

Abstract
Photoinduced controlled radical polymerizations (CRPs) have provided a variety of approaches for the synthesis of polymers possessing targeted structures, compositions, and functionalities with the added capability for spatial and temporal control, presenting the potential for new materials development. However, the scalability and reliability of these systems can be limited as a consequence of dependence on uniform irradiation of the reaction to produce well-defined products. In this perspective, we highlight the utility and promise of photo-CRP approaches through an overview of the adaptation of these methodologies to photo-flow reactor systems. Special emphasis is placed on the current state-of-the-art in polymerization scalability, reactor design, and polymer scope.
Graphical Abstract
1. INTRODUCTION
Polymeric materials have shaped the modern world, becoming integrated into all facets of society. While commodity plastics are undoubtedly important, polymeric materials have also emerged to address challenges in biomedical, electronic, and energy-storage fields.1 The discovery of living anionic polymerization by Szwarc in 1956 enabled precise control over polymer properties, stemming from well-defined polymer compositions, architectures, control over molecular weight growth, and narrow molecular weight distributions.2,3 Other polymerization methodologies with these capabilities have since been developed and include living or controlled coordinative chain-transfer,4 cationic,5 ring-opening,6 ring-opening metathesis,7 group-transfer,8 and controlled radical polymerizations (CRPs).9
Despite the inherent presence of termination events, CRPs have been shown to be effective in their ability to impart a high level of control over desirable polymer characteristics while providing access to a wide scope of materials.10,11 Radical polymerization approaches, including CRPs and “uncontrolled” free radical polymerizations, benefit from functional group tolerance, wide thermal stability, compatibility with a variety of reaction conditions, and simplicity of operation. As such, free radical polymerizations have been broadly implemented industrially,12 but due to pervasiveness of radical termination events, the synthesis of polymers possessing well-defined or complex compositions and architectures is challenging. In CRPs, control over polymer composition and architecture gives rise to unique properties which can be precisely modulated to meet desired applications (Figure 1).13,14 For example, an important asset of CRP systems is the presence of a functional chain-end group, determined by the structure of the initiating species, allowing for facile post-polymerization transformations to install additional functionality as well as sequential additions of monomer to produce block copolymers with distinct chemical domains.
Figure 1.

Overview of the capabilities of controlled radical polymerizations, highlighting their use in diverse applications such as bioconjugates,15 drug delivery,16 photovoltaics,17 batteries,18 coatings,19 and photonics.20
All CRP techniques rely on the formation of a dynamic equilibrium between dormant and active propagating states of the growing polymer chain. Control over radical reactivity is predominantly established either through a “degenerative chain transfer” process or through a “reversible deactivation” mechanism (Figure 2). Degenerative chain-transfer CRPs rely on a fast and selective exchange equilibrium between active radicals and dormant chain-transfer agents, maintaining constant radical concentrations.21 Reversible addition–fragmentation chain transfer (RAFT) is a widely studied degenerative chain-transfer approach.22
Figure 2.

General reaction schemes for CRPs following degenerative chain-transfer (A) and reversible deactivation (B) mechanisms.
In reversible deactivation CRPs, radical concentrations remain low through the reversible activation of the propagating radical species, followed by reformation of a dormant polymer chain through the deactivation by either a reversibly coupling species or through an atom-transfer mechanism (Figure 2B). Traditionally, the dormant polymer species is thermally activated, but can also be activated through the presence of external stimuli, including photo-, electro-, and mechano-approaches. Atom-transfer radical polymerization (ATRP),23 metal-catalyzed living radical polymerization,10 nitroxide-mediated polymerization (NMP),14 and cobalt-mediated radical polymerization (CMRP)24 are the most common examples of reversible deactivation approaches. In both classifications, a controlled polymerization consists of a linear increase of molecular weight (MW) with respect to monomer conversion, dispersity (D̵) less than 1.5, and initiator efficiencies (I*) ideally close to 100%. I* is an important parameter, which compares the theoretical number-average molecular weight (Mn) to the experimentally measured Mn, evidencing the control over the polymerization.
The initial development of CRPs relied on thermally activated catalysts or initiators. In recent years, several photoinduced CRP (photo-CRP) variants have been developed, introducing the added potential for spatial and temporal control over polymerization.25–27 The earliest established methodologies used direct photolysis of an initiating species to begin monomer propagation, for example in NMP,28 organotellurium-mediated radical polymerization,29 RAFT,30 photo-iniferter polymerization,25 and CMRP,31 the latter three of which are discussed in greater detail later in this perspective. Photoredox catalysis,32–35 which has been broadly applied in small molecule transformations, has expanded the capabilities of CRP systems. This focus uses photoredox catalysts (PCs) to mediate a photoredox catalytic cycle, minimizing photo-degradation of dormant polymer species and chain-end groups while introducing an additional avenue to tailored control over polymer properties and functions.
Due to facile reaction conditions and components the development of photo-CRPs has typically been carried out in batch-style reactors with small reaction volumes. In batch reactors, large reaction volumes are difficult to uniformly irradiate, and therefore scalability of these reactions while maintaining a high level of control over polymerization can be difficult and unreliable. For photo-CRP systems, application of these rapidly evolving approaches to the industrially relevant production of materials with tailored properties and applications requires reliable reaction scalability.
Flow reactor systems have been shown to be powerful alternatives to batch reactors in small molecule and polymer synthesis, with original adaptation performed for thermally mediated systems and expanded to include photochemical approaches.36,37 This actualization can be widely accredited to improved scalability,38,39 control over process parameters,40 enhanced reliability,41 and decreased reaction times.42 Furthermore, flow chemistry is viewed as a sustainable approach toward chemical production and processing, offering potential for increased reaction efficiency and limitations in solvent usage and byproduct formations.43 These benefits, while initially applied toward small molecule transformations, have also translated to a large range of thermally mediated polymerization approaches.44–47 While photo-CRP techniques have seen rapid expansion in reaction development and specialized materials design, to fully utilize the capabilities of CRPs expansion into reactor systems capable of refining reaction control and improving product scalability needs to be realized.
In this Perspective, an overview of the development of photo-CRP processes in flow reactor systems is presented, beginning with a description of flow reactor systems and experimental design, and also highlighting useful information for adapting photo-CRPs to flow from batch polymerizations. A thorough description of the current scope of photo-CRP reactions produced in continuous flow is presented, classified by either degenerative chain-transfer or reversible deactivation mechanisms for comparison within similar polymerization approaches. Special emphasis is placed on the effect of polymerization mechanism on reactor design, the current scope of polymer products reported, and future directions to expand the growing field of photo-CRPs.
2. ADVANTAGES AND MOTIVATIONS FOR FLOW REACTORS SYSTEMS
2.1. Key Characteristics of Photochemical Flow Reactors
One of the key advantages of flow reactor systems for photochemically mediated processes stems from the ability to improve the irradiation of the reaction mixture. In a typical batch reactor system, irradiation is attenuated toward the center of the reactor with an increasing path length, as is supported by the Beer–Lambert law,
| (1) |
where ε is the molar absorptivity of the photoactive species, c is the concentration of the photoactive species, and l is the optical path length of the system, or in this case the diameter of the photoreactor. In cases where photochemistry is applied for the synthesis of small molecules, non-uniform irradiation may result in slower reaction rates, non-productive side reactions, and subsequently lower reaction yields. For photo-CRPs, attenuating irradiation in solution causes non-concurrent initiator activation, a subsequent non-uniform formation of polymer chains, and the potential for loss in control over polymer MW and D̵, as well as increased reaction times. In cases where a strong absorber with a high ε is used to mediate polymerization, the effect of reactor diameter can be more dramatic. For example, the light transmission through a solution of the common photoredox catalyst Ru(bpy)3Cl2, (ε = 13 000 cm−1 M−1) was found to decrease by over 50% with an increase in reactor diameter from 0.5 mm to 1.0 mm, which was mitigated by decreasing catalyst concentrations to improve irradiation of the mixture.48 Uniform irradiation, as provided by narrow diameter and high surface-area-to-volume flow reactors, can lessen catalyst and photo-initiator absorption concerns, while providing an avenue for increased scalability, as dictated by the amount of reaction mixture at the reactor source (Figure 3).
Figure 3.

Schematic representations and comparisons of batch and flow reactor systems in photochemically induced polymerizations.
Other important advantages in flow reactor systems stem from the high surface area-to-volume ratio of the reactor itself, facilitating fast reaction times, efficient heat and mass transfer, reduction of batch-to-batch variations, improved safety, and fast mixing.49 As propagation in CRP reactions is thermally controlled while the reactions themselves are exothermic, the isothermal nature of flow systems inherently leads to improved reaction consistency. Furthermore, the mixing behavior in flow systems is typically uniform in comparison to the convective mixing of batch reactors, which is largely driven by mechanical agitation and results in heterogeneous solution movement and nonuniform reaction component distributions.50,51 The characteristics of mixing in a flow reactor are directly dependent on the diameter of the reactor and can be described by a calculation of Reynold’s number, shown by eq 2.
| (2) |
and which relates the flow rate (u) and reactor diameter (L) to the kinematic viscosity (v) of the solution. When Re is less than 1, a laminar flow regime is accessed and viscous forces dominate over inertial forces, making solution mixing dependent on diffusion and preventing the formation of concentration gradients within the reactor, which can result in accessing ideal plug-flow behavior at small reactor diameters. In the context of CRPs, which are reliant on uniform activation to produce well-defined polymers, homogeneous mixing and distribution of the reaction components is important.
Perhaps one drawback to an increased surface-area-to-volume ratio of the flow reactor is the increased presence of wall shear, causing non-uniform polymer chain elongation along the reactor wall (Figure 4). In study of this effect using thermally mediated ATRP, shear rate was found to increase with increasing reactor lengths at constant residence times and increasing residence times using constant reactor lengths.52 Increased polymer elongations results in wider residence time distributions,53 and in CRPs could result in a rise in an increase in D̵ and loss of control over MW if not mitigated.
Figure 4.

Graphical representation of laminar flow regime in flow reactors (A) and the effect of wall shear on polymerizations (B).
The effect of shear on residence time distributions highlights the requirement for additional reactor and reaction considerations when performing a polymerization in a flow reactor system. Besides consideration of reaction stoichiometry, reaction times, and polymer scope, as is typical of any batch reactor system, the added flow reactor component requires additional attention to the effects of solution viscosity, flow regimes, and mixing behavior on polymerization. As the polymer chain length increases during the course of polymerization, all the above parameters will change, introducing additional complexity into comprehensive reaction engineering using photo-CRPs in flow.
2.2. Reactor Design and Development
In practice, there are nearly infinite variations of flow reactors employed for small molecule and macromolecular synthesis in both academic research laboratories and on the industrial scale; however, in the studies discussed in this perspective, there are two principal types of photo-flow reactors employed: glass chip microflow reactors and continuous-flow tubular millireactors.54 The use of glass as a reactor material can be advantageous in terms of low oxygen permeability and high chemical compatibility, but also does not readily allow for customization. The second class of reactors, continuous flow tubular millireactors, also typically with larger diameters and volumes, facilitate reaction scalability while also giving potential for placement of inlets and outlets at any point in the reactor.55 The wide possibilities for reactor configurations gives additional avenues for producing tailored materials using this approach. Commercially available tubing materials, typically made from inert perfluorinated polymeric materials, exhibit an expansive range of optical characteristics, chemical compatibility, and oxygen permeability and can be selected based on the polymerization conditions employed.
Furthermore, straightforward reactor configuration design can be exploited to synthesize complex polymer architectures, including block copolymers. For example, in this Perspective we highlight two principal methods for block copolymer synthesis. These include chain-extension, where an isolated polymer is reintroduced to the flow reactor under polymerization conditions to grow a new polymer domain. Also, use of multi-step continuous reactors has been reported, which perform photo-CRP to high monomer conversions for formation of the first polymer block, followed by addition of a second monomer to form block copolymers with a slight gradient. An additional avenue toward customized reactor design lies in the choice of light source. Diverse light sources have been employed in photo-CRPs, with overlap of light source emission and photoactive species absorption being a key consideration. As such, design of current flow reactor systems primarily encompasses use of commercially available LEDs, fluorescent lamps, or mercury lamps that emit wavelengths of light across the UV and visible regimes.
In a typical photo-CRP performed in flow reactors, a reaction mixture is pushed through a reactor using either a syringe or peristaltic pump. The reaction mixture can either be from one source, or multiple inlets can simultaneously introduce different components into the reactor through a mixing connection. After reaction components are introduced, the mixture is pushed through the reactor and irradiated. Ultimately the reaction is quenched, and the resulting polymer product is collected at the end of the reactor. The reaction time is solution is irradiated. Residence time is calculated as
| (3) |
and can be readily modulated through changes in flow rate, resulting in polymers with different monomer conversions and MWs.
3. DEGENERATIVE CHAIN-TRANSFER-CONTROLLED RADICAL POLYMERIZATIONS IN FLOW
3.1. Degenerative Chain Transfer Controlled through Direct Photolysis
Photo-iniferter controlled radical polymerization (PI-CRP), relies on direct photolysis of a chain-transfer agent to mediate polymerization. These chain transfer agents, such as commonly employed sulfur compounds, can serve as initiator-transfer-agent-terminators, or “iniferters”, and can also be referred to as RAFT agents. In PI-CRP, iniferters are directly activated by irradiation, causing homolytic cleavage of the carbon–sulfur bond and beginning propagation (Figure 5A). Photo-RAFT follows a similar polymerization propagation mechanism but relies on activation by an exogenous photo-initiator to generate the active species (Figure 5B). In either case, the degenerative chain-transfer is dominated by a fast exchange equilibrium between dormant iniferter and growing polymer chain, resulting in control over polymerization. For photocontrol, the active iniferter radical can combine with a propagating polymer chain to produce a dormant chain, which is then reactivated photochemically. Use of photo-RAFT can be advantageous in that it minimizes iniferter decomposition due to high energy irradiation typically required for PI-CRP. However, recent structural modifications of iniferters have extended the absorption profiles of these molecules for PI-CRP under visible light.56 Advantageously, in PI-CRP the lack of initiator leads to lower termination events by unreacted or excess initiator fragments, which can result in products with higher chain-end fidelity and concomitantly lower dispersities.
Figure 5.

General mechanisms for photo-iniferter controlled radical polymerization (A) and photo-RAFT (B).
PI-CRP was first introduced in 1982 using batch conditions for the synthesis of poly(methyl methacrylate) (pMMA) and poly(styrene) using UV light irradiation.57 This technique has been further expanded to include thiocarbonylthio, trithiocarbonates, and disulfides as iniferters capable of mediating PI-CRP.58,25,59 Depending on the structure and relative radical stability of the iniferter, a wide range of monomers with diverse functional groups can be polymerized, including methacrylates, acrylates, acrylamides, and acetates. PI-CRP has been expanded to include the synthesis of telechelic polymers,60 hyper-branched polymers,61 multi-block copolymers,62 ultra-high-MW polymers,63 and brush polymers.64
The first report of a PI-CRP adapted to a flow reactor was demonstrated by Johnson and co-workers in 2015.65 Using a trithiocarbonate iniferter, a successful continuous flow polymerization of acrylamide and acrylate monomers with dispersities ranging from 1.11 to 1.22 was achieved. Previous mechanistic analysis of PI-CRPs in batch systems suggested decreased light intensity may facilitate control over polymerization due to the effect of irradiation on increased bimolecular termination events.66 In that regard, a comparison between reactors with disparate irradiation profiles was performed, finding a reactor with the UV light source placed at 3 cm from the reactor resulted in D̵ lowered to 1.10 from 1.23, produced using a reactor with tubing wrapped directly around the UV lamp (Table 1, entry 1). Poly(dimethylacrylamide) (pDMA) with a range of targeted MWs was synthesized, with MWs as high as Mn = 105.8 kDa and D̵ = 1.22 (Table 1, entry 2). The synthesis of pDMA was scaled to over 2.95 g of isolated material, while also reducing reaction times from 240 min in batch to 60 min in flow for comparable monomer conversions. Block copolymers were successfully synthesized from chain-extension of isolated pDMA synthesized in flow with ethylene glycol methyl ether acrylate (EGMEA), yielding pDMA-b-pEGMEA-b-pDMA with Mn = 34.9 kDa and D̵ = 1.17 (Table 2, entry 1).
Table 1.
Summary of Results of Polymerization Using Degenerative Chain Transfer Approaches Performed in Photo-flow Reactors
| entry | method | irradiation (nm) | monomera | tRb (min) | Mn(theor)c (kDa) | Mn (kDa) | D̵d | ref |
|---|---|---|---|---|---|---|---|---|
| 1 | PI-CRP | 352 | DMA | 40 | 22.0 | 24.9 | 1.11 | 65 |
| 2 | PI-CRP | 352 | DMA | 40 | 109.4 | 105.8 | 1.22 | 65 |
| 3 | PI-CRP | 450 | MMA | 60 | 2.5 | 2.8 | 1.25 | 67 |
| 4e | photo-RAFT | 330–380 | DMA | 30 | 7.4 | 13.9 | 1.30 | 68 |
| 5f | photo-RAFT | 365 | n-BA | 20 | 2.7 | 3.1 | 1.18 | 69 |
| 6 | PI-CRP | 365 | n-BA | 20 | 7.2 | 6.8 | 1.12 | 69 |
| 7 | PET-RAFT | 515–525 | DEA | 60 | 18.7 | 14.2 | 1.06 | 81 |
| 8 | PET-RAFT | 515–525 | DMA | 10.2 | 27.7 | 28.0 | 1.14 | 82 |
| 9 | PET-RAFT | white LEDg | HFBA | 30 | 4.7 | 4.7 | 1.06 | 86 |
DMA = N,N-dimethylacrylamide, MMA = methyl methacrylate, n-BA = butyl acrylate, DEA = N,N-diethylacrylamide, HFBA = hexafluorobutyl acrylate.
Residence time = (reactor volume/flow rate).
Theoretical molecular weights are based off reaction stoichiometry provided in the main source.
D̵= Mw/Mn.
Using 2-benzyl-2-(dimethylamino)-1-[4-(4-morpholinyl)phenyl]-1-butanone as photo-initiator.
Using benzoin as photo-initiator.
White LEDs have broad emission in the visible regime.
Table 2.
Block Copolymers Synthesized in Flow Using Degenerative Chain Transfer CRPs and Reversible End-Capping CRPs
| entry | method | irradiation (nm) | method | compositiona | Mn (kDa) | D̵ | ref |
|---|---|---|---|---|---|---|---|
| 1 | PI-CRP | 352 | chain extension | pDMA-b-pEGMEA-b-pDMA | 34.9 | 1.17 | 65 |
| 2 | PI-CRP | 450 | chain extension | pMMA-b-pEMA-b-pMMA | 7.9 | 1.32 | 67 |
| 3 | PI-CRP | 450 | multi-step continuous | pMMA-b-pHEMA | 5.8 | 1.30 | 67 |
| 4 | photo-RAFT | 365 | chain extension | pBA-b-pMA | 4.0 | 1.30 | 69 |
| 5 | PET-RAFT | 515–525 | multi-step continuous | PDEA-co-PDMA | 22.5 | 1.06 | 81 |
| 6 | PET-RAFT | white | chain extension | PHFBA-b-PMA | 10.8 | 1.07 | 86 |
| 7 | Cu-MRP | 320–500 | chain extension | pMA-b-pBA | 5.0 | 1.16 | 87 |
| 8 | CMRP | 320–500 | statistical copolymers | pVA-b-pOct | 11.9 | 1.83 | 90 |
| 9 | O-ATRP | white | chain-extension | pMMA-b-pBzMA | 102.4 | 1.60 | 117 |
| 10 | O-ATRP | white | chain-extension | pMMA-b-pBA | 75.9 | 1.32 | 117 |
EGMEA = ethylene glycol methyl ether acrylate, EMA = ethyl methacrylate, HEMA = 2-hydroxyethyl methacrylate, HFPA = hexafluorobutyl methacrylate, MA = methyl acrylate, BA = butyl acrylate, VA = vinyl acetate, Oct = 1-Octene, BzMA = benzyl methacrylate. See Table 1 for other details.
Recently, visible-light-induced PI-CRP of methacrylates using a variety of trithiocarbonyl iniferters has been reported.67 Using a continuous flow reactor under oxygen-free conditions at 90 °C, fast reaction times were observed, reaching full monomer conversions at 60 min of residence time with D̵ ranging from 1.20 to 1.30 (Table 1, entry 3). After optimization of reaction conditions, MWs up to 10 kDa of pMMA were successfully synthesized in a controlled fashion. Other methacrylate-based polymers with D̵ ranging from 1.20 to 1.40 were synthesized with well-controlled MW reaching high monomer conversions after 60 min residence time. Multi-block copolymers could also be synthesized through sequential chain-extension reactions, using MMA and EMA (ethyl methacrylate) to ultimately give pMMA-b-pEMA-b-pMMA with Mn = 7.9 kDa and D̵ = 1.32 (Table 2, entry 2). Minimizing the need for tedious purification steps, a multi-step continuous process was employed for the synthesis of diblock copolymers with a small gradient between blocks to realize pMMA-b-pMMA with Mn = 5.3 kDa and D̵ = 1.26 as well as a polymer with a second poly(2-hydroxyethyl methacrylate) block to produce pMMA-b-pHEMA of Mn = 5.8 kDa and D̵ = 1.30 (Table 2, entry 3).
The effect of irradiation conditions for PI-CRP and photo-RAFT in the synthesis of poly(methacrylates) and poly-(acrylamides) was investigated by Gardiner and co-workers using a tubular continuous flow reactor through the systematic modulation of irradiation profiles and intensities while employing conventional photo-initiators, focusing on the ability to achieve decreased reaction times in flow.68 Irradiation between 310 and 380 nm resulted in conversions of 75% after 30 min residence time with D̵ = 1.30 in the polymerization of N,N-dimethylacrylamide (DMA) paired with 2-benzyl-2-(dimethylamino)-1-[4-(4-morpholinyl)phenyl]-1-butanone as photo-initiator (Table 1, entry 4), resulting in a significant decrease in reaction times from batch reactor systems paired with the added benefit of scalability. Additionally, polymerizations with no photo-initiator were performed in a PI-CRP approach, but resulted in an increase in D̵ to above 1.70. This loss of control was attributed to continuous activation of the iniferter, resulting in decomposition and partial end-group removal and further highlights the necessity to consider irradiation conditions for each approach employed in flow.
The ability to precisely control polymer structural fidelity in photo-RAFT systems was further explored in continuous flow by Junkers and co-workers using a glass chip microreactor, focusing on the interplay of stoichiometry, thermal, and irradiation reaction conditions on the performance of this photochemically mediated polymerization.69 An optimized photo-RAFT system using 1 equiv of 2-([(dodecylsulfanyl)-carbonothioyl]sulfanyl)propanoic acid chain transfer agent to 0.25 equiv of photo-initiator benzoin, 365 nm irradiation at 30 mW/cm2 and 60 °C resulted in well-defined poly(butyl acrylate) (pBA) with Mn = 3.1 kDa and D̵ = 1.18 after 20 min residence time (Table 1, entry 5). This system was further used to synthesize block copolymers using chain-extension with methyl acrylate (MA), resulting in pBA-b-pMA with Mn = 4.0 kDa and D̵ = 1.30 (Table 2, entry 4). An examination of first-order kinetic plots for reactions using several photo-initiators, as well as no initiator, revealed the interplay between the photo-RAFT and potential photo-iniferter mechanisms. The removal of the photo-initiator resulted in lower D̵ and slower reaction rates relative to photo-RAFT systems (Table 1, entry 6). Furthermore, it was demonstrated that after initiator consumption the RAFT equilibrium can still occur, concluding that a hybrid between photo-RAFT and photo-iniferter mechanisms must be present.
3.2. Degenerative Chain Transfer through Photo-redox Catalysis
Photoredox catalysis offers opportunities in CRP approaches to improve control over photoactivation through use of a photocatalyst (PC), capable of controlling the necessary activation and deactivation of active radical species for photocontrol. To that end, photoinduced electron transfer RAFT (PET-RAFT) has recently been developed as a method to increase reaction rates while achieving a level of polymerization control characteristic of thermally mediated RAFT systems.58 PET-RAFT relies on the photoexcitation of a ground-state PC to produce an excited state (PC*), which then performs single electron transfer (SET) to reduce the thiocarbonylthio or trithiocarbonate chain transfer agent to give an active propagating species and 2PC•+ paired with the anion of the chain transfer agent. This radical can either initiate polymerization and the RAFT equilibrium or be deactivated by the oxidized 2PC•+ to regenerate PC and the dormant polymer chain, which can reenter the catalytic cycle anew (Figure 6). Advantageously, the formation of a chain transfer agent anion as an intermediate minimizes decomposition byproducts that can be formed through PI-CRP or photo-RAFT approaches.
Figure 6.
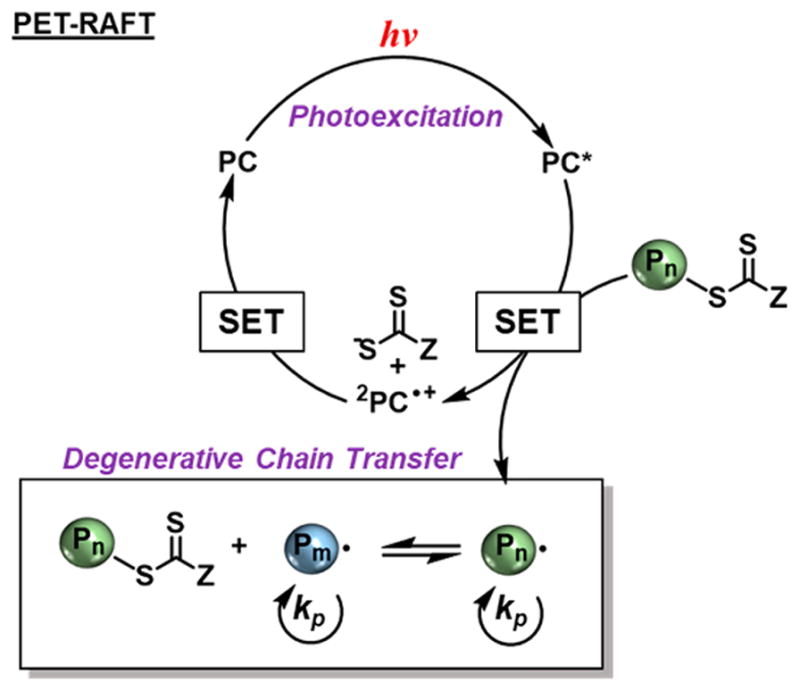
General mechanism of PET-RAFT.
In 2014, PET-RAFT was reported by Boyer et al. using fac-Ir(ppy)3 as PC.70 Since that time, this method has been expanded to include a diverse library of transition-metal and organic71 photoredox catalysts, in some cases capable of absorbing far-red and near-IR irradiation.72 PET-RAFT has also been demonstrated capable of synthesizing a wide scope of polymers and can be readily photocontrolled. Currently, the scope of PET-RAFT has been expanded to include multiblock copolymers,73 stereoregular polymers,74 polymer–protein conjugates,75 functionalized cell surfaces,76 and nanoparticle synthesis,77 as well as oxygen tolerance under the appropriate conditions.78 PET-RAFT has also been combined with other polymerization approaches in orthogonal polymerizations to produced well-defined complex architectures.79
One challenge in CRP systems is reliance on oxygen-free conditions to prevent side reactions, termination, and catalyst quenching in photoredox systems.80 The first report of PET-RAFT in a photo-flow system exemplified the oxygen tolerance of the reaction conditions through the polymerization of N,N-diethylacrylamide (DEA) in continuous flow. This system used zinc tetraphenylporphyrin (Figure 7, PC 1) as PC and 2- (((dodecylthio)carbonylthio)thio)propanoic acid (DTPA) as chain transfer agent under low-energy green light irradiation in a continuous-flow tubular reactor.81 The mechanism of oxygen tolerance is dependent on the generation of triplet–triplet annihilation of ground-state oxygen by photoactivated ZnTPP, followed by consumption of singlet oxygen by the solvent, DMSO. Applied in a photo-flow system, PET-RAFT of DEA targeting several molecular weights, was successfully performed without any previous deoxygenation of reaction components to give polymers with dispersities ranging from 1.05 to 1.12. Block like gradient copolymers, PDEA-co-PDMA (Table 2, entry 5), were also synthesized through addition of the second monomer feedstock directly into the reactor system after reaching <90% conversion of the first block to give a product with Mn = 22.5 kDa and D̵ = 1.06.
Figure 7.
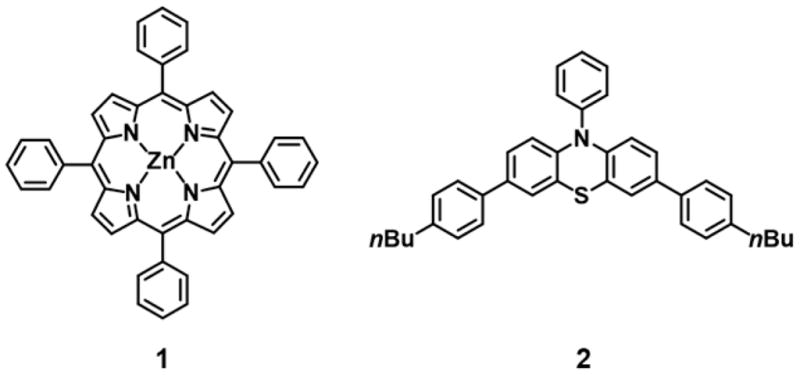
Structures of photoredox catalysts used for PET-RAFT in flow reactors.
Leveraging their previous work, the Boyer group reported a thorough study of the ability to modulate D̵ in continuous flow.82 Control over D̵ imparts control over the properties of the polymeric product83 and is typically achieved through mixing of polymers with different D̵ after production.84 Using DMA as the model monomer, automated in-line D̵ alteration was achieved through the use of gradient residence times, light intensity alterations, and modification of the wavelength of irradiation. For example, when changing the intensity of the irradiation from 0.3 to 3.8 W/m2 during collection, a bimodal distribution of polymer molecular weights was obtained, compared to the typical monomodal distribution seen when one intensity is employed. Interestingly, this work also included an examination of the impact of reactor flow profiles on the results of D̵. Namely, an analysis of the results using different reactor volumes and the introduction of 20 different polymer fractions showed that this system deviates from the posited ideal and predicted plug flow behavior,85 impacting the mixing of the system and the resulting D̵.
Most recently, PET-RAFT in photo-flow systems has been reported by Chen et al. for the polymerization of traditionally challenging semi-fluorinated acrylate monomers.86 These monomers can be used to make materials with unique properties but are challenging to successfully polymerize to high monomer conversions in a controlled fashion. Using a visible light absorbing phenothiazine-based photoredox catalyst (Figure 7, PC 2), the polymerization of several semifluorinated acrylates and methacrylates were studied in a batch reactor, then adapted for the synthesis of block copolymers in continuous flow using white LEDs. Notably, 4.6 g of PHFBA-b-PMA (Table 3, entry 6) with Mn = 10.8 kDa and D̵ = 1.07 was obtained with a decrease in reaction time by 75% in comparison to batch conditions.
Table 3.
Results of Polymerizations in Flow for Reversible Trapping/End-Capping CRPs
| entry | method | irradiation (nm) | catalyst | monomera | tR (min) | Mn (theor) (kDa) | Mn (kDa) | D̵ | ref |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CMRP | 320–500 | 3 | VA | 20 | 5.5 | 6.1 | 1.19 | 90 |
| 2 | CMRP | 320–500 | 3 | VA | 120 | 24.1 | 17.5 | 1.50 | 90 |
| 3 | ATRP | 380 | 4 | MMA | 220 | 4.2 | 5.4 | 1.21 | 97 |
| 4 | ATRP | 380 | 4 | MMA | 220 | 3.1 | 3.3 | 1.23 | 97 |
| 5 | O-ATRP | white | 5 | MMA | 90 | 6.6 | 11.1 | 1.07 | 117 |
| 6 | O-ATRP | white | 6 | MMA | 120 | 4.9 | 6.6 | 1.18 | 117 |
| 7 | O-ATRP | white | 7 | MMA | 90 | 5.0 | 18.9 | 1.10 | 117 |
| 8 | O-ATRP | white | 8 | MMA | 120 | 6.5 | 5.8 | 1.19 | 117 |
| 9 | O-ATRP | 365 | 9 | MAA | 120 | 5.6 | 5.6 | n/a | 118 |
| 10 | O-ATRP | 365 | 9 | MMA | 120 | 5.3 | 4.7 | 2.18 | 118 |
MAA = methacrylic acid. See Table 1 for other details.
4. REVERSIBLE DEACTIVATION CRPS IN FLOW
4.1. Reversible Deactivation through Direct Photolysis
One of the first studies of reversible end-capping CRPs in flow was the examination of photoinduced copper-mediated polymerization (CuMP) of methyl acrylate (MA).87 Improving issues with activator regeneration found in thermal ATRP, Cu(II) is directly photochemically reduced to the active Cu(I) (Figure 8A). Like ATRP, CuMP relies on an equilibrium between oxidation states of copper species to reversibly end-cap the growing polymer chain with a halogen. Similar to thermally mediated ATRP, single electron transfer–living radical polymerization (SET-LRP) operates a copper mediated radical polymerization reliant on reversible activation and deactivation of alkyl halides, but also can be challenging to incorporate into flow systems due to insolubility of copper(II) complexes and salts. These two methodologies yield similar polymer products, but use different reaction conditions, such as solvent polarity and reaction temperature.88
Figure 8.

General mechanisms for copper-mediated polymerization (A) and cobalt-mediated radical polymerization (B).
Using reaction conditions similar to a typical SET-LRP (or supplemental activators and reducing agents-ATRP), Junkers and co-workers investigated the comparison between a small volume (19.5 μ L) glass chip microreactor and a larger volume (11 mL) continuous tubular microreactor system irradiated by UV light under oxygen-free conditions. Using either reactor, CuMP can be performed with the ability to reach high monomer conversions after 20 min of residence time when targeting MWs less than 5 kDa and yielding polymers with D̵ ranging from 1.10 to 1.30. To confirm the ability to maintain the “living-ness” of the CRP, the chain-end group fidelity of the system was analyzed using electrospray ionization mass spectrometry through an analysis of termination products at low polymer MWs. Block copolymers were also synthesized through chain-extension. An isolated PMA macroinitiator with Mn = 3.1 kDa and D̵ = 1.10 synthesized in the tubular reactor was reintroduced to polymerization in the tubular reactor with butyl acrylate (BA) as monomer, resulting in pMA-b-pBA with Mn = 5.0 kDa and D̵ = 1.16 (Table 2, entry 7).
Using a similar CuMP approach, star polymers with four PMA “arms” were synthesized in continuous flow.89 After initial optimization in batch reactors, the study of star polymer synthesis using a core-first approach was performed, focusing on the effect of Cu loading and reaction loading on the formation of star–star coupling termination products. Finding that a diluted system with reactions stopped between 50 and 70% monomer conversion produced the least amount of coupling reactions, with dispersities remaining low at 1.11 for 53% monomer conversion.
Cobalt-mediated radical polymerization (CMRP) has been employed for the polymerization of vinyl acetate in both milli-and microflow reactors.90 CMRP is operated by reversible chain-end trapping by a redox active cobalt complex, which serves as both the light absorbing species and polymer chain-end group (Figure 8B). As a consequence of the photolabile nature of the cobalt–carbon bond, this polymerization can be either thermally or photochemically mediated. In batch conditions, UV irradiation leads to a loss of control over polymer MW and chain architecture.91 CMRP can polymerize a broad monomer scope, including vinyl monomers with unstabilized carbon-centered radicals. Junkers and co-workers investigated the performance of a glass chip milli reactor (19.5 μ L) compared to that of a tubular continuous-flow reactor (11 mL) in the polymerization of vinyl acetate (Table 3, entries 1 and 2) using Co(acac)2 as catalyst (see Figure 10, catalyst 3). In a comparison of thermally driven CMRP and UV supported CMRP at the same temperatures, reaction rates were significantly increased with UV irradiation without loss of polymerization control, reaching 53% monomer conversion after 2 h of residence time compared to 7% conversion with only thermal activation. Using the microreactor, this system was utilized as a tool for screening copolymerizations of vinyl acetate and 1-octene to study relative comonomer reactivity ratios (Table 2, entry 8).
Figure 10.

Structures of catalysts employed in CMRP, ATRP, and O-ATRP in flow reactor systems.
4.2. Reversible Deactivation through Photoredox Catalysis
Another reversible deactivation approach is photo-induced ATRP, which can be mediated by both metal and organic photoredox catalysts. The catalytic cycle can proceed either through reductive or oxidative quenching pathways. Photoredox catalysts such as Ru(bpy)3Cl2,92 Eosin Y,93,94 and other photo-initiators95 can mediate ATRP through a reductive quenching pathway. However, these catalysts are not sufficiently reducing to directly activate an alkyl halide, and a supplemental electron donor is required. Furthermore, this electron donor can induce side reactions,96 which may lead to increased D̵.
The oxidative quenching pathway, which does not rely on any sacrificial electron donors, has been applied to photo-flow systems. In this approach, polymerization proceeds through a photo-redox-oxidative quenching catalytic cycle, where a ground-state PC absorbs light to reach an excited state where it can directly reduce an alkyl halide initiator through a SET event. This reduction generates an active propagating radical as well as 2PC•+, forming an ion-pair with the halide anion. Control over polymerization lies in deactivation, where the active radical is “end-capped” by the halide, returning the growing polymer chain to the dormant state and catalyst to the ground state, completing the catalytic cycle (Figure 9).
Figure 9.
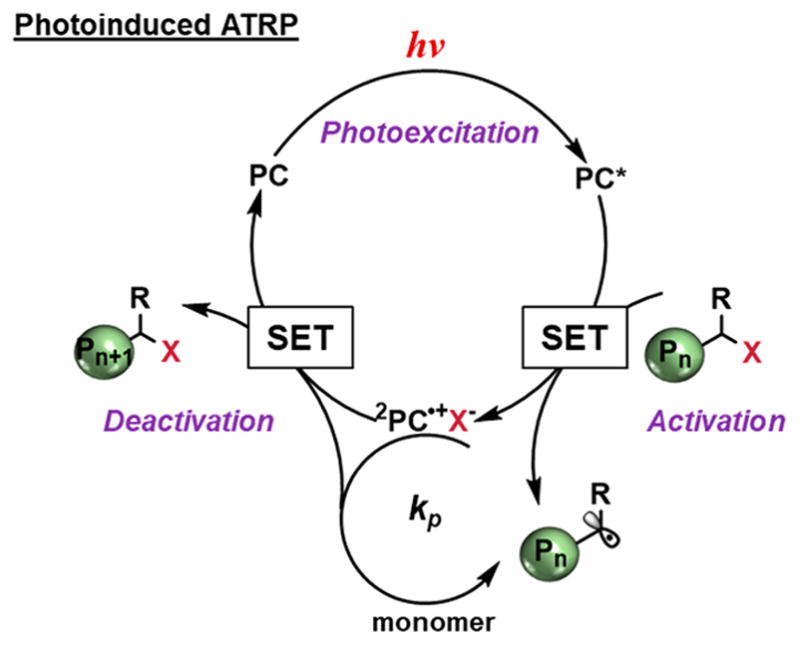
General mechanism for photoredox controlled ATRP proceeding through an oxidative quenching catalytic pathway.
An analysis of the polymerization of MMA using photo-induced ATRP mediated by fac-Ir(ppy)3 (Figure 10, PC 4) under UV irradiation was performed in continuous flow, with emphasis on choice of tubing reactor materials for effective polymerization under oxygen sensitive conditions.97 While an advantageous in terms of uniform irradiation and heat exchange, this can also allow for greater rate of oxygen permeability and subsequent quenching or side reactions. Four different fluorinated tubing materials with differing oxygen permeability and transparency characteristics were investigated, identifying oxygen permeability as an influential factor in imparting reaction control. Ethylene chlorotrifluoroethylene, known as Halar and with oxygen permeability of 25 cm3/100 in.2, was found to perform significantly better in comparison to the more widely used perfluoroalkoxy alkane (PFA) tubing, with an oxygen permeability of 881 cm3/100 in.2. Enhanced performance was demonstrated through a comparison of results after 220 min residence time, producing polymers with Mn = 5.4 kDa and D̵ = 1.21 using Halar and Mn = 3.3 kDa and D̵ = 1.23 with PFA (Table 3, entries 3 and 4). Furthermore, kinetic studies revealed a less controlled increase in MW when PFA was employed with relatively higher D̵, reaching up to 1.90. Further experiments using Halar focused on MW control and on comparison to batch reactions, demonstrating a 50% increase in reaction rate.
A metal-free variant of photoinduced ATRP is organo-catalyzed atom transfer radical polymerization (O-ATRP), which was first reported in 2014.98,99 Removal of the reliance on precious metal PCs allows for improved sustainability and potential expansion of the application scope, for example in electronic fields.100 O-ATRP uses organic PCs capable of directly reducing an alkyl halide initiator, a property that is rare in organic compounds. Catalysts from N,N-diaryl dihydrophe-nazine,101,102 N-arylphenoxazine,103,104 N-arylphenothiazine,99 polycyclic aromatic hydrocarbon,98,105 carbazole,106 and conjugated thienothiophene107 families have been developed. A central focus in the development of O-ATRP to date has centered on mechanistic elucidation108,109 and catalyst design.110–112 However, O-ATRP has also been adapted in batch systems to produce star polymers,113 hyperbranched polymers,114 surface-grafted polymers,115 and application of bio-based monomers for homopolymer synthesis.116
To understand the capabilities of this methodology, the importance of irradiation conditions and O-ATRP catalyst performance was examined in continuous flow conditions, comparing the catalytic performance of three established O-ATRP catalyst families (Figure 10, PCs 5–8) in the polymerization of MMA using visible light in a tubular Halar reactor (Table 3, entries 5–8).117 Generally, polymerization results were found to be superior in flow relative to batch conditions, showing enhanced control over polymerization at low monomer conversions, paired with high I* values. Using the best performing catalyst, 3,7-di(4-biphenyl)-1-naphthalene-10-phenoxazine (Figure 10, PC 8), catalyst loadings could be dramatically reduced to 0.1 mol % and MWs over 20 kDa were synthesized. The monomer scope was extended to a scope of functionalized, hydrophobic and hydrophilic methacrylates with a high degree of control over molecular weight at all monomer conversions. Finally, block copolymers pMMA-b-pBnMA (Mn = 102.4 kDa and D̵ = 1.60) and pMMA-b-pBuMA (Mn = 75.9 kDa and D̵ = 1.32) could be synthesized (Table 2, entries 9 and 10).
O-ATRP has further been applied in continuous flow in a PFA tubing reactor in the polymerization of MMA and methacrylic acid (MAA) using a phenylphenothiazine (Figure 10, PC 9) based photocatalyst under UV light irradiation.118 A loss of chain-end group fidelity was observed in both batch and flow conditions in the polymerization of MMA, as supported by bromine elimination observed using ESI-MS techniques and an increase in D̵. In a comparison between the two methods, a loss of control was observed, with D̵ rising from 1.55 in batch to 2.18 in flow. The polymerization of MAA, a traditionally challenging monomer in many ATRP systems, showed a significantly decreased reaction time in flow systems.
5. SUMMARY AND OUTLOOK
Integrating photo-CRP approaches into flow systems has provided crucial information regarding necessary reactor design and materials, catalytic abilities, and accessible MW ranges as well as highlighting the overall ability to provide improved results of polymerization relative to their batch reactor counterparts. The scope of monomers that have been polymerized include methacrylates, acrylates, acrylamides, and acetates. Block copolymers with a variety of compositions have also been synthesized (Table 3) using chain-extension and inline multistep continuous approaches, already providing to non-experts an avenue to access polymers with a diverse scope of materials properties. However, further work is required as most studies of photo-CRP systems have been performed using model systems, resulting in limited diversity in demonstrated molecular weights, especially above 20 kDa. Additionally, the capabilities to synthesize complex architectures in flow reactor systems using photo-CRPs, including multiblock stars, brushes, and combs has yet to be explored.
To build upon the existing foundation and increase the potential of photo-CRPs, three key areas should be studied. First, adaptation of current approaches to more complex reactor systems with multiple reactors, inlets, outlets, and light sources will enable rapid and readily customizable polymeric materials synthesis. Second, understanding of the effects of photopolymerizations on fluid dynamics in these reactor systems should be established. Viscosity changes from MW growth are well understood in thermally mediated polymer-ization systems, but elucidation of the effect of these changes on catalytic behavior and chain-end group fidelity can have wide consequences for all photo-CRPs. Third, further reactor design and process engineering is required to fully utilize the potential of flow reactor systems in mechanistic elucidation and further polymerization reaction development. Real-time, in situ studies have already been employed for batch photopolymerization systems.66,119 Merging these spectroscopic techniques with the high throughput reaction condition screening possible with flow reactors would greatly facilitate further development of these burgeoning photo-CRP methodologies.
The potential benefits of flow reactor systems in photo-CRPs are numerous. In particular, flow chemistry offers the powerful promise to leverage the “green-ness” of these approaches through solvent reductions, waste prevention, and heightened energy efficiency.43 Certainly, the current scope of work provides a strong foundation for future maturation of photo-CRP systems using flow reactors, offering potential for bridging the gap from an academic pursuit to the industrially relevant production of advanced materials to solve the problems of the modern age.
Acknowledgments
This work was supported by Colorado State University, the American Chemical Society Petroleum Research Fund, and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biographies
Bonnie Buss is a Ph.D. candidate in the chemistry department at Colorado State University in Fort Collins, Colorado. Born in Wichita, Kansas, she earned her B.S. degree in chemistry from the University of Arkansas in 2015 while performing research in glycoside catalysis. Her current research interest in the group of Dr. Garret Miyake focuses on the development of organocatalyzed atom-transfer radical polymerization for application in functional polymer design and synthesis. Furthermore, she is inspired by green chemistry and recently won a Joseph Breen Memorial Fellowship.
Garret M. Miyake earned his B.S. in chemistry from Pacific University (USA). He completed his Ph.D. studies with Eugene Chen at Colorado State University before conducting postdoctoral research with Robert Grubbs at the California Institute of Technology as a Camille and Henry Dreyfus Environmental Chemistry Postdoctoral Fellow. He started his independent career at the University of Colorado–Boulder in 2014 before returning to Colorado State University in 2017. He has been named a Sloan Research Fellow and a Cottrell Scholar and received the 2017 American Chemical Society Division of Polymer Chemistry Mark Young Scholar Award.
Footnotes
This Perspective is part of the Up-and-Coming series.
Notes
The authors declare no competing financial interest.
References
- 1.Hawker CJ, Wooley KL. The Convergence of Synthetic Organic and Polymer Chemistries. Science. 2005;309:1200–1205. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- 2.Szwarc M. “Living” Polymers. Nature. 1956;178:1168–1169. [Google Scholar]
- 3.Hirao A, Goseki R, Ishizone T. Advances in Living Anionic Polymerization: From Functional Monomers, Polymerizations Systems, to Macromolecular Architectures. Macromolecules. 2014;47:1883–1905. [Google Scholar]
- 4.Valente A, Mortreux A, Visseaux M, Zinck P. Coordinative Chain Transfer Polymerization. Chem Rev. 2013;113:3836–3857. doi: 10.1021/cr300289z. [DOI] [PubMed] [Google Scholar]
- 5.Aoshima S, Kanaoka S. A Renaissance in Living Cationic Polymerization. Chem Rev. 2009;109:5245–5287. doi: 10.1021/cr900225g. [DOI] [PubMed] [Google Scholar]
- 6.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled Ring-Opening Polymerization of Lactide and Glycolide. Chem Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 7.Bielawski CW, Grubbs RH. Living ring-opening metathesis polymerization. Prog Polym Sci. 2007;32:1–29. [Google Scholar]
- 8.Webster OW, Hertler WR, Sogah DY, Farnham WB, Rajanbabu TV. Group-Transfer Polymerization. 1. A New Concept for Addition Polymerization with Organo-Silicon Initiators. J Am Chem Soc. 1983;105:5706–5708. [Google Scholar]
- 9.Braunecker WA, Matyjaszewski K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog Polym Sci. 2007;32:93–146. [Google Scholar]
- 10.Kamigaito M, Ando T, Sawamoto M. Metal-Catalyzed Living Radical Polymerization. Chem Rev. 2001;101:3689–3746. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 11.Matyjaszewski K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules. 2012;45:4015–4039. [Google Scholar]
- 12.Vana P, Barnerk-Kowollik C, Davis TP, Matyjaszewski K. Encyclopedia of Polymer Science and Technology. John Wiley and Sons; New York: 2003. Radical Polymerization. [Google Scholar]
- 13.Coessens V, Pintauer T, Matyjaszewski K. Functional polymers by atom transfer radical polymerization. Prog Polym Sci. 2001;26:337–377. [Google Scholar]
- 14.Nicolas J, Guillaneuf Y, Lefay C, Bertin D, Gigmes D, Charleux B. Nitroxide-mediated polymerization. Prog Polym Sci. 2013;38:63–235. [Google Scholar]
- 15.Boyer C, Bulmus V, Davis TP, Ladmiral V, Liu J, Perrier S. Bioapplications of RAFT Polymerization. Chem Rev. 2009;109:5402–5436. doi: 10.1021/cr9001403. [DOI] [PubMed] [Google Scholar]
- 16.Siegwart DJ, Oh JK, Matyjaszewski K. ATRP in the design of functional materials for biomedical applications. Prog Polym Sci. 2012;37:18–37. doi: 10.1016/j.progpolymsci.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moad G, Chen M, Häussler M, Postma A, Rizzardo E, Thang SH. Functional polymers for optoelectronic applications by RAFT polymerization. Polym Chem. 2011;2:492–519. [Google Scholar]
- 18.Long L, Wang S, Xiao M, Meng Y. Polymer electrolytes for lithium polymer batteries. J Mater Chem A. 2016;4:10038–10069. [Google Scholar]
- 19.Edmondson S, Osborne VL, Huck WTS. Polymer brushes via surface-initiated polymerization. Chem Soc Rev. 2004;33:14–22. doi: 10.1039/b210143m. [DOI] [PubMed] [Google Scholar]
- 20.Sveinbjörnsson BR, Weitekamp RA, Miyake GM, Xia Y, Atwater HA, Grubbs RH. Rapid Self-Assembly of Brush Block Copolymers to Photonic Crystals. Proc Natl Acad Sci U S A. 2012;109:14332–14336. doi: 10.1073/pnas.1213055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrier S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules. 2017;50:7433–7447. [Google Scholar]
- 22.Moad G, Chiefari J, Chong BYK, Krstina J, Mayadunne RTA, Postma A, Rizzardo E, Thang SH. Living free radical polymerization with reversible addition-fragmentation chain transfer (the life of RAFT) Polym Int. 2000;49:993–1001. [Google Scholar]
- 23.Matyjaszewski K, Xia J. Atom Transfer Radical Polymerization. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 24.Debuigne A, Poli R, Jerome C, Jerome R, Detrembleur C. Overview of cobalt-mediated radical polymerization: Roots, state of the art and future prospects. Prog Polym Sci. 2009;34:211–239. [Google Scholar]
- 25.Chen M, Zhong M, Johnson JA. Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem Rev. 2016;116:10167–10211. doi: 10.1021/acs.chemrev.5b00671. [DOI] [PubMed] [Google Scholar]
- 26.Pan X, Tasdelen MA, Laun J, Junkers T, Yagci Y, Matyjaszewski K. Photomediated controlled radical polymerization. Prog Polym Sci. 2016;62:73–125. [Google Scholar]
- 27.Tasdelen MA, Uygun M, Yagci Y. Photoinduced Controlled Radical Polymerization. Macromol Rapid Commun. 2011;32:58–62. doi: 10.1002/marc.201000351. [DOI] [PubMed] [Google Scholar]
- 28.Goto A, Scaiano JC, Maretti L. Photolysis of an alkoxyamine using intramolecular energy transfer from a quinoline antenna—towards photo-induced living radical polymerization. Photochem Photobiol Sci. 2007;6:833–835. doi: 10.1039/b705671k. [DOI] [PubMed] [Google Scholar]
- 29.Yamago S, Ukai Y, Matsumoto A, Nakamura Y. Organo-tellurium-Mediated Controlled/Living Radical Polymerization Initiated by Direct C–Te Bond Photolysis. J Am Chem Soc. 2009;131:2100–2101. doi: 10.1021/ja8099689. [DOI] [PubMed] [Google Scholar]
- 30.Quinn JF, Barner L, Barner-Kowollik C, Rizzardo E, Davis TP. Reversible Addition–Fragmentation Chain Transfer Polymerization Initiated with Ultraviolet Radiation. Macromolecules. 2002;35:7620–7627. [Google Scholar]
- 31.Debuigne A, Schoumacher M, Willet N, Riva R, Zhu X, Rutten S, Jerome C, Detrembleur C. new functional poly(N-vinylpyrrolidone) based (co)polymers via photoinitiated cobalt-mediated radical polymerization. Chem Commun. 2011;47:12703–12705. doi: 10.1039/c1cc15471k. [DOI] [PubMed] [Google Scholar]
- 32.Romero NA, Nicewicz DA. Organic Photoredox Catalysis. Chem Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 33.Prier CK, Rankic DA, MacMillan DWC. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narayanam JMR, Stephenson CRJ. Visiblt light photoredox catalysis: applications in organic synthesis. Chem Soc Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 35.Schultz DM, Yoon TP. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tucker JW, Zhang Y, Jamison TF, Stephenson CRJ. Visible-Light Photoredox Catalysis in Flow. Angew Chem, Int Ed. 2012;51:4144–4147. doi: 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garlets ZJ, Nguyen JD, Stephenson CRJ. The Development of Visible Light Photoredox Catalysis in Flow. Isr J Chem. 2014;54:351. doi: 10.1002/ijch.201300136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beatty JW, Douglas JJ, Miller R, McAtee RC, Cole KP, Stephenson CRJ. Photochemical Perfluoroalkylation with Pyridine N-Oxides: Mechanistic Insights and Performance on a Kilogram Scale. Chem. 2016;1:456–472. doi: 10.1016/j.chempr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinbacher JL, McQuade DT. Polymer Chemistry in Flow: New Polymers, Beads, Capsules, and Fibers. J Polym Sci, Part A: Polym Chem. 2006;44:6505–6533. [Google Scholar]
- 40.Diehl C, Laurino P, Azzouz N, Seeberger PH. Accelerated Continuous Flow RAFT Polymerization. Macromolecules. 2010;43:10311–10314. [Google Scholar]
- 41.Fukuyama T, Kajihara Y, Ryu I, Studer A. Nitroxide-Mediated Polymerization of Styrene, Butyl Acrylate, or Methyl Methacrylate by Microflow Reactor Technology. Synthesis. 2012;44:2555–2559. [Google Scholar]
- 42.Bou-Hamdan FR, Seeberger PH. Visible-light-mediated photochemistry: accelerating Ru(bpy)32+-catalyzed reactions in continuous flow. Chem Sci. 2012;3:1612–1616. [Google Scholar]
- 43.Wiles C, Watts P. Continuous flow reactors: a perspective. Green Chem. 2012;14:38–54. [Google Scholar]
- 44.Natalello A, Morsbach J, Friedel A, Alkan A, Tonhauser C, Müller AHE, Frey H. Living Anionic Polymerization in Continuous Flow: Facilitated Synthesis of High-Molecular Weight Poly(2-vinylpyridine) and Polystyrene. Org Process Res Dev. 2014;18:1408–1412. [Google Scholar]
- 45.Wu T, Mei Y, Cabral JT, Xu C, Beers KL. A New Synthetic Method for Controlled Polymerization Using a Microfluidic System. J Am Chem Soc. 2004;126:9880–9881. doi: 10.1021/ja048432n. [DOI] [PubMed] [Google Scholar]
- 46.Enright TE, Cunningham MF, Keoshkerian B. Nitroxide-Mediated Polymerization of Styrene in a Continuous Tubular Reactor. Macromol Rapid Commun. 2005;26:221–225. [Google Scholar]
- 47.Hornung CH, Guerrero-Sanchez C, Brasholz M, Saubern S, Chiefari J, Moad G, Rizzardo E, Thang SH. Controlled RAFT Polymerization in a Continuous Flow Microreactor. Org Process Res Dev. 2011;15:593–601. [Google Scholar]
- 48.Su Y, Straathof NJW, Hessel V, Noel T. Photochemical Transformations Accelerated in Continuous-Flow Reactors: Basic Concepts and Applications. Chem - Eur J. 2014;20:10562–10589. doi: 10.1002/chem.201400283. [DOI] [PubMed] [Google Scholar]
- 49.Hook BDA, Dohle W, Hirst PR, Pickworth M, Berry MB, Booker-Milburn KI. A Practical Flow Reactor for Continuous Organic Photochemistry. J Org Chem. 2005;70:7558–7564. doi: 10.1021/jo050705p. [DOI] [PubMed] [Google Scholar]
- 50.Hartman RL, McMullen JP, Jensen KF. Deciding Whether To Go with the Flow: Evaluating the Merits of Flow Reactors for Synthesis. Angew Chem, Int Ed. 2011;50:7502–7519. doi: 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]
- 51.Baldyga J, Pohorecki R. Turbulent micromixing in chemical reactors-a review. Chem Eng J. 1995;58:183–195. [Google Scholar]
- 52.Parida D, Serra CA, Gomez RI, Garg DK, Hoarau Y, Bouquey M, Muller R. Atom Transfer Radical Polymerization in Continuous Microflow: Effect of Process Parameters. J Flow Chem. 2014;4:92–96. [Google Scholar]
- 53.Danckwerts PV. Continuous flow systems: Distribution of residence times. Chem Eng Sci. 1953;2:1–13. [Google Scholar]
- 54.Cambie D, Bottecchia C, Straathof NJW, Hessel V, Noel T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem Rev. 2016;116:10276–10341. doi: 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- 55.Britton K, Jamison TF. The assembly and use of continuous flow systems for chemical synthesis. Nat Protoc. 2017;12:2423–2446. doi: 10.1038/nprot.2017.102. [DOI] [PubMed] [Google Scholar]
- 56.McKenzie TG, da Costa LPM, Fu Q, Dunstan DE, Qiao GG. Investigation into the photolytic stability of RAFT agents and the implications for photopolymerization reactions. Polym Chem. 2016;7:4246–4253. [Google Scholar]
- 57.Otsu T, Yoshida M. Role of Initiator-Transfer Agent Terminator (Iniferter) in Radical Polymerizations: Polymer Design by Organic Disulfides as Iniferters. Makromol Chem Rapid Commun. 1982;3:127–132. [Google Scholar]
- 58.McKenzie T, Fu Q, Uchiyama M, Satoh K, Xu J, Boyer C, Kamigaito M, Qiao GG. Beyond Traditional RAFT: Alternative Activation of Thiocarbonylthio Compounds for Controlled Polymerization. Adv Sci. 2016;3:1500394. doi: 10.1002/advs.201500394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xi J, Shanmugam S, Corrigan NA, Boyer C. Catalyst-Free Visible Light Induced RAFT Photopolymerization. Controlled Radical Polymerization: Mechanisms ACS Symp Ser. 2015;13:247–267. [Google Scholar]
- 60.Zhou H, Johnson JA. Photo-controlled Growth of Telechelic Polymers and End-linked Polymer Gels. Angew Chem, Int Ed. 2013;52:2235–2238. doi: 10.1002/anie.201207966. [DOI] [PubMed] [Google Scholar]
- 61.Ishizu K, Shibuya T, Kawauchi S. Kinetics on Formation of Hyperbranched Poly(Ethyl Methacrylate) Via a Controlled Radical Mechanism of Photofunctional Inimer. Macromolecules. 2003;36:3505–3510. [Google Scholar]
- 62.You YZ, Hong CY, Bai RK, Pan CY, Wang J. Photo-Initiated Living Free Radical Polymerization in the Presence of Dibenzyl Trithiocarbonate. Macromol Chem Phys. 2002;203:477–483. [Google Scholar]
- 63.Carmean RN, Becker TE, Sims MB, Sumerlin BS. Ultra-High Molecular Weights via Aqueous Reversible-Deactivation Radical Polymerization. Chem. 2017;2:93–101. [Google Scholar]
- 64.Francis R, Ajayaghosh A. Minimization of Homopolymer Formation and Control of Dispersity in Free Radical Induced Graft Polymerization Using Xanthate Derived Macro-Photoinitiators. Macromolecules. 2000;33:4699–4704. [Google Scholar]
- 65.Chen M, Johnson JA. Improving photo-controlled living radical polymerization from trithiocarbonates through the use of continuous-flow techniques. Chem Commun. 2015;51:6742–6745. doi: 10.1039/c5cc01562f. [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Li Q, Dai J, Du F, Zheng H, Bai R. Real-Time and in Situ Investigation of “Living”/Controlled Photopolymerization in the Presence of a Trithiocarbonate. Macromolecules. 2013;46:2576–2582. [Google Scholar]
- 67.Rubens M, Latsrisaeng P, Junkers T. Visible light-induced iniferter polymerization of methacrylates enhanced by continuous flow. Polym Chem. 2017;8:6496–6505. [Google Scholar]
- 68.Gardiner J, Hornung CH, Tsanaktsidis J, Guthrie D. Continuous flow photo-initiated RAFT polymerization using a tubular photochemical reactor. Eur Polym J. 2016;80:200–207. [Google Scholar]
- 69.Wenn B, Junkers T. Continuous Microflow PhotoRAFT Polymerization. Macromolecules. 2016;49:6888–6895. [Google Scholar]
- 70.Xu J, Jung K, Atme A, Shanmugam S, Boyer C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. J Am Chem Soc. 2014;136:5508–5519. doi: 10.1021/ja501745g. [DOI] [PubMed] [Google Scholar]
- 71.Xu J, Shanmugam S, Duong HT, Boyer C. Organo-photocatalyst for photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization. Polym Chem. 2015;6:5615–5624. [Google Scholar]
- 72.Shanmugam S, Xu J, Boyer C. Light-Regulated Polymerization under Near-Infrared/Far-Red Irradiation Catalyzed by Bacteriochlorophyll α. Angew Chem, Int Ed. 2016;55:1036–1040. doi: 10.1002/anie.201510037. [DOI] [PubMed] [Google Scholar]
- 73.Fu C, Xu J, Tao L, Boyer C. Combining Enzymatic Monomer Transformation with Photoinduced Electron Transfer -Reversible Addition-Fragmentation Chain Transfer for the Synthesis of Complex Multiblock Copolymers. ACS Macro Lett. 2014;3:633–638. doi: 10.1021/mz500245k. [DOI] [PubMed] [Google Scholar]
- 74.Shanmugam S, Boyer C. Stereo-, Temporal and Chemical Control through Photoactivation of Living Radical Polymerization: Synthesis of Block and Gradient Copolymers. J Am Chem Soc. 2015;137:9988–9999. doi: 10.1021/jacs.5b05903. [DOI] [PubMed] [Google Scholar]
- 75.Tucker BS, Coughlin ML, Figg CA, Sumerlin BS. Grafting-From Proteins Using Metal-Free PET–RAFT Polymerizations under Mild Visible-Light Irradiation. ACS Macro Lett. 2017;6:452–457. doi: 10.1021/acsmacrolett.7b00140. [DOI] [PubMed] [Google Scholar]
- 76.Niu J, Lunn DJ, Pusuluri A, Yoo JI, O’Malley MA, Mitragotri S, Soh HT, Hawker CJ. Engineering live cell surfaces with functional polymers via cytocompatible controlled radical polymerization. Nat Chem. 2017;9:537–545. doi: 10.1038/nchem.2713. [DOI] [PubMed] [Google Scholar]
- 77.Ng G, Yeow J, Xu J, Boyer C. Application of oxygen tolerant PET-RAFT to polymerization-induced self-assembly. Polym Chem. 2017;8:2841–2851. [Google Scholar]
- 78.Xu J, Jung K, Boyer C. Oxygen Tolerance Study of Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization Mediated by Ru- (bpy)3Cl2. Macromolecules. 2014;47:4217–4229. [Google Scholar]
- 79.Fu C, Xu J, Kokotovic M, Boyer C. One-Pot Synthesis of Block Copolymers by Orthogonal Ring-Opening Polymerization and PET-RAFT Polymerization at Ambient Temperature. ACS Macro Lett. 2016;5:444–449. doi: 10.1021/acsmacrolett.6b00121. [DOI] [PubMed] [Google Scholar]
- 80.Bhanu VA, Kishore K. Role of oxygen in polymerization reactions. Chem Rev. 1991;91:99–117. [Google Scholar]
- 81.Corrigan N, Rosli D, Jones JWJ, Xu J, Boyer C. Oxygen Tolerance in Living Radical Polymerization: Investigation of Mechanism and Implementation in Continuous Flow Polymerization. Macromolecules. 2016;49:6779–6789. [Google Scholar]
- 82.Corrigan N, Almasri A, Taillades W, Xu J, Boyer C. Controlling Molecular Weight Distributions through Photoinduced Flow Polymerization. Macromolecules. 2017;50:8438–8448. [Google Scholar]
- 83.Gentekos DT, Dupuis LN, Fors BP. Beyond Dispersity: Deterministic Control of Polymer Molecular Weight Distribution. J Am Chem Soc. 2016;138:1848–1851. doi: 10.1021/jacs.5b13565. [DOI] [PubMed] [Google Scholar]
- 84.Widin JM, Schmitt AK, Schmitt AL, Im K, Mahanthappa MK. Unexpected consequences of block polydispersity on the self-assembly of ABA triblock copolymers. J Am Chem Soc. 2012;134:3834–3844. doi: 10.1021/ja210548e. [DOI] [PubMed] [Google Scholar]
- 85.Muller M, Cunningham MF, Hutchinson RA. Continuous Atom Transfer Radical Polymerization in a Tubular Reactor. Macromol React Eng. 2008;2:31–36. [Google Scholar]
- 86.Gong H, Zhao Y, Shen X, Lin J, Chen M. Organocatalyzed Photocontrolled Radical Polymerization of Seminfluorinated (Meth)-acrylates Driven by Visible Light. Angew Chem, Int Ed. 2018;57:333–337. doi: 10.1002/anie.201711053. [DOI] [PubMed] [Google Scholar]
- 87.Wenn B, Conradi M, Carreiras AD, Haddleton DM, Junkers T. Photo-induced copper-mediated polymerization of methyl acrylate in continuous flow reactors. Polym Chem. 2014;5:3053–3060. [Google Scholar]
- 88.Konkolewicz D, Wang Y, Zhong M, krys P, Isse AA, Gennaro A, Matyjaszewski K. Reversible-Deactivation Radical Polymerization in the Presence of metallic Copper. A Critical Assessment of the SARA ATRP and SET-LRP Mechanisms. Macromolecules. 2013;46:8749–8772. [Google Scholar]
- 89.Wenn B, Martens AC, Chuang YM, Gruber J, Junkers T. Efficient multiblock star polymer synthesis from photo-induced copper-mediated polymerization with up to 21 arms. Polym Chem. 2016;7:2720–2727. [Google Scholar]
- 90.Kermagoret A, Wenn B, Debuigne A, Jerome C, Junkers T, Detrembleur C. Improved photo-induced cobalt-mediated radical polymerization in continuous flow photoreactors. Polym Chem. 2015;6:3847–3857. [Google Scholar]
- 91.Detrembleur C, Versace DL, Piette Y, Hurtgen M, Jerome C, Lalevee J, Debuigne A. Synthetic and mechanistic inputs of photochemistry into the bis-acetylacetonatocobalt-mediated radical polymerization of n-butyl acrylate and vinyl acetate. Polym Chem. 2012;3:1856–1866. [Google Scholar]
- 92.Zhang G, Song IY, Ahn KH, Park T, Choi W. Free Radical Polymerization Initiated and Controlled by Visible Light Photocatalysis at Ambient Temperature. Macromolecules. 2011;44:7594–7599. [Google Scholar]
- 93.Kutahya C, Aykac FS, Yilmaz G, Yagci Y. LED and visible light-induced metal free ATRP using reducible dyes in the presence of amines. Polym Chem. 2016;7:6094–6098. [Google Scholar]
- 94.Bian C, Zhou YN, Guo JK, Luo ZH. Aqueous Metal-Free Atom Transfer Radical Polymerization: Experiments and Model-Based Approach for Mechanistic Understanding. Macromolecules. 2018;51:2367–2376. [Google Scholar]
- 95.Allushi A, Kutahya C, Aydogan C, Kreutzer J, Yilmaz G, Yagci Y. Conventional Type II photoinitiators as activators for photoinduced metal-free atom transfer radical polymerization. Polym Chem. 2017;8:1972–1977. [Google Scholar]
- 96.Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Visible Light-Mediated Intermolecular H-H Functionalization of Electron-Rich Heterocycles with Malonates. Org Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]
- 97.Melker A, Fors BP, Hawker CJ, Poelma JE. Continuous Flow Synthesis of Poly(methyl methacrylate) via a Light-Mediated Controlled Radical Polymerization. J Polym Sci, Part A: Polym Chem. 2015;53:2693–2698. [Google Scholar]
- 98.Miyake GM, Theriot JC. Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules. 2014;47:8255–8261. [Google Scholar]
- 99.Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read de Alaniz J, Fors BP, Hawker CJ. Metal-Free Atom Transfer Radical Polymerization. J Am Chem Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- 100.Shanmugam S, Boyer C. Organic photocatalysts for cleaner polymer synthesis. Science. 2016;352:1053–1054. doi: 10.1126/science.aaf7465. [DOI] [PubMed] [Google Scholar]
- 101.Theriot JC, Lim CH, Yang H, Ryan MD, Musgrave CB, Miyake GM. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science. 2016;352:1082. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- 102.Ryan MD, Theriot JC, Lim CH, Yang H, Lockwood AG, Garrison NG, Lincoln SR, Musgrave CB, Miyake GM. Solvent effects on the intramolecular charge transfer character of N,N-diaryl dihydrophenazine catalysts for organocatalyzed atom transfer radical polymerization. J Polym Sci, Part A: Polym Chem. 2017;55:3017–3027. doi: 10.1002/pola.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pearson RM, Lim CH, McCarthy BG, Musgrave CB, Miyake GM. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J Am Chem Soc. 2016;138:11399–11407. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McCarthy BG, Pearson RM, Lim CH, Sartor SM, Damrauer NH, Miyake GM. Structure-Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts. J Am Chem Soc. 2018;140:5088–5101. doi: 10.1021/jacs.7b12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Allushi A, Jockusch S, Yilmaz G, Yagci Y. Photoinitiated Metal-Free Controlled/Living Radical Polymerization Using Poly-nuclear Aromatic Hydrocarbons. Macromolecules. 2016;49:7785–7792. [Google Scholar]
- 106.Huang Z, Gu Y, Liu X, Zhang L, Cheng Z, Zhu X. Metal-Free Atom Transfer Radical Polymerization of Methyl Methacrylate with ppm Level of Organic Photocatalyst. Macromol Rapid Commun. 2017;38:1600461. doi: 10.1002/marc.201600461. [DOI] [PubMed] [Google Scholar]
- 107.Kutahya C, Allushi A, Isci R, Kreutzer J, Ozturk T, Yilmaz G, Yagci Y. Photoinduced Metal-Free Atom Transfer Radical Polymerization Using Highly Conjugated Thienothiophene Derivatives. Macromolecules. 2017;50:6903–6910. [Google Scholar]
- 108.Ryan MD, Pearson RM, French TA, Miyake GM. Impact of Light Intensity on Control in Photoinduced Organo-catalyzed Atom Transfer Radical Polymerization. Macromolecules. 2017;50:4616–4622. doi: 10.1021/acs.macromol.7b00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]
- 110.Theriot JC, McCarthy BG, Lim CH, Miyake GM. Organocatalyzed Atom Transfer Radical Polymerization: Perspectives on Catalyst Design and Performance. Macromol Rapid Commun. 2017;38:1700040. doi: 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lim CH, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalyst for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J Am Chem Soc. 2017;139:348–355. doi: 10.1021/jacs.6b11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jockusch S, Yagci Y. The active role of excited states of phenothiazines in photoinduced metal free atom transfer radical polymerization: singlet or triplet excited states? Polym Chem. 2016;7:6039–6043. [Google Scholar]
- 113.Buss BL, Beck LR, Miyake GM. Synthesis of star polymers using organocatalyzed atom transfer radical polymerization through a core-first approach. Polym Chem. 2018;9:1658–1665. doi: 10.1039/C7PY01833A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aydogan C, Yilmaz G, Yagci Y. Synthesis of Hyperbranched Polymers by Photoinduced Metal-Free ATRP. Macromolecules. 2017;50:9115–9120. [Google Scholar]
- 115.Discekici EH, Pester CW, Treat NJ, Lawrence J, Mattson KM, Narupai B, Toumayan EP, Luo Y, McGrath AJ, Clark PG, Read de Alaniz J, Hawker CJ. Simple Benchtop Approach to Polymer Brush Nanostructures Using Visible-Light-Mediated Metal-Free Atom Transfer Radical Polymerization. ACS Macro Lett. 2016;5:258–262. doi: 10.1021/acsmacrolett.6b00004. [DOI] [PubMed] [Google Scholar]
- 116.Wang J, Yuan L, Wang Z, Rahman MA, Huang Y, Zhu T, Wang R, Cheng J, Wang C, Chu F, Tang C. Photoinduced Metal-Free Atom Transfer Radical Polymerization of Biomass-Based Monomers. Macromolecules. 2016;49:7709–7717. [Google Scholar]
- 117.Ramsey BL, Pearson RM, Beck LR, Miyake GM. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization using Continuous Flow. Macromolecules. 2017;50:2668–2674. doi: 10.1021/acs.macromol.6b02791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ramakers G, Krivcov A, Trouillet V, Welle A, Mobius H, Junkers T. Organocatalyzed Photo-Atom Transfer Radical Polymerization of Methacrylic Acid in Continuous Flow and Surface Grafting. Macromol Rapid Commun. 2017;38:1700423. doi: 10.1002/marc.201700423. [DOI] [PubMed] [Google Scholar]
- 119.Aguirre-Soto A, Hwang AT, Glugla D, Wydra JW, McLeod RR, Bowman CN, Stansbury JW. Coupled UV–Vis/ FT–NIR Spectroscopy for Kinetic Analysis of Multiple Reaction Steps in Polymerizations. Macromolecules. 2015;48:6781–6790. [Google Scholar]