradley FT, Palei AC, Granger JP. Immune mechanisms linking obesity and preeclampsia. Biomolecules 5: 3142–3176, 2015. doi: 10.3390/biom5043142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Spradley FT, Tan AY, Joo WS, Daniels G, Kussie P, Karumanchi SA, Granger JP. Placental Growth Factor Administration Abolishes Placental Ischemia-Induced Hypertension. Hypertension 67: 740–747, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Srinivas SK, Edlow AG, Neff PM, Sammel MD, Andrela CM, Elovitz MA. Rethinking IUGR in preeclampsia: dependent or independent of maternal hypertension? J Perinatol 29: 680–684, 2009. doi: 10.1038/jp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes 52: 734–740, 2003. doi: 10.2337/diabetes.52.3.734. [DOI] [PubMed] [Google Scholar]
- 194.Stojanovska V, Holwerda K, van der Graaf AM, Boekschoten M, Faas M, Scherjon S, Plosch T. High sFlt-1 concentrations during pregnancy modulate the ppara promoter methylation in the fetal liver. FASEB J 31: 859.851, 2017. [Google Scholar]
- 195.Sun C, Jiang L, Liu Y, Shen H, Weiss SJ, Zhou Y, Rui L. Adipose Snail1 regulates lipolysis and lipid partitioning by suppressing adipose triacylglycerol lipase expression. Cell Reports 17: 2015–2027, 2016. doi: 10.1016/j.celrep.2016.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sung KC, Kim SH. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J Clin Endocrinol Metab 96: 1093–1097, 2011. doi: 10.1210/jc.2010-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15: 104–116, 2015. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tan SX, Fisher-Wellman KH, Fazakerley DJ, Ng Y, Pant H, Li J, Meoli CC, Coster AC, Stöckli J, James DE. Selective insulin resistance in adipocytes. J Biol Chem 290: 11337–11348, 2015. doi: 10.1074/jbc.M114.623686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Targher G, Valbusa F, Bonapace S, Bertolini L, Zenari L, Rodella S, Zoppini G, Mantovani W, Barbieri E, Byrne CD. Non-alcoholic fatty liver disease is associated with an increased incidence of atrial fibrillation in patients with type 2 diabetes. PLoS One 8: e57183, 2013. doi: 10.1371/journal.pone.0057183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tarry-Adkins JL, Fernandez-Twinn DS, Hargreaves IP, Neergheen V, Aiken CE, Martin-Gronert MS, McConnell JM, Ozanne SE. Coenzyme Q10 prevents hepatic fibrosis, inflammation, and oxidative stress in a male rat model of poor maternal nutrition and accelerated postnatal growth. Am J Clin Nutr 103: 579–588, 2016. doi: 10.3945/ajcn.115.119834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tencerová M, Kračmerová J, Krauzová E, Mališová L, Kováčová Z, Wedellová Z, Šiklová M, Štich V, Rossmeislová L. Experimental hyperglycemia induces an increase of monocyte and T-lymphocyte content in adipose tissue of healthy obese women. PLoS One 10: e0122872, 2015. doi: 10.1371/journal.pone.0122872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Thompson MD, Cismowski MJ, Trask AJ, Lallier SW, Graf AE, Rogers LK, Lucchesi PA, Brigstock DR. Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high-fat diet. Gene Expr 17: 47–59, 2016. doi: 10.3727/105221616X692135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thorn SR, Regnault TR, Brown LD, Rozance PJ, Keng J, Roper M, Wilkening RB, Hay WW Jr, Friedman JE. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces messenger ribonucleic acid translation initiation and nutrient sensing in fetal liver and skeletal muscle. Endocrinology 150: 3021–3030, 2009. doi: 10.1210/en.2008-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tian F, Zhang YJ, Li Y, Xie Y. Celecoxib ameliorates non-alcoholic steatohepatitis in type 2 diabetic rats via suppression of the non-canonical Wnt signaling pathway expression. PLoS One 9: e83819, 2014. doi: 10.1371/journal.pone.0083819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52: 1836–1846, 2010. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 206.Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem 287: 40161–40172, 2012. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Um SH, Sticker-Jantscheff M, Chau GC, Vintersten K, Mueller M, Gangloff YG, Adams RH, Spetz JF, Elghazi L, Pfluger PT, Pende M, Bernal-Mizrachi E, Tauler A, Tschöp MH, Thomas G, Kozma SC. S6K1 controls pancreatic β cell size independently of intrauterine growth restriction. J Clin Invest 125: 2736–2747, 2015. doi: 10.1172/JCI77030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.van Straten EM, Bloks VW, van Dijk TH, Baller JF, Huijkman NC, Kuipers I, Verkade HJ, Plosch T. Sex-dependent programming of glucose and fatty acid metabolism in mouse offspring by maternal protein restriction. Gend Med 9: 166–179.e113, 2012. doi: 10.14814/phy2.13075. [DOI] [PubMed] [Google Scholar]
- 209.Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW, Jurczak MJ, Samuel VT, Shulman GI. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci USA 112: 1143–1148, 2015. doi: 10.1073/pnas.1423952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Visentin S, Lapolla A, Londero AP, Cosma C, Dalfrà M, Camerin M, Faggian D, Plebani M, Cosmi E. Adiponectin levels are reduced while markers of systemic inflammation and aortic remodelling are increased in intrauterine growth restricted mother-child couple. BioMed Res Int 2014: 401595, 2014. doi: 10.1155/2014/401595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Vuguin P, Raab E, Liu B, Barzilai N, Simmons R. Hepatic insulin resistance precedes the development of diabetes in a model of intrauterine growth retardation. Diabetes 53: 2617–2622, 2004. doi: 10.2337/diabetes.53.10.2617. [DOI] [PubMed] [Google Scholar]
- 212.Wang PX, Zhang XJ, Luo P, Jiang X, Zhang P, Guo J, Zhao GN, Zhu X, Zhang Y, Yang S, Li H. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nat Commun 7: 10592, 2016. doi: 10.1038/ncomms10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang Z, do Carmo JM, Aberdein N, Zhou X, Williams JM, da Silva AA, Hall JE. Synergistic interaction of hypertension and diabetes in promoting kidney injury and the role of endoplasmic reticulum stress. Hypertension 69: 879–891, 2017. doi: 10.1161/HYPERTENSIONAHA.116.08560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wankhade UD, Zhong Y, Kang P, Alfaro M, Chintapalli SV, Thakali KM, Shankar K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS One 12: e0175675, 2017. doi: 10.1371/journal.pone.0175675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr 162: 496–500.e491, 2013. doi: 10.1016/j.jpeds.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wikström AK, Gunnarsdottir J, Nelander M, Simic M, Stephansson O, Cnattingius S. Prehypertension in pregnancy and risks of small for gestational age infant and stillbirth. Hypertension 67: 640–646, 2016. doi: 10.1161/HYPERTENSIONAHA.115.06752. [DOI] [PubMed] [Google Scholar]
- 217.Williams L, Seki Y, Delahaye F, Cheng A, Fuloria M, Hughes Einstein F, Charron MJ. DNA hypermethylation of CD3(+) T cells from cord blood of infants exposed to intrauterine growth restriction. Diabetologia 59: 1714–1723, 2016. doi: 10.1007/s00125-016-3983-7. [DOI] [PubMed] [Google Scholar]
- 218.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 157: 570–585, 2016. doi: 10.1210/en.2015-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wójcik M, Ramadori P, Blaschke M, Sultan S, Khan S, Malik IA, Naz N, Martius G, Ramadori G, Schultze FC. Immunodetection of cyclooxygenase-2 (COX-2) is restricted to tissue macrophages in normal rat liver and to recruited mononuclear phagocytes in liver injury and cholangiocarcinoma. Histochem Cell Biol 137: 217–233, 2012. doi: 10.1007/s00418-011-0889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148: 547–555, 2015. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 221.Wood-Bradley RJ, Barrand S, Giot A, Armitage JA. Understanding the role of maternal diet on kidney development; an opportunity to improve cardiovascular and renal health for future generations. Nutrients 7: 1881–1905, 2015. doi: 10.3390/nu7031881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wueest S, Item F, Lucchini FC, Challa TD, Müller W, Blüher M, Konrad D. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes 65: 140–148, 2016. doi: 10.2337/db15-0941. [DOI] [PubMed] [Google Scholar]
- 223.Xia S, Lv J, Gao Q, Li L, Chen N, Wei X, Xiao J, Chen J, Tao J, Sun M, Mao C, Zhang L, Xu Z. Prenatal exposure to hypoxia induced Beclin 1 signaling-mediated renal autophagy and altered renal development in rat fetuses. Reprod Sci 22: 156–164, 2015. doi: 10.1177/1933719114536474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 112: 91–100, 2003. doi: 10.1172/JCI200317797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115: 2656–2664, 2005. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Xu D, Huang XD, Yuan JP, Wu J, Fan Y, Luo HS, Yang YH. Impaired activation of phosphatidylinositol 3-kinase by leptin is a novel mechanism of hepatic leptin resistance in NAFLD. Hepatogastroenterology 58: 1703–1707, 2011. doi: 10.5754/hge11005. [DOI] [PubMed] [Google Scholar]
- 227.Yadav D, Choi E, Ahn SV, Koh SB, Sung KC, Kim JY, Huh JH. Fatty liver index as a simple predictor of incident diabetes from the KoGES-ARIRANG study. Medicine (Baltimore) 95: e4447, 2016. doi: 10.1097/MD.0000000000004447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yamada T, Fukatsu M, Suzuki S, Wada T, Yoshida T, Joh T. Fatty liver predicts impaired fasting glucose and type 2 diabetes mellitus in Japanese undergoing a health checkup. J Gastroenterol Hepatol 25: 352–356, 2010. doi: 10.1111/j.1440-1746.2009.05998.x. [DOI] [PubMed] [Google Scholar]
- 229.Yan F, Wang Q, Xu C, Cao M, Zhou X, Wang T, Yu C, Jing F, Chen W, Gao L, Zhao J. Peroxisome proliferator-activated receptor α activation induces hepatic steatosis, suggesting an adverse effect. PLoS One 9: e99245, 2014. doi: 10.1371/journal.pone.0099245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yan H, Zheng P, Yu B, Yu J, Mao X, He J, Huang Z, Chen D. Postnatal high-fat diet enhances ectopic fat deposition in pigs with intrauterine growth retardation. Eur J Nutr 56: 483–490, 2017. doi: 10.1007/s00394-015-1093-9. [DOI] [PubMed] [Google Scholar]
- 231.Yee JK, Han G, Vega J, Lee WP, Ross MG, Desai M. Fatty acid de novo synthesis in adult intrauterine growth-restricted offspring, and adult male response to a high fat diet. Lipids 51: 1339–1351, 2016. doi: 10.1007/s11745-016-4199-9. [DOI] [PubMed] [Google Scholar]
- 232.Yu EY, Wan EY, Wong CK, Chan AK, Chan KH, Ho SY, Kwok RL, Lam CL. Effects of risk assessment and management programme for hypertension on clinical outcomes and cardiovascular disease risks after 12 months: a population-based matched cohort study. J Hypertens 35: 627–636, 2017. doi: 10.1097/HJH.0000000000001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yzydorczyk C, Armengaud JB, Peyter AC, Chehade H, Cachat F, Juvet C, Siddeek B, Simoncini S, Sabatier F, Dignat-George F, Mitanchez D, Simeoni U. Endothelial dysfunction in individuals born after fetal growth restriction: cardiovascular and renal consequences and preventive approaches. J Dev Orig Health Dis 8: 448–464, 2017. doi: 10.1017/S2040174417000265. [DOI] [PubMed] [Google Scholar]
- 234.Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci 17: 327, 2016. doi: 10.3390/ijms17030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zeng XL, Zhang YF, Tian Q, Xue Y, An RF. Effects of metformin on pregnancy outcomes in women with polycystic ovary syndrome: a meta-analysis. Medicine (Baltimore) 95: e4526, 2016. doi: 10.1097/MD.0000000000004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zezos P, Renner EL. Liver transplantation and non-alcoholic fatty liver disease. World J Gastroenterol 20: 15532–15538, 2014. doi: 10.3748/wjg.v20.i42.15532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhang J, Zhang K, Li Z, Guo B. ER Stress-induced Inflammasome Activation Contributes to Hepatic Inflammation and Steatosis. J Clin Cell Immunol 7: 7, 2016. doi: 10.4172/2155-9899.1000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang L, Shen L, Xu D, Wang L, Guo Y, Liu Z, Liu Y, Liu L, Magdalou J, Chen L, Wang H. Increased susceptibility of prenatal food restricted offspring to high-fat diet-induced nonalcoholic fatty liver disease is intrauterine programmed. Reprod Toxicol 65: 236–247, 2016. doi: 10.1016/j.reprotox.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 239.Zhang X, Han J, Man K, Li X, Du J, Chu ES, Go MY, Sung JJ, Yu J. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol 64: 160–170, 2016. doi: 10.1016/j.jhep.2015.09.005. [DOI] [PubMed] [Google Scholar]