

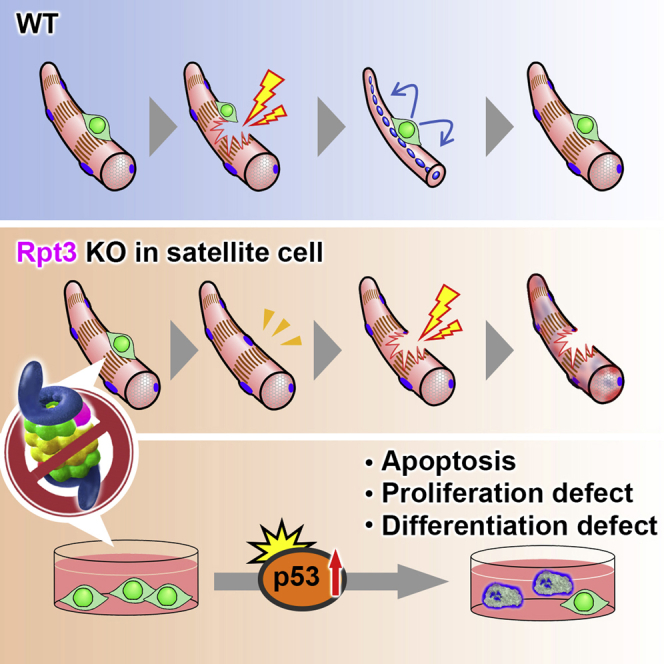
Summary
Adult muscle stem cells (satellite cells) are required for adult skeletal muscle regeneration. A proper balance between quiescence, proliferation, and differentiation is essential for the maintenance of the satellite cell pool and their regenerative function. Although the ubiquitin-proteasome is required for most protein degradation in mammalian cells, how its dysfunction affects tissue stem cells remains unclear. Here, we investigated the function of the proteasome in satellite cells using mice lacking the crucial proteasomal component, Rpt3. Ablation of Rpt3 in satellite cells decreased proteasome activity. Proteasome dysfunction in Rpt3-deficient satellite cells impaired their ability to proliferate, survive and differentiate, resulting in defective muscle regeneration. We found that inactivation of proteasomal activity induced proliferation defects and apoptosis in satellite cells. Mechanistically, insufficient proteasomal activity upregulated the p53 pathway, which caused cell-cycle arrest. Our findings delineate a critical function of the proteasome system in maintaining satellite cells in adult muscle.
Keywords: muscle stem cells, muscle satellite cells, skeletal muscle, muscle regeneration, proteasome dysfunction, Rpt3, p53, proliferation defect, differentiation defect, proteolysis
Graphical Abstract
Highlights
-
•
Ablation of Rpt3 in satellite cells leads to decreased proteasome activity
-
•
Proteasome dysfunction in satellite cells results in defective muscle regeneration
-
•
Proteasome dysfunction induces proliferation defects and apoptosis
-
•
Inhibition of p53 rescues Rpt3-mediated defects in proliferation
It is not clear how proteasome insufficiency affects tissue stem cells. Kitajima et al. demonstrate that the proteasome system is indispensable for the maintenance of muscle stem cells. Proteasome dysfunction in Rpt3-deficient satellite cells impairs their ability to proliferate, survive and differentiate, resulting in defective muscle regeneration.
Introduction
Adult muscle stem cells, also known as muscle satellite cells, which are the resident tissue stem cells of skeletal muscle, provide myonuclei for postnatal muscle growth and for maintenance and regeneration in adults (Blau et al., 2015, Morgan and Partridge, 2003, Relaix and Zammit, 2012, Yin et al., 2013). Satellite cells are located between the basal lamina and the plasmalemma of myofibers (Mauro, 1961). Postnatal myogenesis comprises the activation of quiescent satellite cells, the proliferation of myoblasts (activated satellite cells), and the fusion of myoblasts into multinucleated fibers. Myogenesis is a complex process that is controlled by the spatiotemporal expression of transcription factors including myogenic regulatory factors (MRFs) (Braun and Gautel, 2011, Parker et al., 2003, Wagers and Conboy, 2005).
The ubiquitin-proteasome pathway is responsible for most protein degradation in mammalian cells (Collins and Goldberg, 2017). Proteasomal degradation is mediated by the 26S proteasome, which is an ATP-dependent protease complex that is present in both the cytoplasm and nucleus. The ubiquitin-proteasome pathway has emerged as a central player in the regulation of several diverse cellular processes and functions by catalyzing the selective degradation of short-lived regulatory proteins as well as abnormal proteins. Thus, understanding the role of the proteasome in each cell and tissue type is essential for comprehending the maintenance of homeostasis.
The 26S proteasome is composed of one proteolytically active cylinder-shaped particle (the 20S proteasome) and ATPase-containing complexes (the 19S cap complexes) (Baumeister et al., 1998). The 19S cap complex unfolds ubiquitin-conjugated proteins to allow their entry into the 20S cylindrical particle. It also contains several putative ATPases, such as Rpt1–6. These subunits form a large family with a highly conserved ATPase domain (Sakao et al., 2000). Rpt3, also known as PSMC4, is an essential subunit of the 26S proteasome and is required for the degradation of most proteasomal substrates. In particular, Rpt3-deficient mice die before implantation due to a defect in blastocyst development (Sakao et al., 2000); this indicates that Rpt3 is essential for survival. Interestingly, an insertion/deletion variant in intron 5 of the Rpt3 gene was frequently found in a cohort of patients with Parkinson's disease (Marx et al., 2007). To explore the organ- and cell-specific role of the proteasome, we generated proteasome-deficient mice by targeting Rpt3 (Kitajima et al., 2014, Tashiro et al., 2012). Previously, we found that conditional knockout of Rpt3 in motor neurons results in locomotor dysfunction, which was accompanied by progressive motor neuron loss and gliosis in mice (Tashiro et al., 2012).
Recently, we also reported that muscle-specific Rpt3 knockout mice exhibit proteasome insufficiency, leading to obvious muscle atrophy (Kitajima et al., 2014). Furthermore, centrally nucleated regenerating fibers were observed in muscle-specific Rpt3 knockout mice, indicating the involvement in muscle regeneration. However, it remains unclear how the proteasome system regulates satellite cells. Here, we investigated the pathophysiological effect of proteasome insufficiency induced by depletion of Rpt3 on satellite cells in vivo and in vitro by using satellite cell-specific Rpt3-knockout mice.
Results
Proteasome Activity in Skeletal Muscle during Muscle Regeneration
As the first step to determine proteasome activity in muscle during skeletal muscle regeneration, we induced skeletal muscle injury by injecting cardiotoxin (CTX) into the tibialis anterior (TA) muscle in mice. First, we analyzed the regenerative capacity of C57BL/6J mice over time (Figure S1A). On days 14 and 30 after injury, the weight of CTX-injected muscles was significantly increased compared with that of intact muscles (Figure S1B). At 30 days post injury, the mean TA muscle weight per body weight was also increased compared with that of intact muscle (Figure S1C). Furthermore, histological analysis revealed extensive muscle damage and the infiltration of inflammatory cells into the muscle at 3 days post CTX injection, whereas centrally nucleated regenerating fibers began to be visible at day 7 post injury (Figure S1D). Following muscle injury, satellite cells start to express MyoD and become myoblasts (PAX7+/MYOD + cells) to proliferate (Yin et al., 2013). Quantitative PCR (qPCR) analysis confirmed that mRNA levels of MyoD, Pax7, and Myh3, markers of regenerating fibers, peaked at day 3 post injury (Figure S1E).
We next examined the proteasome system during regeneration after CTX-induced muscle injury. The ubiquitin-proteasome system degrades most long- and short-lived normal and abnormal intracellular proteins (Collins and Goldberg, 2017, Goldberg, 2003). We used lysates from regenerating muscle tissue to evaluate chymotrypsin-like and trypsin-like proteasome activities during muscle regeneration. Chymotrypsin-like and trypsin-like proteasome activities drastically increased to a peak level at 3 days post injury, progressively declined until day 14, and then returned to levels comparable to those observed in uninjured control tissues by 30 post injury (Figures 1A and 1B). Because most proteasomal substrates must be ubiquitinated before degradation (Schrader et al., 2009), we analyzed ubiquitinated proteins during muscle regeneration by immunoblotting. Consistent with the proteasomal activities, we showed that the amount of ubiquitinated proteins increased, peaking at 3 days after injury and then returning to basal levels by 30 days post injury (Figure 1C). Because chymotrypsin-like and trypsin-like proteasome activity dramatically increased to a peak level 3 days after injury (Figure 1B), we then examined proteasome activity in only myocytes, excluding non-muscle cells, such as inflammatory cells, on day 3 post injury. At this time point, chymotrypsin-like and trypsin-like proteasome activities were significantly higher in myoblasts from injured animals, compared with those from control mice (Figures 1D, 1E, S1F, and S1G). These results showed that proteasome activity is dramatically increased in skeletal muscle and myoblasts during the early phase of muscle regeneration.
Figure 1.

Proteasome Activity in Skeletal Muscle during Muscle Regeneration
(A) Time course for CTX treatment and tissue harvesting.
(B) Chymotrypsin-like and trypsin-like proteasome activities (relative to those in intact tissue) in tibialis anterior (TA) muscles at 3, 7, and 14 days after injury are shown. Data represent means ± SEM (t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus intact; n = 3–5 per group). IU, international units.
(C) Immunoblotting analysis of ubiquitin in the TA muscles at 3, 7, and 14 days post injury. Data represent means ± SEM (t test: ∗∗∗p < 0.001 versus intact; n = 3–5 per group).
(D) Time course analysis of satellite cells at 3 days after injury.
(E) Chymotrypsin-like and trypsin-like proteasome activities (relative to those in intact satellite cells) in satellite cells sorted from TA muscles at 3 days post injury. Data represent means ± SEM (t test: ∗∗∗p < 0.001 versus intact satellite cells; n = 4–5 per group). IU, international units.
See also Figures S1.
Satellite Cell-specific Rpt3-Knockout Mice Exhibit No Obvious Phenotype in Muscle
To investigate the function of the proteasome system in satellite cells, we generated satellite cell-specific Rpt3 conditional knockout (Rpt3-scKO) mice by crossing Pax7CreERT2 mice (Lepper and Fan, 2010) with Rpt3f/f mice (Kitajima et al., 2014, Tashiro et al., 2012). Previously, we reported that deficiency of Rpt3 in skeletal muscle or motor neurons causes proteasome insufficiency (Kitajima et al., 2014, Tashiro et al., 2012). First, we examined the expression levels of Rpt3 in muscle stem cells during proliferation and differentiation. Upon activation, muscle satellite cells proliferate, downregulate Pax7, and differentiate (Kitajima and Ono, 2018, Yin et al., 2013). Rpt3 gene expression in satellite cell-derived myoblasts did not differ during the proliferation and differentiation processes (Figure S2). Genetic inactivation of Rpt3 was induced by repeated intraperitoneal injection of tamoxifen (Tmx) into adult Pax7CreERT2/+; Rpt3f/f mice, using Tmx-treated Rpt3f/f littermates as the wildtype control (Figure 2A). Following Tmx treatment in vivo, Rpt3 expression was significantly reduced in satellite cells (Figure 2B). In addition, chymotrypsin-like and trypsin-like proteasome activities were significantly lower in satellite cells from Rpt3-scKO mice (Figure 2C). Moreover, Rpt3 gene knockdown was performed in the C2C12 myoblast cell line (Figure S3A). Two small interfering RNAs (siRNAs) were used and siRNA (#2) resulted in a greater than 90% reduction in Rpt3 expression (Figure S3B), and thus was used in further experiments. Evaluation of proteasome function revealed that chymotrypsin-like and trypsin-like protease activities were significantly decreased 48 and 72 hr after gene knockdown (Figure S3C). These results revealed the efficiency of the satellite cell-specific Rpt3 conditional knockout in our mouse model.
Figure 2.

Satellite Cell-specific Rpt3-Knockout Mice Exhibit No Obvious Skeletal Muscle Phenotype
(A) Time course for tamoxifen (Tmx) treatment and tissue harvesting. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3-knockout mice (Pax7CreERT2/+; Rpt3f/f).
(B) Relative expression of Rpt3 mRNA in freshly isolated satellite cells derived from Con and scKO mice after Tmx injection. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 4 per group). AU, arbitrary units.
(C) Chymotrypsin-like and trypsin-like proteasome activities (relative to Con) in freshly isolated satellite cells derived from Con and scKO mice after Tmx injection. Data represent means ± SEM (t test: ∗∗p < 0.01; n = 4–5 per group). IU, international units.
(D) Change in body weight (g) after Tmx injection. Data represent mean ± SD (NS, statistically nonsignificant, n = 5–10 per group).
(E) Change in tibialis anterior (TA) muscle weight (g) at 2 months after Tmx injection. Data represent mean ± SD (NS, statistically nonsignificant, n = 4–6 per group).
(F) H&E staining of intact TA muscle 2 months after Tmx injection. Scale bar, 50 μm. Also shown in Figure S1D.
(G) Endurance time(s) of Con and scKO mice. Data represent mean ± SD (NS, statistically nonsignificant, n = 4–6 per group).
See also Figures S2–S4.
Next, we investigated the effect of Rpt3 deficiency in satellite cells on skeletal muscle in vivo. There was no change in body weight between control and Rpt3-scKO mice from 1 to 8 months after Tmx treatment (Figure 2D). There was also no difference in TA muscle weight (Figure 2E) and H&E staining of muscle cross sections (Figure 2F) 2 months after Tmx treatment. Other than limb muscles, in the diaphragm, there was no difference in the H&E staining of muscle cross sections and muscle cross-sectional area, 1 month after Tmx treatment (Figures S4A–S4C). Furthermore, when we performed an endurance test to investigate muscle function, there was no significant difference between control and Rpt3-scKO mice (Figure 2G). These results suggest that satellite cell-specific Rpt3 knockout has no obvious effect on intact muscle in mice.
Satellite Cell-specific Rpt3-scKO Mice Are Associated with a Remarkable Defect in Regeneration
To evaluate the role of Rpt3 in satellite cells during muscle regeneration in vivo, we genetically inactivated Rpt3 via five daily intraperitoneal injections of Tmx into Pax7CreERT2/+; Rpt3f/f mice. Intramuscular injection of CTX was performed to induce regeneration of the TA muscle after 2 days of Tmx treatment (Figure 3A). We showed the muscle weight was markedly decreased in Rpt3-scKO mice after regeneration compared with that in control mice at day 7 and 14 post CTX treatment (Figures 3B–3D). Correspondingly, histological analysis confirmed a significant defect in muscle regeneration accompanied by a considerable number of infiltrating inflammatory cells in Rpt3-scKO mice at day 14 after CTX treatment, although efficient muscle regeneration accompanied by centrally nucleated myofibers was observed in control mice (Figure 3E). Immunohistochemistry also revealed incomplete regeneration with marked fibrosis at day 14 post CTX treatment in Rpt3-scKO mice (Figure 3F). In addition, the cross-sectional area of the regenerated fibers was decreased at day 14 post CTX treatment in Rpt3-scKO mice (Figure 3G). To evaluate muscle regeneration in the early phase, we assessed the expression of MYOD, which is a marker of myoblasts. MYOD+ cells were not frequently found in Rpt3-scKO mice 3 days after CTX treatment (Figure 3H). Taken together, our data indicate that the function of Rpt3 in satellite cell is indispensable for muscle regeneration in vivo.
Figure 3.

Satellite Cell-specific Deletion of Rpt3 Prevents Muscle Regeneration
(A) Time course for tamoxifen (Tmx) and cardiotoxin (CTX) treatment. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3 knockout mice (Pax7CreERT2/+; Rpt3f/f).
(B) Representative images of tibialis anterior (TA) muscles in Con and scKO mice.
(C and D) Change in (C) muscle weight (mg) and (D) muscle weight (mg)/body weight (g). Data represent mean ± SEM (t test: ∗p < 0.05, ∗∗p < 0.01 versus Con, n = 5–9 per group).
(E) H&E staining of intact muscles and those injured by CTX injection, analyzed at 3, 7, and 14 days post injury. Scale bar, 50 μm.
(F) Immunostaining of collagen I (red) and DAPI (blue) in TA muscle cryosections from scKO mice 14 days after injury. Scale bar, 100 μm. The y axis shows the mean collagen I fluorescence intensity (ratio). Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 5 per group).
(G) Immunostaining of Laminin (red) and DAPI (blue) in TA muscle cryosections from scKO mice 14 days after injury. Scale bar, 100 μm. The y axis shows the mean cross-sectional area (CSA). Data represent means ± SD (t test: ∗∗p < 0.01; n = 3 per group).
(H) Immunostaining for MYOD (green), Laminin2α, and DAPI (blue) in TA muscle cryosections from scKO mice 3 days after injury. Scale bar, 50 μm. The y axis shows the relative ratio of MYOD-positive areas. Data represent means ± SD (t test: ∗∗p < 0.01; n = 3 per group).
Loss of Rpt3 Leads to a Depletion of the Quiescent Satellite Cell Pool
Previous studies have confirmed the absolute necessity of PAX7-positive satellite cells for muscle regeneration (Lepper et al., 2011, Relaix and Zammit, 2012, Sambasivan et al., 2011, von Maltzahn et al., 2013). Because muscle regeneration was impaired in Rpt3-scKO mice, we next examined how Rpt3 knockout affects quiescent satellite cells in adult resting skeletal muscles. After 5 consecutive days of Tmx injection, we found a surprisingly rapid decline in the number of quiescent satellite cells in freshly isolated extensor digitorum longus (EDL) myofibers from Rpt3-scKO mice, but not in those from control mice (Figures 4A–4C). Specifically, 20% of quiescent satellite cells were lost within 5 days, 80% were lost within 10 days, and 90% were lost within 15 days in Rpt3-scKO mice (Figure 4C). Similarly, immunofluorescence staining for PAX7 and laminin in TA muscle cross sections indicated the robust ablation (90% loss) of PAX7+ quiescent satellite cells in Rpt3-scKO mice at day 15 after Tmx treatment (Figures 4D and 4E). In addition, immunofluorescence staining for M-cadherin, as another satellite cell marker in TA and diaphragm muscles, revealed a robust decrease in satellite cells in Rpt3-scKO mice (Figures 4F, 4G, S4D, and S4E). We further analyzed the number of quiescent satellite cells in limb muscles by flow cytometry. We confirmed the rapid depletion of the satellite cell fraction in Rpt3-scKO mice, consistent with immunohistochemical analysis (Figures 4H, 4I, and S5A–S5D). These results demonstrate that loss of Rpt3 leads to depletion of quiescent satellite cells in adult resting muscle.
Figure 4.

Rpt3 Inactivation Depletes the Satellite Cell Pool
(A) Time course of tamoxifen (Tmx) treatment and tissue harvesting. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3 knockout mice (Pax7CreERT2/+; Rpt3f/f).
(B) Immunostaining for PAX7 (green) and DAPI (blue) in the extensor digitorum longus (EDL) single fiber 5 days after Tmx induction. Arrows indicate satellite cells. Scale bar, 50 μm.
(C) Number of satellite cells per fresh EDL myofiber from Con and scKO mice 0–15 days after Tmx treatment. Data represent mean ± SEM (t test: ∗p < 0.05, ∗∗∗p < 0.001; day 0, n = 5; day 3, n = 4; day 5, n = 5; day 10, n = 5; day 15, n = 4 per group; more than 15 myofibers per animal).
(D) Immunostaining for PAX7 (green), laminin (red), and DAPI (blue) in tibialis anterior (TA) muscle cryosections 5 days after Tmx treatment. Arrows indicate satellite cells. Scale bar, 20 μm.
(E) Average number of satellite cells per 100 fibers. Data represent mean ± SEM (t test: ∗∗p < 0.01; day 5, n = 4; day 15, n = 5 for each group).
(F) Immunostaining for M-cadherin (green), Laminin2α (red), and DAPI (blue) in intact TA muscle cryosections from scKO mice. Arrows indicate satellite cells. Scale bar, 50 μm.
(G) Average number of M-cadherin+ cells per 100 fibers. Data represent means ± SD (t test: ∗∗∗p < 0.001; n = 3 per group).
(H) Fluorescence-activated cell sorting profiles of mononuclear cells derived from Con and scKO mice 2 and 15 days after Tmx treatment. The gated profiles show satellite cell fractions (SM/C-2.6 + CD31− CD45− Sca1−) from Con and scKO mice.
(I) Relative satellite cell fractions from Con and scKO mice. Data represent mean ± SEM (t test: ∗∗∗p < 0.001; day 2, n = 4; day 15, n = 4 per group). SC, satellite cell.
See also Figures S4 and S5.
Deficiency of Rpt3 in Satellite Cells Results in Proliferative Defects during Muscle Regeneration In Vivo
To clarify whether the loss of satellite cells in Rpt3-scKO mice is caused by apoptosis, we performed apoptosis experiments using Cleaved-Caspase 3 in vivo (Figure 5A). Numbers of Cleaved-Caspase 3+ cells relative to PAX7+ cells were significantly increased in the intact TA muscle of Rpt3-scKO mice, 3 days after Tmx treatment (Figure 5B). Next, to elucidate cell proliferation in the satellite cells of Rpt3-scKO mice in vivo, we performed EdU incorporation assays and immunostaining for KI67, a proliferative marker that is not expressed in G0 cells (Figure 5C). The proliferative ability of intact TA muscle in Rpt3-scKO mice was not different from that in control mice (Figures 5D and 5E).
Figure 5.

Deficiency of Rpt3 in Satellite Cells Results in Proliferative Defects during Muscle Regeneration In Vivo
(A) Time course for tamoxifen (Tmx) treatment and tissue harvesting. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3 knockout mice (Pax7CreERT2/+; Rpt3f/f).
(B) Immunostaining for Cleaved-Caspase3 (green), Pax7 (red), and DAPI (blue) in tibialis anterior (TA) muscle cryosections 3 days after Tmx treatment. Arrows indicate satellite cells. Scale bar, 10 μm. The y axis shows the ratio of Cleaved-Caspase3-positive cells to PAX7-positive cells. Data represent means ± SD (t test: ∗∗p < 0.01; n = 3 per group).
(C) Time course for Tmx treatment and tissue harvesting.
(D) Immunostaining for EdU (green), M-cadherin (red), and DAPI (blue) in TA muscle cryosections, 3 days after Tmx treatment. Arrows indicate satellite cells. Scale bar, 10 μm. The y axis shows the ratio of EdU-positive cells to M-cadherin-positive cells. Data represent means ± SD (t test: NS, no significance; n = 3 per group).
(E) Immunostaining for M-cadherin (green), KI67 (red), and DAPI (blue) in TA muscle cryosections, 3 days after Tmx treatment. Arrows indicate satellite cells. Scale bar, 10 μm. The y axis shows the ratio of KI67-positive cells to M-cadherin-positive cells. Data represent means ± SD (t test: NS, no significance; n = 3 per group).
(F) Time course for Tmx treatment and tissue harvesting.
(G) Immunostaining for EdU (green), M-cadherin (red), and DAPI (blue) in TA muscle cryosections, 3 days after injury. Scale bar, 50 μm. The y axis shows the number of EdU-positive and M-cadherin-positive cells per field. Data represent means ± SD (t test: ∗∗p < 0.01; n = 3 per group).
Moreover, to examine the proliferative potential of Rpt3-scKO satellite cells during muscle regeneration, an EdU incorporation assay was performed (Figure 5F). EdU+ M-cadherin+ cells were significantly decreased in Rpt3-scKO mice when compared with the control mice (Figure 5G). These data suggested that the deficiency of Rpt3 in satellite cells results in proliferative defects during muscle regeneration in vivo.
Deletion of Rpt3 in Satellite Cells Induces a Proliferation Defect and Apoptosis
We next focused on how the proliferative state of satellite cells is affected by proteasome dysfunction mediated by Rpt3-deficiency in vitro. Satellite cells isolated from Pax7CreERT2/+; Rpt3f/f mice were plated and treated with 4-hydroxy tamoxifen (4OH-Tmx) for 2 days to genetically delete Rpt3 in satellite cells (Figure 6A). Expression of Rpt3 was suppressed by 85% in Rpt3 KO satellite-cell-derived myoblasts compared with that in control myoblasts (Figure 6B). Cell proliferation was evaluated by performing EdU incorporation assays in vitro. The proliferative ability of Rpt3-deficient primary myoblasts was decreased compared with that in controls (Figure 6C). In addition, to investigate whether cell proliferation defects also occur after Rpt3 knockdown in other cells, Rpt3-knockdown experiments were performed using primary fibroblasts (Figure S6A). Cell proliferation was suppressed in primary fibroblasts with Rpt3-knockdown compared with that in controls (Figures S6B and S6C).
Figure 6.

Deletion of Rpt3 in Satellite Cells Induces a Proliferation Defect and Apoptosis
(A) Time course for tamoxifen (Tmx) treatment and tissue harvesting. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3 knockout mice (Pax7CreERT2/+; Rpt3f/f).
(B) Relative expression of Rpt3 mRNA in Pax7CreERT2/+; Rpt3f/f satellite cells treated with vehicle or 4OH-Tmx. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 3 per group). AU, arbitrary units.
(C) Immunostaining for EdU (green) and DAPI (blue) in satellite cells treated with vehicle or 4OH-Tmx. Scale bar, 50 μm. The y axis shows the relative ratio of EdU-positive cells. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 4 per group).
(D) Immunostaining for TUNEL (green) and DAPI (blue) in satellite cells treated with vehicle or 4OH-Tmx. Arrows indicate TUNEL-positive cells. Scale bar, 100 μm. The y axis shows the ratio of TUNEL-positive cells to DAPI-positive cells. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 3 per group).
(E) Immunoblotting analysis of Cleaved-Caspase3 and GAPDH in satellite cells treated with vehicle or 4OH-Tmx.
(F) Relative ratio of DAPI-positive cells from Con or scKO mice. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 3 per group).
(G) Time course of Tmx treatment and tissue harvesting. Con siRNA indicates control siRNA transfection and Rpt3 siRNA indicates Rpt3 siRNA transfection.
(H) Immunostaining for EdU (green) and DAPI (blue) in C2C12 cells after siRNA transfection. Scale bar, 50 μm. The y axis shows the relative ratio of EdU-positive cells. Data represent means ± SEM (t test: ∗∗p < 0.01; n = 3 per group).
(I) Immunostaining for TUNEL (green) and DAPI (blue) in C2C12 cells after siRNA transfection. Arrows indicate TUNEL-positive cells. Scale bar, 100 μm. The y axis shows the ratio of TUNEL-positive cells to DAPI-positive cells. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 3 per group).
(J) Relative ratio of DAPI-positive cells. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 3 per group).
See also Figures S6 and S7.
We next asked if Rpt3-knockout-satellite cells can undergo myogenic differentiation. Two days after the induction of differentiation, control myoblasts already displayed an obviously elongated morphology, a hallmark of differentiation (Figures S7A and S7B). In contrast, Rpt3-scKO primary myoblasts were mostly spherical (Figure S7B), indicative of a differentiation defect. We further quantified the differentiation index, which measures the fraction of myonuclei that are located in myosin heavy chain (MyHC+)-expressing cells. Although greater than 50% of nuclei in the control were located in MyHC+ cells, only 15% of nuclei in Rpt3-scKO mice were found in MyHC+ cells (Figure S7B).
To determine if the reduction in satellite cell-derived myoblasts proliferation was also due to cell death, apoptosis was evaluated by performing TUNEL assays. TUNEL-positive apoptotic cells were detected more frequently in primary myoblasts of Rpt3-scKO mice (Figure 6D). This was further supported by the upregulation of Cleaved-Caspase 3, an indicator of apoptosis, in Rpt3-inactivated primary myoblasts (Figure 6E). Consistent with these results, the number of cells was reduced in Rpt3-scKO mice compared with that in control mice (Figure 6F). Upon knockdown of Rpt3, proliferation and cell numbers decreased, whereas apoptotic cells were more frequently detected in Rpt3-ablated cells (Figures 6G–6J). These results demonstrate that proteasome dysfunction mediated by Rpt3 deficiency in satellite cells induces a proliferation defect and apoptosis.
p53 Knockdown in Rpt3-Deficient Satellite Cells Rescues the Defect in Proliferation
We next investigated the molecular mechanism underlying Rpt3 deficiency-induced dysfunction in activated satellite cells. To identify the pathways affected by Rpt3 deficiency in an unbiased manner, we performed microarray analysis of quiescent satellite cells from control and Rpt3-scKO mice (accession number GSE114354). We identified 1,254 genes that were up- or downregulated with p values < 0.05 (Table S2). Significantly differentially expressed genes were imported into the DAVID v6.7 annotation tool, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed. The enrichment of specific pathway components into functionally regulated gene groups was characterized with reference to the KEGG pathway database. This analysis identified the top two categories as being related to the proteasome and p53 signaling pathways (Figure 7A).
Figure 7.

p53 Knockdown in Rpt3-Deficient Satellite Cells Rescues the Defect in Proliferation
(A) DAVID v6.7 functional annotation bioinformatics microarray analysis software was used to obtain KEGG pathway functional classification. Only KEGG pathway terms for classification that showed statistically significant differences, in terms of the number of differentially regulated genes (satellite cells from satellite cell-specific Rpt3 mice versus satellite cells from control mice), are shown (p < 0.05). The 1,254 significantly differentially regulated genes are shown in Table S2. The red dash line denotes the significance level of 0.05.
(B) Time course of tamoxifen (Tmx) treatment and tissue harvesting. Con indicates Rpt3f/f mice and scKO indicates satellite cell-specific Rpt3 knockout mice (Pax7CreERT2/+; Rpt3f/f).
(C) Real-time RT-PCR was used to measure the mRNA expression of p53-related genes (p53, p21, Cyclin D1, and Cdk4) in Pax7CreERT2/+; Rpt3f/f satellite cells treated with vehicle or 4OH-Tmx. Data represent means ± SEM (t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 4–5 per group). AU, arbitrary units.
(D) Immunoblotting analysis of relative protein levels of p53 targets in Pax7CreERT2/+; Rpt3f/f satellite cells treated with vehicle or 4OH-Tmx.
(E) Immunostaining for p53 (green) and DAPI (blue) in Pax7CreERT2/+; Rpt3f/f satellite cells treated with vehicle or 4OH-Tmx. Arrows indicate p53-positive cells. Scale bar, 10 μm. The y axis shows the relative ratio of p53-positive cells. Data represent means ± SEM (t test: ∗∗∗p < 0.001; n = 4 per group).
(F) Time course analysis of p53 siRNA knockdown and Tmx treatment.
(G) Immunostaining for EdU (green) and DAPI (blue) in satellite cells treated with vehicle or 4OH-Tmx. Scale bar, 50 μm. The y axis shows the relative ratio of EdU-positive cells. Data represent means ± SEM (one-way ANOVA followed by the Bonferroni post hoc test: ∗∗p < 0.01; NS, no significance; n = 4 per group).
(H) Relative ratio of DAPI-positive cells. Data represent means ± SEM (one-way ANOVA followed by the Bonferroni post hoc test: ∗∗p < 0.01, ∗∗∗p < 0.01; NS, no significance; n = 4–6 per group).
See also Figure S7.
p53 is known to inhibit cell proliferation by both blocking cell-cycle progression and promoting apoptotic cell death (Vousden and Prives, 2009). As shown in Figures 6C, 6D, 6H, and 6I, suppression of cell proliferation and induction of apoptotic cell death was observed in Rpt3-scKO mice. We therefore examined whether the p53 pathway is involved in the regulation of Rpt3-scKO satellite cells. Satellite cells isolated from Pax7CreERT2/+; Rpt3f/f mice were plated and then treated with 4OH-Tmx for 2 days to genetically delete Rpt3 in satellite cells (Figure 7B). Expression of Rpt3 was suppressed by 80% in Rpt3 KO satellite-cell-derived myoblasts compared with that in control myoblasts (Figure 7C). As expected, levels of p53, as well as p21, which is a well-known p53-downstream target, were elevated, whereas Cyclind1 and Cdk4, which are cell-cycle-related genes associated with p53, were suppressed in Rpt3-scKO satellite cell-derived myoblasts (Figure 7C). Consistent with mRNA levels, western blotting also demonstrated a dramatic increase in p53 protein and its phosphorylation (ser392) in Rpt3-scKO satellite cell-derived myoblasts (Figure 7D). p53-related proliferation is regulated by phosphorylated Rb, which was decreased in Rpt3-scKO satellite cell-derived myoblasts (Figure 7D). Moreover, based on immunohistochemistry, a greater than 20-fold increase in the number of p53-positive cells was confirmed in Rpt3 KO satellite-cell-derived myoblasts as compared with those in controls (Figure 7E). These results demonstrate that Rpt3 deletion in satellite-cell-derived myoblasts leads to the activation of p53 signaling in satellite cells.
Finally, to test if activation of p53 signaling is responsible for the inhibition of proliferation in Rpt3-scKO mice, we examined the effect of p53 silencing on Rpt3-null satellite cell-derived myoblasts. p53 knockdown was performed in C2C12 cells (Figure S7C). Two siRNAs were used and siRNA (#2) showed greater than 90% reduction in p53 gene expression (Figures S7D–S7F) and was thus used for further experiments. Satellite cells isolated from Pax7CreERT2/+; Rpt3f/f mice were plated and then treated with 4OH-Tmx for 2 days after p53 siRNA transfection (Figure 7F). Importantly, the proliferative ability as well as the number of cells were both restored in Rpt3-null myoblasts when transfected with p53 siRNA (Figures 7G, 7H, S7G, and S7I), although the differentiation ability was not improved with p53 knockdown (Figures S7J and S7K). Taken together, these results show that knockdown of p53 rescues defective proliferation in Rpt3-deficient primary myoblasts.
Discussion
In this study, we report that the proteasome system is indispensable for the maintenance of muscle stem cells. Ablation of Rpt3 in satellite cells impairs their ability to proliferate, survive, and differentiate, resulting in a severe defect in muscle regeneration. We also found that proteasome inactivation by Rpt3 deficiency in primary myoblasts inhibits cell proliferation and induces apoptosis. Further, proteasome dysfunction conferred by satellite cell-specific Rpt3 knockout induces p53 activation. Previously, it was determined that p53 inhibits cell proliferation by both blocking cell-cycle progression and promoting apoptotic cell death (Vousden and Prives, 2009). We also found that p53 knockdown rescued cell proliferation defects in Rpt3-deficient myoblasts.
The ubiquitin-proteasome system functions to degrade most long- and short-lived normal and abnormal intracellular proteins (Collins and Goldberg, 2017, Goldberg, 2003). Especially in muscle, proteolysis by the ubiquitin-proteasome system is a major mechanism involved in myofibrillar protein degradation (Attaix et al., 1998, Hasselgren et al., 2002). Therefore, when examining proteasome activity during regeneration, we found that both chymotrypsin-like and trypsin-like proteasome activities were increased 3 days after CTX injection, indicating that the early phase of muscle regeneration is associated with the activation of machinery involved in protein degradation. Furthermore, the accumulation of ubiquitinated proteins was also most accelerated on day 3 post injury, indicating that proteasome-related proteolysis was most enhanced at this time point. Interestingly, MYOD, which constitutes an important myogenic transcription factor for muscle regeneration (Charge and Rudnicki, 2004, Sabourin et al., 1999), was also particularly elevated on day 3 post injury. In a previous study, proteasome activity was found to be strongly correlated with proliferating cell nuclear antigen protein levels, suggesting that the proteasome plays a key role in myoblast proliferation (Duguez et al., 2003). Thus, it is suggested that enhancing the proteasome system during the early phase is an important event for normal muscle regeneration.
Previous studies have confirmed the absolute requirement for Pax7-positive satellite cells during muscle regeneration (Lepper et al., 2011, Relaix and Zammit, 2012, Sambasivan et al., 2011, von Maltzahn et al., 2013). Our data support these findings that satellite cells are essential for muscle regeneration. When the number of satellite cells decreases, muscle regeneration fails. These data might indicate that the number of satellite cells is also important. However, other cells. such as mesoangioblasts and PW1+ interstitial cells, which contribute to regeneration, have been identified in muscle tissue (Mitchell et al., 2010, Sampaolesi et al., 2003). Thus, we cannot exclude the possibility that the regenerative defect in Rpt3-scKO mice at day 14 after CTX injection could be improved after a long period of recovery by contributing to the myogenic potential of other cells involved in regeneration (Mitchell et al., 2010, Sampaolesi et al., 2003).
Proteasome insufficiency due to Rpt3 deficiency increased the protein levels of Cleaved-Caspase 3 and caused apoptosis. Furthermore, we performed immunoblotting analysis of Bax, Bak, Bcl-2, and Mcl-1 proteins related to apoptosis, but there was no significant change particularly (data not shown). A previous study reported that the proteasome inhibitor MG132 induces caspase-3-dependent apoptosis, and a caspase-3 inhibitor was found to reduce MG132-induced apoptosis in human mast cells (Westerberg et al., 2012). Our data show that proteasome dysfunction by loss of Rpt3 is accompanied by apoptosis, which is at least due to enhancement of the caspase 3 activity. Therefore, in this study, it is considered that apoptosis in scKO primary myoblasts might be alleviated by the inhibition of Caspase 3.
There have been many previous studies on the association between the proteasome system and p53. Among these, one report suggested that the ubiquitination level of p53 is elevated after treatment with the proteasome inhibitor MG132 (Maki et al., 1996), suggesting that this protein is degraded by the proteasome and that its stability is controlled by ubiquitin-dependent degradation. Consistent with these findings, we demonstrated p53 stability in primary myoblasts exhibiting proteasome dysfunction (via Rpt3 ablation). p53 inhibits cell proliferation by both blocking cell-cycle progression and promoting apoptotic cell death (Vousden and Prives, 2009). In addition, proteasome inhibition has been shown to induce apoptosis in several different cell types (Concannon et al., 2007, Ding et al., 2007, Pandit and Gartel, 2011). Therefore, we hypothesized that proteasome dysfunction, mediated by Rpt3 deletion, results in cell death and the inhibition of cell proliferation due to p53 hyperactivation. Because p53 is upregulated in Rpt3-deficient myoblasts, we examined whether knockdown of p53 rescues the proliferation defects observed in Rpt3-deficient myoblasts. Importantly, we found that inhibition of p53 successfully rescued these effects, indicating that the proteasome system in muscle stem cells regulates cell proliferation at least in part through p53.
Myogenesis is a complex process controlled by the spatiotemporal expression of many MRFs and transcription factors (Braun and Gautel, 2011, Parker et al., 2003, Wagers and Conboy, 2005). As myoblast differentiation proceeds, the timely synthesis and degradation of appropriate myogenic proteins are required, which suggests a significant role for adaptive proteolysis in the myogenic process. Indeed, previous studies have reported that the complex process of myogenic differentiation, which begins with cell-cycle arrest and ends with the fusion of individual myoblasts to form multinucleated myotubes, is related to the proteasome system (Abu Hatoum et al., 1998, Gardrat et al., 1997, Kim et al., 1998). In addition, inhibition or knockdown of the proteasome can block the fusion of myoblasts and inhibit differentiation (Gardrat et al., 1997, Kim et al., 1998). Consistent with previous studies, proteasome dysfunction conferred by Rpt3 deficiency was found to suppress the myogenic differentiation of primary myoblasts, suggesting that proper proteasome function is necessary for myogenic differentiation. In addition, it has been previously shown that p53 can promote muscle differentiation in vitro (Halevy et al., 1995, Porrello et al., 2000, Soddu et al., 1996, Tamir and Bengal, 1998, Weintraub et al., 1991). However, despite the presence of p53 observed in Rpt3-deficient primary myoblasts, Rpt3 ablation was shown to suppress myogenic differentiation. This might be because p53 expression was excessive. Indeed, a 20-fold increase in p53-positive cells was observed in Rpt3-deficient myoblasts, as compared with that in controls, based on immunostaining. A recent study reported that tight control of p53 levels in myoblasts regulates the balance between differentiation and return to quiescence (Flamini et al., 2018), which could indicate appropriate expression of p53 during myogenic differentiation.
Satellite cells provide myonuclei for postnatal muscle growth and for maintenance and regeneration in adults (Relaix and Zammit, 2012, Yin et al., 2013), indicating that the maintenance of satellite cells is essential for the functional homeostasis of skeletal muscle. Moreover, recent studies indicated that skeletal muscle disease is exacerbated by defective satellite cells (Crist, 2017). Importantly, we found that proteasome dysfunction by loss of Rpt3 in satellite cells leads to a dramatic depletion of the quiescent satellite cell pool approximately 2 weeks after knockout induction. Previous studies demonstrated that proteolysis by the proteasome is essential to maintain amino acid pools (Suraweera et al., 2012, Vabulas and Hartl, 2005). Although quiescent satellite cells are thought to be associated with very low turnover in steady state conditions (Lepper and Fan, 2010), minimal amino acid pools obtained from proteolysis by the proteasome system are necessary for the normal maintenance of cells. Therefore, proteasome dysfunction, conferred by Rpt3 deficiency, might have resulted in serious consequences, depriving the cells of pooled resources for cellular maintenance. We hypothesize that satellite cell-specific Rpt3 deficiency might have blocked the cellular “recycling system” that is essential for the maintenance of satellite cells; this question needs to be further examined.
Rpt3 deficiency in primary myoblasts and Rpt3 knockdown in a myoblast cell line resulted in a decrease in proteasome activity, leading to apoptotic cell death. There are several reports showing the relationship between proteasomal activity and aging, including lifespan. During aging, proteasomal activity is decreased in several tissues, such as the brain (Zeng et al., 2005), liver (Dasuri et al., 2009, Hayashi and Goto, 1998), heart (Bulteau et al., 2002), and muscle (Ferrington et al., 2005). Overexpression of proteasome subunits in yeast and Caenorhabditis elegans has been shown to lead to an increase in proteasome activity as well as prolonged lifespan (Chen et al., 2006, Vilchez et al., 2012), whereas flies and mice with a genetic decrease in proteasome activity have a shortened lifespan (Tomaru et al., 2012, Tonoki et al., 2009). Previously, we have also reported a shortened lifespan even in muscle-specific Rpt3-knockout mice (Kitajima et al., 2014). Taken together, these data suggest that there is a close relationship between the proteasome system and aging in vivo and in vitro. Whether adult somatic stem cells also have enhanced proteasome activity remains to be elucidated, but the maintenance of this activity might critically affect organismal aging (Vilchez et al., 2014). Therefore, our findings using muscle stem cells will lead to a detailed understanding of proteolysis and its associated molecular mechanisms in muscle stem cells. Our results show that the ubiquitin-proteasome system is indispensable for the maintenance of muscle stem cells. Further study is needed to understand the role of stem cells in various physiological processes involving the proteasome system; however, this work provides the fundamentals for understanding the regulation of proteolysis in muscle stem cells.
Experimental Procedures
Cell Culture
Satellite cells were cultured in GM (DMEM supplemented with 30% fetal bovine serum, 1% chick embryo extract, 10 ng/mL basic fibroblast growth factor, and 1% penicillin-streptomycin) on culture dishes coated with Matrigel (BD Biosciences). Myogenic differentiation was induced in DM (DMEM supplemented with 5% horse serum and 1% penicillin-streptomycin). For in vitro genetic deletion, 4-OH Tmx (1 μM) was added to culture medium for 2 days to induce Cre-mediated deletion.
Mouse C2C12 myoblasts were cultured in GM (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin). The cells were transfected with 30 nM siRNA using Lipofectamine RNAiMAX (Invitrogen).
Single Fiber Isolation
Individual myofibers were isolated from the EDL muscle as we described previously (Kitajima et al., 2016, Ono et al., 2015). In brief, EDL myofibers were digested using 0.2% type I collagenase (Worthington) in DMEM for 90 min at 37°C with 5% CO2. Muscles were mechanically dissociated and then washed five times to eliminate debris and contaminating cells. For immunohistochemical analysis, EDL myofibers were immediately fixed using 4% paraformaldehyde (PFA, Wako) in PBS.
Author Contributions
Y.K. designed the experiments, performed the experiments, interpreted the data, assembled the input data, and wrote the manuscript. N.S. interpreted the data. A.N., S.O., and K.Y. performed the experiments. Y.T. and R.T. produced the animals. Y.O. interpreted the data, assembled the input data, and wrote the manuscript. M.A. interpreted the data. R.N. interpreted the data and assembled the input data. All authors discussed the results and implications and commented on the manuscript.
Acknowledgments
We thank So-ichiro Fukada (Osaka University, Osaka, Japan) for providing the SM/C-2.6 antibody. This work was supported by a Grant-in-Aid for JSPS Research Fellows (16J00431 to Y.K.). This work was also supported by a Grant-in-Aid for Scientific Research KAKENHI (15K16486 to Y.K., 18H04080 to R.N., and 18K17857 to Y.K.), AMED (16bm0704010h0001 to Y.O. and 18ek0109383h0001 to Y.O.), the Nakatomi Foundation (to Y.K.), and the Uehara Memorial Foundation (to Y.K.).
Published: November 8, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.10.009.
Contributor Information
Yasuo Kitajima, Email: yasuo.kitajima1108@gmail.com.
Yusuke Ono, Email: yusuke-ono@nagasaki-u.ac.jp.
Ryoichi Nagatomi, Email: nagatomi@med.tohoku.ac.jp.
Accession Numbers
The microarray dataset in this study is available from the GEO public depository under the accession number: GEO: GSE114354.
Supplemental Information
References
- Abu Hatoum O., Gross-Mesilaty S., Breitschopf K., Hoffman A., Gonen H., Ciechanover A., Bengal E. Degradation of myogenic transcription factor MyoD by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol. Cell. Biol. 1998;18:5670–5677. doi: 10.1128/mcb.18.10.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attaix D., Aurousseau E., Combaret L., Kee A., Larbaud D., Ralliere C., Souweine B., Taillandier D., Tilignac T. Ubiquitin-proteasome-dependent proteolysis in skeletal muscle. Reprod. Nutr. Dev. 1998;38:153–165. doi: 10.1051/rnd:19980202. [DOI] [PubMed] [Google Scholar]
- Baumeister W., Walz J., Zuhl F., Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- Blau H.M., Cosgrove B.D., Ho A.T. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015;21:854–862. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 2011;12:349–361. doi: 10.1038/nrm3118. [DOI] [PubMed] [Google Scholar]
- Bulteau A.L., Szweda L.I., Friguet B. Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 2002;397:298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- Charge S.B., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- Chen Q., Thorpe J., Dohmen J.R., Li F., Keller J.N. Ump1 extends yeast lifespan and enhances viability during oxidative stress: central role for the proteasome? Free Radic. Biol. Med. 2006;40:120–126. doi: 10.1016/j.freeradbiomed.2005.08.048. [DOI] [PubMed] [Google Scholar]
- Collins G.A., Goldberg A.L. The logic of the 26S proteasome. Cell. 2017;169:792–806. doi: 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon C.G., Koehler B.F., Reimertz C., Murphy B.M., Bonner C., Thurow N., Ward M.W., Villunger A., Strasser A., Kogel D. Apoptosis induced by proteasome inhibition in cancer cells: predominant role of the p53/PUMA pathway. Oncogene. 2007;26:1681–1692. doi: 10.1038/sj.onc.1209974. [DOI] [PubMed] [Google Scholar]
- Crist C. Emerging new tools to study and treat muscle pathologies: genetics and molecular mechanisms underlying skeletal muscle development, regeneration, and disease. J. Pathol. 2017;241:264–272. doi: 10.1002/path.4830. [DOI] [PubMed] [Google Scholar]
- Dasuri K., Nguyen A., Zhang L., Fernandez-Kim O.S., Bruce-Keller A.J., Blalock B.A., Cabo R.D., Keller J.N. Comparison of rat liver and brain proteasomes for oxidative stress-induced inactivation: influence of ageing and dietary restriction. Free Radic. Res. 2009;43:28–36. doi: 10.1080/10715760802534812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W.X., Ni H.M., Chen X., Yu J., Zhang L., Yin X.M. A coordinated action of Bax, PUMA, and p53 promotes MG132-induced mitochondria activation and apoptosis in colon cancer cells. Mol. Cancer Ther. 2007;6:1062–1069. doi: 10.1158/1535-7163.MCT-06-0541. [DOI] [PubMed] [Google Scholar]
- Duguez S., Bihan M.C., Gouttefangeas D., Feasson L., Freyssenet D. Myogenic and nonmyogenic cells differentially express proteinases, Hsc/Hsp70, and BAG-1 during skeletal muscle regeneration. Am. J. Physiol. Endocrinol. Metab. 2003;285:E206–E215. doi: 10.1152/ajpendo.00331.2002. [DOI] [PubMed] [Google Scholar]
- Ferrington D.A., Husom A.D., Thompson L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- Flamini V., Ghadiali R.S., Antczak P., Rothwell A., Turnbull J.E., Pisconti A. The satellite cell niche regulates the balance between myoblast differentiation and self-renewal via p53. Stem Cell Reports. 2018;10:970–983. doi: 10.1016/j.stemcr.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardrat F., Montel V., Raymond J., Azanza J.L. Proteasome and myogenesis. Mol. Biol. Rep. 1997;24:77–81. doi: 10.1023/a:1006877214153. [DOI] [PubMed] [Google Scholar]
- Goldberg A.L. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Halevy O., Novitch B.G., Spicer D.B., Skapek S.X., Rhee J., Hannon G.J., Beach D., Lassar A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Hasselgren P.O., Wray C., Mammen J. Molecular regulation of muscle cachexia: it may be more than the proteasome. Biochem. Biophys. Res. Commun. 2002;290:1–10. doi: 10.1006/bbrc.2001.5849. [DOI] [PubMed] [Google Scholar]
- Hayashi T., Goto S. Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech. Ageing Dev. 1998;102:55–66. doi: 10.1016/s0047-6374(98)00011-6. [DOI] [PubMed] [Google Scholar]
- Kim S.S., Rhee S., Lee K.H., Kim J.H., Kim H.S., Kang M.S., Chung C.H. Inhibitors of the proteasome block the myogenic differentiation of rat L6 myoblasts. FEBS Lett. 1998;433:47–50. doi: 10.1016/s0014-5793(98)00883-7. [DOI] [PubMed] [Google Scholar]
- Kitajima Y., Ogawa S., Ono Y. Visualizing the functional heterogeneity of muscle stem cells. Methods Mol. Biol. 2016;1516:183–193. doi: 10.1007/7651_2016_349. [DOI] [PubMed] [Google Scholar]
- Kitajima Y., Ono Y. Visualization of PAX7 protein dynamics in muscle satellite cells in a YFP knock-in-mouse line. Skelet. Muscle. 2018;8:26. doi: 10.1186/s13395-018-0174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima Y., Tashiro Y., Suzuki N., Warita H., Kato M., Tateyama M., Ando R., Izumi R., Yamazaki M., Abe M. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014;127:5204–5217. doi: 10.1242/jcs.150961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C., Fan C.M. Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis. 2010;48:424–436. doi: 10.1002/dvg.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C., Partridge T.A., Fan C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki C.G., Huibregtse J.M., Howley P.M. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- Marx F.P., Soehn A.S., Berg D., Melle C., Schiesling C., Lang M., Kautzmann S., Strauss K.M., Franck T., Engelender S. The proteasomal subunit S6 ATPase is a novel synphilin-1 interacting protein–implications for Parkinson's disease. FASEB J. 2007;21:1759–1767. doi: 10.1096/fj.06-6734com. [DOI] [PubMed] [Google Scholar]
- Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K.J., Pannerec A., Cadot B., Parlakian A., Besson V., Gomes E.R., Marazzi G., Sassoon D.A. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 2010;12:257–266. doi: 10.1038/ncb2025. [DOI] [PubMed] [Google Scholar]
- Morgan J.E., Partridge T.A. Muscle satellite cells. Int. J. Biochem. Cell Biol. 2003;35:1151–1156. doi: 10.1016/s1357-2725(03)00042-6. [DOI] [PubMed] [Google Scholar]
- Ono Y., Urata Y., Goto S., Nakagawa S., Humbert P.O., Li T.S., Zammit P.S. Muscle stem cell fate is controlled by the cell-polarity protein Scrib. Cell Rep. 2015;10:1135–1148. doi: 10.1016/j.celrep.2015.01.045. [DOI] [PubMed] [Google Scholar]
- Pandit B., Gartel A.L. Proteasome inhibitors induce p53-independent apoptosis in human cancer cells. Am. J. Pathol. 2011;178:355–360. doi: 10.1016/j.ajpath.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M.H., Seale P., Rudnicki M.A. Looking back to the embryo: defining transcriptional networks in adult myogenesis. Nat. Rev. Genet. 2003;4:497–507. doi: 10.1038/nrg1109. [DOI] [PubMed] [Google Scholar]
- Porrello A., Cerone M.A., Coen S., Gurtner A., Fontemaggi G., Cimino L., Piaggio G., Sacchi A., Soddu S. p53 regulates myogenesis by triggering the differentiation activity of pRb. J. Cell Biol. 2000;151:1295–1304. doi: 10.1083/jcb.151.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- Sabourin L.A., Girgis-Gabardo A., Seale P., Asakura A., Rudnicki M.A. Reduced differentiation potential of primary MyoD-/- myogenic cells derived from adult skeletal muscle. J. Cell Biol. 1999;144:631–643. doi: 10.1083/jcb.144.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakao Y., Kawai T., Takeuchi O., Copeland N.G., Gilbert D.J., Jenkins N.A., Takeda K., Akira S. Mouse proteasomal ATPases Psmc3 and Psmc4: genomic organization and gene targeting. Genomics. 2000;67:1–7. doi: 10.1006/geno.2000.6231. [DOI] [PubMed] [Google Scholar]
- Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- Sampaolesi M., Torrente Y., Innocenzi A., Tonlorenzi R., D'Antona G., Pellegrino M.A., Barresi R., Bresolin N., De Angelis M.G., Campbell K.P. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–492. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- Schrader E.K., Harstad K.G., Matouschek A. Targeting proteins for degradation. Nat. Chem. Biol. 2009;5:815–822. doi: 10.1038/nchembio.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soddu S., Blandino G., Scardigli R., Coen S., Marchetti A., Rizzo M.G., Bossi G., Cimino L., Crescenzi M., Sacchi A. Interference with p53 protein inhibits hematopoietic and muscle differentiation. J. Cell Biol. 1996;134:193–204. doi: 10.1083/jcb.134.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A., Munch C., Hanssum A., Bertolotti A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell. 2012;48:242–253. doi: 10.1016/j.molcel.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir Y., Bengal E. p53 protein is activated during muscle differentiation and participates with MyoD in the transcription of muscle creatine kinase gene. Oncogene. 1998;17:347–356. doi: 10.1038/sj.onc.1201929. [DOI] [PubMed] [Google Scholar]
- Tashiro Y., Urushitani M., Inoue H., Koike M., Uchiyama Y., Komatsu M., Tanaka K., Yamazaki M., Abe M., Misawa H. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaru U., Takahashi S., Ishizu A., Miyatake Y., Gohda A., Suzuki S., Ono A., Ohara J., Baba T., Murata S. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012;180:963–972. doi: 10.1016/j.ajpath.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Tonoki A., Kuranaga E., Tomioka T., Hamazaki J., Murata S., Tanaka K., Miura M. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 2009;29:1095–1106. doi: 10.1128/MCB.01227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas R.M., Hartl F.U. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- Vilchez D., Morantte I., Liu Z., Douglas P.M., Merkwirth C., Rodrigues A.P., Manning G., Dillin A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489:263–268. doi: 10.1038/nature11315. [DOI] [PubMed] [Google Scholar]
- Vilchez D., Simic M.S., Dillin A. Proteostasis and aging of stem cells. Trends Cell Biol. 2014;24:161–170. doi: 10.1016/j.tcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- von Maltzahn J., Jones A.E., Parks R.J., Rudnicki M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. U S A. 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wagers A.J., Conboy I.M. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122:659–667. doi: 10.1016/j.cell.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Hauschka S., Tapscott S.J. The MCK enhancer contains a p53 responsive element. Proc. Natl. Acad. Sci. U S A. 1991;88:4570–4571. doi: 10.1073/pnas.88.11.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg C.M., Hagglund H., Nilsson G. Proteasome inhibition upregulates Bim and induces caspase-3-dependent apoptosis in human mast cells expressing the Kit D816V mutation. Cell Death Dis. 2012;3:e417. doi: 10.1038/cddis.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Price F., Rudnicki M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B.Y., Medhurst A.D., Jackson M., Rose S., Jenner P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech. Ageing Dev. 2005;126:760–766. doi: 10.1016/j.mad.2005.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.