

Summary
Mapping the binding sites of DNA- or chromatin-interacting proteins is essential to understanding biological processes. DNA adenine methyltransferase identification (DamID) has emerged as a comprehensive method to map genome-wide occupancy of proteins of interest. A caveat of DamID is the specificity of Dam methyltransferase for GATC motifs that are not homogenously distributed in the genome. Here, we developed an optimized method named MadID, using proximity labeling of DNA by the methyltransferase M.EcoGII. M.EcoGII mediates N6-adenosine methylation in any DNA sequence context, resulting in deeper and unbiased coverage of the genome. We demonstrate, using m6A-specific immunoprecipitation and deep sequencing, that MadID is a robust method to identify protein-DNA interactions at the whole-genome level. Using MadID, we revealed contact sites between human telomeres, repetitive sequences devoid of GATC sites, and the nuclear envelope. Overall, MadID opens the way to identification of binding sites in genomic regions that were largely inaccessible.
Keywords: MadID, M.EcoGII, m6A, LADs, telomeres, nuclear envelope, proximity labeling, methylation
Graphical Abstract
Highlights
-
•
MadID: mapping of protein-DNA interactions in vivo using proximity labeling
-
•
Deeper and unbiased genome-wide coverage using M.EcoGII, a methyltransferase
-
•
Identification of binding sites in previously inaccessible regions of the genome
-
•
Identification of telomere-nuclear envelope contact sites
Mapping the binding sites of DNA- or chromatin-interacting proteins is essential to understanding biological processes. Sobecki et al. developed an optimized method named MadID based on proximity labeling of DNA by the bacterial methyltransferase M.EcoGII. MadID results in deep and unbiased coverage for genome-wide mapping studies.
Introduction
The DamID (DNA adenine methyltransferase identification) proximity labeling technique has emerged as a complementary approach to chromatin immunoprecipitation (ChIP) to map protein-DNA interactions on a genomic scale (Vogel et al., 2007). The popularity of DamID has risen rapidly because of its compatibility with various model organisms and the ability for in vivo detection of both stable and transient interactions without the requirement for ChIP-grade-specific antibodies (Aughey and Southall, 2016). DamID exploits a major difference that exists between prokaryotes and eukaryotes: methylation of adenine is widespread in the former but largely absent from the latter. The technique relies on the targeted expression of the Escherichia coli Dam methyltransferase that catalyzes the methylation of adenine at the N6 position (m6A) of GATC motifs. Methylated GATC sites become DpnI sensitive, a feature used to fragment and detect DNA by various sequencing-, microarray-, or microscopy-based methods. DamID has been used to map the binding sites of various chromatin binding proteins in different organisms; one outstanding example is the identification of lamin-associated domains (LADs) down to single-cell resolution (Kind et al., 2015, Kind et al., 2013, Kind and van Steensel, 2014).
One major caveat of DamID is that it strictly relies on the distribution of the GATC tetrameric recognition site of the Dam methyltransferase. Statistically, this motif occurs every 256 nucleotides, but experimentally, MboI restriction enzyme-sensitive (GATC cutter) sites are found on average every 422 bp in the mouse genome (Sahlén et al., 2015) and close to every 400 bp in humans. However, this particular sequence may not be present at the DNA binding site of a protein of interest, thereby introducing a bias in favor of GATC-rich sequences and preventing the detection of GATC-free regions. Telomeres represent the archetypal DamID-resistant genomic region, because they are composed in mammals of repeated segments of the sequence (TTAGGG)n over several kilobases at the end of linear chromosomes. Other genomic regions are also expected to be challenging for Dam methylation, such as AT-rich regions and centromeres. For example, certain centromeric domains contain alpha-satellite repeats composed of 171-bp repetitive monomers of tandem centromeric protein CENP-A or CENP-B 17 bp boxes (Garavís et al., 2015) or satellite II and III DNA composed of (GGAAT)n motifs (Grady et al., 1992). Although mutations have been introduced in the catalytic pocket of Dam to decrease its specificity for the GATC tetramer, it only partially abrogates site recognition and therefore offers only limited improvement over traditional Dam (Xiao and Moore, 2001).
New bacterial DNA methyltransferases were characterized and their recognition sequences were annotated through the introduction of Pacific Biosciences single-molecule-real-time (SMRT) sequencing that allows the identification of modified template nucleotides such as m6A and 5-methylcytosine (5mC) (Fang et al., 2012). One of these m6A methyltransferases from E. coli, M.EcoGII, was found to be non-specific for all adenine (A) residues and able to methylate close to 100% of adenine residues in a DNA substrate in vitro and more than 85% in vivo (Murray et al., 2018). Here, we exploited the context independence of M.EcoGII to develop MadID (methyl adenine identification), an optimized technique that allows unbiased proximity labeling of adenines in any genomic region. MadID uses antibody-based specific recognition of m6A to identify and characterize methylated sequences using different readouts. We demonstrate the feasibility of this approach in human cells and the advantage of versatilely detecting protein-DNA interactions on a genome-wide scale. Our study also reveals the potential of MadID to study protein-DNA interactions at GATC null repetitive sequences such as human telomeres. Telomeres are recognized by the Shelterin complex, composed of six telomere-specific proteins, that associates with telomeric DNA to protect the ends of the chromosomes from degradation and from end-to-end fusion (de Lange, 2005). In human cells, telomeres are known to be transiently tethered to the nuclear envelope during postmitotic nuclear assembly and to localize close to the nuclear lamina, similar to LADs (Crabbe et al., 2012). MadID allowed us to specifically detect the previously inaccessible telomere-nuclear envelope contact sites in a semiquantitative manner and in asynchronous or synchronized cells.
Results
Design
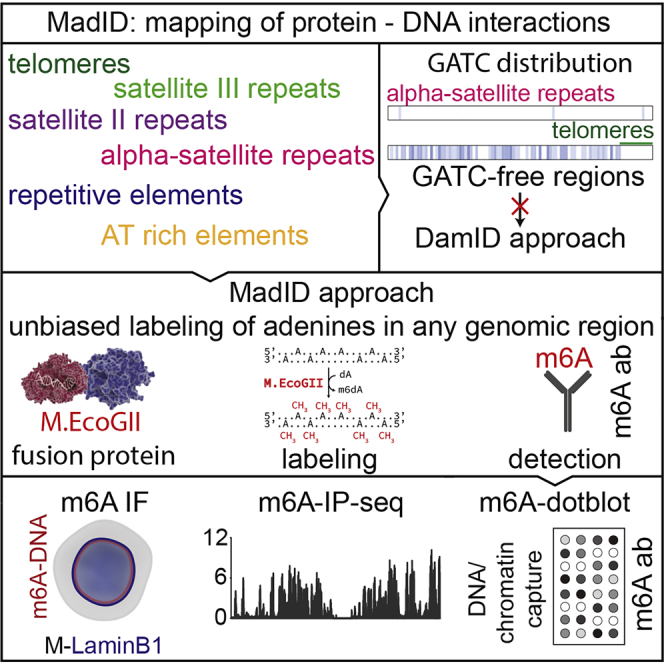
MadID is based on the targeted methylation of adenine residues in genomic DNA by the newly described M.EcoGII methyltransferase from E. coli to specifically map protein-DNA interactions. Unlike previously characterized site-specific methyltransferases showing specific recognition sequences, such as Dam with GATC sites, M.EcoGII methylates adenine residues in any DNA sequence context (Murray et al., 2018). Therefore, MadID circumvents the limitations of the previously characterized DamID, which strictly depends on the GATC distribution in the genome. Many chromatin domains are deprived of GATC sites, such as AT-rich regions, telomeres, or centromeres (Figure 1A) and thus are blind to DamID. However, these regions are fully accessible to MadID and thus constitute an unbiased strategy to map protein-DNA interactions. Another advantage of MadID is the more homogeneous distribution of A/Ts over GATC sites on a genomic scale (Figure 1A), resulting in deeper and unbiased coverage of informative bases, as well as in higher resolution. At a whole-genome level, 29% of the human genome is accessible by MadID, compared to only 0.9% for DamID (Figure 1A). A similar distribution is observed for model organisms such as M. musculus, D. melanogaster and C. elegans, where A/Ts sites offer higher coverage than GATC sites (Figure S1A).
Figure 1.

Schematic Overview of MadID
(A) Left, GATC distribution on human chromosome 1 (hg38 assembly). The blue gradient represents the score for the GATC site within 1 kb genome segments. Magnification from 1 to 0.1 Mb is shown. Green line, telomere T2AG3 sequence; red dashed line, 1 Mb of centromere DNA. Right, smooth scatter graphs of the A+T nucleotide and GATC motif count per 1 kb genome segment (hg38, chromosomes [chr] 1–22, X, and Y).
(B) Experimental setup and detection. (1a) DNA methyltransferases catalyze the transfer of a methyl group to DNA. (1b) M.EcoGII is fused to a destabilization domain (DD) for proteasome degradation unless the compound Shield1 is added to stabilize the protein. M.EcoGII is targeted to the nuclear envelope by fusion with Lamin B1, to telomeres by fusion with telomeric repeat binding factor 1 (TRF1), or to centromeres by fusion with CENP-C. Precise targeting of M.EcoGII causes methylation of DNA in the surrounding regions (m6A). (2a) m6A detection in situ by immunostaining with a m6A antibody. (2b) Genome-wide m6A detection by m6A-specific immunoprecipitation (m6A-IP), followed by whole-genome sequencing. (2c) DNA regions of interest can be purified by chromatin immunoprecipitation or probe-based capture techniques, and m6A can be detected on dot blots using the m6A-specific antibody.
When fused to a protein of interest, M.EcoGII methylates nearby adenines on any DNA motif, including repetitive sequences such as human telomeres. As a proof of principle, we targeted M.EcoGII to telomeres by fusion to the Shelterin protein telomeric repeat binding factor 1 (TRF1), to centromeres with fusion to CENP-C or to the nuclear envelope using fusion to Lamin B1 (Figures 1B and S1B). The resulting DNA methylation is detected using various methods, all based on the use of a commercially available m6A-specific antibody (Figure 1B). M.EcoGII-dependent DNA methylation can be monitored by DNA immunofluorescence to detect methylation in situ, by m6A-specific immunoprecipitation (m6A-IP) to map methylated genomic regions combined to qPCR or whole-genome sequencing, and finally by performing m6A dot blots from genomic DNA extracted before or after m6A-IP. We implemented MadID in asynchronous cells or in a cell-cycle-dependent manner using a prompt-inducible system based on protein stabilization.
Activity of M.EcoGII and Detection of m6A-DNA
N6-methyladenosine (m6A) methylation in RNA is the most prevalent modification in the mRNAs of most eukaryotes. Commercially available antibodies targeting this modification should also be specific for the m6A modification on DNA, as previously suggested (Xiao et al., 2010). To test their specificity, genomic DNA was extracted from E. coli strain K12, which is defective in DNA methylation (ER2796); from a dam−/dcm− strain with an intermediate methylation pattern (ER2925); and from a methylation-proficient wild-type strain (MG1655). The purified DNA was dot-blotted, and a membrane was probed with several m6A-specific antibodies. One of these antibodies, from Synaptic Systems (SYSY, Germany), proved to have the best affinity against m6A DNA modification and did not recognize unmethylated adenine residues (Figure S2). Compared to unmethylated DNA from strain ER2796, we detected an ∼60-fold increase in methylation levels in the strain ER2925 and an ∼100-fold increase in the strain MG1655 (Figure S2). Expression of M.EcoGII in the methylation-defective strain ER2796 was able to promote DNA methylation to the level of the K12 wild-type strain (Figure S2). From these experiments, we conclude that M.EcoGII is an efficient methyltransferase and that m6A modification on DNA can be specifically detected with a m6A-specific antibody.
Next, we performed m6A-specific immunoprecipitation (m6A-IP) on DNA extracted from the E. coli strain K12 MG1655, which is prone to DNA methylation, and from the K12 ER2796 strain, which is defective in DNA methylation. The precipitated DNA was dot-blotted, together with 10% of the starting material as input, and the membrane was probed using the same m6A antibody to detect methylated DNA. We successfully precipitated more than 14% of methylated DNA from the strain K12 MG1655, while no signal was detected for the methylation-defective strain, indicating the specificity of our detection method (Figure 2A). To further characterize the activity of M.EcoGII, we subsequently cloned M.EcoGII into a mammalian expression vector and transfected the expression cassette into human bone osteosarcoma epithelial (U2OS) cells. m6A-IP was performed on either wild-type or cells expressing M.EcoGII (Figure 2A). The m6A antibody specifically precipitated and detected about 11% of methylated DNA from M.EcoGII-expressing cells, confirming that the methyltransferase was active and was able to methylate human genomic DNA. As expected, m6A-IP in wild-type cells failed to recover methylated DNA, consistent with endogenous adenine methylation of DNA being negligible in human cells. The range of detection was assessed by quantifying m6A levels in increasing amounts of DNA extracted from M.EcoGII-expressing cells. Loading of 1.25 ng of genomic DNA was sufficient to specifically detect adenine methylation compared to wild-type cells, and the signal increased proportionally to the amount of loaded DNA (Figure 2B). To assess the potential of MadID to detect different levels of methylation, recombinant M.EcoGII was produced and purified from E. coli and used for in vitro methylation assays. Plasmid DNA extracted from a Dam-positive strain carried basal level of m6A but underwent increased methylation in a dose-dependent manner by M.EcoGII (Figure 2C).
Figure 2.

Activity of M.EcoGII and Detection of m6A-DNA
(A) Dot blot of fragmented genomic DNA (input) or DNA immunoprecipitated with a m6A antibody (m6A-IP) from E. coli ER2796, E. coli MG1655, or U2OS cells expressing or not expressing M.EcoGII. The membranes were probed with a m6A antibody. Quantification of m6A enrichment in the immunoprecipitated fractions relative to the input material is shown from three independent replicates (R1, R2, and R3; mean ± SD).
(B) Dot blot with increasing amounts of sheared genomic DNA extracted from U2OS cells expressing or not expressing M.EcoGII. The membrane was probed with a m6A antibody. The graph represents the mean intensity ± SD of the m6A signal from two independent experiments (R1 and R2).
(C) Dot blot of plasmid DNA in vitro methylated with increasing amounts of M.EcoGII recombinant enzyme. 500 ng of DNA from each reaction was loaded on a membrane probed with a m6A antibody. The normalized intensity relative to the unmethylated plasmid is shown.
Altogether, these results demonstrate that M.EcoGII is active in human cells and can introduce m6A modification on human genomic DNA. This permanent mark can be detected semiquantitatively with a m6A-specific antibody, even in conditions in which a low amount of material is available.
M.EcoGII Methylates GATC-Free DNA Regions
Unlike the Dam methyltransferase, M.EcoGII operates in a sequence-aspecific manner and has the ability to methylate GATC-free regions. To demonstrate the potential of M.EcoGII-based MadID, we evaluated whether M.EcoGII could efficiently methylate DNA sequences devoid of GATC motifs, such as telomeres. We performed in vitro methylation assays using oligonucleotides corresponding to the C-rich strand of telomeric repeats (CCCTAA4, TelC), the G-rich strand of telomeric repeats (TTAGGG4, TelG), or scrambled versions of these sequences. Recombinant M.EcoGII was able to efficiently methylate all these variants, with a level of methylation proportional to the number of target adenines present in the sequence and no bias toward the sequence context (Figure 3A). This is consistent with previous in vitro experiments, in which M.EcoGII activity on duplex DNA substrates rendered them insensitive to cleavage by multiple restriction endonucleases (Murray et al., 2018). To assess M.EcoGII activity on telomeric repeats, we analyzed double-stranded TTAGGG oligonucleotides before and after in vitro methylation by liquid chromatography coupled to high-resolution mass spectrometry (LC-HRMS). Purified deoxyadenosine (dA) and N6-methyl-2-deoxyadenosine (m6dA) were used as standards to determine their relative mass and retention time. We found that M.EcoGII-dependent in vitro methylation of TTAGGG repeats induced a sharp increase in the m6dA signal as expected, with a 800-fold increase in m6dA detection (Figure S3A). To evaluate M.EcoGII activity on telomeric repeats in vivo, the methyltransferase was expressed as a fusion to TRF1 (M-TRF1), a core subunit of the shelterin complex at telomeres (de Lange, 2005). M-TRF1 formed distinct nuclear foci distributed in the nucleus of HeLa 1.2.11, while untethered M.EcoGII was found to be diffusely expressed in the nucleoplasm (Figure 3B). To confirm proper M-TRF1 targeting to telomeres, fluorescence in situ hybridization (FISH) was carried out to visualize telomeres and centromeres. As expected, M-TRF1 foci colocalized with telomeric foci, but not with centromere signals (Figure 3B). The level of M-TRF1 expression varied from one cell to another, including cells in which no signal was detectable, but we confirmed with this staining that unlike untethered M.EcoGII, the protein was mostly localized in these foci and did not freely diffuse in the nucleoplasm. To assess whether M-TRF1 expression induced methylation of telomeres in vivo, we used TeloCapture, an approach to isolate telomeres using biotinylated oligonucleotides (Parikh et al., 2015). In our hands, TeloCapture isolated telomeric DNA more efficiently than regular ChIP protocols, with on average 20 ng of telomeric DNA obtained from 100 μg of HeLa 1.2.11 genomic DNA. Telomeric DNA could be visualized by dot blot after hybridization with a radioactive probe complementary to the TTAGGG sequence (32P:T2AG3) (Figure 3C). The same membrane was then probed with a m6A antibody, which highlighted the presence of adenine methylation specifically at pulled-down telomeres (IB:m6A) (Figure 3C). Recovery of about 15% of telomeric DNA from the input material allowed us to detect methylation, with a m6A signal corresponding to close to 1.5% of the starting material. This suggests that only a portion of telomeres carried the methylation mark, in accordance with the cell-to-cell variability of M-TRF1 expression. To ascertain that methylation is restricted to the site of targeting, M.EcoGII was tethered to centromeres by a fusion with CENP-C (M-CENP-C) (Figure S3B). As expected, this construct failed to promote methylation of telomeric DNA, confirming the specificity of the approach (Figure 3C).
Figure 3.

M.EcoGII Can Methylate GATC-Free DNA Regions and Is Specific to Its Region of Targeting
(A) Dot blot with 500 ng of oligonucleotides corresponding to the C-rich strand of telomeric repeats (TelC), the G-rich strand of telomeric repeats (TelG), and scramble (Scr TelC and Scr TelG) before or after in vitro methylation using recombinant M.EcoGII. The number of adenine present in the sequences is indicated. The membrane was probed with a m6A antibody, and the relative signal intensity was measured as indicated.
(B) Representative immunostaining and FISH of HeLa 1.2.11 cells transduced with the indicated vectors. V5 tag (green); TelC, telomeres (magenta); CEN, centromeres (red), DNA (blue); and merge. Scale bar, 10 μm. The percentage of colocalization is shown.
(C) Representative dot blot of captured telomeric DNA (T) from HeLa 1.2.11 cells expressing the indicated vectors. DNA was probed with a m6A antibody (m6A) and a telomeric probe (T2AG3). The graph represents normalized intensities relative to input (mean ± SD).
(D) DNA immunofluorescence (DNA-IF) of HeLa 1.2.11 cells expressing M-TRF1. m6A (green); TelC, telomeres (magenta); CEN, centromeres (red); DNA (blue); and merge. Scale bar, 10 μm. The percentage of colocalization is shown.
(E) Total number of telomeric reads per million from whole-genome sequencing data obtained from HeLa 1.2.11 transduced with the indicated vectors.
(F) Number of reads per million from whole-genome sequencing data obtained in M-TRF1 cells relative to M.EcoGII along chromosome ends (mean ± SEM).
Next, we tested whether the methylated DNA could be directly visualized in situ using m6A antibody-based immunostaining. A short denaturation step and RNase treatment were added to the protocol of cell fixation and permeabilization to facilitate the access of the m6A antibody to the modified adenines and to prevent recognition of abundant methylated RNAs. Methylated DNA was observed at telomeres when M.EcoGII was fused to TRF1, and this signal colocalized to telomeres, but not to centromeres (Figure 3D). In contrast, the m6A signal was detected at centromeres in M-CENP-C-expressing cells (Figure S3C). To analyze their methylation status, genomic DNA extracted from M-TRF1 cells or cells expressing M.EcoGII only as a control was sheared by sonication into small fragments of 200–400 bp before ligation of indexed Illumina adaptors. The obtained material was denatured and used to perform m6A-IP, and the precipitated material was subjected to whole-genome sequencing. All sequencing reads were normalized to the total number of recovered reads and to the input. We then searched for the total number of telomeric reads per million obtained in both cell types and found that they were highly enriched in M-TRF1 cells (Figure 3E). Reads mapping to combined chromosome ends for M-TRF1 cells were plotted against untethered M.EcoGII. Strong enrichment was observed at chromosome tips, with a gradual decrease of the number of reads as we move from the ends (Figure 3F). A similar analysis was performed on individual chromosomes, using heatmaps to display specific enrichments on each subtelomere. This revealed that most chromosome ends were marked with the m6A signature after M-TRF1 expression (Figure S3D).
Altogether, these result demonstrate that M.EcoGII has the ability to methylate genomic DNA even when targeted to chromatin regions rich in repetitive sequences such as telomeres. M.EcoGII-dependent methylation is constrained to the targeted site, supporting the specificity and robustness of our approach. In addition, methylated DNA can be efficiently detected using m6A-specific immunostaining in situ, on dot blots, and through immunoprecipitation followed by whole-genome sequencing.
MadID Can Identify LADs
Because M.EcoGII is active in vivo and specifically methylates DNA at specific sites upon targeting to genomic regions, we next used MadID to characterize contact sites between the chromatin and the nuclear envelope. These contact sites are known as LADs and were previously mapped in mammals using DamID (Guelen et al., 2008, Kind et al., 2015, Kind et al., 2013, Kind and van Steensel, 2014). In metazoan cells, the nuclear envelope is lined with a thin meshwork composed of intermediate filaments of A- and B-type Lamins, which impart the nucleus with mechanical properties and tether the heterochromatin to the periphery (Naetar et al., 2017). We targeted M.EcoGII to the nuclear envelope by expressing an inducible M.EcoGII-Lamin B1 (M-LB1) fusion protein. M.EcoGII was fused to a destabilization domain (DD) at its N terminus, leading to constant degradation of the M-LB1 protein when expressed in transduced cells, unless stabilized by addition of the Shield compound to the growth medium. This small compound binds the DD and rapidly prevents protein degradation, thus enabling the stabilized M.EcoGII fusion protein to methylate the target sites. Although a basal level of methylation could be detected before induction due to leakage, DNA methylation was strongly enhanced upon Shield1 addition, as visualized on dot blots with genomic DNA extracted from HeLa 1.2.11 and IMR90 expressing human telomerase reverse transcriptase (hTERT) (Figures 4A and S4A). M-LB1 could be stabilized and properly addressed to the nuclear periphery in both cell types after 24 hr of induction, as shown by the v5-tag staining (Figures 4B and S4B). DNA immunostaining using a m6A antibody to detect methylated DNA revealed that the signal was restricted to the nuclear periphery, confirming that DNA methylation occurred locally, where M.EcoGII is targeted after induction (Figures 4B and S4B). As a control, we expressed DD-M.EcoGII alone, without specific targeting, and confirmed that it induced global genomic DNA methylation after induction (Figure 4C). In contrast to what is observed when M-LB1 expression is restricted to the nuclear periphery, such M.EcoGII-expressing cells displayed diffuse nucleoplasm staining of both the methyltransferase and the methylated DNA (Figure 4D).
Figure 4.

Expression of M.EcoGII-Lamin B1 to Target M.EcoGII to the Nuclear Envelope
(A–C) Representative dot blot of genomic DNA from HeLa 1.2.11 cells induced (+) or induced not (−) to express M.EcoGII-v5-Lamin B1 (A) or v5-M.EcoGII (B). The membrane was probed with a m6A antibody. The normalized intensities are shown.
(B and D) Example of immunofluorescence of HeLa 1.2.11 cells induced (+) or not induced (–) to express M.EcoGII-v5-Lamin B1 (B) or v5-M.EcoGII (D). V5 tag (left panel) or m6A (right panel) (red), Lamin A/C (green), DNA (blue), and merge. Scale bar, 10 μm. The enlarged part of the nucleus of m6A and Lamin A/C staining is shown. Scale bar, 1 μm.
To test whether LADs could be identified using M.EcoGII-based approaches, we performed m6A-IP on M-LB1-expressing HeLa 1.2.11 and IMR90 cells after induction, followed by qPCR analysis using primers specific to well-established LADs and inter-LADs regions (Kind et al., 2013). LADs were enriched in both cell lines, while inter-LADs were not represented in the immunoprecipitated methylated DNA fraction (Figures S4C and S4D). We then extended our analysis using whole-genome sequencing following m6A-IP. Genomic DNA extracted from M-LB1 cells or cells expressing M.EcoGII only as a control were subjected to whole-genome sequencing. All sequencing reads were normalized to the total number of recovered reads and to the input and were represented as a ratio to the M.EcoGII control sample. This experiment was performed in two replicates, revealing a high degree of correlation (Spearman’s rank σ = 0.97) (Figure S5A). To facilitate visualization and comparison to published datasets (described later), reads were binned in 100 kb contiguous genomic segments. A ratio value higher than 1 was considered specific to the M-LB1 methylation. We obtained contact maps for each chromosome that clearly identified domain patterns, with strong similarities to domains previously obtained with DamID (Figure S5B). To more systematically analyze the potential correlation between Lamin B1-based MadID and DamID and to gauge the overall quality of our data, we compared it to conventional microarray-based DamID profiles generated from HT1080 cells (Kind et al., 2013). The scores corresponding to the log2 Dam-Lamin B1/Dam ratio from two independent experiments were obtained from the GEO repository and averaged. We obtained highly similar domain patterns (σ = 0.815) despite probing different cell types, which demonstrates that our new protocol was able to capture similar regions of interaction with great specificity (Figures 5A and 5C, left panel). We compared our results to single-cell sequencing DamID experiments performed on KBM7 cells (Kind et al., 2015). The short reads from 118 single-cell samples were obtained from the GEO repository and processed in 100 kb bins, as for our own data. The observed overexpected (OE) score was calculated as described in Kind et al. (2015). The domain patterns were highly similar (σ = 0.854) (Figures 5B and 5C, right panel). Analysis of our MadID data revealed an average number of 1,338 LADs with a median size of 0.5 Mb and coverage close to 37% of the total genome (Figures 5D and S5C). These results are in accordance with previously published data using DamID, supporting the robustness and specificity of MadID.
Figure 5.

Identification of Lamin-Associated Domains Using MadID
(A) Comparison nuclear lamina contact map for chr1 (hg38) with MadID from HeLa 1.2.11 cells (top profile) (ArrayExpress: E-MTAB-6888) and with conventional microarrays DamID from HT1080 cells (bottom profile); y axis, log2. Below the track, graphical representation of identified lamin-associated domains (LADs) as continuous regions in which all 100 kb segments have a score > 0.
(B) Comparison nuclear lamina contact map for chr1 (hg38) with MadID from HeLa 1.2.11 cells (top profile) (ArrayExpress: E-MTAB-6888) and with single-cell DamID sequencing (DamID-seq) of KBM7 cells. An average of 118 single-cell profiles is shown (bottom profile). Below the track, graphical representation of identified LADs as continuous regions in which all 100 kb segments have a score > 1.
(C) Left, log2 score of MadID in individual 100 kb bin (y axis) versus log2 score of conventional DamID in individual 100 kb bin (x axis). Genome-wide Spearman’s ρ = 0.815 between the two methods. Right, score of MadID in individual 100 kb bin (y axis) versus average score of 118 single-cell DamID-seq in individual 100 kb bin (x axis). Genome-wide Spearman’s ρ = 0.854 between the two methods.
(D) Frequency distribution (y axis) of the length of identified LADs (x axis) using MadID (red line), DamID-seq from 118 cells (blue line), and DamID using microarray (green line).
Next, a smaller bin size (5 kb) was used to generate new contact maps within a 4.5 Mb region of chromosome 1, encompassing a LAD and inter-LADs. This binning was incompatible with single-cell DamID data, but it allowed the production of higher-resolution profiles with MadID (Figure S5D). This indicates that sequencing reads generated with MadID give rise to higher signal complexity and resolution and that MadID allows a more detailed view of LAD organization.
MadID Results in Deeper Read Coverage and Higher Resolution
To determine the relative resolution of MadID and DamID, we used a more systematic approach to analyze the sequencing results from cells expressing M.EcoGII alone. First, we determined what we defined as the intrinsic smoothness of both MadID-sequencing (MadID-seq) and DamID-sequencing data using different bin sizes ranging from 1 to 100 kb (Figure 6A). For every fragment of a specific bin size, this test evaluates the variability in signal between neighboring fragments. Because LADs and inter-LADs form large domains, only a few sequences located at the borders of defined peaks should behave differently from their neighbors; therefore, neighboring bins are expected to behave similarly unless the signal is noisy and of poor resolution. Although both techniques had a similar smoothness at large bin sizes, it dropped drastically for DamID as soon as bin sizes below 50 kb were used. DamID smoothness increased again at 1 and 2 kb bins, but this was due to the higher presence of bins with 0 values at this resolution (Figure 6A). In contrast, MadID-intrinsic smoothness remained stable for all bin sizes tested (Figure 6A). This clearly shows that MadID offers a higher resolution and allows the identification of well-defined peaks even at low bin sizes, consistent with what we observed with LADs (Figure S5D).
Figure 6.

Sensitivity and Coverage of MadID
(A) Intrinsic smoothness of MadID versus DamID using sequencing data obtained from cells expressing M.EcoGII. Values of each bin were compared to the two neighboring bins (see STAR Methods) at different bin sizes, ranging from 100 to 1 kb. A smoothness value close to 1 means the lowest experimental bias.
(B) Bias plot of MadID and DamID using sequencing data obtained from cells expressing M.EcoGII. The number of GATC sites or adenine/T nucleotides was calculated in each 1 kb bin and plotted against the number of reads per million. The Spearman correlation is indicated.
We then determined whether the distribution of GATC motifs or A/Ts nucleotides induced a bias in the sequencing data obtained with DamID or MadID, respectively. The number of GATC sites was plotted against the number of reads obtained in 1 kb bins (Figure 6B). A strong correlation was found, with an increase of reads per million as fragments contained a higher number of GATC sites. In addition, as the number of GATC sites reached more than 5 motifs per 1 kb window, the number of reads decreased considerably. Most likely during the digestion of these methylated GATC sites, frequent cutting generates fragments too short to be properly sequenced. In comparison, the number of A/Ts nucleotides per 1 kb bin in MadID did not correlate with a higher number of reads per million (Figure 6B). The bias-plot profile remained mostly equally distributed, suggesting that A/Ts distribution did not skew the datasets as the GATC contents. Altogether, these results demonstrate that MadID has better resolution, had a better signal-to-noise ratio, and is less biased than DamID.
MadID Can Detect Telomere-Nuclear Envelope Contact Sites
A large subset of telomeres in human cells is known to interact with the nuclear envelope during postmitotic nuclear assembly (Crabbe et al., 2012). These telomeres can be visualized using confocal microscopy as overlapping with the nuclear envelope (Figure S6). MadID offers an ideal alternative to analyze the interaction between telomeres and the nuclear envelope, using methylation detection on dot blots. Telomeric DNA was isolated from cells induced to express untethered M.EcoGII, M-TRF1, M-CENP-C, or M-LB1 for 24 hr. We confirmed the presence of adenine DNA methylation in the input fractions of these cells, suggesting that M.EcoGII was able to contact and methylate chromatin in vivo (Figure 7A). Purification of telomeric DNA using TeloCapture was confirmed, and levels of methylation were evaluated in these different settings. Expression of untethered M.EcoGII induced methylation at telomeres, consistent with the methyltransferase diffusing freely in the nucleoplasm (Figures 3B and 4D), and can methylate any accessible DNA (Figure 7A). As we previously observed, telomere methylation also occurred when M.EcoGII was targeted to telomeres, but not to centromeres (Figure 7A). Methylation was found at telomeres in cells expressing M-LB1, which indicated that telomeric chromatin came in close contact with the nuclear periphery, notably the nuclear lamina, and that M.EcoGII was able to catalyze the formation of m6A marks on TTAGGG repeats in vivo (Figure 7A). These results prompted us to analyze the sequencing data obtained from M-LB1 cells and search for specific enrichment at individual chromosome ends. The heatmap obtained revealed that some specific chromosome ends had an enrichment of reads in subtelomeric regions up to 200 kb from the ends, giving a first hint of the identity of tethered chromosomes in these cells (Figure 7B). These regions were described as middle-replicating (3q, 7p, 19p, and 20p) or late-replicating (4p, 11q, 3p, and 2p) telomeres in a previous study, which often correlates with a peripheral nuclear localization (Arnoult et al., 2010).
Figure 7.

M.EcoGII Targeted to the Nuclear Envelope Can Contact and Methylate Telomeric DNA
(A–C) Representative dot blot of captured telomeric DNA from asynchronous HeLa 1.2.11 induced to express the indicated vectors for 24 hr (A) or G1/S-arrested cells induced for 8 hr (C). DNA was probed with a m6A antibody (m6A) and a telomeric probe (T2AG3). The graph represents the normalized intensities relative to input (mean ± SD). (B) Heatmap of the number of reads per million obtained at individual chromosome ends in HeLa 1.2.11 cells expressing M.EcoGII-Lamin B1 (M-LB1). The log2 M.EcoGII-LB1/M.EcoGII ratio is shown. Chromosome ends with positive enrichment are highlighted in red. A box with a cross represents a bin without an associated DNA sequence and therefore excluded from the analysis.
(D) Representative DNA-IF of G1/S-arrested HeLa 1.2.11 cells induced to express the indicated vectors for 8 hr. m6A (green); TelC, telomeres (magenta); CEN, centromeres (red); DNA (blue); and merge. Scale bar, 10 μm.
Implementation of MadID along the Cell Cycle
During open mitosis, the nuclear envelope is broken down, and Lamin filaments are disassembled and released in the cell. We could argue that the M-LB1 construct reached telomeric DNA during this stage, not when telomeres are tethered to the fully functional nuclear envelope. To rule out this possibility, we tested the suitability of MadID for cell-cycle experiments. HeLa 1.2.11 cells were synchronized at the G1/S boundary using thymidine and maintained as arrested at this stage during induction with Shield for 8 hr to transiently express M-LB1. Although telomeres are enriched at the nuclear periphery during postmitotic nuclear assembly, a significant portion of telomeres are still detected at the nuclear rim in interphase and should get methylated (Crabbe et al., 2012). We found that telomeric DNA isolated from M-LB1 arrested in G1/S was decorated with m6A using dot blots and DNA immunofluorescence (DNA-IF) (Figures 7C and 7D), suggesting that telomeres contacted the nuclear lamina outside of mitosis and were not a consequence of nuclear envelope breakdown. Altogether, these results indicate that a short induction of M.EcoGII is sufficient to generate quantifiable methylation levels and, more importantly, that MadID can be implemented to study events that are temporally restricted during the cell cycle.
Discussion
In the present study, we developed the MadID approach to unravel DNA-protein binding in vivo, using proximity labeling of DNA by the newly described bacteria methyltransferase M.EcoGII. We demonstrate the power and robustness of MadID compared to traditional GATC-limited DamID approaches. The heatmap of GATC distribution in the human genome clearly indicates that although GATC motifs are well represented globally, there are major GATC-free and GATC-poor genomic regions that are now accessible to our technology. In addition, the restriction of the Dam methyltransferase to GATC sites limits the analysis to less than 1% of total human genome coverage, thus providing relatively poor information density. As a methyltransferase able to catalyze the addition of a methyl group to adenine residues in any sequence context, M.EcoGII provides higher coverage, reaching almost 30% of the genome. As such, MadID represents an excellent strategy to study DNA-protein interactions at the genome level, including in DNA regions incompatible with DamID. The most compelling evidence was the confirmation of telomere interaction with the nuclear lamina that was previously demonstrated using live-cell confocal microscopy (Crabbe et al., 2012).
The activity of M.EcoGII methyltransferase expressed in human cells allowed for clear enrichment of genomic DNA methylation, which can be semiquantitatively detected on dot blots using m6A-specific antibodies. We used enhanced chemiluminescence detection and exposure to a charge-coupled device camera to obtain a good dynamic range of the signal. In these conditions, we could easily detect variations in the methylation levels of DNA samples blotted on the same membrane, even with a limited amount of material.
The excellent signal-to-noise ratio obtained in cells expressing M.EcoGII compared to controls is consistent with endogenous m6A of DNA being negligible in our system. The presence of m6A modification in higher eukaryotes has been debated, but a methylome analysis clearly indicates that methylation of adenine is more widespread than previously expected and is found in several vertebrates (Koziol et al., 2016). m6A deposition was also found to be correlated with epigenetic silencing in mouse embryonic stem cells (Wu et al., 2016). However, these studies established that unlike prokaryotes, the level of naturally deposited m6A in vertebrates is extremely low, which is consistent with our results.
When targeted to telomeres using a fusion of M.EcoGII to the shelterin protein TRF1, the methyltransferase efficiently methylated telomeric DNA. However, only 1.5% of the m6A signal was found at telomeres relative to input, while about 15% of telomeric DNA was purified (Figure 3C). We observed that once methylated, the affinity of the probes used for purification and detection of telomeres was reduced. Consequently, isolation of unmethylated telomeric DNA might be favored during TeloCapture, which could explain the lower level of m6A detection obtained. Subcloning the cell population to select clones with the optimal M-TRF1 targeting would improve this experiment by decreasing the amount of accessible unmethylated TTAGGG repeats. m6A sequencing (m6A-seq) analysis of M-TRF1-expressing cells also revealed strong over-representation of raw sequences containing the telomeric (TTAGGG)3 motif, like what was obtained after TRF1 ChIP in mice (Garrobo et al., 2014). Enrichment in these reads was higher in wild-type mice that have telomeres on average 5 to 10 times longer than those of human cells. However, TTAGGG-containing read representation after m6A-seq became similar to those obtained after TRF1 chromatin immunoprecipitation sequencing (ChIP-seq) from telomerase mutant Terc−/− G1 and G3 mice that carry shorter telomeres (Garrobo et al., 2014).
The specificity and sensitivity of our approach is evidenced by the mapping of LADs with MadID, combined with m6A-IP and whole-genome sequencing. We identified 1,338 domains with an average size of 0.5 Mb and excellent enrichment ratios over controls. Compared to published DamID results, a few additional LADs that correspond to GATC-poor genomic regions were identified. However, because LADs cover very large domains of about 500 kb, their mapping with Dam or M.EcoGII is similar, as GATC sites will be sufficiently represented in these domains. We provided a comparison between MadID and DamID resolution and could show that MadID resulted in deeper coverage and higher resolution. Similar to conventional ChIP, MadID resolution mostly depends on the average sonication level of the DNA (in our hands, 200–400 bp on average). This is in contrast to the DamID protocol, in which methylated GATCs are recognized by the methylation-sensitive restriction enzyme DpnI to cut fragments and ligate adaptor oligonucleotides for specific amplification (Vogel et al., 2007). Therefore, resolution depends again on the tetramer distribution in the loci of interest and can vary substantially.
An essential control when performing MadID-seq is to compare methylation profiles after expression of M.EcoGII alone (i.e., not fused to a DNA binding protein) to correct for chromatin accessibility and other potential biases. Sensitivity to Dam methylation has previously been used as a tool to monitor chromatin structure in C. elegans, with a strong correlation between methylation and accessibility (Sha et al., 2010). More recently, tissue-specific Dam expression was used to determine chromatin accessibility from Drosophila neural and midgut lineages (Aughey et al., 2018). As discussed in this study, Dam methyltransferase failed to detect loci depleted for GATC when compared to assay for transposase-accessible chromatin (ATAC) or formaldehyde-assisted isolation of regulatory elements (FAIRE) data (Aughey et al., 2018). The authors raised the limitations of the resolution achievable by Dam methylation, consistent with our analysis of GATC distribution in this organism and others (Figure S1A). In our hands, sequencing patterns after m6A-IP sequencing (m6A-IP-seq) from cells expressing M.EcoGII alone highlighted active regions of the genome. MadID therefore represents an attractive alternative to standard methods used for examining nucleosome positioning and regional accessibility, with higher coverage of the genome compared to Dam-based approaches.
Because adenine methylation is a permanent mark, M.EcoGII is able to methylate chromatin loci upon transient contacts with DNA. This is a strong benefit over ChIP-based methods that rely on more stable interactions or transient interactions captured at the time of crosslinking. Care should be taken, however, not to saturate methylation signals due to a high methyltransferase expression level. In the present study, MadID was performed using inducible expression of M.EcoGII based on protein stabilization, which also allowed us to perform experiments in a cell-cycle-dependent manner. Another important aspect to consider is the abundance of m6A-modified RNA in eukaryote cells that can be efficiently detected or precipitated with m6A-specific antibodies. In addition, M.EcoGII can efficiently add methyl groups to single-stranded DNA (ssDNA) and more importantly to single-stranded RNA (ssRNA) (Murray et al., 2018), which may be detected later if care is not taken in removing RNA from the experiment design.
In this study, we targeted M.EcoGII in human cells to telomeres, centromeres, and the nuclear envelope to test its feasibility and sensitivity. M.EcoGII is a protein of ∼44 kDa, slightly bigger than Dam, but fusion proteins previously generated with Dam will most likely be functional with M.EcoGII. We found that adenine residue methylation is restricted to the targeting site, which is essential for the specificity of the experiment. M.EcoGII can therefore be fused to any protein of interest, provided that the fusion protein remains functional, and can even be designed to specifically target organelles within the cell. In addition, because MadID does not rely on ChIP-grade-specific antibodies, it can be implemented in any model organism in which transgenesis in possible. In the case of multicellular organisms, M.EcoGII expression can be driven from tissue-specific promoters to properly understand development or cellular function at a cell-type-specific level.
Altogether, we believe MadID provides an excellent tool to visualize, quantify, and identify binding sites of DNA-interacting proteins, with various experimental setups and in a range of model organisms.
Limitations
Although M.EcoGII can be theoretically fused to any protein of interest, it is important to make sure the targeted protein can be fused in the N or C terminus without compromising its function or localization. This is not specific to MadID but rather is inherent to any technique based on protein fusion, including DamID.
Another limitation for cell-cycle experiments is the minimal time of induction to obtain a sufficient signal to be quantified. In our experimental setup, we chose to regulate the expression of M.EcoGII constructs by protein stabilization, not by transcriptional activation. This system is more rapid, because the protein can supposedly be stabilized within a few hours after the addition of Shield1, but some applications may require even faster inducible systems. This limitation could be overcome by protein sequestration rather than induction. Methods based on split tobacco etch virus (TEV) protease (Williams et al., 2009) and the chemically induced dimerization (CID) system (Rivera et al., 1996) could be combined to release the protein of interest from a plasma membrane anchor and address it within minutes to the designed targeted region. This method could be developed for MadID as a way to rapidly target the methyltransferase to any interest site.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-N6-methyladenosine | Synaptic System | Cat# 202 003 |
| Anti-V5 | Cell Signaling | Cat# 13202; RRID:AB_2687461 |
| Anti-Lamin A/C | Santa Cruz Biot. | Cat# sc-7292; RRID:AB_627875 |
| Anti-TRF2 | Abcam | Cat# ab13579; RRID:AB_300474 |
| Anti-CREST | Immunovision | Cat# HCT-0100; RRID:AB_2744669 |
| Telomere TelC-647 | Panagene INC. | F2003 |
| Centromere CEN-488 | Panagene INC. | F3012 |
| Bacterial and Virus Strains | ||
| E.Coli K-12 MG1655 – F-, λ-, rph-1 | Yoshiharu Yamaichi, I2BC | N/A |
| E.Coli K-12 ER2925 – F-, ara-14, leuB6, fhuA31, lacY1, tsx78, glnV44, galK2, galT22, mcrA, dcm-6, hisG4, rfbD1, R(zgb210::Tn10)TetS, endA1, rpsL136, dam13::Tn9, xylA-5, mtl-1, thi-1, mcrB1, hsdR2 | Yoshiharu Yamaichi, I2BC | N/A |
| E.Coli K-12 ER2796 – F-, fhuA2::IS2, glnX44(AS), λ-, e14-, trp-31, dcm-6, yedZ3069::Tn10, hisG1, argG6, rpsL104, Δdam-16::KanR, xyl-7, mtlA2, metB1, Δ(mcrC-mrr)114:IS10 | Yoshiharu Yamaichi, I2BC | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Shield | Aobious | Cat# AOB1848 |
| Thymidine | Sigma | Cat# T1895 |
| 2-Deoxycytidine | Sigma | Cat# D3897 |
| Critical Commercial Assays | ||
| QIAGEN Blood & Cell Culture DNA Midi Kit | QIAGEN | Cat# 13323 |
| Dynabeads streptavidin | Invitrogen | Cat# M280 |
| Next End-repair module | NEB | Cat# E6050 |
| Next A-tailing module | NEB | Cat# E6053 |
| Next Ligation Module | NEB | Cat# E6056 |
| Gibson Assembly Cloning kit | NEB | Cat# E5510 |
| Deposited Data | ||
| Raw data (MadID) | This paper | ArrayExpress: E-MTAB-6888 |
| DamID microarrays | Kind et al., 2013 | GEO GSM990672 and GSM990672 |
| Single-cell DamID | Kind et al., 2015 | GEO GSE68263 |
| Mendeley dataset | This paper | https://doi.org/10.17632/8j9kmzm4bc.1 |
| Experimental Models: Cell Lines | ||
| IMR90 | ATCC | Cat# CCL-186; RRID:CVCL_0347 |
| U2OS | ATCC | Cat# 300364/p489_U-2_OS; RRID:CVCL_0042 |
| HeLa 1.2.11 | Crabbe et al., 2012 | N/A |
| Oligonucleotides | ||
| Cloning M.EcoGII pLPC | This paper | N/A |
| FW GCGGATCCATGCTTAATACTGTAAAAATATC | ||
| REV GCGAATTCAACGATTAAATCCTGAACTTC | ||
| Cloning M.EcoGII pRetroXPTuner C-tag | This paper | N/A |
| FW CGGGATCCGGTAAGCCTATCCCTAACCCTCTCCT CGGTCTCGATTCTACGCGTACCGGCATGCTTAATAC TGTAAAAATATCC | ||
| REV CGGCGGCCGCTTAAACGATTAAATCCTGAACTTC | ||
| Cloning M.EcoGII pRetroXPTuner N-tag | This paper | N/A |
| FW CGGGATCCGTATGCTTAATACTGTAAAAATATCC | ||
| REV CGCCATGGAACGATTAAATCCTGAACTTC | ||
| Q-PCR primer CFHR3 | Kind et al., 2013 | N/A |
| FW TTGGAAGAAGAGAAAGACAAGG | ||
| REV GCAGTGGATGTTTCTCAGCA | ||
| Q-PCR primer CYP2C19 | Kind et al., 2013 | N/A |
| FW GGATGAGCTTTGCAGGAGAT | ||
| REV AAGCTGTGAGCCTGAGCAGT | ||
| Q-PCR primer CDH12 | Kind et al., 2013 | N/A |
| FW TTTTTCCTCCCAGGTGACAG | ||
| REV TGATAGCACCTGGGTTAGCAC | ||
| Q-PCR primer LAD1 | Kind et al., 2013 | N/A |
| FW CATTGGCTTCTTTGGTGCCAGGT | ||
| REV ACGGTGGAGGCAGTCAAAAGGC | ||
| Q-PCR primer iLAD1 | Kind et al., 2013 | N/A |
| FW GAAGGTTCCCCCACAGAAAT | ||
| REV CTGAGGCAAAGACAGGGAAG | ||
| Q-PCR primer UBE2B | Kind et al., 2013 | N/A |
| FW ACTCAGGGGTGGATTGTTGA | ||
| REV GCCAGAGATTTCAGGGAAAG | ||
| Q-PCR primer STAG2 |
Kind et al., 2013 |
N/A |
| FW GCATTTGGATGCCTTATTGC | ||
| REV GAACATGCTTCCAAAACATCTG | ||
| Recombinant DNA | ||
| pLPC-hTERT | Titia de Lange, Rockefeller University | N/A |
| PRRS GII – M.EcoGII | Yoshiharu Yamaichi, I2BC | N/A |
| pRetroX-PTuner DD-linker-M.EcoGII | This paper | N/A |
| pRetroX-PTuner DD-linker-M.EcoGII-v5-Lamin B1 | This paper | N/A |
| pRetroX-PTuner DD-M.EcoGII-v5-Telomeric repeat-binding factor 1 | This paper | N/A |
| pRetroX-PTuner DD-M.EcoGII-v5-Centromeric protein C | This paper | N/A |
| Software and Algorithms | ||
| bcl2fastq2 V2.15.0 | N/A | N/A |
| Bwa | N/A | http://bio-bwa.sourceforge.net |
| Samtools | N/A | http://www.htslib.org/ |
| Bedtools | N/A | https://bedtools.readthedocs.io/en/latest/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laure Crabbe (laure.crabbe@univ-tlse3.fr).
Experimental Model and Subject Details
Cell lines, culture and treatments
Early passage IMR90 (ATCC) were immortalized using retroviral infection of the catalytic subunit of human telomerase (hTERT) and grown in Glutamax-DMEM (GIBCO) supplemented with 15% (v/v) fetal bovine serum (GIBCO) and non-essential amino acids (GIBCO), at 7.5% CO2 and 5% O2. HeLa 1.2.11 and U2OS were grown in the same conditions except only 10% FBS was supplemented to the medium. Retroviruses were produced and cells were transduced as described (Crabbe et al., 2004). Induction of the pRetroX-PTuner expression vector was achieved by treating the cells with 1μM Shield1 (Aobious) for 24 hours or as indicated. For synchronization, cells were treated for 20 hours with 2mM Thymidine (Sigma), washed with PBS and released in fresh medium supplemented with 10 μmol 2-Deoxycytidine (Sigma).
Cloning
pLPC-hTERT was a gift from Titia de Lange (The Rockefeller University, USA). To generate the pLPC-M.EcoGII construct, M.EcoGII cDNA was amplified by PCR from the PRRS GII vector (gift from Dr. Yoshiharu Yamaichi, I2BC, France) using BamHI and EcoRI restriction sites for further ligation into pLPC vector. pRetroX-PTuner was obtained from Clontech. M.EcoGII was then transferred to the pRetroX-PTuner vector by PCR amplification using BamHI and NcoI restriction sites to generate the pRetroX-PTuner DD-linker-M.EcoGII vector. This vector was used to generate the following constructs: pRetroX-PTuner DD-linker-M.EcoGII-v5-Lamin B1 (M-Lamin B1); pRetroX-PTuner DD-M.EcoGII-v5-Telomeric repeat-binding factor 1 (M-TRF1); pRetroX-PTuner DD-M.EcoGII-v5-Centromeric protein C (M-CENP-C). Lamin B1 cDNA was amplified from the pDEST-Lamin B1 vector, a gift from Martin Hetzer (The Salk Institute for Biological Studies, USA) and cDNA from CENP-C was generated from RNA extracted from human lung fibroblasts (IMR90). These sequences were inserted into the pRetroXpTuner plasmid using Gibson Assembly® (NEB). Cloning primers are described in the Key Resources Table.
Method Details
Genomic DNA and telomere purification
Genomic DNA was isolated using the QIAGEN Blood & Cell Culture DNA Midi Kit (Genomic-tip 100/G) according to the manufacturer’s recommendations together with RNase treatment (200 μg/ml of RNaseA (Sigma) and RNase Cocktail Enzyme Mix (Ambion) (2.5U/ml RnaseA; 100U/ml RnaseT1) at 37°C for 1 hour). Telomere isolation was based on a published method with some modification (Parikh et al., 2015). Double stranded genomic DNA (50 μg) was digested overnight with AluI, HinfI, HphI and MnlI (0.5Umg-1) restriction enzymes in 300 mL reaction volume to release intact telomeric fragments. Reactions were adjusted to 1x SCC and 0.1% Triton X-100, and the digested DNA was then annealed with a biotinylated oligonucleotide (Bio-5′-ACTCC(CCCTAA)3-3′) (3.5 pmol) by controlled stepwise cooling from 80°C to 25°C (1°Cmin-1) using a thermocycler. Then 3 to 10% of samples were collected as an input and streptavidin-coated magnetic beads (18 μl, Invitrogen, M-280) prewashed with 1X PBST and blocked for 1 hour with 5X Denhardt solution (0.1% Ficoll (type 400), 0.1% polyvinylpyrrolidone, and 0.1% bovine serum albumin), were incubated with the annealed samples overnight in a rotator end-over-end at 6 rpm and 4°C. Beads were collected against the side of the tubes by applying a magnet (Invitrogen), and the unbound fraction was collected. The beads were washed four times with 1X sodium chloride–sodium citrate (SSC), 0.1% Triton X-100, and once with 0.2X SSC. Beads were resuspended in 50 μl elution buffer and telomeres were slowly eluted by heating the tubes at 50°C for 20 min. The elution was repeated with 50 μl of elution buffer. To assess telomere capture efficiency, the amount of recovered telomeric DNA was measured using Qubit fluorometric quantitation. An average of 20ng was recovered from 100ug of HeLa 1.2.11 genomic DNA. To control for the purity of telomere capture, these 20ng were blotted next to 20ng of telomeric DNA purified from pSP73.Sty11 plasmid, a gift from Titia de Lange (The Rockefeller University, USA), carrying 800bp of TTAGGG repeats, which gave a similar signal after hybridization with a radioactive probe.
Immunodot blot detection of m6A
Immunodot blots of purified genomic, telomeric and plasmid DNA were performed using the BioRad 96-well Bio-Dot® apparatus. Positively charged Amersham Hybond-N+ membranes (GE Healthcare) and Whatman filter papers (GE Healthcare) preincubated with 2 × SSC buffer were assembled onto the apparatus. Heat-denatured (98 °C, 10 min; on ice, 5min) DNA samples were loaded on the membrane via vacuum blotting, then the wells were washed with 2xSSC. The membrane was denaturated and neutralized sequentially by placing it on top of a Whatmann filter paper (DNA face up) saturated with denaturing solution (1.5M NaCl, 0.5M NaOH) for 10 min at RT and neutralization solution (0.5M Tris-HCl, pH 7.0, 3M NaCl) for 10 min at RT. The membrane was crosslinked with UV at 70000 μJ/cm2 and blocked for 1hr in 5% nonfat dry milk and 0.1% TBST (0.1% Tween-20 in 1xTBS, pH7.4). Subsequently, m6A antibody (Synaptic Systems) was diluted to 1:1000 in 5% nonfat dry milk and 0.1% TBST, and incubated overnight at 4°C. Following 3 washes with 0.1% TBST, a HRP-conjugated secondary antibody was applied for 45min at room temperature. After further 3 washes with 0.1% TBST, the chemiluminescence signal was visualized and quantified using ChemiDoc Imaging System (BioRad).
Detection of telomeric repeat DNA
Telomeric probes 5′-(TTAGGG)4-3′ were radiolabeled by incubating 100 pmoles of each with 50 μCi of 32P-ATP and 50 units of T4 Polynucleotide Kinase (Thermo Fisher Scientific) in 50 μl of 1 × PNK buffer A for 1 h at 37 °C. The reactions were heat inactivated for 10 min at 75 °C and purified using the Micro Bio-Spin P-30 (Bio-Rad). The membranes were incubated for 30 min at 65 °C in Church Mix hybridization buffer (500mM NaPi pH 7.2, 1mM EDTA pH 8.0, 7% SDS, 1% BSA), and then overnight at 65°C with 10 ml of hybridization buffer containing the radiolabeled probe. The membranes were washed 4 times in 2X SSC at 65°C before exposure to a PhosphorImager screen and quantification with ImageJ software.
M.EcoGII purification
M.EcoGII was cloned into pET-28b for His-tagged purification. Bacteria cells transformed with this vector were grown overnight. The preculture was diluted 1/100 in fresh medium and kept until the culture reached OD 0.5. Expression was induced using IPTG 100mM for 3 hours at 30°C. Cells were lysed before purification of His-M.EcoGII using Ni-NTA beads (Invitrogen).
M.EcoGII in-vitro dA methylation
Plasmid or genomic DNA was methylated by incubating 1 μg of DNA with 1μg of home-made or 5U of commercially available M.EcoGII (NEB) in 50 μl of 1 x dam Methyltransferase Reaction Buffer (50 mM Tris-HCl pH 7.5, 5 mM β-ME, 10 mM EDTA) supplemented with 80 μM S-adenosylmethionine (SAM) for 2 h at 37 °C.
Western Blots
Carried out as described in (O’Sullivan et al., 2010). Membranes were overlaid with western blotting substrate for 5 minutes (Clarity, BioRad) before visualization with a ChemiDoc Imaging System (BioRad).
Immunofluorescence, Telomeric and Centromeric FISH
Cells were fixed using 4% PFA for 10 minutes followed by permeabilization using PBS complemented with 0.5% Triton. For m6A-IF detection, samples were first treated with 200 μg/ml of RNaseA (Sigma) and RNase Cocktail Enzyme Mix (Ambion) (2.5U/ml RnaseA; 100U/ml RnaseT1) at 37°C for 1 hour. DNA was then denatured (1.5M NaCl, 0.5M NaOH) for 30 min at RT and neutralized (0.5M Tris-HCl, pH 7.0, 3M NaCl) for 2x5 min at RT. Samples were then washed three times with PBS for 5 minutes at RT before blocking and antibodies incubation. When combined with FISH, cells were fixed again in 4%PFA for 10 minutes, then rinsed with water before ethanol dehydration series (70%, 90%, 100% for 2 minutes). The coverslips were air-dried and the PNA probe was added in hybridization mix (10mM Tris pH7.2, 70% deionized Formamide, 0.5% blocking solution (prepared with blocking reagent from Roche)) at a concentration recommended by the manufacturer. Coverslips were denatured on an 80°C hot plate for 3 minutes before overnight incubation in a humidified chamber. Coverslips were then washed 2 times 15 minutes in 0.1mM Tris pH7.2, 0.1% BSA, 70% Formamide, followed by 3 washes of 5 minutes in 0.1M Tris, 0.15M NaCl, 0.08% Tween. The coverslips were then mounted on microscope slides using mounting medium. Images were acquired on a Leica SP8 laser-scanning confocal microscope driven by LAS X software. Images were captured in the confocal mode with the 63x objective (Leica) and analyzed using ImageJ software. Colocalization analysis was performed using JACoP plugin (Bolte and Cordelières, 2006).
Liquid chromatography coupled to high resolution mass spectrometry (LC-HRMS) analysis
Telomeric DNA was purified from pSP73.Sty11 plasmid, a gift from Titia de Lange (The Rockefeller University, USA), carrying 800bp of TTAGGG repeats, using EcoRI digestion. DNA was hydrolyzed using benzonase, phosphodiesterase I, and alkaline phosphatase for six hours at 37°C to release nucleosides as described previously (Quinlivan and Gregory, 2008). Analysis of nucleoside by narrow bore HPLC was done using a U-3000 HPLC system (Thermo-Fisher). An Accucore RP-MS (2.1 mm X 100 mm, 2.6 μm particle) column (Thermo-Fisher) was used at a flow rate of 200 μl/mn and a temperature controlled 30°C. Mobile phases used were 5 mM ammonium acetate, pH 5.3 (buffer A) and 40% aqueous acetonitrile (Buffer B). A multilinear gradient was used with only minor modification from that described previously (Pomerantz and McCloskey, 1990). A LTQ orbitrap mass spectrometer (Thermo-Fisher) equipped with an electrospray ion source was used for the LC/MS identification of nucleosides. Mass spectra were recorded in the positive ion mode over an m/z range of 100-1000 with a capillary temperature of 300°C, spray voltage of 4.5 kV and sheath gas, auxiliary gas and sweep gas of 40, 12 and 7 arbitrary units, respectively.
m6A-seq
Double stranded genomic DNA (10 μg) was sheared into fragments of 200-400bp using Bioruptor Plus sonicator (Diagenode) – 300 μL of 33,3ng/μl in 1.5ml TPX tube; 40 cycles 30sec/60sec ON/OFF; low power. The temperature was kept at 4°C (using the Diagenode Water cooler, Cat. No. BioAcc-Cool) for optimal shearing results. 1% of sample was taken as an input. Fragmented DNA was end-repaired and A-tailed according to manufacturer recommendations (NEBNext End-repair module (NEB #E6050), NEBNext A-tailing module (NEB #E6053)). Double Stranded TruSeq Illumina adapters were ligated (NEBNext Ligation Module (NEB #E6056). Then, sample were diluted in TE buffer up to 360 μl, denatured for 10 minutes at 95°C and snap cooled on ice for 10 minutes. Samples were supplemented with 10x m6AIP buffer (100mM Na-Phosphate buffer, pH 7.0; 3M NaCl; 0.5% Triton X-100), 2.5 μg m6A antibody (SYSY) and rotated overnight at 4°C. Next 20 uL of protein A/G Dynabeads mix (Thermo Fisher Scientific), pre-blocked for 1 hour (PBS-0.5%BSA-0.1%Tween-20), was added and samples rotated at 4°C for 3 hours. Beads were washed 4 times in 1 mL 1X m6AIP buffer. Beads and input samples were resuspended in 150 μl digestion buffer (50mM Tris, pH 8.0; 10mM EDTA; 0.5% SDS) containing 300 μg/ml proteinase K and incubated for 3 hours at 50°C with shaking. DNA was purified by phenol/chloroform/isoamyl alcohol extraction and resuspended in 21 μl milliQ water. IP efficiency was checked by qPCR using 1μL of IP and INPUT sample and primers listed in the Key Resources Table. DNA recovered from Immunoprecipitation was amplified (10 cycles) using KAPA Hifi DNA polymerase (Kapa Biosystems) using P5 and P7 Illumina primers, and purified with AMPureXB beads (Beckmann Coulter). Libraries were pooled and sequenced in a 2x43pb sequencing run on an Illumina NextSeq500 instrument, using NextSeq 500/550 High Output Kit v2 Kit (75 cycles) according to the manufacturer recommendations. Demultiplexing was performed (bcl2fastq2 V2.15.0) and adapters removed (Cutadapt1.9.1).
Quantification and Statistical Analysis
Processing of m6A-seq
All sequencing reads were aligned using BWA to the human genome (hg38 assembly). Multiple mapped reads and low quality reads (q < 25) were removed using SAMtools. PCR duplicates were removed and reads were binned in 100kb bins using BEDtools. Reads counts in 100kb bins from M-LB1 samples were first normalized by total number of reads and to the input sequencing and represented as a ratio to the control sample (M.EcoGII) that was processed in the same way. Values higher than 1 were consider as specific. Domain calling was done by merging neighboring positive bins (ratios > 1) using bedtools merge function, and averaging their values.
Processing of conventional DamID from HT1080 (Kind et al., 2013)
First, the data representing the scores calculated as the log2 Dam-Lamin B1/Dam ratio obtained from GEO as two replicates: dataset GSM990672 and GSM990672 were averaged. In order to obtain the same resolution as the MadID data, the data was binned into 100kb segments by averaging of all array probes within each segment.
Processing of single-cell DamID sequencing reads from KBM7 (Kind et al., 2015)
The 51 bp reads from 124 single cell Lamin B1 DamID samples obtained from GEO (GSE68263) were trimmed (fastx-trimmer) to remove the first 19 bp containing the Illumina adaptor sequence. Next, reads were filtered to keep reads only starting with GATC. Sequencing reads were aligned using BWA to the human genome (hg38 assembly). Multiple mapped reads, low quality reads (q < 25) were removed using SAMtools. PCR duplicates were removed and reads were binned in 100kb bin using BEDtools. Next, for each 100 kb segment the observed over expected read count (OE) was calculated as was described in Kind et al. (2015). OE higher that 1 were consider as a specific to the Dam-Lamin B1.
Comparison of MadID to conventional and single cell DamID-seq
For MadID-seq comparison to conventional DamID values from 100kb bins or 5kb bins were calculated as log2 and filtered for bins containing probes from array. Comparison of MadID to the conventional DamID and single cell DamID-seq was performed using R and Spearman correlation.
Intrinsic smoothness test
To assess the resolution of sequencing data obtained with DamID and MadID, the values of each bin were compared to the two neighboring bins. For each bin, the following formula was applied, considering that b is the value of the bin, and a and c are the values of the neighbors:
When a, b and c are similar, which is the case if the bin can predict the neighboring values, X value is close to 0. The mean of all values (M) obtained at different binning of the sequencing data (100, 50, 25, 10, 5, 2 and 1kb) was then calculated and plotted as Y = 1/(1+M), representing the intrinsic smoothness of MadID versus DamID. If the data have a high resolution, M is near to 0, and Y is near to 1.
Bias-plot
The number of GATC motifs and A/T nucleotides was calculated for each bin and plotted against the number of reads obtained with these bins. Spearman correlation was performed on these values.
Identification of telomeric reads in m6A-seq
The raw sequencing reads from fastq files were screened for motif composed of (TTAGGG)3. If this motif was found several times in one read, it was counted as one. Number of identified reads was normalized by total number of reads in the sequencing run.
Data and Software Availability
The accession number for the high-throughput sequencing data reported in this paper is ArrayExpress: E-MTAB-6888.
DOI for Mendeley dataset: https://doi.org/10.17632/8j9kmzm4bc.1
Acknowledgments
We thank Dr. Yoshiharu Yamaichi (I2BC, France) for the PRRS-M.EcoGII and the pET-28b vectors and for the BL-21 bacteria strain. We thank all members of the Crabbe laboratory for discussion. We thank Gregory Vert for critical reading of this manuscript. Illustration sources in Figure 1B are from somersault1824 (Creative Commons license CC BY-NC-SA 4.0). This work has benefited from the facilities and expertise of the high-throughput sequencing core facility of I2BC and from the cytometry and imaging facilities of Imagerie-Gif, member of IBiSA, supported by “France-BioImaging” (ANR-10-INBS-04-01) and the Labex “Saclay Plant Science” (ANR-11-IDEX-0003-02). This work was supported by an ATIP starting grant from CNRS in the framework of Plan Cancer 2014–2019 (to L.C.), the ANR Tremplin ERC teloHOOK (ANR-16-TERC-0028-01 to L.C.), and a European Research Council teloHOOK/ERC grant (714653 to L.C.). C.S. and D.N. acknowledge funding by the Agence Nationale de la Recherche (grant ANR-14-ACHN-0009-01).
Author Contributions
M.S. developed MadID technology, performed all experiments except the LC-HRMS, performed data analysis, and created the figures; J.B. cloned M.EcoGII and generated M-LB1 fusion protein; V.G. performed the LC-HRMS; M.S., C.S., and D.N. performed data analysis of whole-genome sequencing; L.C. and M.S. conceived the study and designed the experiments; and L.C. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: December 4, 2018
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.027.
Supplemental Information
References
- Arnoult N., Schluth-Bolard C., Letessier A., Drascovic I., Bouarich-Bourimi R., Campisi J., Kim S.-H., Boussouar A., Ottaviani A., Magdinier F. Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet. 2010;6:e1000920. doi: 10.1371/journal.pgen.1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aughey G.N., Southall T.D. Dam it’s good! DamID profiling of protein-DNA interactions. Wiley Interdiscip. Rev. Dev. Biol. 2016;5:25–37. doi: 10.1002/wdev.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aughey G.N., Estacio Gomez A., Thomson J., Yin H., Southall T.D. CATaDa reveals global remodelling of chromatin accessibility during stem cell differentiation in vivo. eLife. 2018;7:6061. doi: 10.7554/eLife.32341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Crabbe L., Verdun R.E., Haggblom C.I., Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- Crabbe L., Cesare A.J., Kasuboski J.M., Fitzpatrick J.A.J., Karlseder J. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2012;2:1521–1529. doi: 10.1016/j.celrep.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Fang G., Munera D., Friedman D.I., Mandlik A., Chao M.C., Banerjee O., Feng Z., Losic B., Mahajan M.C., Jabado O.J. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012;30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavís M., Escaja N., Gabelica V., Villasante A., González C. Centromeric alpha-satellite DNA adopts dimeric i-motif structures capped by AT Hoogsteen base pairs. Chemistry. 2015;21:9816–9824. doi: 10.1002/chem.201500448. [DOI] [PubMed] [Google Scholar]
- Garrobo I., Marión R.M., Domínguez O., Pisano D.G., Blasco M.A. Genome-wide analysis of in vivo TRF1 binding to chromatin restricts its location exclusively to telomeric repeats. Cell Cycle. 2014;13:3742–3749. doi: 10.4161/15384101.2014.965044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady D.L., Ratliff R.L., Robinson D.L., McCanlies E.C., Meyne J., Moyzis R.K. Highly conserved repetitive DNA sequences are present at human centromeres. Proc. Natl. Acad. Sci. USA. 1992;89:1695–1699. doi: 10.1073/pnas.89.5.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Kind J., van Steensel B. Stochastic genome-nuclear lamina interactions: modulating roles of Lamin A and BAF. Nucleus. 2014;5:124–130. doi: 10.4161/nucl.28825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 2013;153:178–192. doi: 10.1016/j.cell.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Kind J., Pagie L., de Vries S.S., Nahidiazar L., Dey S.S., Bienko M., Zhan Y., Lajoie B., de Graaf C.A., Amendola M. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–147. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol M.J., Bradshaw C.R., Allen G.E., Costa A.S.H., Frezza C., Gurdon J.B. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 2016;23:24–30. doi: 10.1038/nsmb.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray I.A., Morgan R.D., Luyten Y., Fomenkov A., Corrêa I.R., Jr., Dai N., Allaw M.B., Zhang X., Cheng X., Roberts R.J. The non-specific adenine DNA methyltransferase M.EcoGII. Nucleic Acids Res. 2018;46:840–848. doi: 10.1093/nar/gkx1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naetar N., Ferraioli S., Foisner R. Lamins in the nuclear interior—life outside the lamina. J. Cell Sci. 2017;130:2087–2096. doi: 10.1242/jcs.203430. [DOI] [PubMed] [Google Scholar]
- O’Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh D., Fouquerel E., Murphy C.T., Wang H., Opresko P.L. Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat. Commun. 2015;6:8214. doi: 10.1038/ncomms9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz S.C., McCloskey J.A. Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods Enzymol. 1990;193:796–824. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- Quinlivan E.P., Gregory J.F., 3rd DNA digestion to deoxyribonucleoside: a simplified one-step procedure. Anal. Biochem. 2008;373:383–385. doi: 10.1016/j.ab.2007.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera V.M., Clackson T., Natesan S., Pollock R., Amara J.F., Keenan T., Magari S.R., Phillips T., Courage N.L., Cerasoli F., Jr. A humanized system for pharmacologic control of gene expression. Nat. Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- Sahlén P., Abdullayev I., Ramsköld D., Matskova L., Rilakovic N., Lötstedt B., Albert T.J., Lundeberg J., Sandberg R. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol. 2015;16:156. doi: 10.1186/s13059-015-0727-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha K., Gu S.G., Pantalena-Filho L.C., Goh A., Fleenor J., Blanchard D., Krishna C., Fire A. Distributed probing of chromatin structure in vivo reveals pervasive chromatin accessibility for expressed and non-expressed genes during tissue differentiation in C. elegans. BMC Genomics. 2010;11:465. doi: 10.1186/1471-2164-11-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel M.J., Peric-Hupkes D., van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007;2:1467–1478. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- Williams D.J., Puhl H.L., 3rd, Ikeda S.R. Rapid modification of proteins using a rapamycin-inducible tobacco etch virus protease system. PLoS ONE. 2009;4:e7474. doi: 10.1371/journal.pone.0007474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.P., Wang T., Seetin M.G., Lai Y., Zhu S., Lin K., Liu Y., Byrum S.D., Mackintosh S.G., Zhong M. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R., Moore D.D. John Wiley & Sons; 2001. DamIP: Using Mutant DNA Adenine Methyltransferase to Study DNA-Protein Interactions In Vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R., Roman-Sanchez R., Moore D.D. DamIP: a novel method to identify DNA binding sites in vivo. Nucl. Recept. Signal. 2010;8:e003. doi: 10.1621/nrs.08003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.