

Abstract
There is no therapy that promotes maturation and functionality of a dialysis arteriovenous fistula (AVF). The search for such therapies largely relies on evaluation of vascular responses and putative therapies in experimental AVFs. We studied an AVF in mice with chronic kidney disease (CKD). We demonstrate numerous stressors in the vein of the AVF-CKD group, including pathological shear, mitogenic, inflammatory, and hypoxia-reoxygenation stress. Because stress promotes premature senescence, we examined whether senescence is induced in the vein of the AVF-CKD model. We demonstrate a senescence phenotype in the AVF-CKD model, as indicated by increased expression of p16Ink4a, p21Cip1, and p53 and expected changes for certain senescence-associated microRNAs. RNA-sequencing analysis demonstrated differential expression of ~10,000 genes, including upregulation of proinflammatory and proliferative genes, in the vein of the AVF-CKD group. The vein in the AVF-CKD group exhibited telomere erosion and increased senescence-associated β-galactosidase activity and staining. Senescence was induced in the artery of the AVF-CKD group and in the vein of the AVF without CKD. Finally, given the rapidly rising clinical interest in senolytics, we provide proof of concept of senolytics as a therapeutic approach by demonstrating that senolytics decrease p16Ink4a expression in the AVF-CKD model. This study introduces a novel concept underlying the basis for maturational and functional failure in human dialysis AVFs and identifies a new target for senolytic therapy.
Keywords: age, dialysis, fistula, senolytics, vascular stress
INTRODUCTION
The formation of an arteriovenous fistula (AVF) abruptly conjoins vascular segments that became separate and distinct early in vascular development. During embryogenesis, subsets of angioblasts in the primitive capillary plexus move apart and differentiate into what will become arteries and veins (20). Vascular specialization is driven by signaling species from surrounding tissue and angioblasts themselves, the final structures of arteries and veins also shaped by their respective hemodynamic profiles (20). Fully developed arteries are resilient, with high intraluminal pressure, pulsatile flow, and low capacitance. Veins, conversely, are flaccid, their walls are delicate, and they exhibit low intraluminal pressure, low and steady flow, and high capacitance. An AVF, in a certain sense, thus reverses the course of vascular development, thereby subjecting veins to the heightened pressures and flows they were never designed to bear and imposing pathological shear stress on both vessels. The study of the AVF thus offers insights into vascular behavior when the vasculature is uniquely and severely stressed.
The study of the AVF is also of fundamental clinical importance as an AVF, created in superficial, upper extremity vessels, provides a therapeutic device: a high-flow, accessible, venous segment for hemodialysis for end-stage renal disease (ESRD). Indeed, among the accesses, the AVF is the most favored, promoted as it is by the “Fistula-First” initiative (14, 15, 21, 23, 26).
To support intermittent hemodialysis, the AVF must undergo “maturation.” Maturation requires markedly increased blood flow as the artery dilates and both vessels undergo luminal expansion and outward remodeling. Outward remodeling also toughens the vein, allowing it to be repeatedly punctured for dialysis (17, 21, 23). Additionally, maturation and sustained function require suppression of inward remodeling (neointimal hyperplasia and thrombogenesis) (17, 21, 23).
Clinical AVF outcomes, however, are truly grim: 50% of created AVFs may never mature, 30% require instrumentation to mature, and the median survival for AVFs is 7.5 mo, which increases to 62 mo when primary AVF failure is excluded (13). Even after maturation, 15% of created AVFs will be nonfunctional after 1 yr, and 25% will be nonfunctional after 2 yr (23). Dysfunction of AVFs and other accesses markedly contributes to morbidity, mortality, and hospitalization rates (~25%) in patients with ESRD, imposing healthcare costs that may exceed $2.5 billion per year (1).
There is no specific therapy that promotes maturation and functionality of human AVFs; of all the unmet needs in ESRD, this is among the most pressing (15, 23, 26). Strategies targeted to this need largely rely on rodent AVF models for an understanding of the basis for AVF failure. Studies in rodent and human AVFs have revealed an induction of potentially vasculopathic species, but the underlying mechanism that underpins vasculopathic responses remains elusive.
In a previous study, this laboratory refined the clinical fidelity of the murine AVF model (8). First, the AVF is created in the setting of chronic kidney disease (CKD); second, the anastomosis is end vein-to-side artery, as is clinically fashioned (12). In an attempt to elucidate the basis for vasculopathic AVF responses, the present study explored the types of stress imposed on the venous limb of this AVF-CKD model and the attendant consequences of such stress. Uncovered during our studies is a previously unrecognized pathobiological process that may underlie AVF maturational failure and dysfunction: induction of cellular senescence.
MATERIALS AND METHODS
Studies were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic and consistent with National Institutes of Health guidelines. The AVF-CKD model was created in male C57BL/6J mice, as described in our previous study, in which an AVF or a sham AVF was created in mice in which CKD or sham CKD was previously imposed (8). At 1 wk after AVF/sham AVF, the vasculature was harvested for assorted analyses and assessment of senescence-associated β-galactosidase (SA-β-Gal) activity. Ultrasound studies were performed at 2 wk. In selected studies, Po2 was measured by an i-Stat analyzer (Abbott Point of Care, Princeton, NJ) in venous blood samples drawn immediately after AVF/sham AVF. In other studies, mice with AVF-CKD were treated with the senolytic agent combination dasatinib (5 mg/kg) and quercetin (50 mg/kg) or vehicle via gavage at 4 and 5 days after AVF creation (24, 29), and p16Ink4a protein expression was assessed on day 7.
Vascular ultrasound.
Sonography was performed using high-resolution imaging (Vevo 2100 with an MS400 30-MHz transducer, Vevo 3100 with an MX400 38-MHz transducer, FUJIFILM VisualSonics, Toronto, ON, Canada) (28). Blood flow dynamics were three-dimensionally reconstructed from a serial two-dimensional color Doppler ultrasound image at 32-μm intervals (28).
Gene expression by RNA sequencing.
Total RNA was extracted as described elsewhere (8). Our RNA-sequencing (RNA-Seq) samples were sequenced by the Mayo Medical Genome Facility and processed through Mayo Clinic’s MAP-RSeq V2 software (7), a comprehensive RNA-Seq workflow for Illumina-paired end reads. Ensembl’s Mus musculus GRCm38.79 was used for the reference genome and transcriptome. Quality-control metrics from RSeqQC were evaluated to ensure that raw expression values from each sample were reliable and could be collectively used for differential expression analysis (27).
Genes with an average of ≥25 reads were kept for differential expression analysis. The R package edgeR was used to identify which genes were statistically differentially expressed from the group comparisons (22). For each comparison, unsupervised clustering was implemented with the MeV (4) application on genes with a false discovery rate of <0.05 and with an absolute log2 fold change of ≥2. MeV was additionally used to visualize these findings through heat maps.
mRNA and microRNA expression.
mRNA expression was determined as described elsewhere (8, 9) using quantitative real-time RT-PCR; microRNA (miRNA) expression was determined in total RNA using a miRNA isolation kit (mirVana, Thermo Fisher Scientific, Waltham, MA). Quantitation was performed using TaqMan miRNA assays (Applied Biosystems, Foster City, CA).
Western blot analysis.
Vascular protein extracts were prepared as described elsewhere (8). Proteins were separated on TGX Stain-Free gels (Bio-Rad, Hercules, CA), visualized by UV-light activation, and transferred to PVDF membranes. Primary antibodies included p16Ink4a and p21Cip1 (Abcam, Cambridge, MA), p53 (R & D Systems, Minneapolis, MN), proliferating cell nuclear antigen (PCNA; Cell Signaling Technology, Danvers, MA), and β-catenin (BD Biosciences, Franklin Lakes, NJ). Densitometric quantitation of protein expression was normalized to total protein visualized on the PVDF membranes (Gel Doc XR+ imager, Bio-Rad).
SA-β-Gal activity.
SA-β-Gal activity was quantified using a fluorometric assay kit (Enzo Life Sciences, Farmingdale, NY). Staining for SA-β-Gal activity in frozen sections was performed as described elsewhere (18).
Statistics.
Data are expressed as means ± SE and considered statistically significant for P < 0.05. Student’s t-test was used for parametric data, and the Mann-Whitney U-test was employed for nonparametric data.
RESULTS
The AVF exhibits diverse stressors and altered gene expression.
As stress induces vasculopathic responses, we examined whether relevant forms of stress exist in the vein of the AVF. Three-dimensional Doppler ultrasound demonstrates the presence of pathological oscillatory shear stress and turbulent flow (Fig. 1A). Mitogenic and inflammatory stress were also present; the vein of the AVF-CKD group showed markedly upregulated proliferation markers (PCNA), proliferative signaling species (β-catenin) (Fig. 1B), and proinflammatory cytokines (Fig. 1C) compared with the sham group. We also considered whether reoxygenation stress occurs in the AVF. Intact veins are exposed to a low Po2 to which their metabolism is geared. AVF creation immediately exposes the vein to a markedly increased Po2, thereby imposing “hypoxia-reoxygenation” stress on the AVF-CKD (Fig. 1D).
Fig. 1.

Stressors in the vein in the arteriovenous fistula (AVF)-chronic kidney disease (CKD) model. A: 3-dimensional color Doppler ultrasound image of the AVF in the AVF-CKD group. Red and blue indicate cranial and caudal blood flow in the AVF artery and vein, respectively. Oscillatory flow at the anastomosis is revealed by the yellow mosaic pattern, and a mixture of caudal (blue) and cranial-directed (red) flow is seen in the dilated AVF vein. A-V, arteriovenous. B: Western blot analysis of proliferating cell nuclear antigen (PCNA) and β-catenin expression in sham veins and the vein of the AVF-CKD model and normalized expression at 1 wk. Values are means ± SE; n = 5 in each group. *P < 0.05. C: IL-1α, IL-6, and monocyte chemoattractant protein 1 (MCP-1) mRNA expression in sham veins and in the vein of the AVF-CKD model at 1 wk. Values are means ± SE; n = 9 sham and 10 AVF-CKD. *P < 0.01. D: Po2 in blood from sham veins and the vein of the AVF-CKD model after AVF/sham surgery. Values are means ± SE; n = 6 in each group. *P < 0.05. E: heat map of RNA-sequencing analysis of sham veins and the vein of the AVF-CKD model at 1 wk. Map comprises 2,427 genes with a false discovery rate of <0.05 and an absolute log2 fold change of ≥2. Green-to-red coloring was calculated from each sample’s reads per kilobase million (RPKM) value for each gene and then transformed to z scores. Significant transcriptomic landscape differences distinguish each group; unsupervised clustering with the MeV software perfectly separated the sham vein samples from the AVF-CKD samples.
Such stressors were accompanied by altered gene expression in the AVF, as demonstrated by RNA-Seq analyses. Of the ~20,000 genes detected, RNA-Seq revealed differential expression of ~10,000, including the upregulation of proinflammatory and proliferative genes, in the vein of the AVF-CKD group. The heat map revealed clearly visible transcriptomic landscape differences, thereby distinguishing the veins in the AVF-CKD and sham groups (Fig. 1E).
The AVF exhibits a senescence phenotype.
Stress, especially when diverse, induces premature senescence (10, 11, 25). We examined whether a senescence phenotype exists in the AVF, beginning with the senescence drivers p16Ink4a and p21Cip1 (10, 11, 25). Cdkn2a (reflecting mainly p16) and Cdkn1a (reflecting mainly p21) mRNAs were induced in the vein of the AVF and accompanied by increased p16Ink4a and p21Cip1 protein expression (Fig. 2, A and B); induction of p53 protein (upstream of p21Cip1 in the senescence pathway) also occurred (Fig. 2B). The AVF-CKD vein showed expected changes for certain senescence-associated miRNAs; increased expression of miR-21 (19) and decreased expression of miR-92a (3) are implicated in vascular senescence. The AVF-CKD vein exhibited, congruently, increased expression of miR-21 and decreased expression of miR-92a (Fig. 2C).
Fig. 2.
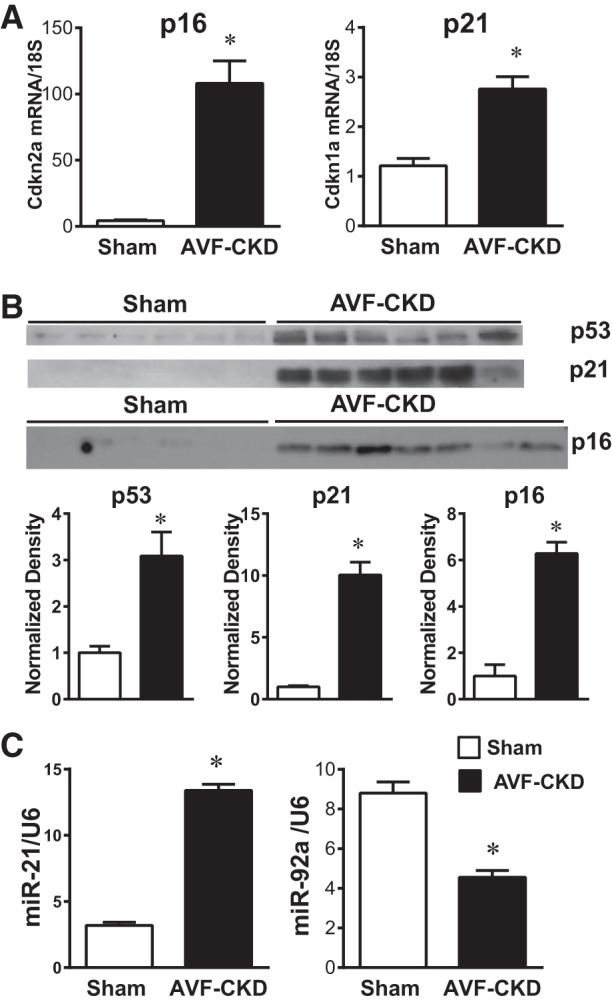
Venous senescence markers in the arteriovenous fistula (AVF)-chronic kidney disease (CKD) model. A: expression of genes encoding for p16Ink4a (Cdkn2a) and p21Cip1 (Cdkn1a) in sham veins and the vein of the AVF-CKD model, measured by quantitative real-time RT-PCR. Values are means ± SE; n = 9 sham and 10 AVF-CKD. *P < 0.0001. B: Western blot analysis of p53, p21Cip1, and p16Ink4a protein expression in sham veins and the vein of the AVF-CKD model and normalized expression. Values are means ± SE; n = 6 in each group for p53 and p21, and 6 sham and 7 AVF-CKD for p16. *P < 0.001. C: miR-21 and miR-92a expression in sham veins and the vein of the AVF-CKD model. Values are means ± SE; n = 8 sham and 10 AVF-CKD. *P < 0.001.
Additional RNA-Seq analyses focused on telomeric regions. The vein in the AVF-CKD group showed ∼900 genes with significantly decreased expression and 66 genes that were essentially undetectable (Fig. 3A). Telomere erosion, a senescence hallmark (10, 11, 25), thus exists in the vein in the AVF-CKD group. Also present at 1 wk was increased SA-β-Gal activity (Fig. 3B). SA-β-Gal (blue) staining was increased in the vein in the AVF-CKD group, with focal cellular staining in the endothelium and tunica media; the control vein was devoid of blue staining (Fig. 3C).
Fig. 3.

Telomere erosion and senescence-associated β-galactosidase (SA-β-Gal) activity/staining in the vein in the arteriovenous fistula (AVF)-chronic kidney disease (CKD) model. A: telomere erosion in the AVF-CKD vein, as shown by a disproportionate loss of gene expression in the telomeric regions, defined as 10 megabases from the start or end of each autosomal chromosome. Differentially expressed genes were defined by a false discovery rate of ≤0.05 when the AVF-CKD group was compared with the sham group. A total of 897 genes in the telomeric regions have decreased expression with a log2 fold change of <0; of these 897 genes, 66 have a log2 fold change less than −4. B: SA-β-Gal activity in sham veins and the vein of the AVF-CKD model at 1 wk. RFU, relative fluorescence units. Values are means ± SE; n = 5 in each group. *P < 0.0001. C: SA-β-Gal activity in frozen sections from a sham vein and the vein of the AVF-CKD model at 1 wk. Blue stain indicates cells with increased SA-β-Gal activity. The sham vein shows no staining, whereas the AVF-CKD model shows blue staining in the endothelium and media. L, lumen.
Senescence changes also occurred in the AVF artery, as revealed by striking p16Ink4a induction in the artery of the AVF-CKD group (Fig. 4A). The AVF itself, in the presence of normal kidney function, evinced p16Ink4a upregulation (Fig. 4B). Finally, because senolytics are emerging as a new therapy, we examined the short-term effect of the senolytics dasatinib and quercetin; dasatinib and quercetin significantly decreased expression of p16Ink4a at 1 wk in the vein in the AVF-CKD group (Fig. 4C).
Fig. 4.
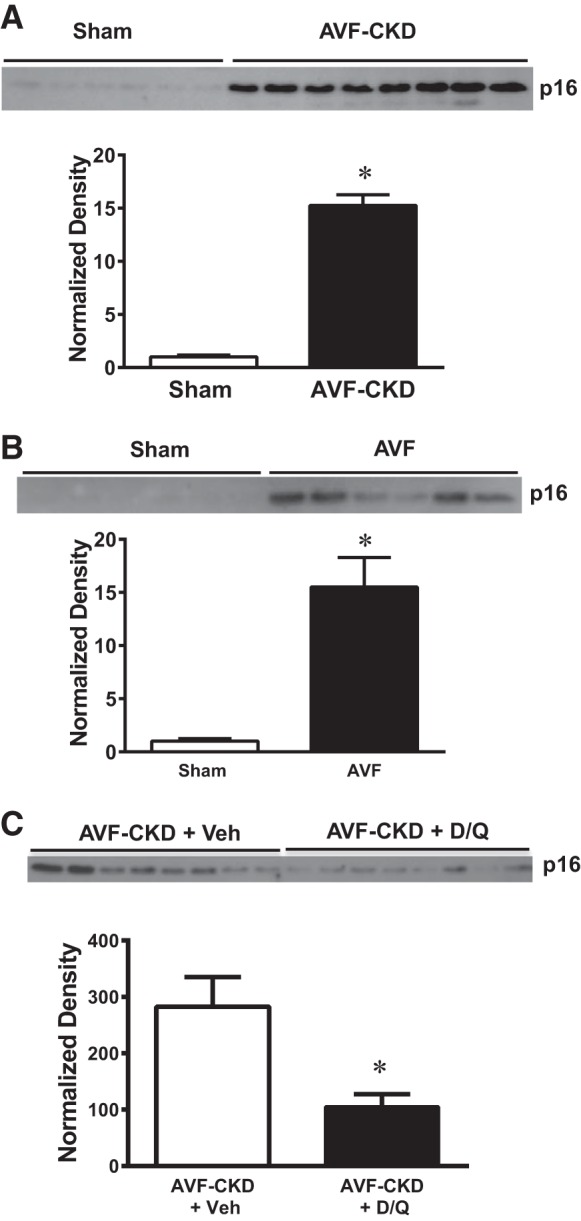
p16Ink4a expression in arteries in the arteriovenous fistula (AVF)-chronic kidney disease (CKD) model, in AVF without CKD, and in the vein in AVF-CKD following administration of senolytics. A–C: p16Ink4a protein expression at 1 wk in sham arteries and the artery of the AVF-CKD model (A), in sham veins and the vein of the AVF in mice with normal kidneys (B), and in the vein of the AVF-CKD model treated with vehicle (Veh) or dasatinib and quercetin (D/Q; C). Normalized expression is shown below each Western blot. Values are means ± SE; n = 6 sham and 8 AVF-CKD (A), 6 sham and 6 AVF-CKD (B), and 8 AVF-CKD + Veh and 8 AVF-CKD + D/Q (C). *P < 0.001 for A and B; *P < 0.05 for C.
DISCUSSION
This study is the first to characterize the range of stress in the AVF. Stressors so uncovered are likely interrelated, in that pathological shear stress induces both mitogenic and inflammatory responses in the venous wall. These responses perturb the endothelium, further disrupting laminar flow, thereby augmenting pathological shear stress. The vein in the AVF is subjected to hypoxia-reoxygenation, the latter exerting a distinct stress, as normal venous metabolism occurs under relatively low Po2. The severity of AVF stress is underscored by the vast number of differentially expressed genes, the latter representing among the highest observed in diseased organs and tissues in vivo.
Severe stress, especially when diverse, can induce premature senescence (10, 11, 25). We provide the first evidence that the vein of the AVF exhibits a senescence phenotype, as substantiated by studies of telomere erosion, expression of p16Ink4a/p21Cip1/p53, SA-β-Gal activity and staining, and specific miRNA expression. Tissue senescence is driven by p16Ink4a- and p21Cip1-expressing senescent cells acting independently or in concert. Such cells exhibit cell cycle arrest, resistance to apoptosis, and a senescence-associated secretory phenotype (SASP) (10, 11, 25). The SASP produces diverse cytokines, chemokines, proteases, growth factors, and prothrombotic and adhesion molecules, among other cytotoxic species. The SASP mediates senescence-associated tissue injury. We thus suggest that 1) upregulation of genes in the AVF may reflect the effects of the SASP and SASP factors, 2) the SASP in the AVF may compromise AVF maturation and function by promoting inward remodeling and compromising outward remodeling, 3) the AVF senescence phenotype may explain how the induction of seemingly disparate vasculopathic species described in the literature are underpinned and linked (2, 5, 6, 8, 16, 23), and 4) these findings may explain why older age invariantly associates with worse human AVF outcomes (23).
This demonstration of an AVF senescence phenotype opens up new therapeutic strategies. Along with their production of SASP factors, p16Ink4a- and p21Cip1-expressing senescent cells concomitantly upregulate survival pathways; such pathways enable these cells to withstand SASP factors that they themselves produce (10, 11, 25). Senolytics inhibit these survival pathways in p16Ink4a- and p21Cip1-expressing senescent cells, thereby killing these senescent cells but leaving nonsenescent cells intact. We demonstrate the efficacy of established senolytics, dasatinib and quercetin, administered to mice with AVF-CKD, in reducing p16Ink4a expression, the latter effect reflecting death of p16Ink4a-expressing cells with attendantly reduced p16Ink4a expression. Death of p16Ink4a-expressing senescent cells would reduce production of SASP factors and, thus, vasculopathic species that may impair AVF maturation and functionality.
SA-β-Gal staining identified such activity in endothelial foci in the AVF-CKD group, a not-unexpected finding, as the endothelium is the lining first exposed to pathological shear stress. Foci of cells in the media of the AVF-CKD group also exhibited such activity. This may reflect elicitation of senescence in the media by SASP factors from senescent endothelial cells diffusing into the media.
Finally, we emphasize three considerations. 1) The cardiovascular and aging fields are in urgent need of clinically relevant models of premature accelerated senescence. Most aging models are chronic, costly, and difficult to study. Many of the models of accelerated senescence may be of uncertain clinical relevance. This AVF model provides a clinically relevant, novel model of accelerated premature vascular senescence. 2) Senolytics offer a therapeutic strategy for a central unmet ESRD need, that is, improving AVF maturation and functionality. 3) As senolytics are being introduced and tested, diseases amenable to local therapy are especially appealing; regionally applied senolytics reduce concerns regarding unexpected systemic toxicity. Human dialysis AVFs are superficial and readily targeted by topical drug delivery. Promotion of AVF maturation and function thus provides an outstanding opportunity to assess the efficacy of local application of senolytics.
GRANTS
These studies are supported by NIH Grants DK-70124 and DK-47060 (K. A. Nath, Z. S. Katusic, and J. P. Grande), HL-134664 (A. Terzic and S. Yamada), and AG-13925 (J. L. Kirkland); the Marriott Family Foundation, Michael S and Mary Sue Shannon Family, Russ and Kathy Van Cleve Foundation, Mayo Clinic Center for Regenerative Medicine, and Regenerative Medicine Minnesota (A. Terzic and S. Yamada); and the Connor Group, Robert J. and Theresa W. Ryan, and the Ted Nash Long Life and Noaber Foundations (J. L. Kirkland).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.A.N. and A.J.C. conceived and designed research; A.J.C., A.W.A., M.C.N., S.Y., and T.T. performed experiments; K.A.N., D.R.O., A.J.C., J.P.G., A.W.A., M.C.N., S.Y., and T.T. analyzed data; K.A.N., D.R.O., A.J.C., J.P.G., A.W.A., M.C.N., S.Y., A.T., T.T., J.L.K., and Z.S.K. interpreted results of experiments; K.A.N., D.R.O., A.J.C., A.W.A., S.Y., and T.T. prepared figures; K.A.N. drafted manuscript; K.A.N., D.R.O., A.J.C., J.P.G., A.W.A., M.C.N., S.Y., A.T., T.T., J.L.K., and Z.S.K. edited and revised manuscript; K.A.N., D.R.O., A.J.C., J.P.G., A.W.A., M.C.N., S.Y., A.T., T.T., J.L.K., and Z.S.K. approved final version of manuscript.
REFERENCES
- 1.Anonymous US Renal Data System 2009 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases, 2009. [Google Scholar]
- 2.Croatt AJ, Grande JP, Hernandez MC, Ackerman AW, Katusic ZS, Nath KA. Characterization of a model of an arteriovenous fistula in the rat: the effect of l-NAME. Am J Pathol 176: 2530–2541, 2010. doi: 10.2353/ajpath.2010.090649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hazra S, Henson GD, Morgan RG, Breevoort SR, Ives SJ, Richardson RS, Donato AJ, Lesniewski LA. Experimental reduction of miR-92a mimics arterial aging. Exp Gerontol 83: 165–170, 2016. doi: 10.1016/j.exger.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howe EA, Sinha R, Schlauch D, Quackenbush J. RNA-Seq analysis in MeV. Bioinformatics 27: 3209–3210, 2011. doi: 10.1093/bioinformatics/btr490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juncos JP, Grande JP, Kang L, Ackerman AW, Croatt AJ, Katusic ZS, Nath KA. MCP-1 contributes to arteriovenous fistula failure. J Am Soc Nephrol 22: 43–48, 2011. doi: 10.1681/ASN.2010040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juncos JP, Tracz MJ, Croatt AJ, Grande JP, Ackerman AW, Katusic ZS, Nath KA. Genetic deficiency of heme oxygenase-1 impairs functionality and form of an arteriovenous fistula in the mouse. Kidney Int 74: 47–51, 2008. doi: 10.1038/ki.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, Middha S, Sicotte H, Thompson AE, Asmann YW, Kocher JP. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics 15: 224, 2014. doi: 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang L, Grande JP, Hillestad ML, Croatt AJ, Barry MA, Katusic ZS, Nath KA. A new model of an arteriovenous fistula in chronic kidney disease in the mouse: beneficial effects of upregulated heme oxygenase-1. Am J Physiol Renal Physiol 310: F466–F476, 2016. doi: 10.1152/ajprenal.00288.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang L, Hillestad ML, Grande JP, Croatt AJ, Barry MA, Farrugia G, Katusic ZS, Nath KA. Induction and functional significance of the heme oxygenase system in pathological shear stress in vivo. Am J Physiol Heart Circ Physiol 308: H1402–H1413, 2015. doi: 10.1152/ajpheart.00882.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine 21: 21–28, 2017. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc 65: 2297–2301, 2017. doi: 10.1111/jgs.14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamoorthy MK, Banerjee RK, Wang Y, Choe AK, Rigger D, Roy-Chaudhury P. Anatomic configuration affects the flow rate and diameter of porcine arteriovenous fistulae. Kidney Int 81: 745–750, 2012. doi: 10.1038/ki.2011.468. [DOI] [PubMed] [Google Scholar]
- 13.Lok CE, Sontrop JM, Tomlinson G, Rajan D, Cattral M, Oreopoulos G, Harris J, Moist L. Cumulative patency of contemporary fistulas versus grafts (2000–2010). Clin J Am Soc Nephrol 8: 810–818, 2013. doi: 10.2215/CJN.00730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehrotra R, Cheung AK, Meyer T, Nath KA. Vascular access for hemodialysis and value-based purchasing for ESRD. J Am Soc Nephrol 28: 395–397, 2017. doi: 10.1681/ASN.2016070769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nath KA, Allon M. Challenges in developing new therapies for vascular access dysfunction. Clin J Am Soc Nephrol 12: 2053–2055, 2017. doi: 10.2215/CJN.06650617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nath KA, Kanakiriya SK, Grande JP, Croatt AJ, Katusic ZS. Increased venous proinflammatory gene expression and intimal hyperplasia in an aorto-caval fistula model in the rat. Am J Pathol 162: 2079–2090, 2003. doi: 10.1016/S0002-9440(10)64339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath KA, Katusic ZS. Predicting the functionality and form of a dialysis fistula. J Am Soc Nephrol 27: 3508–3510, 2016. doi: 10.1681/ASN.2016050569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Barnhoorn S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, Jurk D. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8: 15691, 2017. doi: 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olivieri F, Rippo MR, Monsurrò V, Salvioli S, Capri M, Procopio AD, Franceschi C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev 12: 1056–1068, 2013. doi: 10.1016/j.arr.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol 18: 477–494, 2017. doi: 10.1038/nrm.2017.36. [DOI] [PubMed] [Google Scholar]
- 21.Riella MC, Roy-Chaudhury P. Vascular access in haemodialysis: strengthening the Achilles heel. Nat Rev Nephrol 9: 348–357, 2013. doi: 10.1038/nrneph.2013.76. [DOI] [PubMed] [Google Scholar]
- 22.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy-Chaudhury P, Sukhatme VP, Cheung AK. Hemodialysis vascular access dysfunction: a cellular and molecular viewpoint. J Am Soc Nephrol 17: 1112–1127, 2006. doi: 10.1681/ASN.2005050615. [DOI] [PubMed] [Google Scholar]
- 24.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123: 966–972, 2013. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viecelli AK, Mori TA, Roy-Chaudhury P, Polkinghorne KR, Hawley CM, Johnson DW, Pascoe EM, Irish AB. The pathogenesis of hemodialysis vascular access failure and systemic therapies for its prevention: optimism unfulfilled. Semin Dial 31: 244–257, 2018. doi: 10.1111/sdi.12658. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28: 2184–2185, 2012. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Cao Y, Yamada S, Thirunavukkarasu M, Nin V, Joshi M, Rishi MT, Bhattacharya S, Camacho-Pereira J, Sharma AK, Shameer K, Kocher JP, Sanchez JA, Wang E, Hoeppner LH, Dutta SK, Leof EB, Shah V, Claffey KP, Chini EN, Simons M, Terzic A, Maulik N, Mukhopadhyay D. Cardiomyopathy and worsened ischemic heart failure in SM22-α Cre-mediated neuropilin-1 null mice: dysregulation of PGC1α and mitochondrial homeostasis. Arterioscler Thromb Vasc Biol 35: 1401–1412, 2015. doi: 10.1161/ATVBAHA.115.305566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, Roos CM, Verzosa GC, LeBrasseur NK, Wren JD, Farr JN, Khosla S, Stout MB, McGowan SJ, Fuhrmann-Stroissnigg H, Gurkar AU, Zhao J, Colangelo D, Dorronsoro A, Ling YY, Barghouthy AS, Navarro DC, Sano T, Robbins PD, Niedernhofer LJ, Kirkland JL. The Achilles heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644–658, 2015. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]