

Interleukin 15 (IL-15) is one of the most important cytokines regulating the biology of natural killer (NK) cells1. Here we identified a signaling pathway involving the serine-threonine kinase AKT and XBP1s, a transcription factor that regulates unfolded protein response genes2,3, that was activated in response to IL-15 in human NK cells. IL-15 induced the phosphorylation of AKT, which led to the deubiquitination, increased stability and nuclear accumulation of XBP1s protein. XBP1s bound to and recruited the transcription factor T-BET to the gene encoding granzyme B, leading to increased transcription. XBP1s positively regulated the cytolytic activity of NK cells against leukemia cells and was also required for IL-15-mediated NK cell survival through an anti-apoptotic mechanism. Thus, the newly identified IL-15-AKT-XBP1s signaling pathway contributes to enhanced effector functions and survival of human NK cells.
Unspliced XBP1 mRNA, known as XBP1u, encodes an unstable cytoplasmic protein with no transactivation domains. As a result of unconventional splicing mediated by the serine/threonine-protein kinase/endoribonuclease, IRE1α, mature XBP1 mRNA is converted to XBP1s4. The protein encoded by XBP1s can act as a transcription factor2,3. XBP1s has multiple roles in regulating the immune response. It regulates major histocompatibility complex class II (MHC II) gene transcription in HeLa and COS cells5, as well as the differentiation of plasma cells, eosinophils and CD8+ T cells6–8. XBP1s also modulates anti-tumor immunity by disrupting dendritic cell homeostasis9. We investigated the expression of XBP1s in primary human NK cells purified from the blood of healthy donors in response to interleukin 2 (IL-2), IL-12 or IL-15 for 24 h prior to analysis by flow cytometry or immunoblot. IL-15 induced the expression of XBP1s protein, whereas IL-2 and IL-12 showed reduced effects compared to IL-15 (Fig. 1a,b). Although IL-2 and IL-15 share the cognate receptors IL-2Rβ and IL-2Rγc on NK cells, induction of XBP1s by IL-15 was significantly higher than that triggered by similar concentrations of IL-2 (Fig. 1b and Supplementary Fig. 1a). This suggests that the IL-15Rα chain expressed on NK cells may play a critical role in inducing XBP1s. In addition, the expression of transcripts for XBP1s target genes, including ERDJ4 and SEC61A19, was significantly increased in IL-15-treated primary human NK cells compared to non-treated, IL-2- or IL-12-treated cells (Fig. 1c).
Fig.1: XBP1s is induced by IL-15 at the protein level and controls degranulation in NK cells.
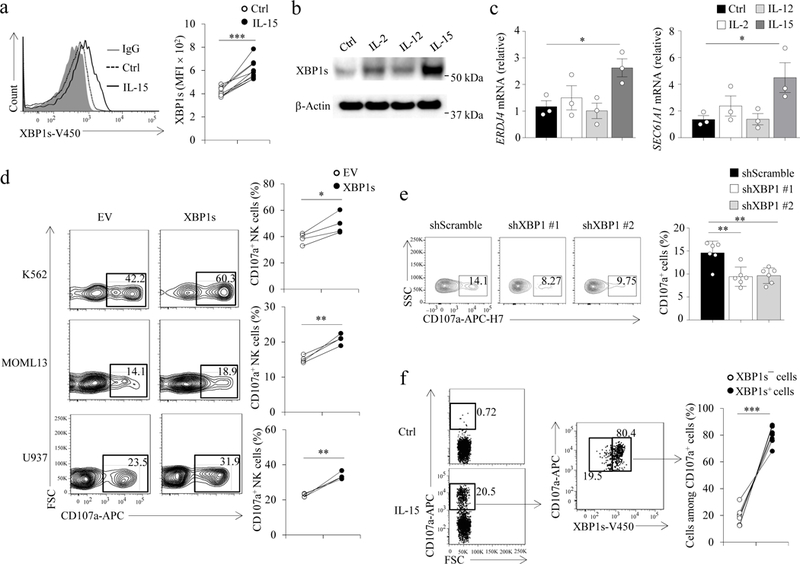
(a,b) NK cells were treated with IL-2 (100 units/ml), IL-12 (10 ng/ml), or IL-15 (100 units/ml) for 24 h for flow cytometric analysis (a, n = 9 donors) and/or immunoblotting (b, n = 4 donors). ***P < 0.001 by two-tailed paired t test. The experiment in (b) was repeated 3 times with similar results; images were cropped, and the full scans are shown in the supplementary figures. c, The expression of XBP1s target genes was assessed by qPCR after NK cells were treated as in (a,b). Bar graphs display mean ± s.e.m. of n = 3 donors. *P < 0.05 by linear mixed model. d, NK cells were transduced with an XBP1s lentiviral construct or empty vector (EV) and 48h later were FACS-sorted for transduced GFP+ cells. Sorted cells were co-cultured with indicated leukemia cells for 4 h, followed by quantifying CD107a+ cells by flow cytometry. n = 4 donors. *P < 0.05, **P < 0.01 by two-tailed paired t test. e, NK cells were transduced with a XBP1 or a scramble shRNA lentiviral construct (pLKO.1) and FACS-sorted for GFP+ cells after 48 h, then co-cultured with the MOML13 leukemia cell line for 4 h, followed by quantification of CD107a+ cells. Bar graphs display mean ± s.d. of n = 6 donors. **P < 0.01 by linear mixed model. f, NK cells were treated with IL-15 for 24 h and co-cultured with the MM1.S cell line for 4 h, followed by evaluation of XBP1s and CD107a expression by flow cytometry. n = 8 donors. ***P < 0.001 by two-tailed paired t test.
We next investigated the effects of XBP1s overexpression on NK cell function. Primary human NK cells transfected with pCDH lentivirus carrying a wild-type XBP1s gene (pCDH-XBP1s) and co-cultured with K562, MOLM-13 or U937 leukemia cell lines had a higher percentage of CD107a+ NK cells compared to NK cells transfected with the lentivirus carrying an empty PCDH vector (pCDH-EV) (Fig. 1d). Upon co-culture with MOML-13 target cells, the percentage of CD107a+ cells in primary human NK cells transduced with pLKO.1 lentivirus carrying XBP1 shRNAs (XBP1-knockdown, KD) was significantly decreased (an approximately 35% reduction) compared to cells transduced with pLKO.1 lentivirus carrying scramble shRNAs (scramble-KD) (Fig. 1e). In addition, primary human NK cell degranulation against multiple myeloma MM.1S cells was observed in IL-15-treated, but not in non-treated primary human NK cells (Fig. 1f). When co-cultured with MM.1S multiple myeloma cells, the percentage of CD107a+ NK cells expressing XBP1s was approximately 4-fold greater than that of CD107a+ NK cells lacking XBP1s (Fig. 1f). Moreover, the expression of XBP1s protein was significantly higher in CD107a+ compared to CD107a─ primary human NK cells co-cultured with MM.1S cells (Supplementary Fig. 1b), indicating that expression of XBP1s correlates with NK cell cytotoxicity against tumor cells. Collectively, our results suggest that IL-15 induces XBP1s protein expression and the expression level of the transcriptional factor directly correlates with cytotoxic activity in human NK cells.
To investigate how XBP1s regulates NK cell function, we analyzed the expression of genes related to NK cell effector functions, including GZMB (granzyme B), IFNG (interferon-γ), and PRF1 (perforin). Expression of GZMB and IFNG but not PRF1 mRNA was higher in pCDH-XBP1s-transduced primary human NK cells compared to pCDH-EV control NK cells (Fig. 1a), along with increased expression of GZMB protein (Fig. 2b,c). Overexpression of the unspliced form of XBP1, XBP1u, which can be processed into XBP1s through IRE1α-mediated mRNA splicing, in primary human NK cells by transduction with pCDH lentivirus carrying a wild-type XBP1u gene (pCDH-XBP1u) also increased the expression of GZMB compared to pCDH-EV NK cells (Fig. 2b,c). Moreover, primary human NK cells treated with thapsigargin (Thap), a chemical drug that induces ER stress and IRE1α catalytic activity10, increased XBP1s protein and GZMB mRNA and protein when compared to NK cells without Thap treatment, in the absence or presence of IL-15 (Supplementary Fig. 2a-c). In addition, downregulation of GZMB protein expression was observed in primary human NK cells transfected with XBP1 siRNAs compared to cells transfected with scramble control siRNAs (Supplementary Fig. 2d,e). We also observed decreased expression of both GZMB and IFNG genes, but not PRF1, in primary human NK cells with XBP1-KD using shRNA, compared to scramble-KD control NK cells (Fig. 1d). Inhibition of XBP1 mRNA splicing in primary human NK cells with 4µ8C, an inhibitor of IRE1α-mediated mRNA splicing11, resulted in decreased expression of XBP1s protein and suppression of IL-15-induced GZMB protein and mRNA compared to the cells without 4µ8C treatment (Supplementary Fig. 2f-h). The expression of XBP1s protein was positively correlated with the mRNA expression of GZMB and IFNG, all of which were induced by treatment with IL-15 at multiple time points in primary human NK cells (Fig. 1e). Thus, XBP1s positively regulates the expression of GZMB and IFN-γ in NK cells.
Fig. 2: XBP1s regulates GZMB and IFNγ expression in NK cells.
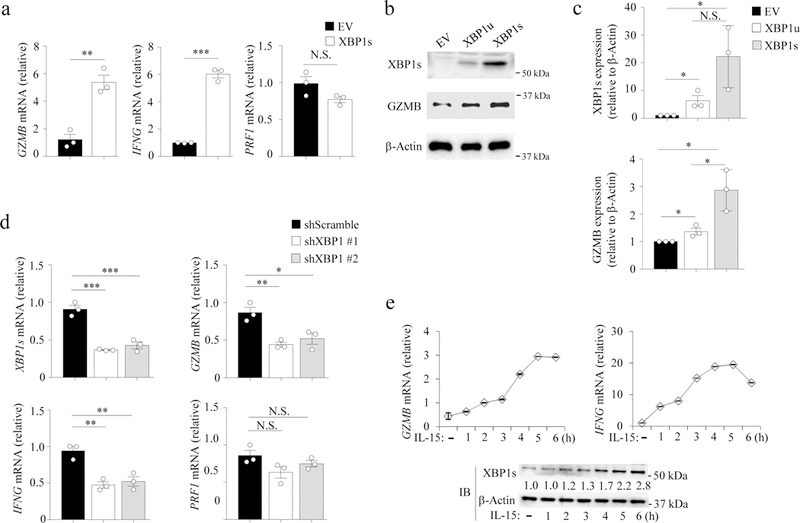
a, NK cells were transduced with an XBP1s lentiviral construct or empty vector (EV) and 48 h later were FACS-sorted for transduced GFP+ cells, which were co-cultured with K562 cells for 4 h, and then fractionated with anti-CD56 magnetic beads, followed by qPCR analysis. Bar graphs display mean ± s.e.m. of n = 3 donors. **P < 0.01, ***P < 0.001 by Student’s two-tailed unpaired t test. b, NK cells were transduced with XBP1u, XBP1s, or pCDH-EV by lentiviral infection and cultured with IL-15 (100 units/ml) for 48 h and then were FACS-sorted for GFP+ cells to determine XBP1s and GZMB protein expression by immunoblotting. The experiment was repeated 3 times with similar results. c, Densitometric quantification of the ratio of the level of XBP1s or GZMB protein to the level of β-Actin protein for (b). Bar graphs display mean ± s.d. of n = 3 donors. *P < 0.05 by linear mixed model. d, NK cells were transduced with a XBP1 or scramble shRNA lentiviral construct (pLKO.1) and 48 h later were FACS-sorted as in (a). FACS-purified cells were co-cultured with K562 cells for 4 h, and then fractionated with anti-CD56 magnetic beads, followed by qPCR analysis. Bar graphs display mean ± s.e.m. of n = 3 donors. *P < 0.05 by linear mixed model. e, NK cells were treated with IL-15, followed by immunoblotting or qPCR analysis. The experiment was repeated independently three times with similar results (b,e). N.S., no significance. Blot images (b,e) were cropped, and the full scans are shown in the Supplementary figures.
We next investigated the cellular localization of XBP1s. Immunoblot analysis of the cytoplasmic and nuclear protein fractions in primary human NK cells treated with IL-15 for 24 h indicated that XBP1s exists almost exclusively in the nucleus following induction by IL-15 (Fig. 2a), consistent with its role in regulating transcription12. Next, we overexpressed T-BET and FLAG-XBP1u or FLAG-XBP1s in 293T cells. Co-immunoprecipitation followed by immunoblot using antibodies against FLAG or T-BET indicated that overexpressed FLAG-XBP1s interacted with T-BET (Fig. 3b,c and Supplementary Fig. 3a), a transcriptional regulator important for NK cell function. T-BET was previously assumed to associate with the GZMB promoter, although no specific binding sites have been identified13,14. Of note, FLAG-XBP1s did not interact with endogenous STAT5 in 293T cells, an important transcriptional factor downstream of IL-15 signaling (Fig. 2c). Using confocal imaging with antibodies identifying both XBP1u and XBP1s, and a T-BET antibody, we observed the co-localization of T-BET and XBP1 in the nuclei of primary human NK cells (Fig. 2d) and in the human NK cell lymphoma cell line NK-92 (Supplementary Fig. 3b). Confocal microscopy indicated that T-BET has an almost exclusively nuclear distribution in NK cells (Fig. 2d), which was validated by staining with an alternative antibody against T-BET in both primary NK cells and NK-92 cells (Supplementary Fig. 3c).
Fig. 3: XBP1s interacts with T-BET and binds to the proximal GZMB promoter.
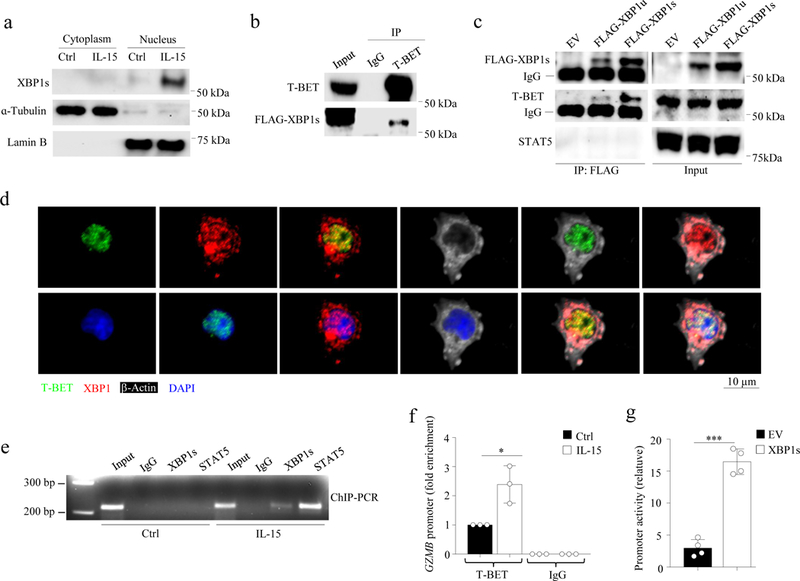
a, Cytoplasmic and nuclear proteins were fractionated from primary human NK cells, followed by immunoblot analysis. b, FLAG-XBP1s and T-BET were co-transfected into 293T cells, followed by immunoprecipitation (IP) and immunoblot (IB) analyses. c, T-BET-expressing 293T cells were transfected with FLAG-XBP1u, FLAG-XBP1s or the control (pCDH) vector, followed by IP and immunoblot analyses. d, NK cells were treated with IL-15 (100 units/ml) for 24 h, followed by immunofluorescent staining with an anti-XBP1 antibody recognizing both XBP1u and XBP1s as well as anti-T-BET and anti-β-Actin antibodies. The nuclei were stained by DAPI (blue). e,f, NK cells were treated with IL-15 for 16 h, followed by ChIP assays performed with specific antibody or control IgG. Precipitated DNA was analyzed by PCR (e) or qPCR (f). Bar graphs display mean ± s.d. of n = 3 donors. *P < 0.05 by Student’s two-tailed unpaired t test. g, XBP1s or control vector was co-transduced with a pGL3 plasmid containing the GZMB promoter and a pRL-TK plasmid (as a control for data normalization) into 293T cells for 48 h. Cells were lysed to determine the promoter activity of GZMB by luciferase reporter assays. Bar graphs display mean ± s.e.m. of n = 4 independently experiments. ***P < 0.001 by Student’s two-tailed unpaired t test. The experiment was repeated independently two times (a,b) or three times (c,d,e) with similar results. Blot (a,b,c) and gel (e) images were cropped, and the full scans are shown in Supplementary figures.
Next, we tested whether XBP1s interacted with its canonical binding motifs (G/C)ACGT15,16 located within the GZMB proximal promoter (Fig. 2e). Chromatin immunoprecipitation (ChIP) indicated that XBP1s and T-BET bound to the same proximal region of the GZMB promoter in primary human NK cells treated with IL-15, but with little or no binding in untreated cells (Fig. 3e,f). In contrast, T-BET did not bind to the GZMB promoter in primary human NK cells treated with 4µ8C (which do not express XBP1s; Supplementary Fig. 2g) in the presence of IL-15, compared to control cells only treated with IL-15 (Supplementary Fig. 3d). Using a luciferase assay to evaluate GZMB promoter activity, we observed that 293T cells transfected with XBP1s had much higher GZMB promoter activity compared to that of the empty vector-transfected cells (Fig. 2g). Of note, we also observed STAT5 binding to the GZMB promoter in primary human NK cells by ChIP assays (Fig. 2e), consistent with previous reports17. STAT5 is known to positively regulate GZMB expression17–19; however, STAT5 did not interact with FLAG-XBP1s in 293T cells by co-immunoprecipitation assays (Fig. 2c). To test whether STAT5 was required for GZMB induction by XBP1s we knocked down the expression of STAT5A or STAT5B using shRNAs in 293T cells that were co-transfected with a pCDH-XBP1s or pCDH-EV and a PGL3 vector carrying the GZMB promoter reporter (PGL3-GZMB) (Supplementary Fig. 4a). The induction of the GZMB promoter reporter by XBP1s overexpression was not inhibited by either STAT5A-KD (279%) or STAT5B-KD (184%) in 293T cells compared to control 293T cells (175%) (Supplementary Fig. 4b), indicating that induction of GZMB promoter activity by XBP1s and T-BET in 293T cells does not require STAT5. Together, our data suggest that XBP1s interacts with T-BET but not STAT5 and regulates the transcriptional activity of GZMB via promoter binding.
XBP1 is a survival gene that protects cells from stress-induced death20,21, and IL-15 is a critical cytokine for NK cell survival1,22. Knockdown of XBP1 by transfection with XBP1 siRNAs resulted in increased expression of cleaved caspase-3 in primary human NK cells compared to cells transfected with scramble siRNAs (Fig. 3a), indicating increased apoptosis in XBP1-KD NK cells. In contrast, pCDH-XBP1u- or pCDH-XBP1s-transduced primary human NK cells showed decreased expression of cleaved caspase-3 compared to pCDH-EV control NK cells (Fig. 3b). Moreover, in IL-15-activated primary human NK cells in which XBP1 splicing was inhibited by treatment with 4µ8C, cleaved caspase-3 was higher and survival was lower compared to similarly activated cells without 4µ8C treatment (Fig. 4c-f). Taken together, these data indicate that XBP1s modulates IL-15-induced survival in human NK cells.
Fig. 4: XBP1s contributes to IL-15-mediated NK cell survival.
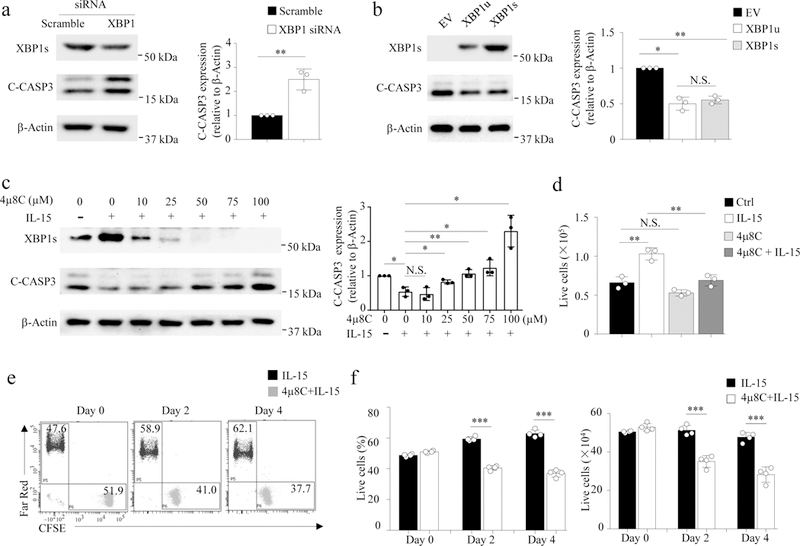
a,b, NK cells were transiently transfected or lentivirally transduced with XBP1 esiRNA, XBP1u, XBP1s, or control [Scramble in (a) and empty vector (EV) in (b), followed by culture for 48 h with IL-15 (100 units/ml). Densitometric quantification shows the ratio of C-CASP3 protein to β-Actin protein (right panel). Bar graphs display mean ± s.d. of n = 3 donors (a,b). *P < 0.05, **P < 0.01 by Student’s two-tailed unpaired t test (a) or linear mixed model (b). c, NK cells were pretreated with 4µ8C for 1 h and then with or without IL-15 for 4 h. Densitometric quantification shows the ratio of C-CASP3 protein to β-Actin protein (right panel). Bar graphs display mean ± s.d. of n = 3 donors. *P < 0.05, **P < 0.01 by linear mixed model. d, NK cells were pre-treated with or without 4µ8C (50 µM) for 1 h, followed by an incubation with IL-15 for 96 h. Bar graphs display mean ± s.d. of n = 3 donors. **P < 0.01 by linear mixed model. e, NK cells were pre-treated with or without 4µ8C (50 µM) for 4 h, then washed and labeled with CFSE or Far Red live staining dye, respectively. Cells were mixed 1:1 and cultured with IL-15. Live cell ratios (CFSE+ vs Far Red+) were analyzed by flow cytometry. The experiment was repeated independently for four donors with similar results. f,g, The percentages and absolute quantities of live cells depicted in (e) are shown in (f) and (g), respectively. Bar graphs display mean ± s.d. of n = 4 donors. ***P < 0.001 by Student’s two-tailed unpaired t test. N.S., no significance. Blot images (a,b,c) were cropped, and the full scans are shown in Supplementary figures.
Next, we investigated the molecular mechanism by which IL-15 regulated the expression of XBP1s protein. IL-15 stimulation of primary human NK cells did not increase the amount of XBP1s mRNA compared to unstimulated NK cells, as evaluated by XBP1 splicing assays and qPCR; however, XBP1s protein accumulated in the nucleus of IL-15-treated primary human NK cells within 2 h of stimulation (Fig. 5a,b). IL-15 stimulation also induced the phosphorylation of the serine-threonine kinase AKT in NK cells primarily in the cytoplasm (Fig. 4b), as previously reported23. Blockade of AKT phosphorylation with the AKT inhibitor AKTi-1/224 in IL-15-stimulated primary human NK cells resulted in decreased expression of XBP1s protein compared to cells treated with IL-15 in the absence AKTi-1/2 (Fig. 4c and Supplementary Fig. 5a). Moreover, the expression of GZMB mRNA and GZMB protein was significantly downregulated in primary NK cells transduced with AKT1-shRNA compared to scramble shRNA (Supplementary Fig. 5b,c). On the other hand, the expression of GZMB mRNA was significantly upregulated and GZMB protein was moderately upregulated in primary NK cells transduced with a constitutively active form of AKT (pCDH-myrAKTΔ4−129, encoding a 14 amino acid src myristoylation signal sequence fused to the N-terminus of AKT delta4–12925), compared to cells transduced with control pCDH-EV (Supplementary Fig. 5d,e). Treatment with AKTi-1/2 also reduced the level of XBP1s protein in IL-15-stimulated NK cells in the presence of cycloheximide (CHX), which blocks de novo protein synthesis and thus prevents any confounding effects of increased protein translation26 (Fig. 4d and Supplementary Fig.6a). These data indicate that AKT plays a role in increasing the IL-15-induced protein levels, but not mRNA expression of XBP1s in primary human NK cells.
Fig. 5: AKT mediates stability of XBP1s.
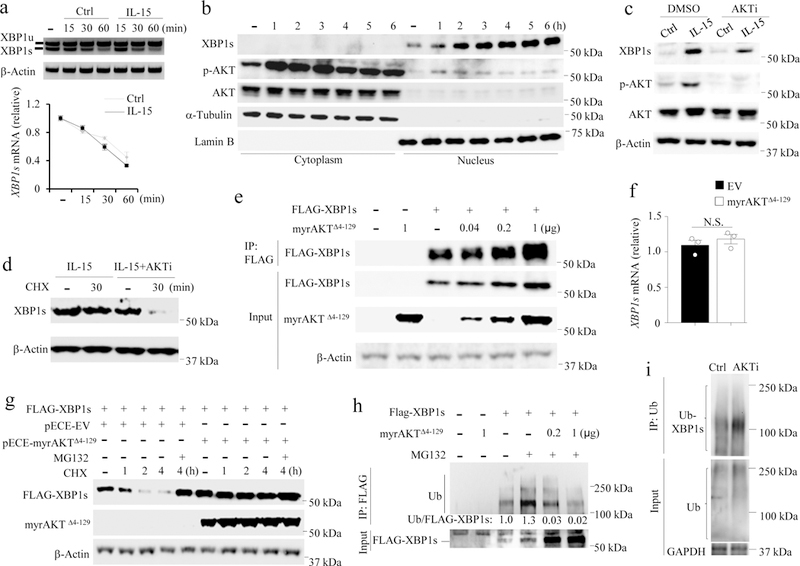
a, XBP1 splicing assays by PCR (upper panel) and quantification of XBP1s by qPCR in NK cells (lower panel). Bar graphs display mean ± s.e.m. of n = 3 donors. b, Immunoblotting of cytoplasmic and nuclear protein fractions of NK cells treated with IL-15 (100 units/ml) for indicated time period. c, NK cells were pre-treated with or without AKTi-1/2 (10 µM) for 30 min, followed by stimulation with IL-15 for 6 h prior to immunoblotting. n = 3 donors. d, NK cells were pre-treated with IL-15 alone or plus AKTi-1/2 for 1 h, then cultured with or without CHX (10 µg/ml) for 30 min prior to immunoblotting. Data are summarized in Supplementary Fig. 6a. n = 3 donors. e, 293T cells were transfected with indicated amount of pECE-myrAKTΔ4−129 vector and/or pCDH-FLAG-tagged XBP1s (FLAG-XBP1s) vector or respective control vector (dash) (1 µg each) and cultured for 24 h. XBP1s protein was analyzed by immunoprecipitation (IP) with anti-FLAG antibody combined with immunoblotting. Data are summarized in Supplementary Fig. 6b. n = 3 independent experiments. f, XBP1s mRNA expression was analyzed by qPCR in the pECE-myrAKTΔ4–129- or pECE control-transfected 293T cells co-transfected with pCDH-FLAG-XBP1s for 24 h (endogenous XBP1s is very low). Bar graphs display mean ± s.d. of n = 3 independent experiments. N.S., not significant, by Student’s two-tailed unpaired t test. g, 293T cells were co-transfected with pCDH-FLAG-XBP1s and pECE-myrAKTΔ4−129 or pECE empty vector (EV) for 24 h and then treated with CHX for the indicated time periods with or without MG132 (10 µM). n = 3 independent experiments. h, myrAKTΔ4−129 or control (dash) was co-transfected with FLAG-XBP1s into 293T cells for 24 h with or without MG132 for 4 h. Protein ubiquitination of XBP1s was analyzed by IP with anti-FLAG antibodies, followed by immunoblotting with densitometric quantification. i, NK cells were pre-treated with or without AKTi-1/2 for 1 h followed by MG132 treatment for 3 h in the presence of IL-15. Ubiquitination of XBP1s was analyzed by IP with anti-ubiquitin antibodies combined with immunoblotting using XBP1s antibodies. The experiment was repeated independently for three times (b,h,i) with similar results. Gel (a) and blot images (b-e, g-i) were cropped, and the full scans are shown in Supplementary figures.
To further test whether AKT is required for maintaining the level of XBP1s protein, we co-transfected various concentrations of pECE-myrAKTΔ4−129 or pECE-EV with pCDH-FLAG-XBP1s or pCDH-EV into 293T cells. Immunoprecipitation experiments indicated that the level of FLAG-XBP1s protein was markedly increased in a dose-dependent manner by myrAKTΔ4−129 overexpression (Fig. 4e and Supplementary Fig. 6b), while XBP1s mRNA was not induced by overexpression of myrAKTΔ4−129 (Fig. 4f), consistent with the idea that IL-15-induced XBP1s upregulation is independent of XBP1s transcription. In addition, co-transduction of pCDH-XBP1s and pECE-myrAKTΔ4−129 enhanced the activity of the GZMB promoter in 293T cells compared to transfection of pCDH-XBP1s alone (Supplementary Fig. 6c). These data indicate that AKT activation is required for maintaining the level of XBP1s protein. As time advanced during a 4-h incubation, a decrease was observed in FLAG-XBP1s protein in 293T cells transfected with pECE-EV, but not with pECE-myrAKTΔ4−129 following CHX treatment (Fig. 4g and Supplementary Fig. 6d), suggesting that signaling downstream of AKT protects XBP1s from degradation. Block of proteosomal degradation with the cell-permeable proteasome and calpain inhibitor MG13227 recovered FLAG-XBP1s protein in pECE-EV- but not pECE-myrAKTΔ4−129-transfected 293T cells treated with CHX (Fig. 4g and Supplementary Fig. 6d), indicating that overexpression of myrAKTΔ4−129 enhanced the protein stability of XBP1s in 293T cells. Ubiquitination of FLAG-XBP1s was reduced following transfection of pECE-myrAKTΔ4−129 compared to pECE-EV in 293T cells (Fig. 4h), while ubiquitination of XBP1s was substantially increased in IL-15-stimulated primary human NK cells treated with the AKT inhibitor AKTi-1/2 compared to untreated cells (Fig. 4i). Our data suggest that AKT controls XBP1s stability, possibly through a mechanism involving the ubiquitination of XBP1s.
Our studies describe a pathway that links IL-15 signaling with intracellular mechanisms that contribute to NK cell cytotoxicity and survival. We show that IL-15 stabilizes XBP1s protein through phosphorylation of AKT, allowing its downstream interaction with T-BET. T-BET activity correlates with GZMB expression14. Yet, there are no direct T-BET binding sites on the GZMB promoter13,14. Based on the presence of an XBP1s binding motif within the GZMB promoter13, we showed that XBP1s mediates the interaction between T-BET and the GZMB promoter, as suggested by the ability of XBP1s to bind the promoter of GZMB and to interact with T-BET.
We showed that AKT is involved in the regulation of XBP1s downstream of IL-15. PI3K-AKT signaling is stimulated by IL-2 and IL-15, with AKT phosphorylation being more sensitive to IL-15 than IL-228, in agreement with our data that IL-15 was the most potent cytokine (among IL-2, IL-12 and IL-15) in increasing XBP1s protein expression and function. AKT is known to regulate both protein ubiquitination and deubiquitination, with distinct mechanisms for each process29,30. Ubiquitination and degradation of PTEN, a natural inhibitor of the PI3K-AKT pathway, in tumor cells require the stabilization of the E3 ligase MKRN1 via AKT-mediated phosphorylation30. However, AKT also induces protein stabilization by activating USP-14, a ubiquitin-specific protease that handles the turnover of short-lived proteins via deubiquitination29. We showed here that AKT signaling caused the deubiquitination and promoted the accumulation of XBP1s. Consistent with our data, deficiency of the PI3K subunits p110γ or p110δ, which regulate AKT activity, has been reported to disrupt NK cell maturation, development and cytotoxicity31,32. Moreover, inhibition of PI3K suppresses GZMB expression in NK cells, and also decreases NK cell cytotoxicity against tumor cells33–35. These observations are consistent with our findings that IL-15 stimulation of NK cells caused an increase of AKT phosphorylation that correlated with decreased degradation of XBP1s, which subsequently enhanced NK cell degranulation following co-culture with tumor cells. Moreover, the active form of AKT, myrAKTΔ4−129, enhanced the XBP1s-induced transcription of GZMB. Notably, myrAKTΔ4−129 alone slightly induced GZMB transcription in 293T cells, but a synergistic effect on GZMB transcription was observed following co-transfection with XBP1s.
IL-15 promotes NK cell survival22. However, the molecular basis of this mechanism remains poorly understood. Our current study supports a model in which XBP1s is located downstream of PI3K-AKT and AKT contributes to the protein stability of XBP1s in NK cells following treatment with IL-15, eventually controlling NK cell survival. Consistent with our data, XBP1s is known to rescue cells from pro-apoptotic processes induced by ER stress, oxidative stress or hypoxia20,21,36. The anti-apoptotic protein Bcl-2 is highly expressed in resting NK cells37 and contributes to NK cell survival in the resting state38,39. However, Bcl-2 appears to be redundant for survival of activated or proliferating NK cells38. Here we found that XBP1s was highly expressed in IL-15-activated NK cells, but its expression was relatively low in resting NK cells. Moreover, inhibition of XBP1s abolished the survival of IL-15-activated NK cells, but had no effect on the survival of resting NK cells. Thus, we speculate that Bcl-2 is responsible for the survival of resting NK cells, while XBP1s is responsible for the survival of IL-15-activated NK cells. In conclusion, we showed that XBP1s acts as an essential transcriptional factor downstream of IL-15 and AKT signaling in controlling two important aspects of NK cell biology: effector functions and survival. The IL-15-AKT-XBP1s axis may offer a potential target to improve the therapeutic efficacy of ex vivo expanded NK cells and/or chimeric antigen receptor (CAR)-modified NK cells40 for the treatment of various cancers.
Methods
Isolation of primary human NK cells.
Leukocyte-enriched peripheral blood samples of human healthy donors were obtained from the American Red Cross. Primary NK cells were isolated using the MACSxpress® NK Cell Isolation Kit (Miltenyi Biotec) and Erythrocyte Depletion Kit (Miltenyi Biotec). Enriched NK cells were approximately 99% pure, which was confirmed by flow cytometry using anti-CD3 and anti-CD56 antibodies (BD Biosciences). Primary human NK cells were used for the experiments in this study unless otherwise indicated that the NK-92 cell line was used for specific experiments. The protocols for human specimen collection were approved by the IRBs of The Ohio State University.
Cell culture.
The K562 and NK-92 cell lines were purchased from ATCC. The U937, MM.1S, and MOLM-13 cell lines were obtained from the Caligiuri laboratory. These cell lines and primary NK cells were cultured in RPMI 1640 medium containing L-glutamine (Sigma) and supplemented with 10% or 20% heat-inactivated fetal bovine serum (FBS; Sigma). The 293T cell line, which was purchased from ATCC, was cultured in Dulbecco’s Modified Eagle’s Medium, DMEM (Sigma), supplemented with 10% FBS. All cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Antibodies and other reagents.
Anti-β-Actin, anti-Lamin B, anti-XBP1, and anti-T-BET antibodies were purchased from Santa Cruz Biotechnology Inc. Anti-p-AKT (S473), anti-AKT, anti-α-Tubulin, anti-FLAG, anti-p-STAT5, anti-STAT5, anti-GZMB, anti-T-BET, anti-ubiquitin (for western), anti-GAPDH, and anti-C-CASP3 antibodies were purchased from Cell Signaling Technology, Inc. (CST). The anti-ubiquitin mAb for IP was purchased from MilliporeSigma. Anti-HA antibody was purchased from Sigma. Anti-XBP1s antibody for immunoblot was purchased from BioLegend. Anti-hCD3-APC-H7 (560176), anti-hCD56-FITC (557699), anti-hCD56-V450 (560360), anti-hCD107a-APC (560664), anti-hCD107a-APC-H7 (561343), anti-XBP1s (563382) and anti-hGZMB (560212) antibodies used for surface and intracellular flow cytometry were purchased from BD Biosciences. Chemical inhibitors (4µ8C, S7272; AKTi-1/2, S7776; MG132, S2619) used in cell treatment experiments were purchased from Selleck Chemicals. CHX (239764) was purchased from Sigma. The CellTrace™ CFSE Cell Proliferation Kit and the Far Red Cell Proliferation Kit were purchased from Thermo Fisher Scientific. The scramble (EHUFLUC) and XBP1 (EHU069131) esiRNAs were purchased from Sigma. The scramble (SHC007), XBP1 (shXBP1 #1, TRCN0000019804; shXBP1 #2, TRCN0000019808), STAT5A shRNA (TRCN0000232134), STAT5B shRNA (TRCN0000232140) and AKT1 shRNA (TRCN0000039797) cloned in the pLKO.1-puro vector were purchased from Sigma. The selection marker, puromycin, in the pLKO.1-puro vector was replaced by GFP for sorting (FACS) of transduced cells. IL-2 (#200–02), IL-12 (#200–12), and IL-15 (#200–15) were purchased from PeproTech.
Transient transfection and lentivirus infection.
293T cells were seeded and incubated at 37°C in a 5% CO2 environment until the cells were 60–80% confluent. The cells were transfected with plasmids using Lipofectamine 3000 Reagent (Fisher Scientific). NK cells were transfected with esiRNA (Sigma) using the nucleofection method (Lonza)41. To generate lentivirus to infect human NK cells, 293T cells were co-transfected with pCDH expressing XBP1u, XBP1s, myrAKTΔ4−129, or pLKO.1 expressing XBP1 or AKT1 shRNA or corresponding control plasmids with the packaging constructs pCMV-VSVG and pCMV-ΔR9 by a ProFection® Mammalian Transfection System (Promega). The protocols for virus production and infection were modified from our previous reports42,43. During the lentiviral infection process, IL-15 was introduced into the cell culture to maintain survival of NK cells.
RT-PCR and XBP1 splicing assay.
Total RNA was extracted with the RNeasy Mini Kit (74106, Qiagen), and cDNA was synthesized using random hexamers and M-MLV reverse transcriptase (Invitrogen). cDNA was amplified by quantitative (q)-PCR with SYBR® Green PCR Master Mix (Applied Biosystems) and gene specific primers. Relative amplification values were normalized to the amplification of β-actin. For the XBP1 splicing assay, cDNA was amplified by PCR with Taq DNA polymerase (Invitrogen) and gene specific primers9. The following primer sequences were used: hXBP1 forward primer: (CCTGGTTGCTGAAGAGGAGG); hXBP1 reverse primer: (CCATGGGGAGTTCTGGAG). The PCR conditions were used: 98°C for 30 sec, followed by 40 cycles of 98°C for 15 sec, 62°C for 30 sec, and 72°C for 60 sec.
Immunoblotting.
Cells were suspended in lysis buffer on ice for 1 h. Equal amounts of protein (~20 μg) were resolved by 5–20% SDS-polyacrylamide gels (Bio-rad) and then transferred onto a PVDF membrane (Fisher Scientific). The membrane was incubated with a primary antibody at 4°C for 16 h and a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature44,45. The immunoblots were visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Fisher Scientific). Densitometric analysis was performed to quantify intensity of gel bands.
Immunoprecipitation (IP).
Cell lysates were prepared with NP40 lysis buffer. For 293T cells, cell lysates were prepared 48 h after plasmid transfection unless otherwise indicated. A beads-antibody complex was prepared using appropriate primary antibodies and Pierce™ Protein G Agarose (Fisher Scientific), followed by IP according to the manufacturer’s protocol and as we previously reported43. The precipitated proteins were detected by immunoblotting.
Chromatin immunoprecipitation (ChIP).
ChIP assays were carried out with a Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (EMD Millipore). Briefly, an equal amount (10 μg) of rabbit anti-XBP1s antibody (BioLegend), rabbit anti-STAT5 antibody (CST), rabbit anti-T-BET antibody (CST) or normal IgG (Santa Cruz) was used to precipitate the cross-linked DNA/protein complexes from 10 × 106 NK cells. After reversal of cross-linking, the precipitated chromatin of the GZMB promoter region was detected by PCR using the following primers: forward primer: (GGGCTCAAACACATACCTGC); reverse primer: (TGACCACATCATCACCCACAG).
Luciferase reporter assay.
Luciferase reporter assays were carried out with a Dual-Luciferase Reporter Assay System (Promega), following our published protocol46 with modifications. After 48 h transfection, cells on a 24-well plate were lysed in 100 µl of 1x Passive Lysis Buffer (Promega). 20 µl of lysates were transferred to a 96-well plate. 100 µl of 1x Glo® luciferase assay substrate (Promega) was added to each well, and firefly luciferase was collected from the transfected pGL3 plasmid using the GloMax® 96 Microplate Luminometer (Promega). Renilla luciferase from the co-transfected pRL-TK plasmid (as a normalized control) was collected after injection with 100 µl of 1x Stop® Substrate (Promega).
Flow cytometry.
Cells were labeled with monoclonal antibodies at room temperature for 15 min and washed with PBS containing 2% BSA prior to analysis using an LSRII flow cytometer (BD Biosciences) to detect surface expression of each antigen. NK cells were gated as CD56+CD3─ lymphocytes. For analysis by intracellular flow cytometric analysis, cells were permeabilized and fixed using a Foxp3/Transcription Factor Fixation/Permeabilization kit (eBioscience).
Statistical analysis.
For continuous, normally-distributed data, two-sample t-tests or paired t-tests were used to compare two independent or two paired groups. Linear mixed model was used to compare three or more groups with a variance-covariance structure due to repeated measures from the same donors. Two-way ANOVA model was applied to the synergistic effect test between two factors. P values were adjusted for multiple comparisons using Holm’s procedure. A P value of 0.05 or less was considered statistically significant.
Reporting Summary.
Further information on experimental design is available in the Nature Research Reporting Summary.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (AI129582, NS106170, J.Y. and CA185301, CA210087, CA068458, M.A.C.), the Leukemia & Lymphoma Society (6503-17, 1364-19, JY), the American Cancer Society (RSG-14-243-01-LIB, J.Y.), the National Key R&D Program (2018YFC1313400, Y.P.), and the Gabrielle’s Angel Cancer Research Foundation (#87, J.Y.).
Footnotes
Conflict-of-interest disclosure
The authors declare no conflicting interests.
Data availability
All summary or representative data generated and supporting the findings of this study are available within the paper. Raw data that support the findings of this study are available upon request.
References
- 1.Becknell B & Caligiuri MA Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv. Immunol 86, 209–239 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Vidal RL & Hetz C Unspliced XBP1 controls autophagy through FoxO1. Cell Res 23, 463–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Uemura A, Oku M, Mori K & Yoshida H Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci 122, 2877–2886 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Clauss IM, Chu M, Zhao JL & Glimcher LH The basic domain/leucine zipper protein hXBP-1 preferentially binds to and transactivates CRE-like sequences containing an ACGT core. Nucleic Acids Res 24, 1855–1864 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettigole SE, et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nature immunology 16, 829–837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamimura D & Bevan MJ Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol 181, 5433–5441 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaffer AL, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81–93 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Cubillos-Ruiz JR, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161, 1527–1538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadighi Akha AA, et al. Heightened induction of proapoptotic signals in response to endoplasmic reticulum stress in primary fibroblasts from a mouse model of longevity. The Journal of biological chemistry 286, 30344–30351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross BC, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 109, E869–878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mai B & Breeden L Xbp1, a stress-induced transcriptional repressor of the Saccharomyces cerevisiae Swi4/Mbp1 family. Mol Cell Biol 17, 6491–6501 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glimcher LH, Townsend MJ, Sullivan BM & Lord GM Recent developments in the transcriptional regulation of cytolytic effector cells. Nature reviews. Immunology 4, 900–911 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Townsend MJ, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 20, 477–494 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 508, 103–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acosta-Alvear D, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27, 53–66 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Verdeil G, Puthier D, Nguyen C, Schmitt-Verhulst AM & Auphan-Anezin N STAT5-mediated signals sustain a TCR-initiated gene expression program toward differentiation of CD8 T cell effectors. J Immunol 176, 4834–4842 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Gotthardt D, et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov 6, 414–429 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Jahrsdorfer B, et al. Granzyme B produced by human plasmacytoid dendritic cells suppresses T-cell expansion. Blood 115, 1156–1165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero-Ramirez L, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer research 64, 5943–5947 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Gupta S, et al. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS biology 8, e1000410 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carson WE, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. The Journal of clinical investigation 99, 937–943 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yano S, Komine M, Fujimoto M, Okochi H & Tamaki K Interleukin 15 induces the signals of epidermal proliferation through ERK and PI 3-kinase in a human epidermal keratinocyte cell line, HaCaT. Biochem Biophys Res Commun 301, 841–847 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, et al. PI3K inhibitor LY294002, as opposed to wortmannin, enhances AKT phosphorylation in gemcitabine-resistant pancreatic cancer cells. Int. J. Oncol 50, 606–612 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Kohn AD, Takeuchi F & Roth RA Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem 271, 21920–21926 (1996). [DOI] [PubMed] [Google Scholar]
- 26.McKeehan W & Hardesty B The mechanism of cycloheximide inhibition of protein synthesis in rabbit reticulocytes. Biochem Biophys Res Commun 36, 625–630 (1969). [DOI] [PubMed] [Google Scholar]
- 27.Rock KL, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Zambricki E, et al. Signaling T-cell survival and death by IL-2 and IL-15. Am J Transplant 5, 2623–2631 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Xu D, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife 4, e10510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee MS, et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat Commun 6, 7769 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo H, Samarakoon A, Vanhaesebroeck B & Malarkannan S The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med 205, 2419–2435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tassi I, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity 27, 214–227 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Ali K, et al. Corrigendum: Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 535, 580 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Nandagopal N, Ali AK, Komal AK & Lee SH The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol 5, 187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zebedin E, et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood 112, 4655–4664 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, et al. Preventing oxidative stress: a new role for XBP1. Cell Death Differ 16, 847–857 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang S, Munker R & Andreeff M Bcl-2 is expressed in human natural killer cells and is regulated by interleukin-2. Nat Immun 15, 312–317 (1996). [PubMed] [Google Scholar]
- 38.Viant C, et al. Cell cycle progression dictates the requirement for BCL2 in natural killer cell survival. The Journal of experimental medicine 214, 491–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minagawa M, et al. Enforced expression of Bcl-2 restores the number of NK cells, but does not rescue the impaired development of NKT cells or intraepithelial lymphocytes, in IL-2/IL-15 receptor beta-chain-deficient mice. J Immunol 169, 4153–4160 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Chu J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 28, 917–927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meinke S & Watzl C NK cell cytotoxicity mediated by 2B4 and NTB-A is dependent on SAP acting downstream of receptor phosphorylation. Front Immunol 4, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu J, et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 3989–4000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng Y, et al. Transcription factor foxo1 is a negative regulator of natural killer cell maturation and function. Immunity 42, 457–470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, et al. CGK733-induced LC3 II formation is positively associated with the expression of cyclin-dependent kinase inhibitor p21Waf1/Cip1 through modulation of the AMPK and PERK/CHOP signaling pathways. Oncotarget 6, 39692–39701 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, et al. PERK/CHOP contributes to the CGK733-induced vesicular calcium sequestration which is accompanied by non-apoptotic cell death. Oncotarget 6, 25252–25265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu J, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity 24, 575–590 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.