

Abstract
The gastrointestinal peptide cholecystokinin (CCK) is released from the duodenum in response to dietary fat to aid in digestion, and plasma CCK levels are elevated with the consumption of high-fat diets. CCK is also a trophic peptide for the pancreas and has also been shown to stimulate growth of pancreatic cancer. In the current investigation, we studied the influence of a diet high in saturated fat on the growth of pancreatic cancer in syngeneic murine models before the mice became obese to exclude the confounding factors associated with obesity. The high-fat diet significantly increased growth and metastasis of pancreatic cancer compared with the control diet, and the stimulatory effect was blocked by the CCK-receptor antagonist proglumide. We then selectively knocked out the CCK receptor on the pancreatic cancer cells using clustered regularly interspaced short palindromic repeats technology and showed that without CCK-receptors, dietary fat was unable to stimulate cancer growth. We next demonstrated that dietary fat failed to influence pancreatic cancer xenograft growth in genetically engineered CCK peptide knockout mice. The tumor-associated fibrosis that is so prevalent in the pancreatic cancer microenvironment was significantly decreased with CCK-receptor antagonist therapy because fibroblasts also have CCK receptors. The CCK-receptor antagonist proglumide also altered tumor metalloprotease expression and increased tumor suppressor genes by a PCR array. Our studies confirm that a diet high in saturated fat promotes growth of pancreatic cancer and the action is mediated by the CCK-receptor pathway.
NEW & NOTEWORTHY Diets high in long-chain saturated fats promote growth of pancreatic cancer independent of obesity. The mechanism through which dietary fat promotes cancer is mediated through the cholecystokinin (CCK) receptor pathway. Therapy with a CCK-receptor antagonist altered the tumor microenvironment by reducing fibrosis, increasing cluster of differentiation 8+ lymphocytes, increasing tumor suppressor genes, and thus decreasing metastases. Use of CCK-receptor antagonist therapy with standard chemotherapy for pancreatic cancer may improve response by altering the tumor microenvironment.
Keywords: CCK, fibrosis, metastases, microenvironment, orthotopic, proglumide
INTRODUCTION
Gastrointestinal (GI) digestion, appetite, motility, meal-related glycemic control, and satiety are tightly regulated by the GI peptides ghrelin, cholecystokinin (CCK), glucagon-like protein-1, and peptide YY 3–36 (77). These peptides have also been linked with obesity and eating disorders (80). Obesity and high body mass index have also been associated with several cancers and are thought to more than double the risk for pancreatic cancer (10, 60). Epidemiological studies in human subjects regarding the relationship between dietary fat and pancreatic cancer have variable results. Some investigations demonstrated that the incidence of pancreatic cancer is increased in countries that consume diets high in fat (25, 33, 49, 79); in particular, saturated fatty acids (FAs) are the type of fat associated with an increase in the risk of pancreatic cancer (30, 88, 89). Alternatively, low-fat diets are associated with a decreased incidence of pancreatic cancer (37), and investigators have shown that diets high in omega-3 FAs can lower the risk of pancreatic carcinogenesis in animal models (78). Part of the reason for the decreased cancer risk with omega-3 FAs has been attributed to the anti-inflammatory effect from eicosanoids derived from eicosapentaenoic acid (46). Increasing evidence suggests that the adverse effects of omega-6 FAs may be largely attributed to the metabolites arachidonic acid and the prostaglandin E2 that result from its metabolism by cyclooxygenase (86).
Other factors may contribute to the risk between pancreatic cancer and obesity. It has been proposed that high-fat diets may increase obesity by altering the intestinal microbiota, leading to increased energy extraction, intestinal permeability, and systemic inflammation (57). Obesity itself can increase inflammation; the expansion of fat tissue involves a complex interaction of adipokines and cytokines (28). In obesity, cytokines and leptin promote monocyte conversion to macrophages creating an environment favorable for carcinogenesis (28, 29). Current mechanisms by which obesity is thought to promote cancer include (1, 64) 1) increased levels of insulin and IGF-1 (42, 62), 2) increased sex steroid hormones such as estrogen (36), 3) altered adipo-cytokine levels such as leptin, adiponectin, and visfatin (20), 4) low-grade inflammation affecting cytokines and immune modulation (16), and 5) altered intestinal microbiome (11). Most studies that have examined the role of inflammation on pancreatic carcinogenesis used animal models of obesity (17, 61, 94).
Under physiological conditions, CCK is produced and released from the I-cells of the duodenum in response to fat and protein (18). The G protein-coupled receptor, GPR40, directly mediates long-chain FA-induced secretion of cholecystokinin (45). Another receptor, immunoglobulin-like domain receptor-1 has been recently described (13), which is selectively expressed in the CCK-producing enteroendocrine cells of the small intestine and modulates FA- and lipoprotein-mediated CCK secretion. In normal human subjects there is a sharp cut-off in FA chain length required for CCK release (18, 52). Saturated FAs with a chain length >C12 are effective releasers (and added chain length has little further effect), whereas C11 or shorter FAs are no different compared with vehicle (52). In addition to chain length, CCK secretion requires a nonesterified FA carboxyl group (53). Roebuck and colleagues (65) showed that CCK blood levels did not increase in rats fed a diet high in polyunsaturated fats (20% corn oil base), supporting the epidemiological human studies that show diets high in polyunsaturated fats do not significantly increase the risk for pancreatic cancer (3, 88). CCK blood levels in human subjects are elevated with chronic fat consumption (27) and also in chronic pancreatitis (67). The actions of CCK are mediated through the CCK receptor (CCK-R), a G protein-coupled receptor (19). Two classic CCK receptors, the CCK-AR and the CCK-BR and one CCK-B splice variant receptor (called the CCK-CR; 72) have been identified. The predominant receptor in the normal mouse pancreas is the CCK-AR and the predominant receptor in the human pancreas is the CCK-BR; this topic was recently reviewed (71). Several CCK-R antagonists have been developed that have specificity for either the CCK-AR or CCK-BR (6). Proglumide is a nonselective CCK-R antagonist that inhibits the actions of CCK at both the CCK-AR and CCK-BR (32). CCK-R stimulation results in the release of digestive enzymes from the pancreas (74), contraction of the gall bladder, regulation of insulin release from islet beta cells, and mediation of satiety in the brain (55, 56).
Apart from being involved in normal digestion, CCK is known to be a trophic peptide regulating pancreatic growth in both adult (75, 76, 93) and neonatal animals (93). In addition to its trophic effect, CCK has also been shown to play an important role in pancreatic regeneration (21) after pancreatitis or surgical resection (43). Furthermore, CCK also stimulates the growth of human pancreatic cancer through its interactions with the CCK-BRs that are over-expressed in pancreatic cancer cells (70) and in precancerous pancreatic intraepithelial neoplastic cells (69). Because CCK has mitogenic effects through the CCK-R (50, 76) and is elevated in animals consuming high-fat diets (51), we propose that CCK is responsible for mediating the growth stimulatory effects on pancreatic cancer from a high-fat diet.
In this investigation, we used three different strategies and two different murine models to test our hypothesis that signaling of the CCK-R pathway is a key mechanism in regards to how dietary fat increases the risk and growth of pancreatic cancer. In the first model, “pharmacological blockade,” we used Panc02 murine pancreatic cancer cells (subcutaneous xenografts) with CCK-ARs in syngeneic C57BL/6 mice that received either a high-fat or control diet and a CCK-R antagonist. In the second model [receptor absence/clustered regularly interspaced short palindromic repeats (CRISPR) knockout], we selectively knocked out the CCK-AR from Panc02 cells and tested the effects of CCK and dietary fat on growth. In the third model (peptide absent; a genetically engineered CCK peptide-deficient transgenic mouse), we evaluated if dietary fat would stimulate pancreatic cancer growth in the absence of CCK. Lastly, we studied the role of CCK-R antagonism on tumor progression and metastasis using murine mT5-2D (mT5) cells (orthotopic model in pancreas) with CCK-BRs. The mechanism regulating metastasis was assessed by PCR array to study tumor-associated genes altered by dietary fat and influenced by CCK-R antagonist therapy,
MATERIALS AND METHODS
Cell lines.
Two different murine pancreatic cancer cells were used in this investigation. Both cell lines were analyzed by IMPACT II PCR Profile before use and found to be pathogen free. The murine Panc02 pancreatic cancer cell line was a gift from Dr. Corbett (15) and is syngeneic to the immune competent C57BL/6 mouse and is ductal in appearance, invasive, metastatic, and is associated with a dense fibrotic tumor microenvironment. This cell line has wild-type Kras and CCK-ARs (51).
The second cell line used in this investigation, mT5, was a gift from Dr. Tuveson and is a murine pancreatic cancer cell line that was developed from organoids isolated from mutant KrasLSL-G12D, Pdx1-Cre mouse pancreatic cancer lesions (9) and has mutant Kras and CCK-BRs.
Gene editing by CRISPR of the CCK-R.
Panc02 cells were genetically manipulated to knockout the CCK-AR with CRISPR-Cas9 technology using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and the CRISPR vector pSpCas9 BB-2A-GFP PX485 (GeneScript, Piscataway, NJ). Clonal cells were selected based upon their green fluorescent protein fluorescence. RNA was extracted from both wild-type (Panc02-WT) cells and CCK-AR knockout (KO) cells (receptornull). Total RNA was extracted with an RNeasy Plus Mini Kit (Qiagen, Germantown, MD), and 1 μg was subjected to RT-PCR in a SimpliAmp Thermal Cycler (Applied Biosystems, Carlsbad, CA) under the following conditions: reverse transcription was followed by 95°C × 30 s, 60°C × 1 min and 72°C for 30 s × 35 cycles using murine CCK-AR cDNA-validated primers located at codon 79-99 and 194-174 to yield a 110-bp PCR product using: 5′-CTTTTCTGCCTGGATCAACCT-3′ (forward); 5′- ACCGTGATAACCAGCGTGTTC-3′ (reverse). HPRT was used as a reference gene. The HPRT murine cDNA validated primers were located at codon 324-344 and 465-445 to yield a 142-bp PCR product using: 5′-TCAGTCAACGGGGGACATAAA-3′ (forward); and 5′-GGGGCTGTACTGCTTAACCAG-3′ (reverse). PCR products were evaluated by gel electrophoresis in a 2% agarose gel.
Similarly, the CCK-BR in the mT5 cells was selectively knocked out with CCK-BR CRISPR Guide RNA or crRNA 1 (GeneScript). RNA was extracted from clonal cells and evaluated for CCK-BR expression by RT-PCR using validated cDNA primers were proprietary and purchased from GeneCopeia (Rockville, MD).
In vitro growth study.
Panc02-WT cells and receptornull cells (30,000 each) were plated in 12-well plates in media with 10% FBS. The next day, medium was replaced with media containing 2% FBS alone, and cells were treated with PBS (control) or cerulein (Sigma, St. Louis, MO), a CCK analogue, at a final concentration of 0.1, 1.0, or 10 nM per well (n = 6 wells per treatment). After 72 h, the old medium was removed and discarded. Fresh medium with specified cerulein concentrations (as above) was reapplied to cells in the respective wells. The cells were then incubated for an additional 48 h before harvesting the cells with trypsin containing 0.025% EDTA. Manual viable cell counts were performed using the trypan blue exclusion technique.
Growth of two mT5-KO clones was evaluated in vitro starting with 25,000 cells/well (n = 6 wells each), wild-type cells were plated at 5,000 cells/well. Cells were treated with cerulein (0.1 to 1.0 nM) for 72 h, and manual trypan blue exclusion technique cell counts were done for viable cells.
Animal models.
Male C57BL/6 mice aged 6 wk were used in these studies and were purchased from Charles Rivers. In the CCK-KO experiment, CCK peptide heterozygous mice (B6.129-Cck<tm1Lcs>/J – 017710) were obtained from Jackson Laboratories (Bar Harbor, ME), bred to obtain a colony of mice genetically null of the CCK-peptide (CCK-KO). Both male and female mice were used from the CCK-peptide-KO colony. Mice were housed with five per cage with filter-top cages. All mouse studies were performed in an ethical fashion and approved by the Institutional Animal Care and Use Committee for animal research by the Georgetown University Comparative Medicine Department.
Study design.
There were four different approaches to study the role of dietary fat on activating the CCK peptide/CCK-R pathway (Table 1). In the first series of experiments, pharmacological blockade, a CCK-R antagonist proglumide was used. Panc02 cells (2 × 106) were injected subcutaneously in a volume of 100 μl. In each experiment, 40 mice were used, and half of the mice (n = 20) received a high-fat diet, and the other half (n= 20) received control diet. And then half on each specific diet (n = 10) were treated with either the CCK-R antagonist proglumide or untreated drinking water (regular water). This experiment with subcutaneous Panc02 tumors was terminated after 42 days before the mice became obese.
Table 1.
Study design
| Study: Experiment | Cell Line | Therapy | Duration | Diet |
|---|---|---|---|---|
| 1. CCK-receptor pharmacological blockade | Panc02 (sc) & WT (n = 40; 10 per group) | CCK-receptor antagonist proglumide or Water | 42 days | High fat or control |
| 2. CCK-A receptor KO (on cancer cells) | Panc02-WT & Panc02 CCK-A receptornull (n = 40; 10 per group) | None | 42 days | High fat or control |
| 3. CCK peptide absence (CCK-KO) | Panc02-WT (n = 16; 8 per group) | None | 37 days | High fat or control |
| 4. Orthotopic | mT5 (orthotopic) n = 32; 8 per group) | CCK-receptor antagonist proglumide or water | 21 days | High fat or control |
The outline of each of the four experimental models is shown. CCK, cholecystokinin; KO, knockout; mT5, murine mT5-2D cells; receptornull, CCK-AR knockout cells; WT, wild-type; sc, subcutaneous.
In the next experiment, we investigated the role of dietary fat on pancreatic cancer growth in tumor cells lacking the CCK-R. In the “receptor knockout” experiments, half (n = 20) of the mice received a subcutaneous inoculum of Panc02-WT cells (2 × 106) with normal CCK-R expression, and the other half (n = 20) received Panc02 CCK-receptornull cells (2 × 106). As above, these mice were then also placed on the high-fat or control diet and all mice had regular, untreated drinking water.
In the third experiment both male and female mice were bred and genotyped to establish a colony that was CCK-peptidenull. These CCK-KO mice (both males and females) were injected with 2 × 106 Panc02-WT cells subcutaneously and the mice were placed on either the high-fat or control diet with regular untreated drinking water. Tumor growth was measured over time, and animal weights were measured.
In each of the three studies, half of the mice were fed the high-fat diet, and the other half received the control diet. In all experiments, the diet and treatment were initiated after tumor inoculation, and mice were divided into groups of equal weight. Mice were weighed, and subcutaneous tumor volumes were measured weekly with calipers with the formula length × width2 × 0.5. At the termination of each experiment, the mice were ethically euthanized with CO2 asphyxiation followed by cervical dislocation. Blood was collected by pericardial puncture for glucose and CCK measurements from experiment 1. CCK blood levels were measured using a Human/Mouse/Rat Cholecystokinin EIA Kit (RayBio, Norcross, GA). Glucose (nonfasting) was measured with a blood Precision Xtra glucometer (Abbott Laboratories, Chicago, IL). Tumors were removed and weighed, and the mouse pancreas were dissected and weighed from control and high-fat diet mice. The overall study design is shown in Table 1.
In the fourth experiment, mT5 cells (100,000) in 50 μl of PBS were orthotopically implanted into the tail of the pancreas of syngeneic C57BL/6 male mice (n = 32). Before surgery, the mT5 cells were transfected with luciferase so that tumor growth could be monitored in an IVIS imaging system (Xenogen Corp, Alameda, CA). Luciferin (Nanolight Technology) (35 mg/kg ip, in a volume of 100 μl) was administered to mice to image tumors weekly. Mice were divided into four groups of eight mice each: 1) high-fat diet/regular water, 2) high-fat diet/proglumide water, 3) control diet/regular water, and 4) control diet/proglumide water. At the termination of the experiment, mice were ethically euthanized, and the primary tumors were removed, weighed, and frozen for RNA extraction and PCR array (below). The number of metastases was counted and confirmed by histology.
Diet.
Animals were fed one of two diets (Table 2). A rodent high-fat diet (cat. no. 58-Y1, blue) was purchased from TestDiet (St. Louis, MO). This diet was composed of 34.9% fat from lard, and with this diet animals obtained 60% of their energy in kilocalories from fat. The main protein source was from casein. The control mice received a regular fat diet (cat. no. 5001) also from TestDiet with similar but slightly less kcal (5.0 vs. 4.1). The diet was stored in dry-refrigerated conditions at 4°C. Food was weighed weekly to assure mice in both groups were consuming equal volumes of food.
Table 2.
Diet composition of the control and high-fat diet
| Component | Control Diet |
High-Fat Diet |
||
|---|---|---|---|---|
| %weight | %kcal | %weight | %kcal | |
| Protein, % | 25 | 30 | 23.1 | 18 |
| Fat, % | 11.4 | 14 | 34.9 | 60 |
| Cholesterol | 209 | 301 | ||
| Saturated FA | 1.48 | 13.68 | ||
| Unsaturated FA | 1.62 | 14 | ||
| Carbohydrates | 47.5 | 57 | 26 | 20.3 |
| Fiber | 5.3 | 6.5 | ||
| Other (minerals) | 7 | 6.5 | ||
| Energy, kcal/g | 4.1 | 5.0 | ||
Treatment.
In the pharmacological blockade studies, 10 mice on each of the diets received water containing the CCK-R antagonist proglumide (32; Tocris Bioscience, Bristol, UK) at a concentration of 0.1mg/ml and the estimated amount consumed per mouse was 30 mg·kg·−1day−1 (69).
Quantitative fibrosis analysis and immunohistochemistry.
Tumors were fixed in 4% paraformaldehyde and paraffin embedded, and 8-μm sections were cut and mounted. Tissue sections were stained for fibrosis with Masson’s trichrome. Orthotopic tumors were evaluated for immunoreactivity to alpha smooth muscle actin (αSMA; cat. no. ab-124964, Abcam, Cambridge, MA) at 1:3,500. Fibrosis quantitative scores were analyzed by a computer program using ImageJ. CD8+ lymphocytes were stained in the tumor microenvironment with CD8 antibodies (1:75 titer; eBioscience, San Diego, CA), and CD8+ cells were manually counted.
Tumor metastasis PCR array.
A mouse tumor metastases PCR array (cat. no. PAMM-028ZC-2 Qiagen) was used to study differentially expressed genes from the mouse orthotopic tumors. RNA was extracted from mT5 orthotopic tumors, checked for RNA integrity with RIN values, and converted to cDNA for the array. Analysis of the array results and comparison between groups was performed using the array software provide on the Qiagen website.
Statistical analysis.
Statistical analyses were done with GraphPad Prism V.6.0 or Minitab. Tumor data were compared using pairwise Mann-Whitney tests on groups as indicated. Effects of dietary fat on tumor growth were analyzed using linear regression and both parametric and nonparametric (Spearman) correlation coefficients. Two-sided statistical tests were performed at the 0.05 significance level to assess the significance of the coefficients.
RESULTS
Food intake, blood glucose, pancreas size, and CCK levels.
There were no statistical differences in the amount of food consumed based upon food weight in the mice with Panc02 tumors in the CCK-R pharmacological blockade study (experiment 1). The mice on the high-fat diet and regular water ate the least amount with a mean of 10.3 g/day. The average food intake for mice on the high-fat diet but with proglumide water was 11.4 g/day. Control diet mice ate slightly (but not significant) more food at 12 g/day and 11.7 g/ day, respectively, for the regular water and proglumide groups. The food was measured because CCK is known to interact with the CCK-Rs in the brain and induce satiety (12, 56), and there was also a concern regarding potential increase in food intake on those receiving the CCK-R antagonist. There was no significant difference in nonfasting blood glucose levels between mice receiving the high-fat and control diet (Fig. 1A). The proliferative effects of CCK on the normal mouse pancreas (50), were confirmed; mice receiving the high-fat diet without proglumide had significantly heavier pancreata per animal body weight than the mice on the control diet (Fig. 1B). CCK blood levels were significantly higher in mice consuming a high-fat diet compared with those on the control diet (Fig. 1C). As expected, there was no change in the CCK blood levels with the CCK-R antagonist for the 42 days of this study because the antagonist was only blocking the interaction of CCK with its receptor and the CCK level was increased by the high-fat diet.
Fig. 1.
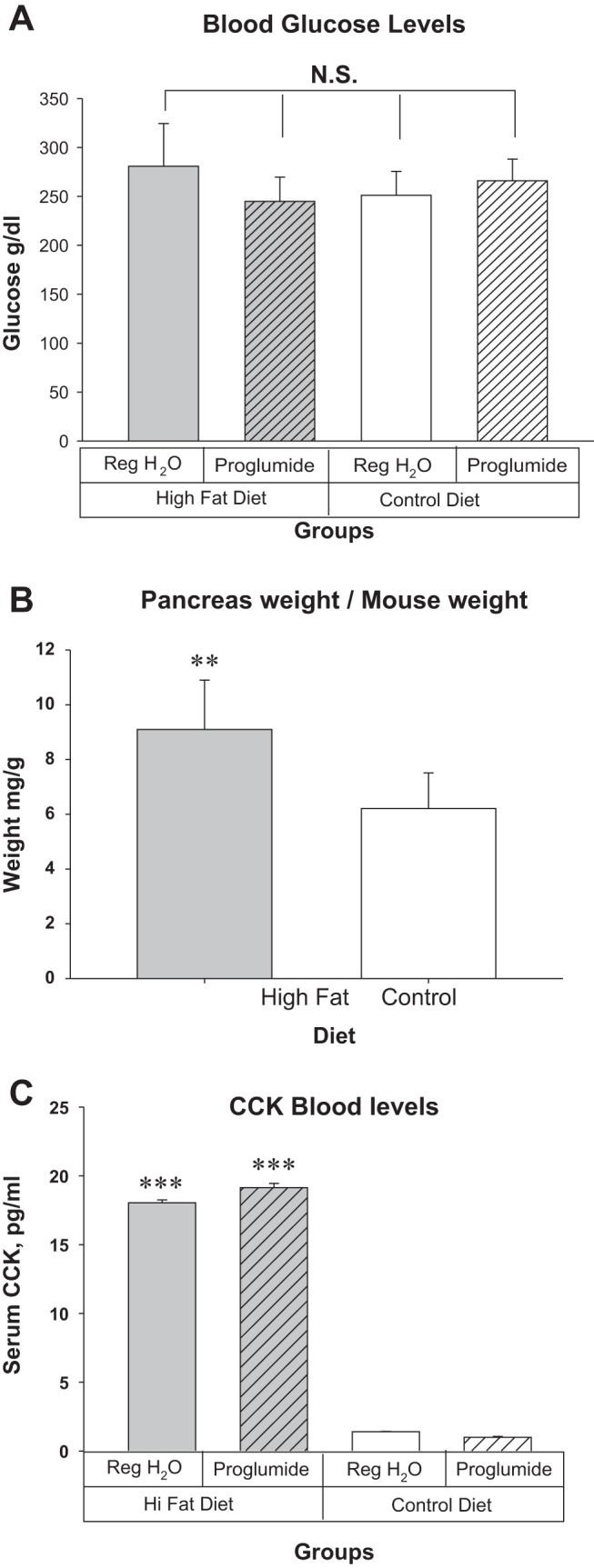
Effects of dietary fat on nonfasting blood glucose, pancreas weight, and CCK blood levels after 42 days and at the termination of experiment 1. A: there were no significant differences in blood glucose levels between the mice on the high-fat and the control diet. Proglumide therapy had no effect on the nonfasting blood sugar. B: mouse pancreas weight in mg per gram mouse weight was significantly greater in the mice on the high-fat diet compared with control diet. C: CCK blood levels are markedly elevated in mice fed a high-fat saturated diet. **P = 0.004, ***P = 0.001). CCK, cholecystokinin; NS, not significant.
CCK-R antagonist (proglumide) therapy blocks dietary fat-stimulated pancreatic cancer growth; pharmacological blockade, Panc02 tumors.
The experiment was purposely terminated before any of the mice became obese (35 g) to eliminate any confounding factors related to obesity. In this study, there were no significant differences in weights between the mice on the high-fat diet compared with the control diet (Fig. 2A). Final Panc02 tumor volumes were 62% larger in mice fed a high-fat diet compared with the control diet, and this effect was completely blocked by proglumide (Fig. 2B). Mice on the high-fat diet had tumors that were 52% larger by weight than mice on the control diet (Fig. 2C). Proglumide therapy blocked the effects of dietary fat on tumor weight such that the tumor weight was comparable to those tumors of the mice on the control diet. CCK-R blockade did not alter tumor weight when mice were fed a control diet (Fig. 2C).
Fig. 2.

Receptor antagonist (pharmacological blockade) study with proglumide; Panc02 tumors. A: final animal body weights. There was no statistical difference in body weights between the treatment and diet groups (n = 10 mice per group) at the termination of the experiment on day 42. The mice were all less than 35 g. B: final tumor volumes. Mice on the high-fat diet had tumors that were larger but not significant (P = 0.057) than those on the control diet. CCK-R blockade with proglumide significantly reduced the tumor size of mice on the high-fat diet (*P = 0.038). Proglumide therapy did not change the tumor size in mice receiving the control diet. C: final tumor weights. Mice on the high-fat diet had tumors that were 52% larger than those on the control diet. Proglumide therapy significantly decreased the tumor weight in spite of being on the high-fat diet (*P = 0.032). D: calculated fibrosis score. Fibrosis was significantly reduced in mice treated with proglumide whether on a high-fat diet (*P = 0.03) or in mice receiving the control diet (*P = 0.017). E-1: a representative tumor from an animal on the high-fat diet reacted with Masson’s trichrome stain, showing marked intratumoral fibrosis. E-2: a representative tumor from a mouse also on the high-fat diet but treated with proglumide and reacted with Masson’s trichrome stain revealing the marked decrease in fibrosis. F-1: paucity of intratumoral CD8+ lymphocytes by immunohistochemistry in a mouse on the high-fat diet. F-2: CD8+ immunoreactivity increased in mice receiving proglumide therapy. CCK-R, cholecystokinin receptor; CD, cluster of differentiation; NS, not significant.
CCK-R antagonist decreases fibrosis of the tumor microenvironment.
One characteristic of pancreatic cancer is the dense fibrosis of the surrounding tumor microenvironment (2, 23). Quantitative fibrosis scores from the tumors from mice treated with the CCK-R antagonist, proglumide, were significantly decreased in both the high-fat cohort and the control cohort (Fig. 2D). Histological evaluation with Masson’s trichrome stain showed that Panc02 tumors from mice on untreated water exhibited extensive fibrosis surrounding the cancer epithelial cells (Fig. 2, E-1). In contrast, tumors from mice treated with the CCK-R antagonist proglumide had significantly less fibrosis even while maintained on a high-fat diet (Fig. 2, E-2).
Immunohistochemical staining for tumor infiltrating CD8+ lymphocytes in mice on the high-fat diet and receiving untreated water showed a paucity of tumor infiltrating lymphocytes (Fig. 2, F-1). In contrast, the number of CD8+ lymphocytes increased in the tumors of mice on the high-fat diet treated with proglumide (Fig. 2, F-2) although the difference did not reach significance.
CCK analogue only stimulates growth of pancreatic cancer cells with CCK-Rs in vitro and not in CCK-receptornull cancer cells.
CCK-AR expression from Panc02-WT and Panc02 CCK-receptornull cancer cells was evaluated. RT-PCR and gel electrophoresis demonstrated the absence of a PCR product from Panc02 receptornull cells. The appropriate PCR product size of 104 bp was detected for the murine CCK-AR in Panc02-WT cells (Fig. 3A). The integrity of RNA was confirmed with reference HPRT gene (Fig. 3A). Panc02 receptornull cells treated with the CCK analogue cerulein in vitro showed no response or proliferation (Fig. 3B). In contrast, Panc02-WT cells that express the CCK-AR increased in cell number at a concentration of cerulein (1.0 nM) consistent with the physiological binding affinity (Kd) (Fig. 3C). These results support the fact that CCK mediates growth of murine pancreatic cancer through the CCK-AR because the same Panc02 cells void of the receptor fail to respond to the proliferative effects of CCK in culture. Similarly, the CCK-B receptor was knocked down in the mT5 pancreatic cancer cells and confirmed by gel electrophoresis (Fig. 3D). The mT5 cells with CCK-BR knock-down did not proliferate when stimulated with cerulein (0.1 to 10 nM), (Fig. 3E). However, the wild-type mT5 cells with CCK-BRs increased in growth when treated with cerulein (Fig. 3F).
Fig. 3.

Results of in vitro experiments with CCK-R gene editing with CRISPR. A: gel electrophoresis demonstrates PCR products from RT-PCR for the CCK-AR in Panc02 cells. In cells transfected with the CRISPR plasmid to knockout the CCK-AR (receptornull) there is no band consistent with the loss of the CCK-AR mRNA expression. Control untransfected wild-type Panc02 cancer cells (Panc-02-WT) reveal the expected RT-PCR product of 104 bp. The HPRT control PCR product was present in both Panc02-WT and Panc02 Receptornull cells showing the integrity of the RNA. B: growth of Panc02 receptornull cells in vitro exhibited no effect when treated with the CCK analogue cerulein (N.S). C: cerulein expectedly stimulated growth of Panc02-WT cells after 72 h with the greatest proliferative effect occurring with a concentration of 1 nM, which is compatible with the binding affinity (Kd) of CCK at its receptor (*P = 0.032). D: gel electrophoresis shows the decreased CCK-BR in the mT5 clone-1 and clone-2 knockdown cells with HPRT reference gene as control. E: when these mT5-KO cells were treated with cerulein, there was no effect on cell growth. F: cerulein treatment in vitro significantly increased the number of WT mT5 cells. CCK-R, cholecystokinin receptor; CRISPR, clustered regularly interspaced short palindromic repeats; KO, knockout; mT5, murine mT5-2D cells; NS, not significant; receptornull, CCK-AR knockout cells; WT, wild-type.
Pancreatic tumors lacking CCK-Rs in vivo do not respond to dietary fat.
In vivo Panc02 receptornull cells that lacked the CCK-AR failed to respond to dietary fat. In contrast, the high-fat diet significantly stimulated growth (Fig. 4, A and B) of Panc02 tumors with CCK-WT receptors compared with mice on the control diet.
Fig. 4.

Panc02 tumor cells lacking CCK-ARs do not respond to dietary fat in mice fed a high-fat diet. A: tumor volumes were significantly greater in mice on the high-fat diet with CCK-WT receptors compare with mice on the high-fat diet without the CCK-ARs (*P = 0.038; n = 10 mice per group) at the termination of the experiment on day 42. Receptornull tumor volumes were also smaller in mice on the control diet than the mice with intact CCK-R on the control diet (**P = 0.009). B: weights of WT Panc02 tumors of mice on a high-fat diet were 6-fold greater than tumors from the mice on the high-fat diet lacking the CCK-AR (**P = 0.002) and 4.5-fold greater than mice on the control diet (**P = 0.01). C-1: trichrome stain shows extensive intratumoral fibrosis from a representative tumor of a mouse on the high-fat diet with WT CCK-ARs. C-2: significant fibrosis in stroma of the CCK-Receptornull tumors from a mouse on the high-fat diet. D: fibrosis score: With objective analysis, there was no difference between the fibrosis in the tumors regardless of cancer cell CCK-R status. Bar = 100 μm. CCK-R, cholecystokinin receptor; receptornull, CCK-AR knockout cells; WT, wild-type.
Unlike the changes in fibrosis seen with pharmacological blockade, in this experiment the tumors of the mice in all four treatment groups showed extensive fibrosis by trichrome stain (Fig. 4C), and no differences by quantitative analysis for fibrosis were found between any of the treatment groups (Fig. 4D). The lack of effect on fibrosis in the tumor microenvironment in this experimental model is because only the CCK-Rs expression on the tumor epithelial cells were altered, not the CCK-Rs on the fibroblasts.
Dietary fat does not stimulate pancreatic cancer growth in CCK-KO transgenic mice.
Panc02-WT tumors grown in mice genetically void of the CCK peptide showed no difference in tumor volumes over time whether on a high-fat diet or control diet (Fig. 5A). Also the final Panc02 tumor volumes and tumor mass measured after 37 days (Fig. 5B) showed no difference in CCK-KO mice whether they were fed a high-fat diet or the control diet. These results demonstrate that the CCK peptide is necessary to stimulate cancer growth. Lo and colleagues (48) have previously shown that CCK-KO mice do not gain weight on a high-fat diet, and they concluded that CCK is involved in regulating the metabolic rate and is important for lipid absorption.
Fig. 5.
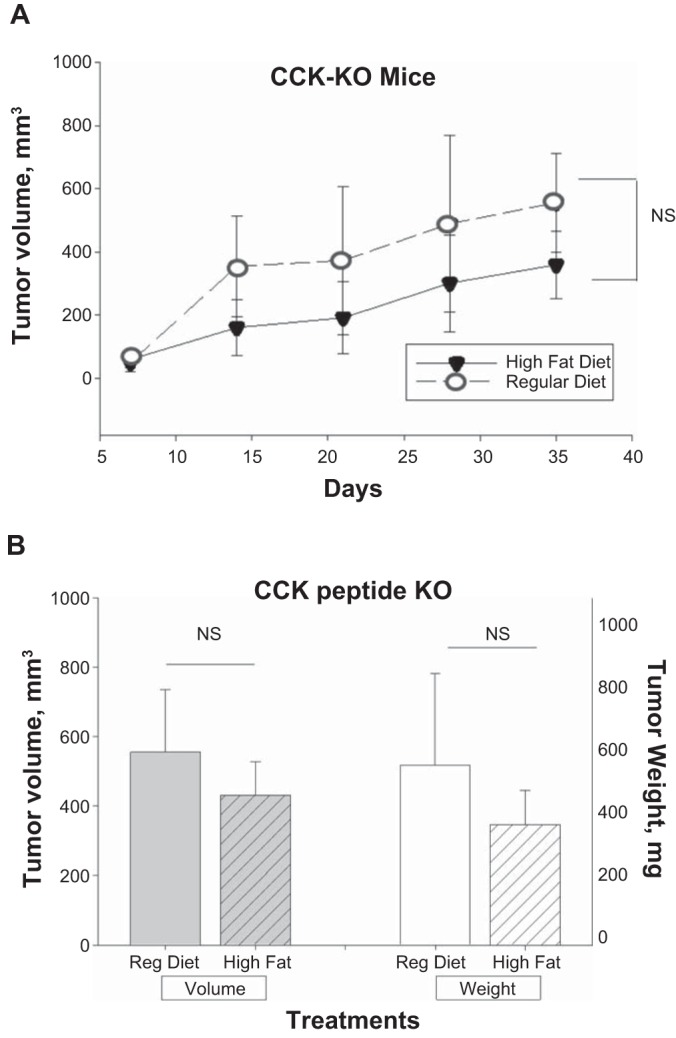
Dietary fat does not stimulate tumor growth in CCK-KO mice. A: mean tumor volumes ± SE in mm3 measured weekly show a steady increase in volumes but revealed no statistical difference between the mice on the high-fat diet and the control diet (n = 8 mice per group) at the termination of the experiment after 37 days. B: final tumor volumes and tumor mass were measured at the termination of the experiment and no difference was found between the CCK-KO mice on the high-fat diet and the control diet. CCK-KO, cholecystokinin knockout.
CCK-R blockade with proglumide decreases metastases and tumor fibrosis in an orthotopic model (mT5 cells).
Using the orthotopic model and mT5 cells, we examined the role of dietary fat and proglumide on tumor growth and metastases. mT5 cells are very aggressive and after 3 wk the mice on the high-fat diet with regular drinking water were becoming moribund, and we were required to terminate the experiment prematurely. mT5 tumors in the mice on the high-fat diet were significantly larger than those tumors of mice on the control diet (Fig. 6A). The tumors of proglumide-treated mice were smaller but did not reach statistical significance after just 21 days. Dietary fat also significantly increased the mean number of metastases per mouse compared with the number of metastases found in the mice on the control diet (Fig. 6B). The metastases were significantly reduced in mice treated with proglumide in spite of the high-fat diet (Fig. 6B). Most of the metastases were peritoneal, lymph node, and liver metastases and these were confirmed with histology. Masson’s trichrome stain from a representative tumor from each group is shown in Fig. 6C. Similar to the Panc02 tumor subcutaneous experiment, quantitative analysis of the fibrosis again shows significantly less fibrosis in mT5 tumors of mice treated with proglumide (Fig. 6D). Tumors from mice fed the high-fat diet and regular drinking water were compared with tumors of mice on the high fat diet treated with proglumide with regard to αSMA immunoreactivity (Fig. 6E). Computer analysis failed to show statistical differences in αSMA between the groups. To understand the mechanism by which CCK antagonism interacts with the tumor microenvironment to prevent metastases and reduce fibrosis, a Tumor Metastasis RT2 Profiler PCR Array (Qiagen) was performed to identify differentially expressed genes in tumors from mT5 tumors of mice on a high-fat diet compared with tumors from mice on the control diet. Significant genes altered by the high-fat diet that changed at least twofold compared with the control diet are shown (Fig. 6F). Among the top genes identified included matrix metalloprotease (Mmp) genes (Mmp7, Mmp9, Mmp13, and Mmp10). MMPs comprises several tightly regulated classes of proteases that play important roles in tumor progression and the metastatic process by facilitating extracellular matrix degradation (38). Gastrin has been shown to stimulate MMP7 secretion (81) and can stimulate epithelial growth factor receptor (EGFR) by transactivation, and gastrin activates the MMP7 promoter (54). Timp4 is an inhibitor of the MMPs that is associated with cancer (8); it is of interest that Timp4’s expression is the opposite of the MMP’s. Lpar6 gene is the receptor for Lysophosphatidic acid and is involved in cell migration, proliferation, and differentiation (83). Chemokine (C-C motif) ligand 7 is a chemokine associated with immune cells in the microenvironment. IL1β is a cytokine associated with angiogenesis and tumor metastases and invasion (85). The Fat-1 gene encodes a tumor suppressor essential for controlling cell proliferation and prevents epithelial mesenchymal transition via the MAPK/ERK signaling pathway (35). Next, we evaluated the changes in differentially expressed genes from tumors of mice on the high-fat diet that received the CCK-R antagonist to understand how proglumide altered fibrosis and metastases. Proglumide therapy decreased the gene expression of all the metalloproteases with the exception of Mmp7 (Table 3), which correlates with the changes in the fibrosis. Genes that were significantly increased with proglumide included Elane (or Elastase neutrophil-expressed gene), Nme2, Tnsf10, NR4a3, Fxyd5, and Fat-1. Tnfs10 or TNF-related apoptosis-inducing ligand is an apoptosis gene, and Fat-1 is a tumor suppressor gene. NR4a3 is increased with T cell-induced apoptosis, and its elevation in tumors of mice treated with proglumide may correlate with the influx of CD8+ cells found.
Fig. 6.

Proglumide decreases metastases in the mT5 orthotopic mouse model (n = 8 per group) after 21 days. A: final mT5 tumor weights after 21 days: Mean tumor weights ± SE of orthotopic mT5 tumors show that dietary fat significantly increases the size of tumors in mice on a high-fat diet compared with a regular diet (*P = 0.023). The tumors of mice treated with proglumide (prog) were smaller than the mice on untreated (reg) water but this did not reach significance. B: mean number of metastases per mouse was significantly increased in mice fed a high-fat diet compared with those on a regular diet (**P = 0.007). Proglumide significantly reduced the number of metastases in mice on a high-fat diet (***P = 0.004). C: Masson’s trichrome stain in representative tumor from each treatment group shows increased fibrosis in mice on either high-fat or control (cont) diet but with untreated drinking water (left). Proglumide treatment decreased the fibrosis in both groups of mice receiving either high-fat or control diets (right). D: quantitative analysis of fibrosis scoring shows significantly decreased fibrosis in both groups of mice treated with proglumide (*P < 0.02) compared with mice on regular untreated drinking water. E: immunoreactivity for αSMA is shown (cat. no. Ab-124964, Abcam, Cambridge, MA; 1:3,500) in a representative tumor from a mouse on the high fat diet and regular drinking water (bottom left, n = 10) and a representative image showing αSMA immunoreactivity in tumors of mice on the high-fat diet that received proglumide in the drinking water (bottom right, n = 10). Quantitative analysis of αSMA-positive tissue does not reveal any significant difference in tumors of mice on proglumide (P = 0.288). F: results of metastases PCR array showing genes from tumors with significant fold-changes when mice were placed on a high-fat diet compared with regular diet. αSMA, alpha smooth muscle actin. αSMA, alpha smooth muscle actin; mT5, murine mT5-2D cells; NS, not significant.
Table 3.
Genes with significant fold change in tumors of mice on the high-fat diet compared with control diet and the fold-change in these same genes in tumors of mice on the high-fat diet that were treated with proglumide
| Gene | High-Fat Diet/Reg. Water | High-Fat Diet/Proglumide Water |
|---|---|---|
| Mmp7 | 16.59 | 47.69 |
| Ilβ-1 | 4.39 | 2.27 |
| Elane | −4.03 | 17.25 |
| Nme2 | 1.39 | 3.86 |
| Tnfs10 | 1.41 | 3.61 |
| Nr4a3 | −1.6 | 3.0 |
| Fxyd5 | −1.08 | 2.39 |
| Lpar6 | 4.11 | 1.54 |
| Mmp10 | 2.51 | 1.24 |
| Mmp9 | 3.47 | 0.94 |
| Mmp13 | 3.96 | 2.03 |
| Fat1 | −10.3 | −2.12 |
| Fgfr4 | −3.51 | 1.52 |
DISCUSSION
Our studies confirm that a diet high in saturated fat promotes growth of pancreatic cancer in mice. In contrast to other’s studies, the effects of dietary fat in this investigation was independent of obesity because these experiments were terminated before the mice became obese. Because the Panc02 tumors were subcutaneous and remote from the GI tract, this study supports the concept that something in the circulation was responsible for the proliferative effects of the fat. Our studies support that the responsible peptide is most likely CCK, because the effects were abolished with an antagonist that blocks CCK’s interaction with its receptor and were confirmed by elevation of CCK in the blood of mice on the high-fat diet. It is possible that circulating gastrin may have also played a role in the mT5 murine tumor model because proglumide is a nonselective antagonist and blocks both CCK and gastrin at the CCK-AR and CCK-BR. Further evidence that the effects of dietary fat are mediated through the CCK:CCK-R axis is that pancreatic cancer cells genetically modified to lack CCK-Rs failed to respond to the stimulatory effects of dietary fat or to cerulein in culture. Lastly, a transgenic mouse engineered to be void of CCK did not respond to dietary fat with increased tumor proliferation.
The diet we used in our investigation was high in saturated fats compared with the control diet, and the saturated fats with long-chain FAs were most likely responsible for the release and increased CCK blood levels observed. Others have shown that diets high in saturated FAs increase pancreatic cancer growth and metastases (89), but this is the first paper, to our knowledge, to demonstrate that CCK is the responsible peptide for the tropic effects related to saturated dietary fat. This same diet was utilized in a study by Philip et al. (61) when examining pancreatic carcinogenesis in LSL-Kras/Ela-CreERT mice. Although the authors try to correlate cyclooxygenase 2 expression to the high-fat diet, cyclooxygenase 2 is generally activated by n-6 polyunsaturated fatty acids and reduced by n-3 polyunsaturated fatty acids and not affected by a diet high in saturated fats. In this same study, the authors failed to measure CCK levels or discuss the potential role of the trophic and inflammatory actions of CCK on the pancreas.
We found that CCK blood levels were elevated in mice on the high-saturated fat diet even though the exposure to the high-fat diet was for a fairly short duration. Previously we found that CCK blood levels were 10-fold higher in C57BL/6 mice on the same diet but for a longer duration (8 wk; 51). In this former study we waited until the mice became obese to operate and inject Panc02 cells orthotopically. Rather than proglumide, mice in this prior study were treated with a selective CCK-AR antagonist devazepide or L364,718 (14; 4 mg·kg−1·day−1 ip), and we did not find any reduction in primary tumor size compared with PBS-treated controls. One reason that proglumide may have been more effective than devazepide in the former studies is because it was administered in the drinking water, which provided continuous receptor blockade rather than the intermittent blockade by intraperitoneal injections.
Proglumide is a nonselective CCK receptor antagonist with affinity at both the CCK-A and CCK-B receptors. This current investigation showed that proglumide blocked the effects of dietary fat in mice with tumors expressing CCK-ARs (Panc02 tumors) and also decreased metastasis in mice with tumors expressing CCK-BRs (mT5 tumors). Because the mT5 tumors grew much faster, the mice had less exposure to proglumide. Perhaps a higher dose, longer treatment, or with smaller starting tumor size, we would have seen statistical change also in the mT5 primary tumor size with proglumide.
Inflammation and immunity are also important in carcinogenesis and for this reason we chose to conduct our investigation in immune competent mouse models. One of the mechanisms thought to play a role in cancer and obesity is cytokines that are produced from adipose tissue and recruit immune cells resulting in a proinflammatory state (28). An animal model to study acute and chronic pancreatitis has been well established (44) and involves the repeated injection of the CCK analogue cerulein (41). One could speculate that a diet high in saturated fat that raises CCK levels could also induce inflammation in the mouse pancreas. Human subjects with chronic pancreatitis have been found to have high CCK blood levels (67), and this condition is known to increase the risk for pancreatic cancer (87). Apart from its normal physiological role in digestion and proliferative effects on cancer epithelial cells, CCK peptide has also been found to play a role as an immunomodulatory peptide. Furthermore, immune cells also have CCK-Rs (90) that respond to CCK-R blockade by changing their cytokine expression signature (90). CCK peptide differentially affects the development and function of CD4+ T cell populations, with a negative influence on T helper (Th)1 and Th17 cells and positive regulatory effect on inducible T regulatory cells (90). Notably, CCK administration suppresses Th1 although slightly enhances Th2 development and cytokine production. Similarly, CCK treatment has been shown to promote Foxp3 expression. These immunomodulatory effects are mediated via CCK receptors on immune cells and these effects of CCK on immune cells are blocked by CCK-R antagonists (90). Zheng et al. (92) recently reviewed the role of immune cells in the pancreatic tumor microenvironment and how the immune cell type relates to outcome. The actions of proglumide on the immune cells of the tumor microenvironment may have contributed to the influx of CD8+ lymphocytes we observed in our study. Intratumoral CD8+ T-effector cells have been associated with a better outcome in pancreatic cancer. In our investigation, the tumor sizes and metastases were less in proglumide-treated mice, and the influx of CD8+ T cells may have contributed to this improved outcome. We have previously shown that CCK-R blockade enhances the efficacy of immune checkpoint antibodies by decreasing the immunosuppressive T-regulatory (Foxp3+) cells and inducing an influx of CD8+ T cell lymphocytes into pancreatic cancer (73).
We purposefully designed our study so that the experiments were terminated before the mice developed obesity to eliminate all the confounding factors associated with obesity and cancer. In fact, none of our mice became obese (>35 g), and the mice on the high-fat diets showed no statistical difference in body weights compared with mice on the control diet. There are a number of other factors, including leptin, glucose, and insulin involved with obesity and cancer risk (29). CCK-R signaling may also play a role in obesity. Obesity-induced epigenetic changes may remove DNA methylation marks from the CCK-BR, leading to its upregulation (24, 66, 84, 91).
A very interesting finding of our investigation was that the CCK-R antagonist significantly decreased tumor-associated fibrosis. CCK-Rs have also been identified on tissue fibroblasts (68) and pancreatic stellate (7) cells and when these receptors are activated, they produce desmoplastic stroma characteristic of the microenvironment of chronic pancreatitis and pancreatic cancer (2). CCK-Rs play a role in the dense fibrosis in the Kras murine model for pancreatic cancer and proglumide inhibits the fibrosis of the microenvironment in this model of carcinogenesis (69). Perhaps the decreased fibrosis allowed for better penetration of the drug to the epithelial cancer cells and thus was responsible for fewer metastases in mice treated with proglumide in spite of being on a high-fat diet. Cancer-associated fibroblasts secrete proteins of the extracellular matrix and soluble factors that stimulate cancer progression (82). Strategies to decrease the inflammatory fibroblasts and fibroblast-activated protein are associated with decreased cancer growth and metastases (22). There are many factors that contribute to fibrosis, including αSMA, collagen1-a, fibronectin, transforming growth factor-β, periostin, osteopotin, type IV collagen, and hyaluronan (47). There are many cells of the tumor microenvironment that are responsible for the dense fibrosis, including cancer associated fibroblasts, αSMA+ myofibroblasts, and mesenchymal stem cells. Rhim et al. (63) showed that in a genetically engineered animal model void of sonic hedgehog (shh) that αSMA-associated fibrosis was decreased and tumors more readily metastasized. This study suggested that the stroma components such as αSMA appeared to restrain neoplastic growth and progression. Other investigations have shown that a reduction in the tumor-associated fibrosis prevents metastases. Several strategies are been used in an attempt to decrease tumor associated fibrosis including blocking the activity of fibroblast activated protein (47), interrupting activity of the macrophage induced chemokine (C-C motif) ligand 2-CCR2 chemokine axis (58), or inhibition of hyaluronan with hyaluronic acid (34). In our investigation, we found less tumor-associated fibrosis and fewer metastases in mice treated with proglumide without a reduction in αSMA. These results would imply that CCK-R blockade does not affect αSMA but must be acting through another antifibrotic mechanism. In fact, when pancreatic stellate cells in culture were treated with a long-chain saturated FA (palmitic acid) comparable to the high-fat diet used in our studies, no change in αSMA was found (4). The favorable result we found was a decrease in metastases.
Most patients with advanced pancreatic cancer succumb to the disease because of metastases; therefore, a compound that blocks metastases, even when the primary tumor size is large, may have clinical significance. Our results indicate that genes involved in the extracellular matrix as well as some tumor suppressor genes were altered with CCK-R antagonist therapy, and these changes may have mediated the decrease in metastases noted. Hallmarks of cancer include angiogenesis, invasion, migration, and metastasis, and these processes only occur through changes in the microenvironment with remodeling of the extracellular matrix. Matrix metalloproteinases participate in cancer progression by exerting their proteolytic activity to degrade barriers and promote metastases (26). We found that Mmp gene expression was increased in mice on the high-fat diet, and these mice indeed had markedly increased metastases; the expression of Mmps decreased in mice treated with proglumide. Another gene that significantly increased in the mice on a high-fat diet mice treated with proglumide was Elane. Typically, this neutrophil-associated gene has been studied in fibrotic conditions such as chronic obstructive pulmonary disease and cystic fibrosis (39). Recently, studies have shown that subjects with pancreatic cancer that have a high neutrophil-to- lymphocyte ratio have prolonged survival with fewer metastases (59). Increased expression of the Elane gene in the mice treated with proglumide could be related to the breakdown of fibrosis in the microenvironment from neutrophil elastase resulting in fewer metastases. Our metastasis array also identified tumor suppressor genes that were increased by proglumide, including Fat-1 and Nme2, suggesting that interruption of CCK-R signaling activates genes involved with the suppression of cancer growth. Lastly, proglumide significantly increased the tumor expression of Tnfsf10, a TNF-related apoptosis-inducing ligand that induces cancer cell death by accelerating apoptosis (31). There has been a recent interest in developing TNF-related apoptosis-inducing ligand receptor agonists to treat pancreatic cancer (40). Changes in these genes indicate that proglumide therapy, by interrupting signaling at the CCK receptor, results in intracellular and genetic alterations in pancreatic tumor cells leading to decreased tumor progression and metastases through several mechanisms.
Dietary fat and cancer is a very complex issue and numerous mediators have been described. This multifaceted system involves growth factors, immune cells, adipocytes, and even possibly an altered intestinal microbiome (5). Our investigation shows that an important mechanism involved in the cancer-promoting effects of dietary fat involves CCK. Because CCK-Rs are present on the cancer epithelial cells, the fibroblasts, and the immune cells, CCK-R blockade may play a role in the treatment and prevention of pancreatic cancer. CCK-R antagonist blockade may potentially improve therapy for pancreatic cancer by decreasing the fibrosis of the tumor microenvironment and preventing metastases.
GRANTS
This study was supported by National Institutes of Health Grants CA-194745, TL-1-TR-001431, and CA-051008.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.P.S. conceived and designed research; S.N., J.B., A.A.-S., G.I., J.W., R.D.T., M.E.Z., W.B., N.S., and J.P.S. performed experiments; S.N., J.B., A.A.-S., G.I., J.W., M.E.Z., N.S., and J.P.S. analyzed data; S.N., J.B., A.A.-S., G.I., J.W., R.D.T., W.B., N.S., and J.P.S. interpreted results of experiments; S.N., J.B., G.I., J.W., M.E.Z., W.B., and J.P.S. prepared figures; J.P.S. drafted manuscript; S.N., J.B., A.A.-S., G.I., J.W., R.D.T., N.S., and J.P.S. edited and revised manuscript; S.N., J.B., A.A.-S., G.I., J.W., R.D.T., M.E.Z., W.B., N.S., and J.P.S. approved final version of manuscript.
REFERENCES
- 1.Alemán JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of obesity-induced gastrointestinal neoplasia. Gastroenterology 146: 357–373, 2014. doi: 10.1053/j.gastro.2013.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, Keogh G, Merrett N, Pirola R, Wilson JS. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29: 179–187, 2004. doi: 10.1097/00006676-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Arem H, Mayne ST, Sampson J, Risch H, Stolzenberg-Solomon RZ. Dietary fat intake and risk of pancreatic cancer in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Ann Epidemiol 23: 571–575, 2013. doi: 10.1016/j.annepidem.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Harosh Y, Anosov M, Salem H, Yatchenko Y, Birk R. Pancreatic stellate cell activation is regulated by fatty acids and ER stress. Exp Cell Res 359: 76–85, 2017. doi: 10.1016/j.yexcr.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Berger NA. Obesity and cancer pathogenesis. Ann N Y Acad Sci 1311: 57–76, 2014. doi: 10.1111/nyas.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem 7: 1211–1231, 2007. doi: 10.2174/156802607780960519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berna MJ, Seiz O, Nast JF, Benten D, Bläker M, Koch J, Lohse AW, Pace A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem 285: 38905–38914, 2010. doi: 10.1074/jbc.M110.125534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bister V, Skoog T, Virolainen S, Kiviluoto T, Puolakkainen P, Saarialho-Kere U. Increased expression of matrix metalloproteinases-21 and -26 and TIMP-4 in pancreatic adenocarcinoma. Mod Pathol 20: 1128–1140, 2007. doi: 10.1038/modpathol.3800956. [DOI] [PubMed] [Google Scholar]
- 9.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, Wilson JP, Feigin ME, Öhlund D, Handly-Santana A, Ardito-Abraham CM, Ludwig M, Elyada E, Alagesan B, Biffi G, Yordanov GN, Delcuze B, Creighton B, Wright K, Park Y, Morsink FH, Molenaar IQ, Borel Rinkes IH, Cuppen E, Hao Y, Jin Y, Nijman IJ, Iacobuzio-Donahue C, Leach SD, Pappin DJ, Hammell M, Klimstra DS, Basturk O, Hruban RH, Offerhaus GJ, Vries RG, Clevers H, Tuveson DA. Organoid models of human and mouse ductal pancreatic cancer. Cell 160: 324–338, 2015. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog 51: 53–63, 2012. doi: 10.1002/mc.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandi G, De Lorenzo S, Candela M, Pantaleo MA, Bellentani S, Tovoli F, Saccoccio G, Biasco G. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis 38: 231–240, 2017. doi: 10.1093/carcin/bgx007. [DOI] [PubMed] [Google Scholar]
- 12.Camilleri M. Peripheral mechanisms in appetite regulation. Gastroenterology 148: 1219–1233, 2015. doi: 10.1053/j.gastro.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra R, Wang Y, Shahid RA, Vigna SR, Freedman NJ, Liddle RA. Immunoglobulin-like domain containing receptor 1 mediates fat-stimulated cholecystokinin secretion. J Clin Invest 123: 3343–3352, 2013. doi: 10.1172/JCI68587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang RS, Lotti VJ, Chen TB, Kunkel KA. Characterization of the binding of [3H]-(+/-)-L-364,718: a new potent, nonpeptide cholecystokinin antagonist radioligand selective for peripheral receptors. Mol Pharmacol 30: 212–217, 1986. [PubMed] [Google Scholar]
- 15.Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP Jr, Schabel FM JR. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res 44: 717–726, 1984. [PubMed] [Google Scholar]
- 16.Coussens LM, Werb Z. Inflammation and cancer. Nature 420: 860–867, 2002. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, Lugea A, Gukovskaya AS, Li G, Hines OJ, Rozengurt E, Eibl G. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 6: 1064–1073, 2013. doi: 10.1158/1940-6207.CAPR-13-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dockray GJ. Luminal sensing in the gut: an overview. J Physiol Pharmacol 54, Suppl 4: 9–17, 2003. [PubMed] [Google Scholar]
- 19.Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev 86: 805–847, 2006. doi: 10.1152/physrev.00014.2005. [DOI] [PubMed] [Google Scholar]
- 20.Dutta D, Ghosh S, Pandit K, Mukhopadhyay P, Chowdhury S. Leptin and cancer: Pathogenesis and modulation. Indian J Endocrinol Metab 16, Suppl 3: S596–S600, 2012. doi: 10.4103/2230-8210.105577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elsässer HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas 1: 421–429, 1986. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Fan MH, Zhu Q, Li HH, Ra HJ, Majumdar S, Gulick DL, Jerome JA, Madsen DH, Christofidou-Solomidou M, Speicher DW, Bachovchin WW, Feghali-Bostwick C, Puré E. Fibroblast activation protein (FAP) accelerates collagen degradation and clearance from lungs in mice. J Biol Chem 291: 8070–8089, 2016. doi: 10.1074/jbc.M115.701433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res 18: 4266–4276, 2012. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrante SC, Nadler EP, Pillai DK, Hubal MJ, Wang Z, Wang JM, Gordish-Dressman H, Koeck E, Sevilla S, Wiles AA, Freishtat RJ. Adipocyte-derived exosomal miRNAs: a novel mechanism for obesity-related disease. Pediatr Res 77: 447–454, 2015. doi: 10.1038/pr.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghadirian P, Lynch HT, Krewski D. Epidemiology of pancreatic cancer: an overview. Cancer Detect Prev 27: 87–93, 2003. doi: 10.1016/S0361-090X(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 26.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278: 16–27, 2011. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 27.Gibbons C, Finlayson G, Caudwell P, Webb DL, Hellström PM, Näslund E, Blundell JE. Postprandial profiles of CCK after high fat and high carbohydrate meals and the relationship to satiety in humans. Peptides 77: 3–8, 2016. doi: 10.1016/j.peptides.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med 64: 45–57, 2013. doi: 10.1146/annurev-med-121211-091527. [DOI] [PubMed] [Google Scholar]
- 29.Goodwin PJ, Stambolic V. Impact of the obesity epidemic on cancer. Annu Rev Med 66: 281–296, 2015. doi: 10.1146/annurev-med-051613-012328. [DOI] [PubMed] [Google Scholar]
- 30.Gordon-Dseagu VL, Thompson FE, Subar AF, Ruder EH, Thiébaut AC, Potischman N, Stolzenberg-Solomon R. A cohort study of adolescent and midlife diet and pancreatic cancer risk in the NIH-AARP diet and health study. Am J Epidemiol 186: 305–317, 2017. doi: 10.1093/aje/kwx036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffith TS. Induction of tumor cell apoptosis by TRAIL gene therapy. Methods Mol Biol 542: 315–334, 2009. doi: 10.1007/978-1-59745-561-9_17. [DOI] [PubMed] [Google Scholar]
- 32.Hahne WF, Jensen RT, Lemp GF, Gardner JD. Proglumide and benzotript: members of a different class of cholecystokinin receptor antagonists. Proc Natl Acad Sci USA 78: 6304–6308, 1981. doi: 10.1073/pnas.78.10.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinen MM, Verhage BA, Goldbohm RA, van den Brandt PA. Meat and fat intake and pancreatic cancer risk in the Netherlands Cohort Study. Int J Cancer 125: 1118–1126, 2009. doi: 10.1002/ijc.24387. [DOI] [PubMed] [Google Scholar]
- 34.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, Raghunand N, Dychter S, Jiang P, Shepard HM, Devoe CE. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res 22: 2848–2854, 2016. doi: 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu X, Zhai Y, Kong P, Cui H, Yan T, Yang J, Qian Y, Ma Y, Wang F, Li H, Cheng C, Zhang L, Jia Z, Li Y, Yang B, Xu E, Wang J, Yang J, Bi Y, Chang L, Wang Y, Zhang Y, Song B, Li G, Shi R, Liu J, Zhang M, Cheng X, Cui Y. FAT1 prevents epithelial mesenchymal transition (EMT) via MAPK/ERK signaling pathway in esophageal squamous cell cancer. Cancer Lett 397: 83–93, 2017. doi: 10.1016/j.canlet.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 36.Iyengar NM, Hudis CA, Dannenberg AJ. Obesity and cancer: local and systemic mechanisms. Annu Rev Med 66: 297–309, 2015. doi: 10.1146/annurev-med-050913-022228. [DOI] [PubMed] [Google Scholar]
- 37.Jiao L, Chen L, White DL, Tinker L, Chlebowski RT, Van Horn LV, Richardson P, Lane D, Sangi-Haghpeykar H, El-Serag HB. Low-fat dietary pattern and pancreatic cancer risk in the women’s health initiative dietary modification randomized controlled trial. J Natl Cancer Inst 110: 49–56, 2018. doi: 10.1093/jnci/djx117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kapoor C, Vaidya S, Wadhwan V, Hitesh, Kaur G, Pathak A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Ther 12: 28–35, 2016. doi: 10.4103/0973-1482.157337. [DOI] [PubMed] [Google Scholar]
- 39.Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62: 726–759, 2010. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kretz AL, von Karstedt S, Hillenbrand A, Henne-Bruns D, Knippschild U, Trauzold A, Lemke J. Should we keep walking along the trail for pancreatic cancer treatment? revisiting TNF-related apoptosis-inducing ligand for anticancer therapy. Cancers (Basel) 10: 77, 2018. doi: 10.3390/cancers10030077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol 373: 97–117, 1977. doi: 10.1007/BF00432156. [DOI] [PubMed] [Google Scholar]
- 42.Lashinger LM, Harrison LM, Rasmussen AJ, Logsdon CD, Fischer SM, McArthur MJ, Hursting SD. Dietary energy balance modulation of Kras- and Ink4a/Arf+/--driven pancreatic cancer: the role of insulin-like growth factor-I. Cancer Prev Res (Phila) 6: 1046–1055, 2013. doi: 10.1158/1940-6207.CAPR-13-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehv M, Fitzgerald PJ. Pancreatic acinar cell regeneration. IV. Regeneration after resection. Am J Pathol 53: 513–535, 1968. [PMC free article] [PubMed] [Google Scholar]
- 44.Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology 144: 1180–1193, 2013. doi: 10.1053/j.gastro.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 45.Liou AP, Lu X, Sei Y, Zhao X, Pechhold S, Carrero RJ, Raybould HE, Wank S. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140: 903–912.e4, 2011. doi: 10.1053/j.gastro.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Ma DW. The role of n-3 polyunsaturated fatty acids in the prevention and treatment of breast cancer. Nutrients 6: 5184–5223, 2014. doi: 10.3390/nu6115184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, Durham AC, Buza EL, Vonderheide RH, June CH, Albelda SM, Puré E. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res 75: 2800–2810, 2015. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo CM, King A, Samuelson LC, Kindel TL, Rider T, Jandacek RJ, Raybould HE, Woods SC, Tso P. Cholecystokinin knockout mice are resistant to high-fat diet-induced obesity. Gastroenterology 138: 1997–2005, 2010. doi: 10.1053/j.gastro.2010.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 20: 197–209, 2006. doi: 10.1016/j.bpg.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Mainz DL, Black O, Webster PD. Hormonal control of pancreatic growth. J Clin Invest 52: 2300–2304, 1973. doi: 10.1172/JCI107418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matters GL, Cooper TK, McGovern CO, Gilius EL, Liao J, Barth BM, Kester M, Smith JP. Cholecystokinin mediates progression and metastasis of pancreatic cancer associated with dietary fat. Dig Dis Sci 59: 1180–1191, 2014. doi: 10.1007/s10620-014-3201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLaughlin J, Grazia Lucà M, Jones MN, D’Amato M, Dockray GJ, Thompson DG. Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology 116: 46–53, 1999. doi: 10.1016/S0016-5085(99)70227-1. [DOI] [PubMed] [Google Scholar]
- 53.McLaughlin JT, Lomax RB, Hall L, Dockray GJ, Thompson DG, Warhurst G. Fatty acids stimulate cholecystokinin secretion via an acyl chain length-specific, Ca2+-dependent mechanism in the enteroendocrine cell line STC-1. J Physiol 513: 11–18, 1998. doi: 10.1111/j.1469-7793.1998.011by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra P, Senthivinayagam S, Rana A, Rana B. Glycogen synthase kinase-3beta regulates Snail and beta-catenin during gastrin-induced migration of gastric cancer cells. J Mol Signal 5: 9, 2010. doi: 10.1186/1750-2187-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moran TH. Gut peptides in the control of food intake. Int J Obes 33, Suppl 1: S7–S10, 2009. doi: 10.1038/ijo.2009.9. [DOI] [PubMed] [Google Scholar]
- 56.Moran TH, Sawyer TK, Seeb DH, Ameglio PJ, Lombard MA, McHugh PR. Potent and sustained satiety actions of a cholecystokinin octapeptide analogue. Am J Clin Nutr 55, Suppl: 286S–290S, 1992. doi: 10.1093/ajcn/55.1.286s. [DOI] [PubMed] [Google Scholar]
- 57.Mulders RJ, de Git KC, Schéle E, Dickson SL, Sanz Y, Adan RA. Microbiota in obesity: interactions with enteroendocrine, immune and central nervous systems. Obes Rev 19: 435–451, 2018. doi: 10.1111/obr.12661. [DOI] [PubMed] [Google Scholar]
- 58.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim KH, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Goedegebuure SP, Linehan DC. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17: 651–662, 2016. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park I, Choi SJ, Kim YS, Ahn HK, Hong J, Sym SJ, Park J, Cho EK, Lee JH, Shin YJ, Shin DB. Prognostic factors for risk stratification of patients with recurrent or metastatic pancreatic adenocarcinoma who were treated with gemcitabine-based chemotherapy. Cancer Res Treat 48: 1264–1273, 2016. doi: 10.4143/crt.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel AV, Rodriguez C, Bernstein L, Chao A, Thun MJ, Calle EE. Obesity, recreational physical activity, and risk of pancreatic cancer in a large U.S. Cohort. Cancer Epidemiol Biomarkers Prev 14: 459–466, 2005. doi: 10.1158/1055-9965.EPI-04-0583. [DOI] [PubMed] [Google Scholar]
- 61.Philip B, Roland CL, Daniluk J, Liu Y, Chatterjee D, Gomez SB, Ji B, Huang H, Wang H, Fleming JB, Logsdon CD, Cruz-Monserrate Z. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 145: 1449–1458, 2013. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem 114: 63–70, 2008. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 63.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25: 735–747, 2014. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med 61: 301–316, 2010. doi: 10.1146/annurev.med.080708.082713. [DOI] [PubMed] [Google Scholar]
- 65.Roebuck BD, Kaplita PV, Edwards BR, Praissman M. Effects of dietary fats and soybean protein on azaserine-induced pancreatic carcinogenesis and plasma cholecystokinin in the rat. Cancer Res 47: 1333–1338, 1987. [PubMed] [Google Scholar]
- 66.Shi C, Zhang M, Tong M, Yang L, Pang L, Chen L, Xu G, Chi X, Hong Q, Ni Y, Ji C, Guo X. miR-148a is associated with obesity and modulates adipocyte differentiation of mesenchymal stem cells through Wnt signaling. Sci Rep 5: 9930, 2015. doi: 10.1038/srep09930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shiratori K, Takeuchi T, Satake K, Matsuno S; Study Group of Loxiglumide in Japan . Clinical evaluation of oral administration of a cholecystokinin-A receptor antagonist (loxiglumide) to patients with acute, painful attacks of chronic pancreatitis: a multicenter dose-response study in Japan. Pancreas 25: e1–e5, 2002. doi: 10.1097/00006676-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Singh P, Owlia A, Espeijo R, Dai B. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J Biol Chem 270: 8429–8438, 1995. doi: 10.1074/jbc.270.15.8429. [DOI] [PubMed] [Google Scholar]
- 69.Smith JP, Cooper TK, McGovern CO, Gilius EL, Zhong Q, Liao J, Molinolo AA, Gutkind JS, Matters GL. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 43: 1050–1059, 2014. doi: 10.1097/MPA.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith JP, Liu G, Soundararajan V, McLaughlin PJ, Zagon IS. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol 266: R277–R283, 1994. doi: 10.1152/ajpregu.1994.266.1.R277. [DOI] [PubMed] [Google Scholar]
- 71.Smith JP, Solomon TE. Cholecystokinin and pancreatic cancer: the chicken or the egg? Am J Physiol Gastrointest Liver Physiol 306: G91–G101, 2014. doi: 10.1152/ajpgi.00301.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med 10: 689–694, 2002. doi: 10.3892/ijmm.10.6.689. [DOI] [PubMed] [Google Scholar]
- 73.Smith JP, Wang S, Nadella S, Jablonski SA, Weiner LM. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol Immunother 67: 195–207, 2018. doi: 10.1007/s00262-017-2077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Solomon TE. Control of exocrine pancreatic secretion. In: Physiology of the Gastrointestinal Tract, edited by Johnson LR. New York: Raven Press, 1994, p. 1499–1530. [Google Scholar]
- 75.Solomon TE, Petersen H, Elashoff J, Grossman MI. Interaction of caerulein and secretin on pancreatic size and composition in rat. Am J Physiol Endocrinol Metab Gastrointest Physiol 235: E714–E719, 1978. doi: 10.1152/ajpendo.1978.235.6.E714. [DOI] [PubMed] [Google Scholar]
- 76.Solomon TE, Vanier M, Morisset J. Cell site and time course of DNA synthesis in pancreas after caerulein and secretin. Am J Physiol Gastrointest Liver Physiol 245: G99–G105, 1983. doi: 10.1152/ajpgi.1983.245.1.G99. [DOI] [PubMed] [Google Scholar]
- 77.Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, Geary N. Ghrelin, CCK, GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev 97: 411–463, 2017. doi: 10.1152/physrev.00031.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Strouch MJ, Ding Y, Salabat MR, Melstrom LG, Adrian K, Quinn C, Pelham C, Rao S, Adrian TE, Bentrem DJ, Grippo PJ. A high omega-3 fatty acid diet mitigates murine pancreatic precancer development. J Surg Res 165: 75–81, 2011. doi: 10.1016/j.jss.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thiébaut AC, Jiao L, Silverman DT, Cross AJ, Thompson FE, Subar AF, Hollenbeck AR, Schatzkin A, Stolzenberg-Solomon RZ. Dietary fatty acids and pancreatic cancer in the NIH-AARP diet and health study. J Natl Cancer Inst 101: 1001–1011, 2009. doi: 10.1093/jnci/djp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tong J, D’Alessio D. Eating disorders and gastrointestinal peptides. Curr Opin Endocrinol Diabetes Obes 18: 42–49, 2011. doi: 10.1097/MED.0b013e328341e12b. [DOI] [PubMed] [Google Scholar]
- 81.Varro A, Kenny S, Hemers E, McCaig C, Przemeck S, Wang TC, Bodger K, Pritchard DM. Increased gastric expression of MMP-7 in hypergastrinemia and significance for epithelial-mesenchymal signaling. Am J Physiol Gastrointest Liver Physiol 292: G1133–G1140, 2007. doi: 10.1152/ajpgi.00526.2006. [DOI] [PubMed] [Google Scholar]
- 82.Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C. Role of tumor microenvironment in tumorigenesis. J Cancer 8: 761–773, 2017. doi: 10.7150/jca.17648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Willier S, Butt E, Grunewald TG. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol Cell 105: 317–333, 2013. doi: 10.1111/boc.201300011. [DOI] [PubMed] [Google Scholar]
- 84.Wu L, Dai X, Zhan J, Zhang Y, Zhang H, Zhang H, Zeng S, Xi W. Profiling peripheral microRNAs in obesity and type 2 diabetes mellitus. APMIS 123: 580–585, 2015. doi: 10.1111/apm.12389. [DOI] [PubMed] [Google Scholar]
- 85.Xu J, Yin Z, Cao S, Gao W, Liu L, Yin Y, Liu P, Shu Y. Systematic review and meta-analysis on the association between IL-1B polymorphisms and cancer risk. PLoS One 8: e63654, 2013. doi: 10.1371/journal.pone.0063654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu Y, Qian SY. Anti-cancer activities of ω-6 polyunsaturated fatty acids. Biomed J 37: 112–119, 2014. doi: 10.4103/2319-4170.131378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144: 1252–1261, 2013. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yao X, Tian Z. Saturated, monounsaturated and polyunsaturated fatty acids intake and risk of pancreatic cancer: evidence from observational studies. PLoS One 10: e0130870, 2015. doi: 10.1371/journal.pone.0130870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu M, Liu H, Duan Y, Zhang D, Li S, Wang F. Four types of fatty acids exert differential impact on pancreatic cancer growth. Cancer Lett 360: 187–194, 2015. doi: 10.1016/j.canlet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 90.Zhang JG, Liu JX, Jia XX, Geng J, Yu F, Cong B. Cholecystokinin octapeptide regulates the differentiation and effector cytokine production of CD4(+) T cells in vitro. Int Immunopharmacol 20: 307–315, 2014. doi: 10.1016/j.intimp.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 91.Zhao E, Keller MP, Rabaglia ME, Oler AT, Stapleton DS, Schueler KL, Neto EC, Moon JY, Wang P, Wang IM, Lum PY, Ivanovska I, Cleary M, Greenawalt D, Tsang J, Choi YJ, Kleinhanz R, Shang J, Zhou YP, Howard AD, Zhang BB, Kendziorski C, Thornberry NA, Yandell BS, Schadt EE, Attie AD. Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mamm Genome 20: 476–485, 2009. doi: 10.1007/s00335-009-9217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 144: 1230–1240, 2013. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zucker KA, Adrian TE, Bilchik AJ, Modlin IM. Effects of the CCK receptor antagonist L364,718 on pancreatic growth in adult and developing animals. Am J Physiol Gastrointest Liver Physiol 257: G511–G516, 1989. doi: 10.1152/ajpgi.1989.257.4.G511. [DOI] [PubMed] [Google Scholar]
- 94.Zyromski NJ, Mathur A, Pitt HA, Wade TE, Wang S, Nakshatri P, Swartz-Basile DA, Nakshatri H. Obesity potentiates the growth and dissemination of pancreatic cancer. Surgery 146: 258–263, 2009. doi: 10.1016/j.surg.2009.02.024. [DOI] [PubMed] [Google Scholar]