

Abstract
Phosphate homeostasis is primarily maintained in the renal proximal tubules, where the expression of sodium/phosphate cotransporters (Npt2a and Npt2c) is modified by the endocrine actions of both fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH). However, the specific contribution of each regulatory pathway in the proximal tubules has not been fully elucidated in vivo. We have previously demonstrated that proximal tubule-specific deletion of the FGF23 coreceptor Klotho results in mild hyperphosphatemia with little to no change in serum levels of FGF23, 1,25(OH)2D3, and PTH. In the present study, we characterized mice in which the PTH receptor PTH1R was specifically deleted from the proximal tubules, either alone or in combination with Klotho (PT-PTH1R−/− and PT-PTH1R/KL−/−, respectively). PT-PTH1R−/− mice showed significant increases in serum FGF23 and PTH levels, whereas serum phosphate levels were maintained in the normal range, and Npt2a and Npt2c expression in brush border membrane (BBM) did not change compared with control mice. In contrast, PT-PTH1R/KL−/− mice displayed hyperphosphatemia and an increased abundance of Npt2a and Npt2c in the renal BBM, along with increased circulating FGF23 levels. While serum calcium was normal, 1,25(OH)2D3 levels were significantly decreased, leading to extremely high levels of PTH. Collectively, mice with a deletion of PTH1R alone in proximal tubules results in only minor changes in phosphate regulation, whereas deletion of both PTH1R and Klotho leads to a severe disturbance, including hyperphosphatemia with increased sodium/phosphate cotransporter expression in BBM. These results suggest an important interplay between the PTH/PTH1R and FGF23/Klotho pathways to affect renal phosphate handling in the proximal tubules.
Keywords: 1,25(OH)2D3; mineral metabolism; parathyroid hormone; renal Npt2a
INTRODUCTION
The maintenance of serum phosphate is critical for many biological processes, including bone mineralization and cell function. Hyperphosphatemia is linked to vascular calcifications and decreased life expectancy (22, 26, 42, 45), whereas continued hypophosphatemia leads to bone demineralization and an increased risk of renal stone formation (49, 50).
The kidney plays a pivotal role in the maintenance of phosphate homeostasis. It adjusts urinary phosphate excretion to balance the quantity of phosphate absorbed in the intestine or the amount stored by cells and bone (3, 5, 48). The phosphate freely filtered at the glomerulus is mainly (70%–85%) reabsorbed in the renal proximal tubule (PT) through the type 2 sodium/phosphate cotransporters (Npt2a and Np2c), and phosphate regulation in the remainder of the nephron plays only a minor role (5, 6, 19, 34, 48, 58). Npt2a is expressed in the S1, S2, and S3 segments of apical brush border membranes (BBMs) of superficial and juxtamedullary nephrons with gradually decreasing expression along the PTs, whereas Npt2c is mainly expressed in the S1 segment (3, 5, 6, 19, 47). Parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23) are two phosphaturic hormones regulating the expression of these transporters (3, 5, 19). However, their respective roles in regulating renal phosphate reabsorption have not been fully elucidated.
Parathyroid hormone 1 receptor (PTH1R) is a seven-transmembrane protein that acts as the receptor for PTH and PTH-related protein. PTH1R is abundantly expressed in kidney and bone, where it plays an essential regulatory role in mineral ion homeostasis (17, 18, 55). In the kidney, PTH1R is expressed in the PTs, the cortical portion of thick ascending limbs, and the distal convoluted tubules, corresponding to the locations targeted by PTH (36, 39, 41). In the renal PTs, PTH1R is located in both the basolateral and apical side of the membrane. PTH suppresses reabsorption of phosphate by internalizing Npt2a and Npt2c in the BBM (10, 40, 46) through interaction with Na+/H+-exchanger isoform 3 regulatory factor (NHERF)-1 and by increasing serine phosphorylation of NHERF1 (13). PTH is also purported to enhance the synthesis of active vitamin D by increasing the activating enzyme Cyp27b1 and decreasing the inhibiting enzyme Cyp24a1 (11, 15, 16, 21).
Klotho is a transmembrane protein that is highly expressed in the renal distal tubules (44) and, to a much lesser extent, in the basolateral membrane and apical brush border as well as in the cytoplasm of the PTs (23, 24). Klotho functions as an obligatory coreceptor for FGF23, which acts primarily through a Klotho-FGFR1 receptor signaling mechanism (30, 54). In addition, FGF23 can act via an FGFR4 receptor complex in the heart in the absence of Klotho (20). Like PTH, FGF23 reduces apical membrane abundance of Npt2a and Npt2c in the renal BBM and thereby enhances phosphate excretion (1, 2, 32). Contrary to PTH, FGF23 downregulates Cyp27b1 expression while stimulating Cyp24a1 expression and, consequently, inhibits the production of active vitamin D (11, 21, 53). Deleting Klotho from the whole nephron leads to severe disturbances in mineral ion metabolism, including hyperphosphatemia and high serum levels of 1,25(OH)2D3 (37). In contrast, deleting Klotho from either the proximal or distal tubules alone results in a mild hyperphosphatemic phenotype (24, 44). This discrepancy suggests the presence of additional mechanisms that can largely compensate for the loss of Klotho from either the proximal or distal tubules.
In the current study, we investigated the combined functions of PTH1R and Klotho in the proximal renal tubules to address the complexity of hormonal renal phosphate regulation and possible interactions between PTH1R and FGF23/Klotho signaling. We first generated a novel mouse model in which PTH1R is specifically deleted from the PTs using Cre recombinase driven by the Npt2a (Slc34a1) promoter. We then extended this model by generating a transgenic mouse line with both PTH1R and Klotho ablated from the PTs. Our findings suggest that PTH and FGF23 act in a combined manner to regulate phosphate reabsorption in the PTs.
MATERIAL AND METHODS
Animal experiments.
Slc34a1-Cre (sodium phosphate cotransporter-2a) mice were kindly provided by Benjamin D. Humphreys (Washington University, St. Louis, MO). PTH1Rfl/fl mice (28) and Klohtofl/fl mice (44) were described previously. All mice were kept in a C57BL6 background. To analyze the recombination specificity and efficiency, a tdTomato reporter strain [B6.Cg-Gt(ROSA)26Sortm14 (CAG-tdTomato)Hze/J; 007914, Jackson Laboratory, Bar Harbor, ME] was used. With these mice, we successfully generated Slc34a1-Cre; PTH1Rfl/fl, Tomatofl/+ (PT-PTH1R−/−) and Slc34a1-Cre; PTH1Rfl/fl, Klothofl/fl, Tomatofl/+ (PT-PTH1R/KL−/−) mice. Slc34a1-Cre; Tomatofl/+ (Control) mice were generated as controls. Routine polymerase chain reaction (PCR) was performed to genotype the mice. The specific primer sets for the target gene used are listed as follows: Klotho-LoxP: [forward (F)] 5′-TTG TCA ATA TGT AAA TAA TTT GAG CAG TAG GG, [reverse (R)] 5′-GTT GTT GAA AGA GGG AGC TAG TGG TAG TTA- 3′; PTH1R-LoxP: (F) 5′-TGG ACG CAG ACG ATG TCT TTA CCA- 3′, (R) 5′-ACA TGG CCA TGC CTG GGT CTG AGA- 3′; tdTomato: (F) 5′-GGC ATT AAA GCA GCG TAT CC- 3′, (R) 5′-CTG TTC CTG TAC GGC ATG G- 3′; and Slc34a1Cre: (F) 5′-TTG CCT GCA TTA CCG GTC GAT GCA ACG AGT- 3′, (R) 5′-CCT GGT CGA AAT CAG TGC GTT CGA ACG CTA- 3′. Tamoxifen (Sigma-Aldrich, St. Louis, MO) was dissolved in corn oil (Sigma-Aldrich) at a concentration of 10 mg/ml. Tamoxifen (3mg) was injected intraperitoneally per day for 5 consecutive days to induce activation of Cre recombinase. Control, PT-PTH1R−/−, and PT-PTH1R/KL−/− mice were injected at 6 wk old and were euthanized for analysis at 9 wk. The high phosphate (HP) loading experiment was performed as previously described (24). Briefly, Mice were given ad libitum access to HP drinking water [×5 phosphate-buffered saline (PBS) dissolved in distilled water, pH 7.4, [Pi] = 50 mM) for 24 h on the day before euthanization. The amount of HP water intake was measured in each mouse. All animal studies performed were approved by regional ethical committees.
X-ray radiology.
X-ray and florescent images were taken by Bruker MS FX PRO in vivo imaging system (Bruker, Hamburg, Germany).
Kidney histology.
Kidneys were fixed with 10% buffered formalin, dehydrated in ethanol, and embedded with paraffin. Paraffin-embedded kidneys were sectioned 5-μm thick. For immunofluorescent studies, sections were incubated with red fluorescence protein antibody (1:100, Rockland; 600-406-379) overnight at 4°C and fluorescein labeled Lotus tetragonolobus lectin (LTL; 1:100, FL-1321; Vector Laboratories, Burlingame, CA) for 1 h at 37°C. Red-fluorescent conjugated (Alexa Fluor 594, Invitrogen, Carlsbad, CA) secondary antibody was then used. Nuclei were stained by DAPI (D9542; Sigma-Aldrich) for 5 min at room temperature. For immunohistochemical studies, sections were stained with anti-Klotho monoclonal antibody (1:100, rat monoclonal, Km2076; Trans Genic), anti-phospho-CREB antibody (1:100, rabbit monoclonal; Cell Signaling Technology, Danvers, MA), or anti-Cyp27b1 antibody (1:100, rabbit monoclonal, ab206655; Abcam, Cambridge, UK). A DAB substrate kit (Vector Laboratories) was later used to detect primary antibodies. Nuclei were counterstained by Mayer’s hematoxylin solution. The slides were observed on a Keyence florescent microscope (BZ-X700).
Proximal tubular cell isolation and culture.
Proximal tubular cells (PTCs) were isolated from kidneys of Control, PT-PTH1R−/−, and PT-PTH1R/KL−/− mice using a previously described procedure with some modifications (27). Briefly, kidney cortices were minced into small pieces and incubated with collagenase type 2 (2 mg/ml) for 45 min at 37°C. The tube was shaken at 200 revolutions/min. The cellular digest was filtered through sterile 70-μm nylon mesh, centrifuged at 300 g for 5 min, and washed twice in PBS. Medium was added, and cells were used for magnetic bead cell separations or plated on collagen-coated culture plates and then incubated for 24 h at 37°C in 5% CO2. On the second day of cell culture, supernatant was transferred to a new dish and left for 48 h at 37°C in 5% CO2. The cell pellet was resuspended in medium and used for experiments. Each experiment required PTCs generated from a single animal, and cells were not passaged. Cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (GIBCO). For magnetic bead separation of PT segments, at first, the S3 segment was isolated by positive selection using SGLT1 antibody (1:100, rabbit polyclonal, no. 07–1417; EMD Millipore) (57). The S1/S2 segments were substantially isolated from bead-free PTCs using Npt2a (1:100, rabbit polyclonal, no. A6742; ABclonal Technology, Woburn, MA). Briefly, PTC suspension was incubated with SGLT1 antibody for 30 min at 4°C with rotator, and then Dynabeads (M-280) were added and coated with sheep anti-rabbit IgG (25-μl aliquot per 5-ml tube) for 30 min at 4°C with rotator. With the magnet detached, the bead-bound cells were recovered (positive selection), washed five times with ×1 PBS containing 0.5mM MgCl2 and 0.5mM CaCl2. Supernatants (bead-free PTCs) were collected and centrifuged and washed twice in PBS. The cell pellet was resuspended in PBS and then used for the S1/S2 segment selection by Npt2a antibody (positive selection).
Quantitative real-time PCR.
Total RNA was extracted from the renal cortex using Trizol (Invitrogen), according to the manufacturer’s protocol. Cortical parts of the kidneys were collected by shaving the outer parts of the kidneys. The cortical part of the kidney and the fraction of S1/S2 segments or S3 segment of the PTCs were used for the quantitative PCR analysis. cDNA was generated with QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Quantitative real-time PCR with SYBR Green was performed for kidney using the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). GAPDH was used as a reference gene to normalize the expression of target genes in kidney. The specific primer sets for the target gene used are listed as follows: PTH1R: (F) 5′-TTT CCC GGT GCC TTC TCT TTC- 3′, (R) 5-CAG GCG CAA TGT GAC AAG C- 3′; Klotho: (F) 5′-TCT CAA GAA TTC ATA ATG GAA A- 3′, (R) 5′-CAG AAA GTC AAC GTA GAA GAG TCC T- 3′; Npt2a: (F) 5′-GGG AGA AGC TAT CCA GCT CA- 3′, (R) 5′-GTA CTT GGC GGT GCG TTT- 3′; Npt2c: (F) 5′-TTC TTG GGT TCC AAC ATC G- 3′, (R) 5′-GAA GAA GTG GAT GAG AGC AAC C- 3′; and GAPDH: (F) 5′-ACT GAG GAC CAG GTT GTC- 3′, (R) 5′-TGC TGT AGC CGT ATT CAT TG- 3′.
Flow cytometry.
Flow cytometry was performed using a method previously described (24). Briefly, kidneys were dissected and cut into small pieces using scissors and blades and then digested with collagenase type 2 (1 mg/ml) for 1 h at 37°C with shaking (150 revolutions/min). Digested tissue was filtered through a 70-μm cell strainer, spun down with serum-including medium (1,000 revolutions/min, 5 min), and then washed twice with PBS with 5% FBS. Isolated cells were incubated with the following primary antibodies: anti-phospho CREB antibody (1:100, rabbit monoclonal; Cell Signaling Technology), anti-early growth-responsive 1 (Egr-1) antibody (1:100, rabbit polyclonal; Santa Cruz Biotechnology, Dallas, TX) at room temperature for 60 min. Samples were then washed, followed by staining with appropriate secondary antibody at room temperature for 30 min and analyzed by the FACSCalibur (BD Biosciences, San Jose, CA) and Cell Quest software (BD Biosciences). Each data set was analyzed by FlowJo software (FLOWJO, Ashland, OR).
Biochemical analyses.
Serum phosphorus, calcium, and creatinine were determined using Stanbio kits (Stanbio Laboratory, Boerne, TX), according to the manufacturer’s instructions. Serum intact PTH-(1–84) concentrations were measured with ELISA kits (Quidel, San Diego, CA). Serum concentration of intact FGF23 was measured by Kainos intact FGF23 ELISA kits (Kainos Laboratories, Tokyo, Japan). Serum 1,25-dihydroxy vitamin D and cross-linked C telopeptide (CTX) levels were determined by an EIA kit (Immunodiagnostics Systems, Scottsdale, AZ).
Western blot analysis.
For Western blot analysis, isolated PTs were lysed in radioimmunoprecipitation assay buffer (Sigma-Aldrich, R0278) containing protease inhibitors (no. 04693159001; Roche) for 15 min on ice. Protein concentrations were determined using bicinchoninic acid assay. Equal amounts of protein were separated on SDS-polyacrylamide gels, blotted onto PVDF membranes, and blocked in 5% nonfat dry milk in Tris-buffered saline, 0.1% Tween 20. The membranes were further treated with antibodies against Cyp27b1 (1:1,000, rabbit monoclonal, ab206655; Abcam) and Cyp24a1 (1:1,000, rabbit polyclonal, GTX-105884; Genetex, Irvine, CA) overnight at 4°C and β-actin (1:5,000, mouse monoclonal, A5316; Sigma-Aldrich) for 1 h at room temperature. To assess Npt2a and Npt2c expression, we isolated protein from the BBM by a published Ca2+ precipitation method with some modifications (4), as described previously (24). The membranes were incubated overnight at 4°C with antibodies against Npt2a (1:1,000, rabbit polyclonal, no. A6742; ABclonal) or Npt2c (1:1,000, rabbit polyclonal, no. ab83093; Abcam). The membranes were then washed with Tris-buffered saline, 0.1% Tween 20 buffer and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG used as the secondary antibody (1:10,000; Cell Signaling Technology) for 1 h at room temperature. After washing, the membranes were detected with the ECL detection reagent (Pierce ECL Western Blotting, no. 32209; Thermo Fisher Scientific, Waltham MA).
Statistical analysis.
Data were expressed as the mean ± SD. Statistical analysis was performed by Student’s t-test or one way-ANOVA followed by Tukey's post hoc test using Prism 7 (GraphPad, San Diego, CA). Values of P < 0.05 were considered statistically significant.
RESULTS
Generation and validation of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice.
We generated new transgenic mouse lines with either deletion of PTH1R alone or in combination with Klotho (PTH1R/KL), specifically from the PTs by crossing mice expressing Cre recombinase under the Slc34a1 promoter with PTH1R floxed or PTH1R/Klotho double-floxed mice. As reported previously (31), these mice had efficient recombination in the S1 and S2 segments of proximal renal tubules upon tamoxifen administration. This is the main site of phosphate uptake in the kidney. To help visualize the site of Cre expression, we also introduced a tdTomato reporter gene (SLC34a1cre; PTH1Rfl/fl, Tomatofl/+: PT-PTH1R) or (SLC34a1cre; PTH1Rfl/fl, Klotho fl/fl, Tomatofl/+: PT-PTH1R-KL). Mice expressing only Cre recombinase and the reporter gene (SLC34a1cre; Tomatofl/+: Control) served as controls. First, tdTomato expression was confirmed to be restricted to the kidneys by in vivo imaging (Fig. 1A). Kidney sections were stained with Lotus tetragonolobus lectin, which is specific for the PTs, and red fluorescence protein (for tdTomato). The double-stained area was mainly observed in the convoluted PTs and not in the pars recta (Fig. 1B). PT-PTH1R−/− mice displayed no growth or body weight abnormalities as compared with Control mice in both genders, whereas the body weight was significantly decreased in PT-PTH1R/KL−/− mice compared with Control mice in both males (13% reduction compared with Control mice) and females (18% reduction compared with Control mice) (Fig. 1C). PT-PTH1R/KL−/− female mice had significantly reduced body weight compared with PT-PTH1R−/− female mice (13% reduction), a finding that was not observed in male mice. Next, using magnetic proximal cell separation (S1/S2 and S3 segments), deletion efficiency of the PTH1R and Klotho genes in S1/2 and S3 segments was determined by quantitative PCR (Fig. 1D). Gene expression of PTH1R was significantly reduced in the S1/2 segment of PT-PTH1R−/− mice (71% deletion) and PT-PTH1R/KL−/− mice (80% deletion) as compared with Control mice. Klotho mRNA expression was also significantly reduced in the S1/2 segment of PT-PTH1R/KL−/− mice (78% deletion) compared with Control mice (Fig. 1D). Although, there was no difference in gene deletion of PTH1R in the S3 segment among the three groups or of Klotho in the S3 segment between Control mice and PT-PTH1R/KL−/− mice. (Fig. 1D). Deletion of target genes was also confirmed by immunohistochemistry in PT-PTH1R/KL−/− mice (Fig. 1E) and flow cytometry in PT-PTH1R/KL−/− mice and PT-PTH1R−/− mice (Fig. 1, F and G). The expressions of Klotho and phospho-CREB (a downstream target of PTH1R signaling) were examined by flow cytometry, and a significant reduction was observed in PT-PTH1R/KL−/− mice compared with Control mice (53% and 47% reduction, respectively) (Fig. 1F). In PT-PTH1R−/− mice, the expression of phospho-CREB was also significantly reduced compared with Control mice (33% reduction) (Fig. 1G).
Fig. 1.

Generation and validation of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice. A: X-ray analyses of 9-wk-old Control (Slc34a1-Cre; Tomatofl/+) mice with (right) or without (left) tamoxifen were performed and show Slc34a1-Cre expression. B: kidney sections of Control mice were double-stained with Lotus tetragonolobus lectin (LTL) and an anti- red fluorescence protein (RFP) antibody (scale bar = 500 μm). C: body weight was measured in Control mice (male: n = 16 and female: n = 20), PT-PTH1R−/− mice (male: n = 21 and female: n = 17), and PT-PTH1R/KL−/− mice (male: n = 28 and female: n = 16) at 9 wk. D: PTH1R mRNA and Klotho mRNA were determined by quantitative PCR from magnetic beads isolated in S1/S2 and S3 segments in proximal tubular cells of Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice (n = 5–8). E: representative images of Klotho and phospho-CREB (pCREB) immunostaining are shown in the kidney of PT-PTH1R/KL−/− mice and Control mice (scale bar = 50 µm). F: quantification of pCREB or Klotho and tdTomato double-positive cells in the kidney was achieved by flow cytometry in PT-PTH1R/KL−/− mice (n = 3–4). G: quantification of pCREB and tdTomato double-positive cells was achieved by flow cytometry in PT-PTH1R−/− mice (n = 3–4). Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data represent the means ± SD.
Characterization of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice.
We then measured serum and urinary parameters to determine whether systemic mineral ion homeostasis is influenced by the lack of PTH1R signaling or by the combined lack of PTH1R and FGF23/Klotho signaling in the renal PTs (Fig. 2A). Serum phosphate levels were significantly increased only in PT-PTH1R/KL−/− mice. There were no differences in serum calcium or fractional phosphate excretion among any of the groups (Fig. 2A). Both PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice had significantly increased serum-intact FGF23 levels (1.77-fold and 1.94-fold, respectively) and PTH levels (5.9-fold and 11.6-fold, respectively) compared with Control mice. The circulating levels of 1,25(OH)2D3 were significantly decreased in PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice compared with Control mice (0.74-fold and 0.72-fold, respectively) (Fig. 2B). Western blot analysis revealed that Cyp27b1 expression was significantly decreased in PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice compared with Control mice, whereas Cyp24a1 expression was significantly increased in PT-PTH1R−/− mice compared with Control mice. This was not observed in PT-PTH1R/KL−/− mice (Fig. 2C). Immunohistochemical staining for Cyp27b1 was also performed in all three groups. Consistent with Western blot analysis, Cyp27b1 expression was significantly decreased in PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice compared with Control mice (Fig. 2D). Renal function as measured by serum creatinine was similar between the groups (Fig. 2E). The mRNA expression of Npt2a, and Npt2c was significantly increased only in PT-PTH1R/KL−/− mice (Fig. 2F). Similarly, Western blot analysis revealed that PT-PTH1R/KL−/− mice did have a higher protein level of Npt2a and of Npt2c in the BBM, which was not observed in PT-PTH1R−/− mice (Fig. 2F).
Fig. 2.

Characterization of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice. A: Serum and urinary biochemistry. Serum calcium levels (n = 11–14), phosphate levels (n = 9–10), and fractional phosphate excretion (n = 6–9) were measured in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice. B: regulatory factors iFGF23 (n = 9–13), parathyroid hormone (PTH) (n = 13–16), and 1,25(OH)2D3 (n = 10–13) were measured in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice. C: Cyp27b1 and Cyp24a1 protein expression from isolated proximal tubules of Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice were determined by Western blot analysis (n = 3). D: representative images of Cyp27b1 immunostaining are shown in the kidney of PT-PTH1R/KL−/− mice and Control mice (scale bar = 100 µm). E: serum creatinine levels were measured in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice (n = 9–15). F: relative mRNA levels of renal transcripts (Npt2a and Npt2c) were measured by quantitative PCR in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice (n = 5–7). Npt2a and Npt2c protein expression were detected by Western blot analysis (n = 4). Brush border membrane was isolated from the kidney of Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice. β-actin was used as internal control. Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data represent the means ± SD.
Disruption of compensatory mechanism for the phosphate handling in PT-PTH1R/KL−/− mice.
As described above, PT-PTH1R/KL−/− mice had severe mineral disorder, whereas both PT-PTH1R−/− and PT-KL−/− single knockout (KO) mice (24) had milder phenotypes. We next evaluated a potential compensatory mechanism where the lack of PTH signaling could be compensated by increased sensitivity to FGF23 signaling. For this purpose, we examined the response to FGF23 on Egr-1 in PTs by flow cytometry analysis (Fig. 3). In the absence of PTH1R, Egr-1 levels were higher than in controls, whereas in the absence of PTH1R and Klotho, Egr-1 was not “activated,” despite the significant increase in FGF23. These data indicate an FGF23 resistance state in the PT-PTH1R/KL−/− mice and a compensatory response by FGF23 signaling in the PT-PTH1R−/− mice (Fig. 3).
Fig. 3.
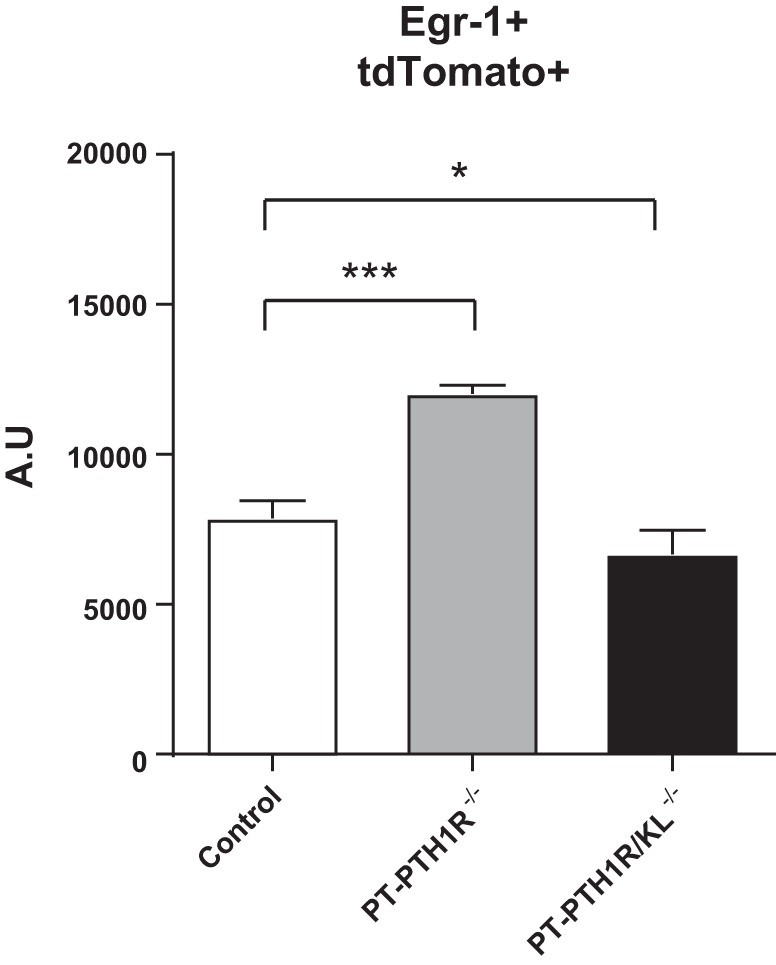
Comparison of early growth-responsive 1 (Egr-1) signaling in proximal tubules of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice. Quantifications of Egr-1 and tdTomato double-positive cells in the kidney were determined by flow cytometry in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice (n = 3–4). Values of P < 0.05 were considered statistically significant. *P < 0.05 and ***P < 0.001. Data represent the means ± SD. A.U., arbitrary units.
Next, we examined the bone phenotype to assess secondary effects of PTH1R deletion alone or in combination with Klotho in PTs. We measured serum CTX (C-terminal telopeptides of type I collagen) levels, a bone resorption marker, and found that they were significantly increased in the PT-PTH1R/KL−/− mice compared with Control mice and PT-PTH1R mice (Table 1).
Table 1.
The comparison of serum CTX (bone resorption biomarker) levels
| Genotype |
One-Way ANOVA |
||||
|---|---|---|---|---|---|
| Control (n = 25) | PT-PTH1R−/− (n = 14) | PT-PTH1R/KL−/− (n = 13) | F | P value | |
| CTX, ng/ml | 30.94 ± 2.26 | 31.48 ± 2.18 | 45.66 ± 2.34a,b | 10.1 | 0.0002 |
Data represent the means ± SD. Serum CTX was measured by ELISA in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice at 9 wk of age. CTX, cross-linked C telopeptide.
P < 0.001, Control vs. PT-PTH1R/KL−/−,
P < 0.01, PT-PTH1R−/− vs. PT-PTH1R/KL−/−.
PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice with HP challenge.
We continued to explore potential systemic compensatory effects by challenging Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− mice with HP in their drinking water starting at 9 wk of age for 24 h. The amount of HP drinking water ingested by each group was comparable (Table 2). Serum phosphate levels in PT-PTH1R−/− mice and Control mice did not significantly change upon HP, whereas the levels were significantly increased in PT-PTH1R/KL−/− mice (Fig. 4A). In contrast to PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice, the fractional excretion of phosphate (FEPO4) significantly increased only in Control mice (Fig. 4A). However, we could observe a slight, but nonsignificant, increase in FEPO4 in PT-PTH1R−/− mice, whereas the FEPO4 remained unchanged in PT-PTH1R/KL−/− mice (Fig. 4A). Serum calcium levels and fractional excretion of calcium remained unchanged in the three groups in both normal phosphate and HP situations (Fig. 4B).
Table 2.
The amount of high phosphate drinking water ingested by each group
| Genotype |
One-Way ANOVA |
||||
|---|---|---|---|---|---|
| Control (n = 6) | PT-PTH1R−/− (n = 4) | PT-PTH1R/KL−/− (n = 5) | F | P value | |
| HP water, ml/day | 4.26 ± 1.23 | 3.25 ± 0.71 | 3.53 ± 0.84 | 1.411 | 0.28 |
Data represent the means ± SD. The 24-h HP drinking water challenge was performed at the age of 9 wk. HP, high phosphate.
Fig. 4.

High phosphate (HP) challenge of PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice. Levels of serum phosphate and fractional phosphate excretion (A) and levels of serum calcium and fractional calcium excretion (B) were measured in Control mice, PT-PTH1R−/− mice, and PT-PTH1R/KL−/− on normal phosphate (NP) and HP (50 mM phosphate) drinking water for 24 h (n = 6–9). Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, and ***P < 0.001. Data represent the means ± SD.
DISCUSSION
In this study, we found a phosphate dysregulation phenotype with graded severity based on PTH1R deletion and combined deletion with Klotho in PTs without affecting the distal tubules or collecting ducts. This demonstrates distinct effects that are synergistic and partially overlapping. We previously demonstrated that PT-specific Klotho-deletion mice displayed a mild phenotype in general (24). Together with our previous report, we show that deletion of either PTH1R alone or Klotho alone in PTs results in only minor changes in phosphate regulation (Table 3).
Table 3.
Phenotype comparison between the proximal tubule-specific deletion of Klotho, PTH1R, and PTH1R/Klotho
| PT-KL−/−* | PT-PTH1R−/− | PT-PTH1R/KL−/− | |
|---|---|---|---|
| Serum | |||
| Calcium | ↑ | — | — |
| Phosphate | ↑ | — | ↑ |
| FGF23 | — | ↑ | ↑ |
| PTH | — | ↑ | ↑↑ |
| 1,25(OH)2D3 | — | ↓ | ↓ |
| Urine | |||
| Ca | — | — | — |
| Pi | ↓↓ | — | — |
| BBM | |||
| NaPi2a | ↑ | — | ↑ |
Each strain is compared with Control mice (Slc34a1-Cre; Tomatofl/+). —, unchanged and/or normal; BBM, brush border membrane; FGF23, fibroblast growth factor 23; PTH, parathyroid hormone.
Reference 24.
It has been shown that FGF23 plays a critical role in maintaining normal serum phosphate levels by fine adjustment of renal phosphate excretion (1, 2, 32). Elimination of PTH action by deleting the PTH1R in the PTs could therefore initially lead to increased phosphate reabsorption accompanied by higher serum phosphate, which in turn triggers the secretion of FGF23 to maintain serum phosphate in the normal range. We suggest that serum phosphate remained stable in the PTH1R single KO mouse because of a likely compensatory increase in FGF23 acting through the FGFR1/Klotho pathway. To investigate the compensatory mechanisms between the PTH/PTH1R and FGF23/Klotho pathways, which could lead to the attenuated phenotypes observed in PTH1R or Klotho single KO mice, we generated a transgenic mouse line lacking both PTH1R and Klotho in the PTs. These mice express Cre recombinase under control of the tamoxifen-inducible Slc34a1 promoter, a transport protein expressed mainly in the proximal S1/S2 segments and, to a lesser extent, in the S3 segment of the PTs; although, gene deletion was not efficient in the S3 segment in this mouse model (Fig. 1D), as others have also reported (25, 31). Nonefficient gene deletion of the S3 segment might be a weakness in this mouse model; however, the majority of the phosphate reabsorption occurs in the S1/S2 segments in PTs (60%–70%), with the S3 segment being only responsible for 10%–15% (8, 12). In addition, PTH1R and Klotho mRNA expressions in the S3 segment were significantly lower than in the S1/S2 segments in Control mice (Fig. 1D), which was also observed in a previous study (35). Based on these reports, the impact of mouse-model limitation was minimal.
Interestingly, double KO mice displayed significantly higher serum phosphate levels compared with Control mice, whereas there were no differences between Control mice and PT-PTH1R−/− mice. Increased serum phosphate levels are consistent with our hypothesis and could be explained in two nonexclusive ways. First, the double KO mouse model had elevated levels of Npt2a and Npt2c in the renal BBM compared with controls, whereas the levels were not significantly increased in the PTH1R single KO. This resulted in enhanced renal phosphate reabsorption followed by an increase in serum phosphate, indicating the disruption of a compensatory mechanism that maintained serum phosphate levels within the levels observed in Control mice and PTH1R single KO mice. Second, we observed that double KO mice had significantly increased levels of CTX, which reflects a high bone turnover consistent with a huge increase in serum PTH. This led to a large amount of phosphate being released from bone into circulation. Of note, this increase in the filtered load of phosphate could explain why urinary phosphate excretion did not decrease in double KO mice as seen in Klotho single KO mice (24), despite the increase in sodium/phosphate cotransporter expression in the BBM in double KO mice.
However, the serum phosphate levels in Klotho/PTH1R double KO mice did not reach the levels observed in Klotho null mice (13–15 mg/dl) (29, 43). This could be explained by the circulating form of soluble Klotho that may maintain the phosphate levels at around 10–11 mg/dl in our model of Klotho deletion from the PT. Recently, Chen et al. (9) revealed the molecular structure of α-Klotho. One of the main findings from their work is that the circulating form of soluble Klotho can act as a coreceptor for FGF23 signaling. This could be the reason why the hyperphosphatemic phenotype is milder in PT-specific Klotho single KO mice and in Klotho/PTH1R double KO mice compared with Klotho null mice. Since the distal tubule is the main site of Klotho expression, it might function as a reservoir for soluble Klotho in the PT-specific Klotho single KO, enabling FGF23 to act on the PT. The absence of soluble Klotho may also explain why phosphate levels observed in Klotho null mice are higher.
As expected, deleting both PTH1R and Klotho led to a marked increase in serum PTH and FGF23 levels. This is different than in mice with Klotho ablated alone, which showed no changes in serum PTH or FGF23 levels when compared with control (24). PTH1R single KO mice did not display hyperphosphatemia, whereas serum PTH and FGF23 levels were markedly increased compared with Control mice. PTH1R deletion in the PTs could be associated with an increase in serum PTH that could in turn participate in the increase in circulating FGF23 observed in the PTH1R single KO. Our data could support previous studies that proposed a direct effect of PTH on FGF23 secretion (14, 33, 38, 51). The double KO mouse model appears to reveal compensatory effects that are active in the single KO mouse models but blunted in the double KO models. It therefore suggests that PTH1R and Klotho signaling interact in the proximal renal tubules to regulate phosphate homeostasis. However, further investigations are required to fully explain the exact underlying mechanisms.
Furthermore, we examined the FGF23 downstream signaling pathway in all groups and found that PTH1R single KO mice showed a significant increase in Egr-1 in PTCs when compared with controls. However, it was diminished in double KO mice even though they had very high levels of circulating FGF23. These data indicate the disruption of the compensatory mechanism and the presence of FGF23 resistance in double KO mice.
We supported our findings by performing an HP challenge using HP drinking water for 24 h in Control mice, PT-PTH1R−/− mice, PT-PTH1R/KL−/− mice. In PTH1R single KO mice, urinary phosphate excretion showed a trend to increase, although statistical significance was not reached. Noteworthy, these mice had normal serum phosphate levels, probably because of the presence of intact FGF23/Klotho signaling. Interestingly, HP challenge induced hyperphosphatemia only in mice with PTH1R and Klotho double deletion in PTs. Control mice responded with a significant increase in fractional phosphate excretion, a process that was totally blunted in PTH1R and Klotho double mutant mice. The results of the HP challenge reinforce our findings that deletion of a single pathway is largely compensated for, but dual deletions are detrimental. Serum calcium levels were in the normal range in both PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice at the basal condition. Serum calcium levels and fractional calcium excretion remained unchanged in PT-PTH1R−/− mice and PT-PTH1R/KL−/− mice in both normal phosphate and HP situations, suggesting that PTH1R and Klotho play only a minor role in calcium regulation in the PTs. As previously shown by us and others, the calcium regulating actions of PTH are present in the thick-ascending limb of Henle (52) and distal convoluted tubules (7, 56).
PTH and FGF23 have opposite regulatory effects on 1,25(OH)2D3 synthesis. PTH1R signaling in the PTs induces active vitamin D production by increasing Cyp27b1 and decreasing Cyp24a1 mRNA expression, whereas FGF23/Klotho signaling in the PTs prevents active vitamin D formation by suppressing renal Cyp27b1 mRNA expression and stimulating Cyp24a1 mRNA (11, 21, 59). As expected, the protein expression of Cyp27b1 was significantly decreased and Cyp24a1 was significantly increased in the PTH1R single KO mutants. In PTH1R and Klotho double KO mice, the protein expression of renal Cyp27b1 was also significantly decreased compared with Control mice, whereas Cyp24a1 protein expression was not changed. The significant decrease in Cyp27b1 protein expression in the double KO implies that the PTH1R signaling pathway is predominant over the FGF23/Klotho signaling pathway in regulating Cyp27b1 protein. This is consistent with the decreased serum calcitriol levels observed in the double KO mice. Unchanged Cyp24a1 protein expression in the double KO mice could be explained by the lower serum 1,25(OH)2D3 levels and the lack of induction by the FGF23/Klotho signaling.
In conclusion, we have demonstrated that deletion of PTH1R alone from the renal PTs results in a mild phenotype, indicating the existence of a direct or indirect interaction between the PTH1R and FGF23/Klotho pathways. In contrast, deletion of both PTH1R and Klotho from the PTs leads to severe disturbances in phosphate metabolism under both normal and HP conditions. This suggests that the deletion of a single pathway is largely compensated for, but dual deletions are detrimental. These data indicate that the interplay between the PTH1R and FGF23/Klotho pathways is crucial to maintain proper renal phosphate handling in proximal renal tubules.
GRANTS
This work was supported by Department of Defense Grant no. PR120411 (to B. Lanske).
DISCLOSURES
Tobias E. Larsson is an employee at Astellas Pharma. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
N.I., J.H., and B.L. conceived and designed research; N.I., R.Y., and J.H. performed experiments; N.I. and J.H. analyzed data; N.I., M.C., H.O., T.E.L., J.H., and B.L. interpreted results of experiments; N.I. prepared figures; N.I. and R.Y. drafted manuscript; N.I., M.C., H.O., M.J.D., T.E.L., J.H., and B.L. edited and revised manuscript; N.I. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Tatsuya Kobayashi (Massachusetts General Hospital, Boston, MA) for providing PTH1R flox and Benjamin D. Humphreys (Washington University, St. Louis, MO) for providing Slc34a1-Cre (sodium phosphate cotransporter-2a1) mice.
REFERENCES
- 1.Bai X, Miao D, Li J, Goltzman D, Karaplis AC. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145: 5269–5279, 2004. doi: 10.1210/en.2004-0233. [DOI] [PubMed] [Google Scholar]
- 2.Baum M, Schiavi S, Dwarakanath V, Quigley R. Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney Int 68: 1148–1153, 2005. doi: 10.1111/j.1523-1755.2005.00506.x. [DOI] [PubMed] [Google Scholar]
- 3.Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol 75: 535–550, 2013. doi: 10.1146/annurev-physiol-030212-183748. [DOI] [PubMed] [Google Scholar]
- 4.Biber J, Stieger B, Stange G, Murer H. Isolation of renal proximal tubular brush-border membranes. Nat Protoc 2: 1356–1359, 2007. doi: 10.1038/nprot.2007.156. [DOI] [PubMed] [Google Scholar]
- 5.Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol 10: 1257–1272, 2015. doi: 10.2215/CJN.09750913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaine J, Weinman EJ, Cunningham R. The regulation of renal phosphate transport. Adv Chronic Kidney Dis 18: 77–84, 2011. doi: 10.1053/j.ackd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Bouhtiauy I, Lajeunesse D, Brunette MG. The mechanism of parathyroid hormone action on calcium reabsorption by the distal tubule. Endocrinology 128: 251–258, 1991. doi: 10.1210/endo-128-1-251. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter TO. Primary disorders of phosphate metabolism. In: Endotext, edited by De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A. South Dartmouth, MA: MDText.com, 2000. [Google Scholar]
- 9.Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, Moe OW, Liang G, Li X, Mohammadi M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553: 461–466, 2018. doi: 10.1038/nature25451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi NW. Kidney and phosphate metabolism. Electrolyte Blood Press 6: 77–85, 2008. doi: 10.5049/EBP.2008.6.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metabol Clin North Am 39: 243–253, 2010. doi: 10.1016/j.ecl.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damjanov I. Pathophysiology. Philadelphia, PA: Saunders/Elsevier, 2008, chapt. 12, p. 418. [Google Scholar]
- 13.Déliot N, Hernando N, Horst-Liu Z, Gisler SM, Capuano P, Wagner CA, Bacic D, O’Brien S, Biber J, Murer H. Parathyroid hormone treatment induces dissociation of type IIa Na+-Pi cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am J Physiol Cell Physiol 289: C159–C167, 2005. doi: 10.1152/ajpcell.00456.2004. [DOI] [PubMed] [Google Scholar]
- 14.Fan Y, Bi R, Densmore MJ, Sato T, Kobayashi T, Yuan Q, Zhou X, Erben RG, Lanske B. Parathyroid hormone 1 receptor is essential to induce FGF23 production and maintain systemic mineral ion homeostasis. FASEB J 30: 428–440, 2016. doi: 10.1096/fj.15-278184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraser DR, Kodicek E. Regulation of 25-hydroxycholecalciferol-1-hydroxylase activity in kidney by parathyroid hormone. Nat New Biol 241: 163–166, 1973. doi: 10.1038/newbio241163a0. [DOI] [PubMed] [Google Scholar]
- 16.Garabedian M, Holick MF, Deluca HF, Boyle IT. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc Natl Acad Sci USA 69: 1673–1676, 1972. doi: 10.1073/pnas.69.7.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardella TJ, Jüppner H. Interaction of PTH and PTHrP with their receptors. Rev Endocr Metab Disord 1: 317–329, 2000. doi: 10.1023/A:1026522619828. [DOI] [PubMed] [Google Scholar]
- 18.Gardella TJ, Jüppner H. Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metab 12: 210–217, 2001. doi: 10.1016/S1043-2760(01)00409-X. [DOI] [PubMed] [Google Scholar]
- 19.Gattineni J, Friedman PA. Regulation of hormone-sensitive renal phosphate transport. Vitam Horm 98: 249–306, 2015. doi: 10.1016/bs.vh.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, Martin A, Fornoni A, Di Marco GS, Kentrup D, Reuter S, Mayer AB, Pavenstädt H, Stypmann J, Kuhn C, Hille S, Frey N, Leifheit-Nestler M, Richter B, Haffner D, Abraham R, Bange J, Sperl B, Ullrich A, Brand M, Wolf M, Faul C. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 22: 1020–1032, 2015. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry HL. Regulation of vitamin D metabolism. Best Pract Res Clin Endocrinol Metab 25: 531–541, 2011. doi: 10.1016/j.beem.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int 74: 148–157, 2008. doi: 10.1038/ki.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24: 3438–3450, 2010. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ide N, Olauson H, Sato T, Densmore MJ, Wang H, Hanai JI, Larsson TE, Lanske B. In vivo evidence for a limited role of proximal tubular Klotho in renal phosphate handling. Kidney Int 90: 348–362, 2016. doi: 10.1016/j.kint.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Kefaloyianni E, Muthu ML, Kaeppler J, Sun X, Sabbisetti V, Chalaris A, Rose-John S, Wong E, Sagi I, Waikar SS, Rennke H, Humphreys BD, Bonventre JV, Herrlich A. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 1: e87023, 2016. doi: 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kendrick J, Chonchol M. The role of phosphorus in the development and progression of vascular calcification. Am J Kidney Dis 58: 826–834, 2011. doi: 10.1053/j.ajkd.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kie JH, Kapturczak MH, Traylor A, Agarwal A, Hill-Kapturczak N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J Am Soc Nephrol 19: 1681–1691, 2008. doi: 10.1681/ASN.2007101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T, Chung UI, Schipani E, Starbuck M, Karsenty G, Katagiri T, Goad DL, Lanske B, Kronenberg HM. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 129: 2977–2986, 2002. [DOI] [PubMed] [Google Scholar]
- 29.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390: 45–51, 1997. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 30.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281: 6120–6123, 2006. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci USA 111: 1527–1532, 2014. doi: 10.1073/pnas.1310653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Jüppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145: 3087–3094, 2004. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 33.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 299: F882–F889, 2010. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 34.Lederer E. Renal phosphate transporters. Curr Opin Nephrol Hypertens 23: 502–506, 2014. doi: 10.1097/MNH.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K, Brown D, Ureña P, Ardaillou N, Ardaillou R, Deeds J, Segre GV. Localization of parathyroid hormone/parathyroid hormone-related peptide receptor mRNA in kidney. Am J Physiol Renal Physiol 270: F186–F191, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Östman Wernerson A, Lanske B, Olauson H, Larsson TE. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol 25: 2169–2175, 2014. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.López I, Rodríguez-Ortiz ME, Almadén Y, Guerrero F, de Oca AM, Pineda C, Shalhoub V, Rodríguez M, Aguilera-Tejero E. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int 80: 475–482, 2011. doi: 10.1038/ki.2011.107. [DOI] [PubMed] [Google Scholar]
- 39.Lupp A, Klenk C, Röcken C, Evert M, Mawrin C, Schulz S. Immunohistochemical identification of the PTHR1 parathyroid hormone receptor in normal and neoplastic human tissues. Eur J Endocrinol 162: 979–986, 2010. doi: 10.1530/EJE-09-0821. [DOI] [PubMed] [Google Scholar]
- 40.Malmström K, Murer H. Parathyroid hormone regulates phosphate transport in OK cells via an irreversible inactivation of a membrane protein. FEBS Lett 216: 257–260, 1987. doi: 10.1016/0014-5793(87)80701-9. [DOI] [PubMed] [Google Scholar]
- 41.Mannstadt M, Jüppner H, Gardella TJ. Receptors for PTH and PTHrP: their biological importance and functional properties. Am J Physiol Renal Physiol 277: F665–F675, 1999. doi: 10.1152/ajprenal.1999.277.5.F665. [DOI] [PubMed] [Google Scholar]
- 42.Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol 20: 1453–1464, 2009. doi: 10.1681/ASN.2008070692. [DOI] [PubMed] [Google Scholar]
- 43.Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J 23: 433–441, 2009. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, Larsson TE. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol 23: 1641–1651, 2012. doi: 10.1681/ASN.2012010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palit S, Kendrick J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr Pharm Des 20: 5829–5833, 2014. doi: 10.2174/1381612820666140212194926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfister MF, Ruf I, Stange G, Ziegler U, Lederer E, Biber J, Murer H. Parathyroid hormone leads to the lysosomal degradation of the renal type II Na/Pi cotransporter. Proc Natl Acad Sci USA 95: 1909–1914, 1998. doi: 10.1073/pnas.95.4.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, Biber J, Wagner CA. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch 460: 677–687, 2010. doi: 10.1007/s00424-010-0841-1. [DOI] [PubMed] [Google Scholar]
- 48.Prasad N, Bhadauria D. Renal phosphate handling: physiology. Indian J Endocrinol Metab 17: 620–627, 2013. doi: 10.4103/2230-8210.113752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prié D, Beck L, Silve C, Friedlander G. Hypophosphatemia and calcium nephrolithiasis. Nephron, Exp Nephrol 98: e50–e54, 2004. doi: 10.1159/000080256. [DOI] [PubMed] [Google Scholar]
- 50.Prié D, Huart V, Bakouh N, Planelles G, Dellis O, Gérard B, Hulin P, Benqué-Blanchet F, Silve C, Grandchamp B, Friedlander G. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med 347: 983–991, 2002. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 51.Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, Bellido T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49: 636–643, 2011. doi: 10.1016/j.bone.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sato T, Courbebaisse M, Ide N, Fan Y, Hanai JI, Kaludjerovic J, Densmore MJ, Yuan Q, Toka HR, Pollak MR, Hou J, Lanske B. Parathyroid hormone controls paracellular Ca2+ transport in the thick ascending limb by regulating the tight-junction protein Claudin14. Proc Natl Acad Sci USA 114: E3344–E3353, 2017. doi: 10.1073/pnas.1616733114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19: 429–435, 2004. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 54.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770–774, 2006. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 55.Ureña P, Kong XF, Abou-Samra AB, Jüppner H, Kronenberg HM, Potts JT Jr, Segre GV. Parathyroid hormone (PTH)/PTH-related peptide receptor messenger ribonucleic acids are widely distributed in rat tissues. Endocrinology 133: 617–623, 1993. doi: 10.1210/endo.133.2.8393771. [DOI] [PubMed] [Google Scholar]
- 56.van Abel M, Hoenderop JG, van der Kemp AW, Friedlaender MM, van Leeuwen JP, Bindels RJ. Coordinated control of renal Ca(2+) transport proteins by parathyroid hormone. Kidney Int 68: 1708–1721, 2005. doi: 10.1111/j.1523-1755.2005.00587.x. [DOI] [PubMed] [Google Scholar]
- 57.Van der Hauwaert C, Savary G, Gnemmi V, Glowacki F, Pottier N, Bouillez A, Maboudou P, Zini L, Leroy X, Cauffiez C, Perrais M, Aubert S. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS One 8: e66750, 2013. doi: 10.1371/journal.pone.0066750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner CA, Hernando N, Forster IC, Biber J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch 466: 139–153, 2014. doi: 10.1007/s00424-013-1418-6. [DOI] [PubMed] [Google Scholar]
- 59.Zierold C, Mings JA, DeLuca HF. Regulation of 25-hydroxyvitamin D3-24-hydroxylase mRNA by 1,25-dihydroxyvitamin D3 and parathyroid hormone. J Cell Biochem 88: 234–237, 2003. doi: 10.1002/jcb.10341. [DOI] [PubMed] [Google Scholar]