

Abstract
The mechanisms of cardiovascular and renal protection observed in clinical trials of sodium-glucose cotransporter 2 (SGLT2) inhibitors (SGLT2i) are incompletely understood and likely multifactorial, including natriuretic, diuretic, and antihypertensive effects, glomerular pressure reduction, and lowering of plasma and interstitial fluid volume. To quantitatively evaluate the contribution of proposed SGLT2i mechanisms of action on changes in renal hemodynamics and volume status, we coupled a mathematical model of renal function and volume homeostasis with clinical data in healthy subjects administered 10 mg of dapagliflozin once daily. The minimum set of mechanisms necessary to reproduce observed clinical responses (urinary sodium and water excretion, serum creatinine and sodium) was determined, and important unobserved physiological variables (glomerular pressure, blood and interstitial fluid volume) were then simulated. We further simulated the response to SGLT2i in diabetic virtual patients with and without renal impairment. Multiple mechanisms were required to explain the observed response: 1) direct inhibition of sodium and glucose reabsorption through SGLT2, 2) SGLT2-driven inhibition of Na+/H+ exchanger 3 sodium reabsorption, and 3) osmotic diuresis coupled with peripheral sodium storage. The model also showed that the consequences of these mechanisms include lowering of glomerular pressure, reduction of blood and interstitial fluid volume, and mild blood pressure reduction, in agreement with clinical observations. The simulations suggest that these effects are more significant in diabetic patients than healthy subjects and that while glucose excretion may diminish with renal impairment, improvements in glomerular pressure and blood volume are not diminished at lower glomerular filtration rate, suggesting that cardiorenal benefits of SGLT2i may be sustained in renally impaired patients.
Keywords: chronic kidney disease, computational modeling, heart failure, renal function, SGLT2 inhibition
INTRODUCTION
Recent cardiovascular outcomes trials demonstrate that therapies that inhibit sodium glucose cotransporter (SGLT)2 may reduce heart failure morbidity and mortality and slow decline in renal function (7, 8, 23, 42, 43). Multiple mechanisms have been proposed to explain these effects (6, 29, 41). Proximal mechanisms include natriuresis due to direct inhibition of sodium reabsorption through SGLT2, natriuresis due to indirect inhibition of sodium reabsorption through the interlinking of SGLT2 and Na+/H+ exchanger 3 (NHE3) (4, 9, 25), natriuresis as a consequence of lowering insulin, which has sodium-retaining effects in the proximal tubule (PT) (2, 17), and the osmotic diuretic effect of unreabsorbed glucose remaining in the tubule. These natriuretic and diuretic effects have consequent effects on renal and systemic hemodynamics. SGLT2 inhibitors (SGLT2i) suppress glomerular hyperfiltration, likely reflecting reduced intraglomerular pressure in response to reduced PT Na+ reabsorption (38, 39). SGLT2i also indirectly reduces blood pressure and blood volume (21). Mathematical analyses suggest that the excess electrolyte-free water clearance associated with osmotic diuresis may result in greater clearance of interstitial fluid compared with other forms of diuretics (14). Other proposed mechanisms, such as improvements in fuel metabolism from fat oxidation to ketones (6) or reduced arterial stiffness (3, 26, 33), have a less clear link to these renal actions of SGLT2i.
The relative contributions of these various potential mechanisms are not quantitatively understood. Mechanism-based mathematical modeling provides a tool for evaluating the physiological consequences of mechanisms in complex systems when data are sparse and for predicting responses under conditions for which data are not yet available. This facilitates a deeper quantitative understanding of drug mechanisms and can motivate and focus future data collection.
We previously used a mechanistic mathematical model of renal function and volume homeostasis to demonstrate the role of a primary increase in PT Na+ reabsorption in diabetic hyperfiltration and glomerular hypertension (13). In this study, we extended that model to explicitly account for glucose and Na+ reabsorption through SGLT2 and SGLT1 in the PT. We coupled mathematical modeling with clinical measurements of urinary and serum biomarkers in healthy subjects in the response to dapagliflozin to quantify the contribution of proposed proximal mechanisms of SGLT2i to the consequent effects (reductions in intraglomerular and systemic blood pressure, reduced blood and interstitial fluid volume) that may improve renal and cardiovascular outcomes. After identifying a set of mechanisms capable of reproducing the observed response in healthy subjects, we then conducted simulations in diabetic states with normal and impaired renal function to assess the impact of kidney disease on the hemodynamic response to SGLT2i.
METHODS
Mathematical Model
The base model of renal function is summarized in Fig. 1 and has been described in detail previously (11, 12). Full model equations are provided in the Supplemental Material for this article (Supplemental Material for this article is available online at the Journal website). Briefly, this model describes key physiological processes involved in renal function and in maintaining Na+ and water homeostasis. It describes blood flow, resistance, and pressures through the renal vasculature; filtration of water and Na+ through the glomerulus; Na+ and water reabsorption, flow rates, and pressures in each tubular segment; Na+ and water excretion; and Na+ and water balance and its effects on interstitial fluid volume and blood pressure. Key regulatory feedback mechanisms are incorporated in the model, including the renin-angiotensin-aldosterone system (RAAS), tubuloglomerular feedback (TGF), myogenic autoregulation, effects of renal interstitial hydrostatic pressure on regulation of tubular Na+ reabsorption, vasopressin regulation of tubular water reabsorption, and local blood flow autoregulation. For this study, we have extended this model as described below to account for 1) glucose filtration, reabsorption through SGLT1 and 2, and excretion; 2) the coupling of Na+ with glucose reabsorption through SGLT1 and 2; 3) the osmotic effects of glucose along the tubule; and 4) filtration, reabsorption, and excretion of albumin.
Fig. 1.

Base model of renal function. Top left: the renal vasculature is modeled by a single preafferent resistance vessel flowing into N parallel nephrons. Bottom left: glomerular filtration is modeled according to Starling’s law. Na+ and water are reabsorbed at different fractional rates in the proximal tubule, loop of Henle, distal convoluted tubule, and connecting tubule/collecting duct. Glucose and Na+ reabsorption are coupled through sodium-glucose cotransporter (SGLT)2 and SGLT1 in the proximal tubule. Top right: the balance between Na+ and water excretion and intake determines blood volume and Na+ concentration. Na+ and water move between the blood and interstitial fluid across a concentration gradient, and Na+ may be sequestered nonosmotically in a peripheral storage compartment. Blood volume determines venous return and cardiac output, which together with total peripheral resistance determine mean arterial pressure and subsequently renal perfusion pressure, closing the loop. Bottom right: multiple regulatory mechanisms, including the renin-angiotensin-aldosterone system and tubuloglomerular feedback (TGF), provide feedback on model variables to maintain or return homeostasis. ACE, angiotensin-converting enzyme. [Adapted from Hallow et al. (13) with permission.] See equations for definitions.
Single-nephron glomerular filtration rate (SNGFR) is defined according to Starling’s equation, where Kf is the glomerular ultrafiltration coefficient, Pgc is glomerular capillary hydrostatic pressure, PBow is pressure in Bowman’s space, and πgo-avg is average glomerular capillary oncotic pressure.
| (1) |
Total GFR is then the SNGFR times the number of nephrons:
| (2) |
Glucose filtration, reabsorption, and excretion.
Glucose is filtered freely through the glomerulus, so that single-nephron filtered glucose load is
| (3) |
where Cglu is the plasma glucose concentration.
Glucose reabsorbed in the S1 and S2 segments is given by
| (4) |
where Rglu,S12 is the rate of glucose reabsorption per unit length of the S1 and S2 segments together and Lpt,S12 is the length of the PT S1 and S2 segments together. Similarly, glucose reabsorbed in the S3 segment is given by
| (5) |
Any unreabsorbed glucose then flows through the rest of the tubule and is ultimately excreted, so that the rate of urinary glucose excretion (RUGE) is
| (6) |
SGLT2 in the S1 and S2 segments of the PT reabsorbs 90–97% of filtered glucose, whereas SGLT1 in the S3 segment reabsorbs the remaining 3–10% (10, 24, 27, 36, 37). At high plasma glucose concentrations, filtered glucose can exceed the kidney’s capacity for reabsorption. Rglu,S12 and Rglu,S3 represent the number and function of SGLT2 and SGLT1 transporters, respectively. The values were determined such that 95% of filtered glucose is reabsorbed in the S1 and S2 segments while the remaining glucose was reabsorbed in the S3 segment and so that all glucose is reabsorbed and urinary glucose excretion (UGE) is zero for blood glucose concentrations up to 9 mmol/l (5).
Na+ filtration and reabsorption in PT.
Na+ is freely filtered across the glomerulus, so that the single-nephron filtered Na+ load is given by
| (7) |
where CNa is the plasma Na+ concentration.
The rate of Na+ reabsorption through SGLT2 equals the rate of glucose reabsorption in the S1 and S2 segments, since SGLT2 reabsorbs Na+ and glucose at a 1-to-1 molar ratio:
| (8) |
The rate of Na+ reabsorption through SGLT1 is twice the rate of glucose reabsorption in the S3 segment, since SGLT1 reabsorbs Na+ and glucose at a 2-to-1 molar ratio:
| (9) |
Total PT Na+ reabsorption is then given by
| (10) |
where ηNa,reabs-PT,NHE3 and ηNa,reabs-PT,other are the fractional rates of PT Na+ reabsorption through NHE3 and through mechanisms other than SGLT2 and NHE3, respectively. Na+ flow rate out of the PT is then
| (11) |
Na+ reabsorption along the rest of the tubule is modeled as in Ref. 13.
Water reabsorption along the tubule.
Water reabsorption in the PT is isosmotic. Therefore, water leaving the PT and entering the loop of Henle (LoH) is given by
| (12) |
where DCT is the distal convoluted tubule and filtered osmolytes include both Na+ and glucose:
| (13) |
| (14) |
In the LoH, water is reabsorbed in the water-permeable descending LoH (DLH) because of the osmotic gradient created by actively pumping Na+ out of the water-impermeable ascending limb (ALH). Here we adapted our previous description of this countercurrent mechanism (13) to include the osmotic activity of both Na+ and glucose. The osmolality along the length of the DLH (OsmDLH), which is assumed in equilibrium with the osmolality in the surrounding interstitium (OsmIS), is given by
| (15) |
where RALH is the rate of Na+ reabsorption per unit length in the ALH. Water flow through the DLH is given by
| (16) |
The ALH and the DCT are modeled as impermeable to water, so that flow through these segments equals flow out of the DLH:
| (17) |
In the collecting duct (CD), water reabsorption is driven by the osmotic gradient between CD tubular fluid and the interstitium and is modulated by vasopressin:
| (18) |
where the osmolality in the CD [OsmCD(L)] accounts for Na+ reabsorbed in the CD:
| (19) |
The effect of vasopressin on water reabsorption is modeled as a sigmoidal relationship, such that ηvasopressin is 1 and water flows freely across the osmotic gradient when vasopressin levels are very high and ηvasopressin is 0 and no water is reabsorbed when vasopressin is absent:
| (20) |
Then, single-nephron water excretion rate is given by
| (21) |
and urine flow rate is
| (22) |
SGLT2 inhibition.
The direct effect of dapagliflozin on SGLT2 was modeled as an inhibitory effect on the glucose reabsorption rate per unit length through SGLT2 in the S1 and S2 segments (Eq. 23, utilized above in Eq. 4). The clinical data utilized in this analysis showed that after the initial peak in 24-h UGE after SGLT2i, UGE decreased slightly from its peak over the next several days. This effect is assumed at least in part to be due to compensation, as SGLT1 and 2 are upregulated. Here we assumed that unreabsorbed glucose is the signal for upregulation of SGLT. To model this, a compensatory factor Xsglt, which is normally zero, increases over time when RUGE is nonzero, up to some maximum limit Xsglt,max and with a time constant Tx,sglt.
| (23) |
| (24) |
| (25) |
In addition to direct effects on SGLT2, SGLT2i may reduce reabsorption through NHE3 (4, 9, 25). To investigate this potential effect, we allowed an inhibitory effect of SGLT2i on reabsorption through NHE3:
| (26) |
We made no a priori assumption about the mechanism or degree of inhibition of NHE3. The degree of inhibition, SGLT2i_NHE3_inhibition, was initially set to 0. Its value was only increased when components of the observed response could not be explained by direct inhibition of SGLT2 or by osmotic diuresis.
Peripheral Na+ storage.
We also extended the model to allow potential peripheral nonosmotic storage of Na+. There has been growing appreciation over the last few years for the role of peripheral tissues, in particular the skin, muscle, and bone, in dynamically storing and regulating body Na+ (15, 34, 35). Modeling the same clinical data utilized here, we have recently used a three-compartment model of blood, interstitium, and peripheral Na+ storage to show that peripheral Na+ storage may play a key role not only in buffering blood Na+ concentration but also in regulating differential changes in blood and interstitial volume in response to diuretic therapy (14). Here we adapted the model to include a peripheral Na+ storage compartment, to allow evaluation of the potential role of this compartment in the renal response to dapagliflozin. The equations for this portion of the model are shown in Eqs. 27–30. Na+ concentrations in the blood and interstitial compartments are assumed to equilibrate quickly. When interstitial Na+ concentration ([Na]IF) exceeds the normal equilibrium level ([Na]IF,ref), Na+ moves out of the interstitium and is sequestered in the peripheral Na+ compartment, at a rate of ΦNa,stored, where it is osmotically inactive. Na+ cannot be stored indefinitely, and thus there is a limit Nastored,max on how much Na+ can be stored. The peripheral Na+ compartment can be effectively removed from the model by setting QNa,stored to 0.
| (27) |
| (28) |
| (29) |
| (30) |
Dapagliflozin pharmacodynamic study.
Data from the dapagliflozin arm of a previously conducted drug-drug interaction study were used post hoc to inform the model. In this study, healthy subjects [n = 42, 10 women, 32 men, aged 19–45 yr (mean age 30 yr), 28 Caucasian and 14 black, with body mass index values of 20.0–31.2 kg/m2 (mean body mass index: 26.5 kg/m2)] gave written informed consent to participate in the Institutional Review Board-approved study. The subjects were randomized in this open label study to dapagliflozin (10 mg/day) for 7 days (additional study arms with bumetanide and the combination of bumetanide and dapagliflozin were not utilized in this analysis). Subjects were confined in a clinical trial unit for the duration of the study and received a fixed diet with ~110 mmol/day of Na+ and 60 mmol/day of potassium for 2 days before dosing and throughout dosing. Pooled 24-h urine samples were collected at baseline and throughout the study and analyzed for volume, osmolality, and total amounts of Na+, glucose, creatinine, and others. Fasting serum samples were obtained at baseline and throughout the study for determination of Na+, creatinine, and other biomarkers. Of note, the salt tablets provided in this study to precisely control Na+ intake were poorly tolerated and associated with frequent adverse events including nausea, vomiting, and dizziness. For the purposes of the analyses described here, data from dapagliflozin given alone for the first 7 days of the study were used. Observed time courses of changes in urinary and serum Na+, urinary volume, urinary glucose, and serum creatinine were used for model fitting.
Model implementation.
The model was implemented in free open-source programming software (R 3.1.2). It utilizes the RxODE package (40). Model code is provided in the Supplemental Material for this article.
Modeling fitting and simulation approach.
We assume that SGLT2i produces three proximal effects and that any additional effects are the consequence of these mechanisms: 1) direct inhibition of glucose and Na+ by SGLT2; 2) indirect reductions in water reabsorption due to glucose-driven osmotic diuresis; and 3) reductions in Na+ reabsorption through NHE3 (we do not differentiate whether this is a direct effect or the consequence of reducing insulin). In addition, we also considered the possibility of a third, nonosmotic extrarenal sodium compartment.
We first estimated SGLT2 percent inhibition, as well as the two parameters governing the magnitude and time course of SGLT compensation (see Eq. 25), by fitting the observed UGE response to dapagliflozin.
We then compared the resulting model-predicted changes in urine water and sodium and serum creatinine and sodium with the observed 7-day time courses of serum creatinine, urine volume, urine Na+, and plasma Na+ under different assumptions about the above mechanisms: 1) SGLT2i acts only through direct effects on SGLT2 Na+ reabsorption; 2) SGLT2i acts through both direct SGLT2 inhibition and indirect NHE3 inhibition of Na+ reabsorption, but with no osmotic diuresis effect; 3) SGLT2i acts through the above natriuretic mechanisms as well as through glucose-driven osmotic diuresis; and 4) SGLT2i acts through the above mechanisms, there exists an extrarenal compartment where Na+ may be stored nonosmotically, and sodium can move into/out of this compartment in response to changes in interstitial sodium concentration.
For each combination of mechanisms, we simulated the response to SGLT2i over 7 days and compared it with the clinically observed changes in UGE, serum creatinine, urine volume, urine Na+, and plasma Na+. For cases 2 and 4, we used nonlinear least squares to estimate relevant parameters (SGLT2i_NHE3_inhibition from Eq. 26 and QNa,stored from Eq. 27) to best fit the observed data.
RESULTS
Evaluating Ability of SGLT2 Inhibition Mechanisms to Explain Clinical Response
Case 1: Direct effect of dapagliflozin on SGLT2 only.
Figure 2 shows simulated and observed 24-h UGE in healthy individuals under different values for SGLT2% inhibition and maximum SGLT compensation Xsglt,max. The optimal fit was achieved at 85.3% inhibition and 30% maximum compensation. Using the fitted parameter values for SGLT2 inhibition and compensation, Fig. 3 compares the simulated and observed responses to 7 days of dapagliflozin treatment. In addition, the simulated values for important unmeasured variables are shown. In this case, we ignored the osmotic effects of tubular glucose (glucose components of Eqs. 13 and 14 set to 0). We assumed no effect of SGLT2i on Na+ reabsorption through NHE3 (SGLT2iNH3inhibition = 0) and no peripheral Na+ storage (QNa,stored = 0).
Fig. 2.
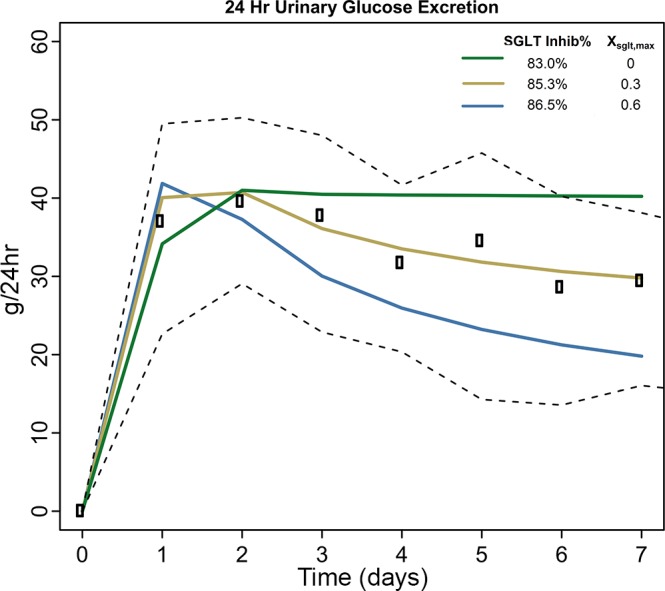
Urinary glucose excretion in response to dapagliflozin for different values of sodium-glucose cotransporter (SGLT)2 inhibition and compensatory upregulation of SGLT (Xsglt,max). Rectangles, median observed value; black dashed lines, 25th and 75th percentiles of observed data.
Fig. 3.

Simulated and observed responses to dapagliflozin under 4 different sets of assumptions. Case 1: direct effect of dapagliflozin on sodium-glucose cotransporter 2 (SGLT2) only; case 2: direct SGLT inhibitors (SGLT2i) + osmotic diuresis due to unreabsorbed glucose; case 3: direct SGLT2i + osmotic diuresis + SGLT2-driven Na+/H+ exchanger 3 (NHE3) inhibition; case 4: direct SGLT2i + osmotic diuresis + NHE3 inhibition + allowance for peripheral Na+ storage. Only case 4, in which the model accounts for direct reductions in glucose and Na+ reabsorption due to SGLT2 inhibition as well as osmotic diuresis effects of glucose, SGLT2-associated NHE3 inhibition, and peripheral Na+ storage, is able to reproduce all features of the data. Gray dashed line, model baseline; circles, median observed value; black dashed lines, 10th and 90th percentiles of observed data. GFR, glomerular filtration rate.
This model case was completely unable to explain the data and in fact predicted small decreases in serum creatinine, opposite of those observed. Figure 4 helps understand this prediction. At baseline, nearly all glucose reabsorption occurs through SGLT2, and reabsorption through SGLT1 is minimal. Dapagliflozin inhibition of SGLT2 allows additional glucose (and Na+) to reach SGLT1, so that while SGLT2 reabsorption decreases, reabsorption through SGLT1 increases, and the total decrease in glucose reabsorption is much less than the decrease in SGLT2 (Fig. 4A). SGLT2 reabsorbs glucose and Na+ at a 1-to-1 molar ratio, while SGLT1 reabsorbs at a 1-to-2 ratio. Thus as glucose reabsorption through SGLT1 increases, the molar rate of Na+ reabsorption through SGLT1 increases twice as much. The result is a net increase in Na+ reabsorption (Fig. 4B). As shown in Fig. 4C, Na+ reabsorption through both SGLT1 and SGLT2 accounts for only 5.8% of all Na+ reabsorption in normoglycemic conditions [as we have shown previously (13)]. The model predicts that if direct SGLT2 inhibition were the only consequence of dapagliflozin treatment, total PT Na+ reabsorption would slightly increase. Fractional PT Na+ reabsorption is also predicted to increase slightly in this case (Fig. 4D). As we have previously modeled, increases in PT Na+ reabsorption cause GFR to increase (13) and serum creatinine to decrease, as predicted here. But, of course, such behavior was not observed clinically, and this case cannot represent the full effects of SGLT inhibition.
Fig. 4.

Effect of sodium-glucose cotransporter (SGLT)2 inhibitors (SGLT2i) on proximal tubule (PT) glucose and Na+ reabsorption at normal blood glucose concentrations. A: reduced SGLT2 glucose reabsorption through SGLT2 increases glucose delivery to and reabsorption by SGLT1 (all cases). B: SGLT2 Na+ reabsorption is reduced, but since SGLT1 reabsorbs Na+ and glucose at a 2-to-1 ratio, SGLT1 Na+ reabsorption is greatly increased, so that there is a net Na+ increase through SGLT (all cases). C and D: in cases 1 and 2, total PT Na+ reabsorption is nearly unchanged. At normal glucose levels, SGLT accounts for only a small fraction of PT Na+ reabsorption, so the effect of SGLT2i on total PT Na+ reabsorption is small. Na+/H+ exchanger 3 (NHE3) accounts for the bulk of PT Na+ reabsorption. E and F: in cases 3 and 4, when SGLT2 inhibition also triggers NHE3 inhibition, total and fractional PT Na+ reabsorption is reduced, primarily because of reduced reabsorption through NHE3.
Case 2: Direct and osmotic diuresis effects of SGLT2 inhibition.
We then adapted the model to include tubular osmotic effects of glucose, again assuming no effect on NHE3 and no peripheral Na+ storage. As shown in Fig. 3, prediction of water excretion improved and the dynamics of serum creatinine were more similar to the observed data, but serum creatinine still decreased overall and the model still poorly fit the data.
Case 3: Direct SGLT2 inhibition + osmotic diuresis + NHE3 inhibition.
We then added an additional inhibitory effect on NHE3 Na+ reabsorption, again assuming no peripheral Na+ storage. As shown in Fig. 3, this model case was able to reproduce the observed response quite well, with the exception of plasma Na+ concentration, for which the model predicted a transient increase that was not observed. The degree of NHE3 inhibition required to optimally fit the data was estimated to be 7%, a relatively small change. As shown in Fig. 4, E and F, reabsorption through NHE3 accounts for the bulk of PT Na+ reabsorption, and inhibiting NHE3 by just 7% resulted in a similar reduction in total PT Na+ reabsorption, with the direct effect of dapagliflozin on Na+ reabsorption through SGLT2 having a negligible effect. The addition of an effect of SGLT2 inhibition on NHE3 in the model produced a transient increase in Na+ excretion on day 1 that returned to baseline soon after (Fig. 3, middle row, left panel).
Case 4: Direct SGLT2 inhibition + osmotic diuresis + NHE3 + peripheral Na+ storage.
Case 3 explained changes in serum creatinine and water excretion well but predicted a transient increase in plasma Na+ that was not observed. We have previously utilized a compartmental model of blood and interstitial Na+ (14) and showed that for the electrolyte-free water clearance observed in this study conservation of mass indicates that either the blood volume should have increased or Na+ must have left the blood through some mechanism other than renal excretion. In light of the growing understanding of the role of nonosmotic Na+ storage in peripheral tissue (15, 34), we hypothesized that a third compartment of nonosmotically active Na+ exists, and that Na+ can move into or out of this compartment as the interstitial fluid Na+ concentration changes. In case 4, we “turned on” this peripheral compartment by making QNa,stored from Eq. 27 nonzero. As Fig. 3 shows, with this addition the model reproduced the observed data well.
Predicted Effect of Dapagliflozin on Pressure and Volume Changes
We then used the model to evaluate the effect of dapagliflozin on variables not easily measurable clinically but that may be important in kidney disease and heart failure. Glomerular hypertension is a driving factor for progression of diabetic kidney disease (1, 16, 32), and reduction in glomerular capillary hydrostatic pressure slows the rate of kidney disease progression. As shown in Fig. 3, bottom, under cases 3 and 4 the model predicts that SGLT2i reduces glomerular pressure, even in healthy subjects. Cases 1 and 2, which do not include effects on Na+ reabsorption through NHE3, do not show a glomerular pressure reduction, indicating that the predicted glomerular pressure reduction is primarily due to reductions in PT Na+ reabsorption through NHE3.
Heart failure is characterized by volume overload, congestion, and excess interstitial fluid. Treatments that reduce blood volume and interstitial congestion are beneficial in heart failure, although excessive reduction in blood volume can be detrimental (30, 31). The model predicts an initial reduction in blood volume in cases 3 and 4, although it returns to near baseline over time. In case 4, the model also predicts a substantial reduction in interstitial fluid volume. This suggests that the capacity to store Na+ peripherally is associated with an ability to clear interstitial fluid, as we have described previously (14). Peripherally stored Na+ was increased by 130 mEq.
Simulation of Diabetic Virtual Patients With and Without Renal Impairment
Using the model established above (case 4), we next altered model parameters to describe states of diabetes, with and without renal impairment.
Diabetes with hyperfiltration.
Diabetes was induced in the model by increasing plasma glucose from 5.5 to 9 mmol/l. This resulted in an increase in baseline GFR from 105 to 120 ml/min, as a consequence of increased PT Na+ reabsorption though SGLT2 and NHE3 (13).
Diabetes with moderate renal function.
GFR declines with age, and type 2 diabetes patients tend to be older with lower GFRs in the range of 70–100 ml/min. These patients are still hyperfiltering at the single-nephron level but have lost sufficient nephrons to lower GFR. To simulate this, we reduced the number of nephrons to 60% of normal, which reduced baseline GFR to 87 ml/min.
Diabetes with impaired renal function.
To simulate patients with chronic kidney disease (CKD), the number of nephrons was further reduced to 25% of normal. To represent glomerulosclerosis commonly found in CKD, Kf was reduced by 25%. These changes lowered baseline GFR to 40 ml/min.
These changes, introduced before simulating drug treatment, produced a new “baseline” steady state in each case. Both GFR and SNGFR were increased in the hyperfiltering diabetic state (Fig. 5A). In the diabetic state with moderate renal function, GFR was in the normal range (87 ml/min) but SNGFR increased further to 73 nl/min. In the diabetic state with renal impairment, GFR was reduced to 40 ml/min but SNGFR increased even further, to 81 nl/min. It should be noted that for the model to reach a new steady state sodium balance must be achieved. As nephron number is reduced, the initial sodium imbalance is corrected by both an increase in single-nephron filtration and a decrease in tubular sodium reabsorption (due to pressure-natriuresis) so that glomerotubular balance is lost but sodium balance is restored.
Fig. 5.

Simulated baseline renal function under normal (healthy) and diabetic (DB) conditions with hyperfiltration, normal glomerular filtration rate (GFR), or chronic kidney disease (CKD). A: GFR and single-nephron GFR (SNGFR). B: proximal tubule (PT) glucose reabsorption. C: PT Na+ reabsorption through SGLT2. D: total PT Na+ reabsorption. SNGFR and PT Na+ reabsorption increased from the healthy to the diabetic state and increased further as kidney function was reduced. Total GFR and total Na+ reabsorption increased in diabetes but decreased as renal function was reduced.
Increasing blood glucose from the healthy to the diabetic state (normal renal function) caused a small increase in glucose reabsorption through SGLT2 (from 0.54 to 0.75 mEq/min) but a 10-fold increase in glucose reabsorption through SGLT1 (from 0.03 to 0.3 mEq/min) (Fig. 5B), In the healthy state SGLT1 accounted for 5.7% of glucose reabsorption, but in all three diabetic states it accounted for 29%. In the diabetic virtual patients with moderate or impaired renal function, total glucose reabsorption decreased relative to the hyperfiltering case, because of the reduced filtered load with fewer nephrons. However, single-nephron total glucose reabsorption rose further as renal function fell.
Na+ reabsorption through SGLT was doubled in the diabetic hyperfiltering virtual patient compared with normal (Fig. 5C). Most of this increase was due to increased SGLT1 reabsorption. In the diabetic virtual patients with moderate and impaired renal function, Na+ reabsorption through SGLT followed trends similar to glucose, decreasing overall compared with the hyperfiltering case but increasing at the single-nephron level. In the healthy state SGLT (1, 2) accounted for 5.8% of total PT Na+ reabsorption, but in all three diabetic states the contribution of SGLT was increased to 11% (Fig. 5D). In the hyperfiltering diabetic state, total PT Na+ reabsorption through all mechanisms increased 18% relative to the healthy state, in line with measured levels of increased PT Na+ reabsorption in diabetes. Again, total PT Na+ reabsorption declined as renal function declined, but single-nephron PT Na+ reabsorption increased further.
Simulated Response to Dapagliflozin in Diabetic Virtual Patients With and Without Renal Impairment
Figure 6 shows the simulated response to dapagliflozin in three virtual patients: normal, diabetic with moderate renal function, and diabetic with CKD. For the virtual patient with moderate renal function, predicted changes in GFR, mean arterial pressure (MAP), and blood volume were compared with clinical results obtained from a similar population in response to dapagliflozin (21). In the absence of renal impairment, UGE was higher in the hyperglycemic than in the normoglycemic case. With renal impairment, UGE was lower and was similar to the normoglycemic case. These trends are in agreement with clinical findings (19, 28).
Fig. 6.

Simulated response to dapagliflozin in 3 virtual patients: nondiabetic with normal renal function, diabetic (DB) with moderate renal function, and diabetic with chronic kidney disease (CKD). Glomerular filtration rate (GFR; E), mean arterial pressure (MAP; H), and blood volume (I) changes were compared with observed results from Lambers Heerspink et al. (21), in which diabetic patients with normal renal function were treated with dapagliflozin. SNGFR, single-neuron GFR; NH3, Na+/H+ exchanger 3; PG, plasma glucose.
In all cases, the reduction in GFR/increase in serum creatinine with treatment was explained by a reduction in glomerular pressure. In the diabetic virtual patient with moderate renal function, GFR decreased by 12 ml/min, in agreement with reductions observed by iohexol measurements by Lambers Heerspink et al. (21). In the renally impaired diabetic virtual patient, GFR reduction was much smaller, 4 ml/min. Still, importantly, the glomerular pressure reduction was larger in both diabetic virtual patients than in the normoglycemic case, and renal impairment did not ameliorate the reduction.
Blood volume was reduced similarly in both diabetic cases and larger than in the normoglycemic case. Interstitial fluid volume reduction was larger in the diabetic virtual patient with preserved renal function compared with the healthy state, but renal impairment limited the interstitial volume reduction. MAP was reduced by 3.5 mmHg in both diabetic virtual patients, in agreement with clinical observations of a mild antihypertensive effect of SGLT2i (21).
DISCUSSION
We demonstrated mathematically that multiple mechanisms are required to quantitatively explain the renal hemodynamic effects of SGLT2i inhibition: 1) direct blocking of Na+ and glucose reabsorption through SGLT2, 2) SGLT2-driven inhibition of NHE3 Na+ reabsorption, and 3) osmotic diuresis coupled with peripheral Na+ storage. The analysis further indicates that the consequences of these mechanisms include lowering of glomerular pressure, reduction of blood and interstitial fluid volume, and a mild reduction in blood pressure (in agreement with clinical observations). In addition, these simulations suggest that these effects are more significant in diabetic patients than in healthy subjects and that while glucose excretion may be diminished with renal impairment, improvements in glomerular pressure and blood volume are not expected to be diminished at lower GFR.
Because Na+ and glucose reabsorption are coupled, it has been suggested that reduction in Na+ reabsorption may also explain the renal hemodynamic response to SGLT2i, with increased distal Na+ delivery leading to lowering of glomerular pressure through both TGF and reduced volume resulting from increased Na+ excretion. However, this analysis suggests that reduced Na+ reabsorption through SGLT2 is completely compensated for by increased SGLT1 reabsorption. Thus SGLT2i likely does not directly increase Na+ excretion. Rather, this analysis adds to the growing body of evidence that SGLT2i may also reduce Na+ reabsorption indirectly through effects on NHE3. SGLT2, but not SGLT1, colocalizes with NHE3 in the rat PT; phlorizin decreases NHE3 activity even in the absence of glucose (25); and empagliflozin increases NHE3 phosphorylation in mice, suggesting reduced NHE3 activity (9). Including relatively weak (5–9%) SGLT2-linked suppression of NHE3 activity allowed the model to reproduce the transient increase in Na+ and water excretion in the first 24 h as well as the observed changes in serum creatinine.
This analysis supports decreased PT Na+ reabsorption as the primary mechanism responsible for the acute decrease in GFR and shows that this drop results from a decrease in glomerular pressure. We have previously demonstrated mathematically how increases in PT Na+ reabsorption can cause increases in glomerular pressure and glomerular filtration, with the extent of the increase depending on the degree to which the distal nephron is able to compensate for the increase in Na+ reabsorption (13). Increased Na+ reabsorption induces a Na+ imbalance that must be corrected to return homeostasis, through increased filtration rate, decreased distal reabsorption rate, or both. TGF provides a partial correction by increasing GFR (although hyperfiltration will occur even in the absence of TGF) (38), but if the pressure-natriuresis mechanism does not sufficiently adjust distal Na+ reabsorption, any remaining Na+ imbalance will cause additional fluid retention, increases in renal perfusion pressure, and increasing glomerular pressure and GFR until Na+ excretion again equals intake. Here we demonstrated mathematically that inhibition of PT Na+ reabsorption by SGLT2i produces the opposite effect—a reduction in glomerular pressure and glomerular hyperfiltration. Increased Na+ excretion following initiation of treatment produces a Na+ imbalance that must be restored, and a reduction in glomerular pressure and GFR helps achieve this, ultimately returning Na+ excretion to baseline but at a lower glomerular pressure.
In agreement with recent simulations by Layton and Vallon, our model predicts that the natriuretic and diuretic effect of SGLT2 inhibition is weaker but sustained in normoglycemia compared with diabetes and with renal impairment compared with normal renal function (22). We further predicted that reductions in glomerular pressure and SNGFR were larger in diabetes than in normoglycemia and nearly independent of renal function. However, total GFR reduction was weaker with renal impairment than in preserved renal function. This is because in the diabetic state nephrons are hyperfiltering, and as nephrons are lost the degree of hyperfiltration in the remaining nephrons increases further. Thus in diabetes with renal impairment, remaining nephrons are filtering more Na+ and more glucose (because of both hyperfiltration and hyperglycemia). SGLT2 is reabsorbing at or near capacity, and SGLT1 is already contributing more to reabsorption. So when SGLT2 is inhibited there is a greater reduction in Na+ and glucose reabsorption and SGLT1 is less able to compensate, resulting in an overall increase in Na+ and glucose excretion at the single-nephron level. However, because there are many fewer nephrons, the total reduction in GFR is less than in the case of preserved renal function. This is important because a reduction in glomerular pressure is likely to be renoprotective, but large initial reductions in renal function could be detrimental in patients whose renal function is already low. This study indicates that SGLT2i may lower intraglomerular pressure in diabetic kidney disease with a relatively small cost in terms of initial GFR reduction. If the renoprotective effects of SGLT2i observed in patients with preserved renal function are indeed due to lowered glomerular pressure (and associated reductions in proteinuria), these findings suggest that these protective effects will extend to patients with CKD. In addition, the finding of glomerular pressure reduction even in nondiabetic subjects (although smaller than in diabetic patients) suggests that SGLT2i could be renoprotective in nondiabetic CKD. These results support further clinical investigation of effects of SGLT2i on CKD progression.
Although treatment-induced reductions in PT Na+ reabsorption explained observed increases in serum creatinine and decreases in GFR, the diuresis associated with this natriuretic response was not sufficient to explain observed increases in water excretion with dapagliflozin. Inclusion of glucose-driven tubular osmotic diuretic effect was necessary to reproduce this portion of the response. In addition, as we have described previously (14), the clinically observed degree of water excretion relative to Na+ excretion (the electrolyte-free water clearance) would be expected to increase blood Na+ concentration unless Na+ is removed from the blood in some other way. Inclusion of a peripheral storage compartment allowed the model to explain the large increases in water excretion but unchanged blood Na+ concentration.
Inclusion of this mechanism is supported by the growing understanding of the role of nonosmotic Na+ storage in skin, muscle, and other tissue in regulating Na+ (34, 44). Tissue Na+ is increased in salt-induced hypertension and heart failure (15, 20). Measured Na+ concentrations in muscle and skin range from 17 and 22 mEq/l in control subjects to 35 and 45 mEq/l in heart failure patients, respectively (15). Changes in peripheral stored Na+ of 130–200 mEq predicted in the present analysis would represent at most a 7–10% change. Diuretic treatment of patients with acute heart failure reduces tissue Na+ by 20–26% (15). A recent clinical trial showed a modest decrease in skin Na (5.8%) and no change in muscle Na+ in diabetic patients treated with dapagliflozin for 6 wk (18). It is possible that reduced tissue osmolality due to lowering of glucose levels over 6 wk contributed to this response.
Including osmotic diuresis and peripheral Na+ storage mechanisms in the model also impacted predicted effects of SGLT2i on plasma and interstitial fluid volume. Without these mechanisms, blood and interstitial fluid volume changes were transient. However, with these mechanisms, the model predicted sustained reductions in interstitial fluid volume. The reduction in blood volume predicted in diabetic patients with normal renal function is consistent with clinical measurements (Fig. 6) (21). Clinical data on the effects of SGLT2i on interstitial fluid volume are currently not available. However, given that excessive congestion is a primary cause of heart failure hospitalization and mortality, reductions of interstitial fluid volume with SGLT2i may provide an explanation for the observed protective effects in heart failure and should be investigated further.
This study has several limitations. The primary data used to inform the effects of SGLT2i were obtained in healthy subjects, and the analysis was conducted post hoc. While predicted changes in UGE, GFR, and blood volume in diabetes with normal renal function were consistent with clinical observations, data could not be collected for validating predicted changes in interstitial fluid volume. In addition, few data are available on the response in CKD patients. Thus the model provides prospective predictions that require validation as additional data on these end points and populations are collected going forward.
The model utilized a simplified representation of the renal vasculature and other components. There are certainly more detailed models out there of various aspects of renal physiology, but the present model attempts to strike a balance that sufficiently represents key physiological behaviors while being simple enough to allow integrative dynamic simulations and to isolate the consequences of the mechanisms considered in this analysis—thus we believe the current model is “fit for purpose” for the analysis conducted here. It is possible that these simplifications may have some effect on specific numerical results, but we do not expect that they impact the overall trends and conclusions.
Diabetic nephropathy was also modeled as a decrease in nephron number and Kf in a setting of high glucose. We recognize that nephropathy is complicated and that other factors (strength of TGF, the myogenic response, neurohumoral activation) may change. There are conflicting literature reports on the magnitude and direction of many of these changes, and thus consideration of these effects is not a trivial task. Those factors may be considered in future analyses, but we believe that the present analysis provides important and valid insights on the effect of reduced nephron number and Kf on the response to SGLT2i. Although the model had the capability to explore the role of the RAAS or the impact of comedication with commonly used antihypertensives like angiotensin-converting enzyme inhibitors or diuretics, we have left these important questions for future work as well.
In conclusion, although the pharmacological action of SGLT2i in the PT at first appears straightforward, the consequences of this inhibition involve multiple mechanisms, including NHE3 suppression and osmotic diuresis, which subsequently lead to reduced glomerular hydrostatic pressure, reduced blood volume, and clearance of interstitial fluid. This combination of mechanisms may explain the unexpected benefits of SGLT2i on cardiac and renal function observed in recent cardiovascular outcomes trials.
GRANTS
This research was funded by AstraZeneca.
DISCLOSURES
K. M. Hallow and H. J. L. Heerspink have received grant funding from AstraZeneca. G. Helmlinger, D. W. Boulton, P. J. Greasley, and L. Chu are employed by AstraZeneca. K. M. Hallow has received research funding from Pfizer, Merck, and Takeda.
AUTHOR CONTRIBUTIONS
K.M.H., P.J.G., G.H., H.J.H., and D.W.B. conceived and designed research; K.M.H., P.J.G., G.H., and H.J.H. performed experiments; K.M.H. analyzed data; K.M.H., P.J.G., G.H., L.C., H.J.H., and D.W.B. interpreted results of experiments; K.M.H. prepared figures; K.M.H. and D.W.B. drafted manuscript; K.M.H., P.J.G., G.H., L.C., H.J.H., and D.W.B. edited and revised manuscript; K.M.H., P.J.G., G.H., L.C., H.J.H., and D.W.B. approved final version of manuscript.
REFERENCES
- 1.Anderson S, Brenner BM. The role of intraglomerular pressure in the initiation and progression of renal disease. J Hypertens Suppl 4: S236–S238, 1986. [PubMed] [Google Scholar]
- 2.Brands MW, Manhiani MM. Sodium-retaining effect of insulin in diabetes. Am J Physiol Regul Integr Comp Physiol 303: R1101–R1109, 2012. doi: 10.1152/ajpregu.00390.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, Johansen OE. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab 17: 1180–1193, 2015. doi: 10.1111/dom.12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coady MJ, El Tarazi A, Santer R, Bissonnette P, Sasseville LJ, Calado J, Lussier Y, Dumayne C, Bichet DG, Lapointe JY. MAP17 is a necessary activator of renal Na+/glucose cotransporter SGLT2. J Am Soc Nephrol 28: 85–93, 2017. doi: 10.1681/ASN.2015111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, Morrow LA, Leslie BR, Boulton DW, Ching A, LaCreta FP, Griffen SC. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 36: 3169–3176, 2013. doi: 10.2337/dc13-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME Trial: a “thrifty substrate” hypothesis. Diabetes Care 39: 1108–1114, 2016. doi: 10.2337/dc16-0330. [DOI] [PubMed] [Google Scholar]
- 7.Fitchett D, Butler J, van de Borne P, Zinman B, Lachin JM, Wanner C, Woerle HJ, Hantel S, George JT, Johansen OE, Inzucchi SE; EMPA-REG OUTCOME trial investigators . Effects of empagliflozin on risk for cardiovascular death and heart failure hospitalization across the spectrum of heart failure risk in the EMPA-REG OUTCOME trial. Eur Heart J 39: 363–370, 2018. doi: 10.1093/eurheartj/ehx511. [DOI] [PubMed] [Google Scholar]
- 8.Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE; EMPA-REG OUTCOME trial investigators . Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME trial. Eur Heart J 37: 1526–1534, 2016. doi: 10.1093/eurheartj/ehv728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu Y, Gerasimova M, Mayoux E, Masuda T, Vallon V. SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: possible implications for the prevention of glomerular hyperfiltration (Abstract) Diabetes 63: A132, 2014. [Google Scholar]
- 10.Gorboulev V, Schürmann A, Vallon V, Kipp H, Jaschke A, Klessen D, Friedrich A, Scherneck S, Rieg T, Cunard R, Veyhl-Wichmann M, Srinivasan A, Balen D, Breljak D, Rexhepaj R, Parker HE, Gribble FM, Reimann F, Lang F, Wiese S, Sabolic I, Sendtner M, Koepsell H. Na(+)-d-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61: 187–196, 2012. doi: 10.2337/db11-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallow KM, Gebremichael Y. A quantitative systems physiology model of renal function and blood pressure regulation: application in salt-sensitive hypertension. CPT Pharmacometrics Syst Pharmacol 6: 393–400, 2017. doi: 10.1002/psp4.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallow KM, Gebremichael Y. A quantitative systems physiology model of renal function and blood pressure regulation: model description. CPT Pharmacometrics Syst Pharmacol 6: 383–392, 2017. doi: 10.1002/psp4.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallow KM, Gebremichael Y, Helmlinger G, Vallon V. Primary proximal tubule hyperreabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: a modeling analysis. Am J Physiol Renal Physiol 312: F819–F835, 2017. doi: 10.1152/ajprenal.00497.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallow KM, Helmlinger G, Greasley PJ, McMurray JJ, Boulton DW. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes Metab 20: 479–487, 2018. doi: 10.1111/dom.13126. [DOI] [PubMed] [Google Scholar]
- 15.Hammon M, Grossmann S, Linz P, Kopp C, Dahlmann A, Garlichs C, Janka R, Cavallaro A, Luft FC, Uder M, Titze J. 23Na magnetic resonance imaging of the lower leg of acute heart failure patients during diuretic treatment. PLoS One 10: e0141336, 2015. doi: 10.1371/journal.pone.0141336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol 8: 293–300, 2012. doi: 10.1038/nrneph.2012.19. [DOI] [PubMed] [Google Scholar]
- 17.Irsik DL, Blazer-Yost BL, Staruschenko A, Brands MW. The normal increase in insulin after a meal may be required to prevent postprandial renal sodium and volume losses. Am J Physiol Regul Integr Comp Physiol 312: R965–R972, 2017. doi: 10.1152/ajpregu.00354.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karg MV, Bosch A, Kannenkeril D, Striepe K, Ott C, Schneider MP, Boemke-Zelch F, Linz P, Nagel AM, Titze J, Uder M, Schmieder RE. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc Diabetol 17: 5, 2018. doi: 10.1186/s12933-017-0654-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasichayanula S, Liu X, Pe Benito M, Yao M, Pfister M, LaCreta FP, Humphreys WG, Boulton DW. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol 76: 432–444, 2013. doi: 10.1111/bcp.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Müller DN, Schmieder RE, Cavallaro A, Eckardt KU, Uder M, Luft FC, Titze J. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 61: 635–640, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00566. [DOI] [PubMed] [Google Scholar]
- 21.Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab 15: 853–862, 2013. doi: 10.1111/dom.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Layton AT, Vallon V. SGLT2 inhibition in a kidney with reduced nephron number: modeling and analysis of solute transport and metabolism. Am J Physiol Renal Physiol 314: F969–F984, 2018. doi: 10.1152/ajprenal.00551.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR; CANVAS Program Collaborative Group . Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377: 644–657, 2017. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 24.Novikov A, Vallon V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: an update. Curr Opin Nephrol Hypertens 25: 50–58, 2016. doi: 10.1097/MNH.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25: 2028–2039, 2014. doi: 10.1681/ASN.2013060588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfeifer M, Townsend RR, Davies MJ, Vijapurkar U, Ren J. Effects of canagliflozin, a sodium glucose co-transporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: a post hoc analysis. Cardiovasc Diabetol 16: 29, 2017. doi: 10.1186/s12933-017-0511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, Thomson SC, Koepsell H, Vallon V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol 306: F188–F193, 2014. doi: 10.1152/ajprenal.00518.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarashina A, Ueki K, Sasaki T, Tanaka Y, Koiwai K, Sakamoto W, Woerle HJ, Salsali A, Broedl UC, Macha S. Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of empagliflozin, a sodium glucose cotransporter 2 inhibitor, in Japanese patients with type 2 diabetes mellitus. Clin Ther 36: 1606–1615, 2014. doi: 10.1016/j.clinthera.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Schneider MP, Raff U, Kopp C, Scheppach JB, Toncar S, Wanner C, Schlieper G, Saritas T, Floege J, Schmid M, Birukov A, Dahlmann A, Linz P, Janka R, Uder M, Schmieder RE, Titze JM, Eckardt KU. Skin sodium concentration correlates with left ventricular hypertrophy in CKD. J Am Soc Nephrol 28: 1867–1876, 2017. doi: 10.1681/ASN.2016060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrier RW. Body fluid volume regulation in health and disease: a unifying hypothesis. Ann Intern Med 113: 155–159, 1990. doi: 10.7326/0003-4819-113-2-155. [DOI] [PubMed] [Google Scholar]
- 31.Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol 18: 2028–2031, 2007. doi: 10.1681/ASN.2006111302. [DOI] [PubMed] [Google Scholar]
- 32.Simons JL, Provoost AP, Anderson S, Rennke HG, Troy JL, Brenner BM. Modulation of glomerular hypertension defines susceptibility to progressive glomerular injury. Kidney Int 46: 396–404, 1994. doi: 10.1038/ki.1994.287. [DOI] [PubMed] [Google Scholar]
- 33.Solini A, Giannini L, Seghieri M, Vitolo E, Taddei S, Ghiadoni L, Bruno RM. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: a pilot study. Cardiovasc Diabetol 16: 138, 2017. doi: 10.1186/s12933-017-0621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Titze J. Sodium balance is not just a renal affair. Curr Opin Nephrol Hypertens 23: 101–105, 2014. doi: 10.1097/01.mnh.0000441151.55320.c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Titze J. Water-free sodium accumulation. Semin Dial 22: 253–255, 2009. doi: 10.1111/j.1525-139X.2009.00569.x. [DOI] [PubMed] [Google Scholar]
- 36.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74: 351–375, 2012. doi: 10.1146/annurev-physiol-020911-153333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60: 215–225, 2017. doi: 10.1007/s00125-016-4157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Hallow KM, James DA. A tutorial on RxODE: simulating differential equation pharmacometric models in R. CPT Pharmacometrics Syst Pharmacol 5: 3–10, 2016. doi: 10.1002/psp4.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wanner C. EMPA-REG OUTCOME: the nephrologist’s point of view. Am J Med 130, 6 Suppl: S63–S72, 2017. doi: 10.1016/j.amjmed.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B; EMPA-REG OUTCOME Investigators . Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375: 323–334, 2016. doi: 10.1056/NEJMoa1515920. [DOI] [PubMed] [Google Scholar]
- 43.Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George JT, Woerle HJ, Broedl UC, von Eynatten M, Zinman B; EMPA-REG OUTCOME Investigators . Empagliflozin and clinical outcomes in patients with type 2 diabetes, established cardiovascular disease and chronic kidney disease. Circulation 137: 119–129, 2018. doi: 10.1161/CIRCULATIONAHA.117.028268. [DOI] [PubMed] [Google Scholar]
- 44.Wiig H, Luft FC, Titze JM. The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol (Oxf) 222: e13006, 2018. doi: 10.1111/apha.13006. [DOI] [PubMed] [Google Scholar]