

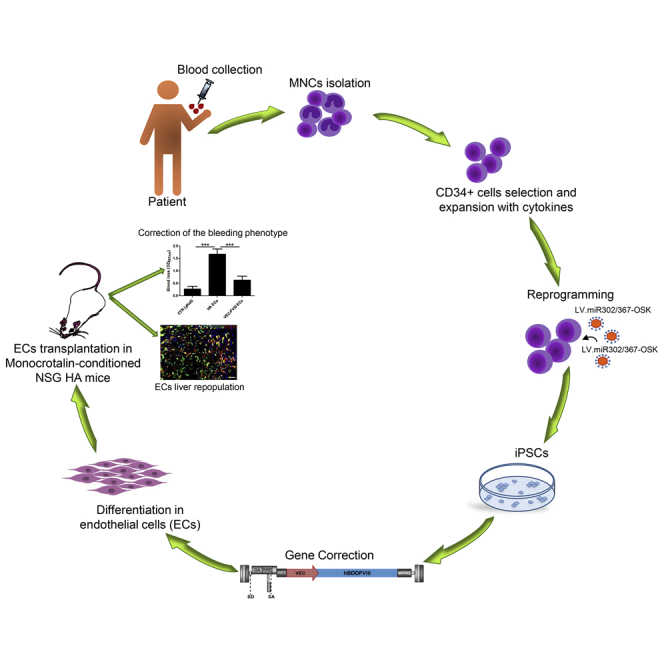
Summary
We generated patient-specific disease-free induced pluripotent stem cells (iPSCs) from peripheral blood CD34+ cells and differentiated them into functional endothelial cells (ECs) secreting factor VIII (FVIII) for gene and cell therapy approaches to cure hemophilia A (HA), an X-linked bleeding disorder caused by F8 mutations. iPSCs were transduced with a lentiviral vector carrying FVIII transgene driven by an endothelial-specific promoter (VEC) and differentiated into bona fide ECs using an optimized protocol. FVIII-expressing ECs were intraportally transplanted in monocrotaline-conditioned non-obese diabetic (NOD) severe combined immune-deficient (scid)-IL2rγ null HA mice generating a chimeric liver with functional human ECs. Transplanted cells engrafted and proliferated in the liver along sinusoids, in the long term showed stable therapeutic FVIII activity (6%). These results demonstrate that the hemophilic phenotype can be rescued by transplantation of ECs derived from HA FVIII-corrected iPSCs, confirming the feasibility of cell-reprogramming strategy in patient-derived cells as an approach for HA gene and cell therapy.
Keywords: hemophilia A, induced pluripotent stem cells (iPSCs), endothelial cells, cell and gene therapy, FVIII, chimeric vasculature, lentiviral vectors
Graphical Abstract
Highlights
-
•
Endothelial cells were differentiated from iPSCs obtained from hemophilic donors
-
•
LV transduced-hemophilic endothelial cells secreted FVIII in vitro and in vivo
Follenzi and colleagues show that endothelial cells can be obtained starting from blood CD34+ cells of both healthy and hemophilic donors passing through iPSC generation. During the differentiation process the cells can be genetically modified by LV to express FVIII; corrected endothelial cells secrete FVIII in vitro and ameliorate the hemophilic phenotype in a mouse model of the disease.
Introduction
Hemophilia A (HA) is a rare recessive X-linked bleeding disorder and is caused by mutations and/or deletions in the F8 gene, encoding the coagulation factor VIII (FVIII) (Bolton-Maggs and Pasi, 2003, Graw et al., 2005). On the basis of FVIII plasma activity, three forms of HA are recognized: severe (<1%), moderate (1%–5%), and mild (5%–40%) (Bolton-Maggs and Pasi, 2003). The current therapy consists of a repetitive infusion of recombinant or plasma-derived FVIII. This replacement therapy, however, does not represent a definitive cure and, moreover, 20%–40% of the treated patients develop anti-FVIII neutralizing antibodies (Den Uijl et al., 2011). Several alternatives are proposed, such as the drug emicizumab, a prophylactic therapy for adult and pediatric patients (Lenting et al., 2017, Shima et al., 2016), and some approaches of gene and cell therapy. Indeed, in HA patients the restoration of FVIII activity above 2%–5% could ameliorate the patients' quality of life. Gene transfer approaches for hemophilic patients have been running for almost 20 years (Nathwani et al., 2017). Presently, adeno-associated viruses (AAVs) are used for hemophilia gene therapy due to their relative safety, simplicity, and high liver tropism (Naso et al., 2017) but, despite remarkable results obtained with HB (Nathwani et al., 2011) and HA (Rangarajan et al., 2017), pre-existing immunity to AAVs and their long-term efficacy in young patients are still a concern. Several approaches for hemophilia gene therapy using lentiviral vectors (LVs) were developed and tested in preclinical models showing promising results (Cantore et al., 2015, Merlin et al., 2017). Nevertheless, a combined approach of gene and cell therapy deserves further consideration to develop a safe cell-based treatment.
Previous studies have demonstrated that transplanted liver sinusoidal endothelial cells (LSECs) from a healthy donor can correct the bleeding phenotype of HA mice (Follenzi et al., 2008, Yadav et al., 2012), and indeed LSECs are recognized as key FVIII producers (Fomin et al., 2013, Shahani et al., 2014, Zanolini et al., 2015). Recent data have proven that FVIII expression under an endothelial specific promoter using LV, is able to induce immunotolerance to FVIII and correct the bleeding phenotype of the disease (Merlin et al., 2017). In addition, successful transplantation of human LSECs isolated from adult liver has recently been reported (Filali et al., 2013, Fomin et al., 2013). Accordingly, HA cell therapy must rely on LSECs or immature endothelial progenitors as sources of transplantable cells, which are not always readily available. As such, other stem cell (SC) and progenitor cell populations have been evaluated either for their potential for in vivo FVIII production, or as carrier cells engineered for ectopic FVIII expression (Harb et al., 2009, Wang et al., 2012a, Wang et al., 2012b).
The discovery of induced pluripotent stem cells (iPSCs) has revolutionized regenerative medicine due to their potential for self-renewal, growth, and differentiation capacity. In particular, reprogramming of genetically corrected somatic cells can be used to generate autologous, disease-free iPSCs, which can be then differentiated into progenitor cells relevant for gene and cell therapy applications, e.g., endothelial cells (ECs) (Garcon et al., 2013, Raya et al., 2009, Wang et al., 2012c). iPSCs can be generated starting from several cell types, including fibroblasts (Takahashi et al., 2007), peripheral blood (PB) mononuclear cells (MNC), mobilized CD34+ cells from PB (Loh et al., 2009) and, interestingly, from non-mobilized cells enriched from small volumes of PB (Merling et al., 2013).
In the present study, we generated iPSCs from PB CD34+ cells of healthy donors and hemophilic patients, using two polycistronic, LoxP-flanked LVs expressing OCT4, SOX2, and KLF4 with or without miRNAs 302/367 cluster to enhance the reprogramming efficiency (Barroso-del Jesus et al., 2009).
The iPSCs were subsequently differentiated into functional ECs according to a newly optimized differentiation protocol and were corrected by gene transfer using a LV. Upon injection into a mouse model of HA, the FVIII-expressing HA-iPSC-derived ECs were able to produce and secrete functional FVIII, restoring therapeutic levels of FVIII activity.
Results
Generation of iPSCs from Fibroblasts and CD34+ Cells from Cord Blood
To generate iPSCs we used two floxable LVs at MOI = 5, one expressing OCT4, SOX2, and KLF4 (LV.SFFV.OSK; Figure S1A) from a single cistron, and another expressing the same reprogramming cassette together with the human miR302/367 cluster (LV.SFFV.OSK.miR302/367, Figure S1B). We obtained good quality iPSCs by reprogramming both patient skin fibroblasts and CD34+ cells from cord blood (CB) of healthy donors (Figure S2). iPSC colonies from the reprogrammed fibroblasts appeared after 2 months (Table S1). Obtained iPSCs showed ESC-like morphology (Figure S2A), were positive for alkaline phosphatase (AP) (Figure S2B) and expressed the endogenous factors (Figure S2C), while the exogenous factors were silenced (Figure S2D). Immunofluorescence (IF) showed the expression of stem cell markers (OCT4, SOX2, TRA1-81, and SSEA-4; Figure S2E), and NANOG promoter analysis demonstrated that 40% of CpG analyzed were unmethylated (Figure S2F). In parallel, we generated iPSCs from CB-derived CD34+ cells, colonies appeared after 20 days and we obtained over 20 bona fide iPSC clones (Figures S3A–S3E). Compared with fibroblasts, CD34+ cells from PB are easier to collect, because the blood can be recovered during routine blood sample collection, thus avoiding an invasive risky biopsy procedure for HA patients. On this basis, we opted to carry out our study using PB CD34+ cells from healthy donors and HA patients.
Generation of iPSCs from CD34+ Cells of Healthy and HA Donors
In ten independent experiments (five healthy donors, one heterozygous control, and four HA patients), we isolated 3.3 ± 1.18 × 105 non-mobilized CD34+ cells from 27.8 ± 11 mL of PB (Table 1). As described previously (Merling et al., 2013), expanded CD34+ cells were transduced with both reprogramming LVs at MOI = 5 and colonies appeared after 20 days. From each LV.SFFV.OSK reprogramming experiment, we obtained about 20 clones (data not shown), most of which degenerated over time, while, from the reprogramming with LV.SFFV.OSK.miR302/367 of healthy and HA CD34+ cells, we obtained 41.8 ± 16 and 18.2 ± 6.5 clones, respectively, which reached advanced passages in culture. The iPSC colonies were picked based on ESC-like morphology (Figures 1A and 1C). We established 10–15 clones for each donor (Table S1). We observed that the highest quality clones from both healthy and HA iPSC clones, had one to two copies of integrated LV in the genome, as determined by real-time PCR analysis (Table S2). All iPSC clones were characterized for pluripotency markers including AP positivity (Figures 1B and 1D), expression of endogenous (Figures 1E and 1F) and exogenous reprogramming factors (Figures 1G and 1H), and SC markers (OCT4, SOX2, TRA1-81, and SSEA-4; Figures 1I and 1J). The iPSC clones reached >100 passages (P) and maintained a stable karyotype (Figures 1K and 1L). NANOG promoter methylation profile showed that 63% of analyzed CpG islands were unmethylated both in healthy and HA iPSCs. In contrast, in CD34+ cells from healthy and HA donors 92% and 95% of sites were methylated, respectively (Figures 1M and 1N).
Table 1.
List of Hemophilic Patients and Healthy Donors from Whom iPSCs Were Generated
| Number of Isolation | Source | Donor | Mutation | mL of Starting Peripheral Blood | Number of CD34+ Cells Isolated | Number of Obtained Colonies |
|---|---|---|---|---|---|---|
| 1 | peripheral blood | healthy | none | 50 | 450,000 CD34+ |
30 iPSC colonies |
| 2 | peripheral blood | healthy | none | 25 | 300,000 CD34+ |
19 iPSC colonies |
| 3 | peripheral blood | healthy | none | 50 | 600,000 CD34+ |
40 iPSC colonies |
| 4 | peripheral blood | healthy | none | 22 | 350,000 CD34+ |
60 iPSC colonies |
| 5 | peripheral blood | healthy | none | 21 | 400,000 CD34+ |
60 iPSC colonies |
| 6 | cord blood | healthy | none | 35 | 400,000 CD34+ |
25 iPSC colonies |
| 7 | cord blood | healthy | none | 40 | 500,000 CD34+ |
35 iPSC colonies |
| 8 | peripheral blood | heterozygous | c.2244C > G, exon 14 |
24 | 250,000 CD34+ |
22 iPSC colonies |
| 9 | peripheral blood | severe HA | intron 22 inversion | 21 | 200,000 CD34+ |
6 iPSC colonies |
| 10 | peripheral blood | severe HA | unknown | 23 | 300,000 CD34+ |
18 iPSC colonies |
| 11 | peripheral blood | severe HA | c.2244C > G, exon 14 |
21 | 300,000 CD34+ |
20 iPSC colonies |
| 12 | peripheral blood | severe HA | c.6273G > A, exon 21 |
21 | 180,000 CD34+ |
25 iPSC colonies |
| 13 | skin biopsy | severe HA | c.6273G > A, exon 21 |
11 iPSC colonies |
Figure 1.

Characterization of Healthy and Hemophilic CD34+-Derived iPSCs
Representative phase-contrast microscopy showing ESC-like morphology of both healthy (clone HD 1.5) (A) and hemophilic (clone HA 3.1) (C) CD34+ cell-derived iPSCs. Positivity for alkaline phosphatase staining of healthy (B) and HA iPSCs (D). Scale bars, 200 μm. RT-PCR for endogenous SC markers (OCT4, SOX2, and KLF4) on healthy (E) and HA (F) iPSCs. RT-PCR for exogenous factors (G and H). CD34+ cells were used as negative control and HEK293T cells transduced with the reprogramming vector was used as positive control. SC marker expression at the protein level was detected by immunofluorescence in healthy (I) and HA iPSCs (J). Markers used were: OCT4 (green), SOX2 (green), SSEA4 (green), TRA1-81 (red), and DAPI (blue). Scale bars, 100 μm. Data are representative of three independent experiments. Cytogenetic analysis of healthy and HA CD34+-iPSCs demonstrated normal karyotypes (K and L, respectively). NANOG promoter was in a methylated status in isolated CD34+ cells and unmethylated in iPSCs (M and N). Telomere length analysis showed an increase during passages both in healthy and HA iPSCs (O and P, respectively). iPSCs, cultured in low adhesion plate, gave rise to EBs that expressed markers of the three germ layers (Q): NES, NCAM, and OTX2 (ectoderm), ACTA2, TBXT, and TBX6 (mesoderm), AFP, FOXA2, and SOX17 (endoderm), and showed adipose (R), osteogenic (S), and chondrogenic (T) potential. All data are representative of three independent experiments. Scale bars, 100 μm.
Moreover, telomere lengths increased between P5 and P20 (the longest cell passage analyzed), demonstrating a reactivation of the telomerase complex (Figures 1O and 1P).
Finally, iPSCs were able to generate embryoid bodies (EBs) which expressed the three germ layers markers (NES, NCAM, and OTX2 for ectoderm; ACTA2, TBXT, and TBX6 for mesoderm; AFP, FOXA2, and SOX17 for endoderm; Figures 1Q) and could be differentiated into adipogenic, osteogenic, and chondrogenic cells (Figures 1R–1T). Collectively, these results showed that PB-CD34+ cells were successfully reprogrammed into iPSCs expressing SC markers with a good differentiation potential.
Differentiation of CD34+-iPSCs into ECs
We differentiated CD34+-iPSCs into ECs using two different protocols, one involving BMP4 and VEGF and the other VEGF only. During differentiation, EBs acquired an endothelial-like morphology with both protocols (Figures 2A and 2B). RT-PCR analyses on ECs obtained from differentiation at days 10 and 20 showed an increase of endothelial markers, both early stage, such as KDR and TEK, and late stage, such as PECAM1 and VEC (Figure 2C). Noteworthy, ECs differentiated with the BMP4 protocol, expressed endothelial markers in a comparable manner to human umbilical vein endothelial cells (HUVECs), which were used as positive control. During the differentiation protocol, healthy EBs showed the acquisition of the endothelial phenotype that correlate with the gradual increase of F8 expression (Figure 2C). We further confirmed endothelial markers expression by flow cytometry analyses on ECs differentiated with BMP4 protocol. ECs were positive for CD31, VEC, VWF, and KDR, and were negative for CD45 (Figure 2D). Human blood outgrowth endothelial cells (BOECs) and fibroblasts were used as positive and negative controls, respectively (Figure S4). These findings were corroborated by the fact that ECs showed a decrease in telomere length when compared with parental iPSCs (Figure 2E), were heavily methylated (97%; Figure 2F) and showed a stable karyotype (Figure 2G). In conclusion, ECs can be obtained from CD34+-derived iPSCs with higher efficiency using the BMP4-inclusive protocol.
Figure 2.

Endothelial Differentiation of Healthy CD34+-Derived iPSCs, Clone HD 1.5
Representative phase-contrast microscopy showing the acquired endothelial morphology after 10 and 20 days of iPSC differentiated with BMP4 (A) and VEGF protocols (B). RT-PCR for endothelial markers on ECs after 10 and 20 days of differentiation with the two different protocols (C). Data are representative of three independent experiments. CD34+-derived iPSCs were used as negative control, HUVECs as positive control. LSECs were used as positive control only for F8. Representative flow cytometry plot (D) revealing the acquired EC phenotype of wild-type ECs at passage 8 post differentiation. The histogram overlays show the expression of different endothelial markers (CD31, VEC, KDR, and VWF) and of CD45, used as negative marker. Non-labeled cells were used as negative control in each plot (black line). Numbers are mean percentage ± SD of three independent experiments (n = 3). Telomere length analysis showed a progressive shortening in differentiated cells at different passages (E). NANOG promoter returned to a methylated state in ECs (F). Cytogenetic analysis of healthy endothelial cells (G). Scale bars, 200 μm.
Endothelial Phenotype of iPSC-Derived ECs
As a further demonstration of the quality of obtained ECs, we proceeded in the analysis of specific markers. In particular, a specific gene expression profile was previously described for blood endothelial cells (BECs) and lymphatic endothelial cells (LECs) (Furuhata et al., 2007, Nelson et al., 2007). Thus, we investigated whether these genes were expressed by iPSC-derived ECs using HUVECs, BOECs, HIMECs (human islet microvascular endothelial cells), and HDLECs (human dermal LECs) as comparison. RT-PCR analysis using BECs-specific markers (Figure 3A) showed similar expression among iPSC-derived ECs, BOECs, and HUVECs, while LEC-specific markers were not expressed in differentiated ECs (Figure 3B), demonstrating that iPSC-derived ECs acquired the phenotype of microvascular ECs with our new optimized differentiation protocol. Previous works demonstrated that ECs of different origin can express F8 at different levels (Pan et al., 2016). We were able to obtain higher levels of F8 at the mRNA level in healthy iPSC-derived ECs through the differentiation in BECs demonstrating that they are more efficient, compared with LECs, in F8 expression in vitro (Figure 3C).
Figure 3.

Markers for Endothelial Specification and Acquired Functionality of Healthy ECs, Clone HD 1.5
RT-PCR for endothelial markers specific for BECs on healthy ECs (A). RT-PCR for endothelial markers specific for LECs on healthy ECs. HUVECs, BOECs, HIMECs, and HDLECs were used as positive controls, water was used as negative control (B). RT-PCR for F8 and VWF in different endothelial cell types. ACTB correlates to the previous RT-PCR (C). Matrigel assay showing the ability of differentiated cells to form tubules in vitro (D). Scale bars, 25 μm. ECs transduced with LV expressing GFP under the control of endothelial-specific promoters FLK1, TIE2, and VEC (E). Dot plots are representative and numbers are mean percentage ± SD of three independent experiments.
Furthermore, we proved the acquired functionality of ECs performing tubulogenesis assay, showing that ECs were able to form complete and well-defined vessel-like tubular structures when cultured in Matrigel (Figure 3D).
To further confirm terminal differentiation of the iPSC-derived ECs, we transduced these cells with LVs carrying GFP under the control of endothelial-specific promoters FLK1, TIE-2, and VEC (LV.FLK1.GFP, LV.TIE-2.GFP, and LV.VEC.GFP). The ubiquitous PGK promoter (LV.PGK.GFP) was used as positive control, while hepatocyte- and myeloid-specific promoters (TTR and CD11b) were used as negative controls. Flow cytometry analyses showed that 72% ± 6.5%, 61% ± 2.5%, and 50% ± 4% of transduced cells were GFP+ under FLK1, TIE-2, and VEC promoters, respectively, while only 3% ± 1.6% and 3% ± 1% were GFP+ under TTR and CD11b promoters (Figure 3E). These data proved that iPSC-derived ECs were bona fide ECs. The differences in transgene (GFP) expression reflect the heterogeneous populations obtained with the endothelial differentiation protocol used. Indeed, KDR and TIE-2 are early-stage EC markers of endothelial differentiation, while VEC is a late-stage marker.
Finally, ECs were further transduced with an integrase defective (ID)-LV carrying the Cre recombinase to excise the reprogramming vector, thus avoiding reactivation of the exogenous reprogramming factors in terminally differentiated iPSC-derived ECs. The recombinase efficiently excised the reprogramming LoxP-flanked LV cassette, since the copy-number analysis revealed a mean of 0.05 integrated copy/cell, while the parental EC population contained 2.5 copies/cell (Table S3). In conclusion, we demonstrated that Cre expression successfully removed LV-exogenous reprogramming factors in differentiated ECs incrementing the safety of the cells to be used in vivo.
Hemophilic CD34+-iPSC-Expressing FVIII Can Be Differentiated into ECs
Before endothelial differentiation, HA-CD34-iPSCs were genetically corrected by LV.VEC.FVIII transduction (Figure S1C); LV.VEC.GFP served as control. EBs were generated from both FVIII- expressing and not expressing (NE) HA-CD34+-iPSCs. Expressing and NE cells acquired endothelial-like morphology and demonstrated endothelial marker expression by RT-PCR (Figures 4A, S5A, and S5B) and flow cytometry analyses (Figure 4B for NE and 4c for FVIII-expressing ECs). BOECs and fibroblasts were used as positive and negative controls, respectively (Figure S4). IF staining revealed the expression of CD31 in both corrected and NE ECs (Figures 4D and 4E, respectively, and Figures S5C and S5D), highlighting their distribution at cellular junction level and FVIII expression only in FVIII-transduced cells (Figure 4E). Interestingly, von Willebrand factor (VWF), an endothelial marker functioning as FVIII carrier in the bloodstream, appeared during the differentiation process (Figures 4A–4C). As previously described for healthy ECs, telomere lengths and NANOG methylation profile were analyzed and showed the mature stage of differentiation reached by HA iPSC-derived ECs (Figures 4F and 4G). Furthermore, FVIII-expressing ECs showed a stable karyotype (Figure 4H). We performed in vitro tubulogenesis assay and VEC-FVIII-ECs gave rise to a complex tubule network, like healthy ECs (Figures 4I, S5E, and S5F).
Figure 4.

Endothelial Differentiation of Hemophilic CD34+-Derived iPSCs, Clones HA 2.2, HA 3.1, and HA 4.9
HA iPSCs were corrected with an LV.VEC-hBDDFVIII. LV.VEC-GFP was used as control of transduction. Representative RT-PCR for endothelial markers on FVIII-expressing and not expressing ECs (A). HA-CD34+-derived iPSCs were used as negative control, HUVECs as positive control. HEK293T cells transduced with VEC.FVIII were used as positive control only for F8. Data are representative of three independent experiments. Representative FACS analysis of HA- (B) and FVIII-expressing ECs (C) acquired at passage 7 and 8, respectively. The histogram overlays show the expression of different endothelial markers (CD31, VEC, KDR, and VWF) and of CD45, used as negative marker. Non-labeled cells were used as negative control in each plot (black line). Numbers are mean percentage ± SD of three independent experiments (n = 3). Immunofluorescence staining with CD31 (red), FVIII (green), and DAPI (blue) on HA ECs (D) and VEC.FVIII ECs (E). Scale bars, 25 μm. Telomeres length showed a progressive shortening in differentiated cells at different passages (F). NANOG promoter returned to a methylated state in HA ECs (G). Cytogenetic analysis of VEC-FVIII-expressing ECs demonstrated a normal karyotype (H). VEC-FVIII-ECs formed tubule networks when cultured in Matrigel (I). Scale bars, 200 μm. VEC-FVIII ECs were able to secrete FVIII. FVIII antigen assay showed FVIII expression only by healthy and VEC.FVIII-transduced ECs (J). aPTT analysis showed FVIII activity in culture media only from VEC.FVIII and healthy ECs and not from HA-ECs (K). ∗∗∗p < 0.001. (A and D–I) Data were obtained from clone HA 3.1. (B, C, J, and K) Average of three biological replicates (means ± SD) from clones HA 2.2, HA 3.1, and HA 4.9.
Finally, to assess if FVIII was efficiently secreted, FVIII antigen and activated partial thromboplastin time aPTT assays were performed on the culture medium from both FVIII-expressing and HA cells. Antigen assay showed the secretion of 60 ± 3.4 ng/mL of FVIII by FVIII-expressing cells; healthy iPSC-derived ECs were used as positive control showing the secretion of 28 ± 2.9 ng/mL of FVIII, while FVIII was undetectable for HA-ECs (Figure 4J). VEC-FVIII-EC-conditioned medium displayed a shorter aPTT when compared with culture medium from HA-ECs (Figure 4K). Collectively, these results demonstrate that genetically corrected HA-CD34-iPSCs acquired the morphology, gene expression pattern, and functionality of mature ECs.
Transplanted iPSC-Derived ECs Engraft and Proliferate in the Mouse Liver
The FVIII expression and secretion was evaluated in vivo using a previously described mouse model of HA and LSEC transplantation (Follenzi et al., 2008). To assess the feasibility of this approach, we initially transplanted 2 × 106 healthy GFP+ iPSC-derived ECs into the liver of monocrotaline (MCT)-conditioned NSG mice to evaluate their engraftment and proliferation (n = 6). Cells engrafted in liver parenchyma and proliferated up to 12 weeks after transplantation, the longest time point analyzed (Figure 5A). The co-staining with human CD31 and the cell morphology confirmed the endothelial phenotype of transplanted cells (Figure 5A). Moreover, they formed vessel-like structures in the host liver demonstrating that healthy iPSC-derived ECs were able to engraft and proliferate successfully in the mouse liver after transplantation.
Figure 5.

Endothelial Transplantation by Portal Vein Injection of Healthy and Hemophilic CD34+-Derived ECs
(A–C) Healthy ECs (clone HD 1.5) were transplanted by portal vein injection in MCT-conditioned NSG mice (n = 6). Representative immunofluorescence on liver sections showing the presence of the co-staining between GFP and hCD31 up to 12 weeks after transplantation (A). FVIII-expressing and not expressing HA-ECs (clone HA 3.1) were transplanted in MCT-conditioned NSG-HA mice (n = 8 each condition). Representative immunofluorescence on liver sections of mice transplanted with GFP+ -VEC-FVIII-ECs confirmed the co-staining between GFP and hCD31 (B). Representative flow cytometry plots revealing the percentage of GFP+ and hCD31+ cells among liver NPCs (C). aPTT on plasma of transplanted mice, performed from 3 to 12 weeks after transplantation, showing an increase in mice transplanted with VEC-FVIII-ECs up to 5.64 ± 0.84 after 12 weeks. Untreated mouse was used as negative control.
(D and E) Mice transplanted with VEC-FVIII-ECs showed a decrease in bleeding volume (D) compared with HA-ECs transplanted mice (E) (n = 8 each condition) (means ± SD) (∗∗p < 0.01, ∗∗∗p < 0.001). Scale bars, 50 μm.
In Vivo FVIII Expression and Correction of the Hemophilic Phenotype after Transplantation of Genetically Corrected iPSC-Derived ECs
To further evaluate FVIII secretion by FVIII-expressing cells, we transplanted GFP+ VEC-FVIII- ECs in MCT-conditioned NSG-HA mice (n = 8 each condition). The engraftment was assessed by IF staining and flow cytometry analyses 12 weeks after GFP+ VEC-FVIII-ECs transplantation. Cells proliferated and repopulated up to 30% of non-parenchymal cells (NPC) in recipient mice livers and were present for up to 3 months (Figure 5B). The expression of hCD31 confirmed the endothelial phenotype of transplanted cells (Figures 5B and 5C). No GFP+ hepatocytes were detected (Figure S5A). Transplanted cells were observed only in liver sections of transplanted mice and were not detected in other organs such as spleen, kidney, and lung, as shown by IF (Figure S6B). The absence of positive cells in these organs was also confirmed by PCR on genomic DNA (gDNA) (Figure S6C). aPTT assay was performed 3, 6, 9, and 12 weeks after transplantation. The relative FVIII activity in mice transplanted with VEC-FVIII-ECs was 3.36% ± 0.6% after 3 weeks, which increased to 4.51% ± 0.3% after 6 weeks, and remained stable at 9 and 12 weeks (5.21% ± 0.38% and 5.77% ± 0.52%, respectively), while mice transplanted with uncorrected cells had no detectable FVIII activity (Figure 5D). At 12 weeks, bleeding assay on transplanted mice confirmed aPTT results. Indeed, mice transplanted with HA-ECs did not show a therapeutic correction, unlike the VEC-FVIII-EC-transplanted mice (Figure 5E). Taken together, these results demonstrate that the hemophilic phenotype can be rescued in HA mice by transplantation of ECs derived from HA-iPSCs and corrected by LVs carrying FVIII under the control of the endothelial-specific VEC promoter.
Intraperitoneal Injection of iPSC-Derived ECs in a Mouse Model of HA Secreted FVIII
Finally, we assessed whether ECs injected in the peritoneal cavity can survive and secrete FVIII at therapeutic levels when combined with microcarrier beads supporting their viability, a safer therapeutic strategy than using MCT to condition the recipient that has a tumorigenic potential. Ten million GFP+ healthy iPSC-derived ECs, hemophilic GFP+ ECs, and GFP+-VEC-FVIII-ECs were injected in NSG-HA mice in association with microcarrier beads (n = 4 each condition). Three days after transplantation FVIII activity levels in mice receiving healthy iPSC-derived ECs and VEC- FVIII-ECs were 3.24% ± 0.6% and 5.88% ± 1.5%, respectively, while in controls (beads only and HA ECs) FVIII activity was undetectable (Figure 6A). One week later, mice injected with healthy iPSC-derived ECs showed 2.52% ± 0.6% FVIII activity, and VEC-FVIII-ECs transplanted mice showed 6.0% ± 0.96% FVIII activity. Three weeks after injection, FVIII activity levels in mice receiving healthy iPSC-derived ECs and VEC-FVIII-ECs were 1.28% ± 0.18% and 4.82% ± 0.14%, respectively (Figure 6A). These results demonstrate that VEC-FVIII-ECs were able to secrete FVIII at therapeutic levels in vivo and at higher level than healthy iPSC-derived ECs. IF staining and PCR on gDNA on recovered beads after 3 weeks demonstrated that GFP+ cells were still associated to the beads (Figures 6B and 6C). The co-staining of GFP and hCD31 confirmed the presence and the endothelial phenotype of the transplanted cells. These findings suggest that iPSC-derived ECs can survive and secrete functional FVIII when injected into the peritoneal cavity associated to a scaffold supporting their cell viability and functionality.
Figure 6.

Intraperitoneal Implantation of Endothelial Cells with Cytodex Micro-Carrier Beads
GFP+-VEC-FVIII-ECs, HA-ECs (clone HA 3.1), and healthy ECs (clone HD 1.5) were injected intraperitoneally in NOD/SCID HA mice in association with Cytodex micro-carrier beads (n = 4 each condition). aPTT performed 3 days, 7 days, and 3 weeks after injection showed that LV-FVIII- corrected ECs restored FVIII activity (5.88% ± 1.5% on day 3; 6.0% ± 0.96% on day 7; 4.82% ± 0.14% after 3 weeks), more than healthy ECs (3.24% ± 0.6% on day 3; 2.52% ± 0.6% on day 7; 1.28% ± 0.18% after 3 weeks) (means ± SD) (∗p < 0.05) (A). Representative immunofluorescence on beads showed the co staining of GFP and hCD31 confirming the endothelial phenotype of the cells (B). PCR on gDNA for integration on recovered beads (C). LV.VEC.FVIII was used as positive control. Scale bar, 50 μm.
Discussion
New strategies for a definitive cure of HA are needed, and gene and cell therapy could represent a powerful therapeutic approach. Thus, the possible use of genetically corrected autologous iPSCs is appealing. However, several issues need to be solved, such as the identification of the best cell source to be reprogrammed or the best cellular target to use after iPSC differentiation. We generated iPSCs from skin-derived fibroblasts and CD34+ cells of CB and PB. The CD34+ cells gave rise to iPSCs earlier (approximately 3 weeks) compared with fibroblasts (2 months) in all reprogrammed samples. This observation highlighted that the differentiation stage of the starting cells is critical for the return to a pluripotent state (Eminli et al., 2009). We identified CD34+ cells from PB as the safest and easiest-to-access cell source to be reprogrammed in HA patients compared with skin biopsy. The iPSCs can be derived through several strategies and by several factors. For safety concerns, we generated iPSCs omitting c-Myc, therefore resulting in a reduced reprogramming efficiency (Nakagawa et al., 2008). By including the miRNAs302/367 into LoxP-flanked LVs carrying OCT4, SOX2, and KLF4 we generated iPSCs from PB CD34+ cells in a time range and efficiency comparable with a reported study using all four factors (Merling et al., 2013). This is due to the interplay between miRNAs302/367 clusters and OCT4 and SOX2 in the induction and maintenance of pluripotency (Card et al., 2008). We demonstrated that the LV expression cassette was spontaneously silenced in established iPSC clones. However, the integrative nature of LVs required the excision of transgenes after reprogramming to avoid the reactivation of exogenous factors that could increase iPSC oncogenic potential and interfere with the differentiation and maintenance of the differentiated state (Chakraborty et al., 2013). Hence, we used Cre recombinase carried by an IDLV to remove the LoxP-flanked reprogramming cassette in ECs thus allowing the maintenance of the acquired phenotype for over 30 passages. While successful, this proof-of-concept study could be improved in the future in terms of efficacy and safety by applying state-of-the art methods for cell reprogramming. For instance, the stable integration approach can be substituted by an efficient non-integrating method such as Sendai virus vectors (Fusaki et al., 2009).
It is well known that liver is the main organ producing FVIII. In particular, among the liver cell types LSECs are the main source of FVIII (Everett et al., 2014, Fahs et al., 2014, Shahani et al., 2014), and LSEC transplantation in the liver of HA mice can correct the bleeding phenotype (Follenzi et al., 2008, Kumaran et al., 2005). In 2009, endothelial progenitors were generated from murine iPSCs and were able to correct the HA phenotype when injected into the liver parenchyma (Xu et al., 2009). Moreover, Park et al., 2014, Park et al., 2015 created model cell lines by using TALEN or CRISPR-Cas9 technology to invert/revert a big portion of the chromosomal segment that spans the portion of F8 gene in iPSCs. ECs obtained from the inversion-corrected iPSCs expressed F8 and were able to rescue the phenotype in short-term experiments when injected in HA mice (Park et al., 2015).
In the present investigation, we differentiated iPSCs into ECs using two different protocols enlightening the endothelial differentiation efficiency. The improved differentiation protocol involving BMP4, besides VEGF, pushed cells through a mesoderm transition before definitive endothelial differentiation, allowing the formation of functional and stable microvascular endothelial cells able to secrete FVIII for long term. These ECs acquired an endothelial gene expression pattern specific for blood endothelial markers more than lymphatic endothelial markers. Obtaining ECs from iPSCs with this protocol could be useful for disease modeling, cell therapy, and tissue engineering applications in which it is important to obtain high amounts of ECs. For HA cell therapy, we successfully corrected ECs by LV expressing a functional form of FVIII under the control of the endothelial-specific VEC (cadherin 5) promoter. The use of the VEC promoter allowed us to restrict FVIII expression in the desired cell type and to assess that iPSC-derived differentiated cells were effectively ECs (Merlin et al., 2017). In fact, following differentiation, ECs were able to express and secrete FVIII after genetic correction, demonstrating the transcriptional activation of the endothelial-specific promoter in vitro and in vivo. The iPSC-derived ECs transplanted into the liver of MCT-conditioned NSG-HA mice repopulated the parenchyma, forming new vessels expressing hCD31. More importantly, EC transplantation rescued the hemophilic phenotype both in the short and long term, resulting in up to 6% FVIII activity. We demonstrated that iPSC-derived ECs expressed the FVIII protein effectively, engrafted within the hepatic parenchyma, and provided the phenotypic correction of HA mice. Envisioning clinical application of EC transplantation into the liver, it would be important to select the right drug because it should not be detrimental for the recipient while selectively damaging the hepatic endothelium to improve EC engraftment and proliferation.
Importantly, ECs restored FVIII activity not only when transplanted directly into the liver but also when intraperitoneally injected in association with micro-carrier beads.
This result is exciting from a basic and translational perspective. As basic perspective, it is crucial to obtain human ECs that could engraft in a mouse generating a chimeric liver in which it would be possible to study the endothelial functions during cell-cell interactions in a controlled environment during liver repopulation. Ideally, as a translational outlook, this strategy can be effective because ex-vivo corrected cells need to be expanded without losing transgene expression and to be embedded into a medical device. While confining and protecting transplanted cells into desired anatomical sites, this structure allows the release of the therapeutic product over time. Such an approach could be translated in children, where direct gene therapy is still a concern for the hemophilia community.
Moreover, to overcome safety issues related to LV random integration, it would be better to rely on the use of AAV- or LV-mediated genome-editing strategies to introduce the FVIII coding sequence into putative safe harbor loci, such as AAVS1 or CCR5 (Oceguera-Yanez et al., 2016, Sebastiano et al., 2014, Wallen et al., 2015).
At present, a phase I clinical trial for type 1 diabetes is ongoing in which pancreatic progenitor cells are encapsulated in a device, which prevents immunoreactions (http://www.eurostemcell.org/commentanalysis/making-insulin-producing-beta-cells-stem-cells-how-close-are-we). Further, it has recently been published that macro-encapsulation devices loaded with neonatal pancreatic tissue and transplanted into RIP-LCMV.GP mice prevented disease onset in a model of virus-induced diabetes mellitus (Boettler et al., 2016). As such, the prospect of placing FVIII-secreting ECs into a device or scaffold is feasible and clinically attractive for future therapeutic approaches to patients affected with HA and other diseases.
Experimental Procedures
CD34+ Cell Purification and Culture
Non-mobilized PB was collected from seven healthy donors, and one heterozygous donor, and four patients. The study for the use of samples of human origin was approved by the Ethical Committee of Novara, Italy. MNCs were purified by gradient centrifugation on Ficoll (GE Healthcare) and CD34+ cells were isolated using the MACS CD34+ MicroBeadKit (Miltenyi Biotec) according to the manufacturer's protocol. Isolated cells were cultured for 4 days in HPGM medium (Lonza) supplemented with 1% human serum albumin (Sigma-Aldrich), 50 ng/mL of hSCF, hFlt3-ligand, hTPO, and hIL-3 (Immunotools).
Vector Transduction for Reprogramming and FVIII Correction
Five days after isolation, both healthy and hemophilic CD34+ cells from PB and CB and fibroblasts from skin biopsies were transduced with LV.SFFV.OSK and an additional Cre-excisable polycistronic LV carrying miRNA cluster 302\367 followed by OSK cassette (LV.SFFV.miR- 302\367.OSK) by a single spinoculation at MOI = 5–10 at 300 g for 1 hr. Two days later, the cells were seeded on a human foreskin fibroblast (HFF) feeder layer in HPGM, medium was replaced with HES medium after 2 days. Individual iPSCs colonies were passaged by mechanical dissociation. HA-CD34+-derived iPSCs were genetically corrected by an LV carrying the hBDD.FVIII under the control of endothelial-specific VEC promoter (LV.VEC.hBDDFVIII). LV.VEC.GFP was used as a control.
iPSC Culture and EB Generation
iPSCs were cultured and characterized using standard techniques on irradiated HFFs in HES medium (Okada et al., 2010). For EB generation, iPSCs were disintegrated mechanically and plated onto low attachment plates for 5 days in HPGM. Then EBs were used for ECs differentiation and adipogenic, osteogenic and chondrogenic differentiation.
Endothelial Cell Differentiation
For endothelial differentiation, EBs were generated and differentiated in ECs using two different protocols the VEGF protocol and the BMP4 protocol. For the VEGF protocol, formed EBs were directly plated on 0.1% gelatin-coated six-well plates in KO-DMEM+10% FBS (EC medium) with 50 ng/mL of VEGF (Immunotools) for 20 days. For the BMP4 protocol, EBs were grown for 2 days in EC medium alone, on day 4 BMP4 (Immunotools) was added (20 ng/mL). On day 6, EBs were cultured in EC medium with 20 ng/mL of basic FGF and 20 ng/mL of BMP4. On day 8 EBs were plated on 0.1% gelatin-coated six-well plates in EC medium with 20 ng/mL bFGF and 50 ng/mL of VEGF. At day 10, medium containing only 50 ng/mL of VEGF was added and maintained until day 20. Cells were maintained in EC medium with 50 ng/mL of VEGF for up to 25–30 passages.
LV Transduction of ECs
ECs were transduced with LVs containing GFP under the control of endothelial-specific promoters TIE-2, VEC, and FLK1. As a positive control, LVs expressing GFP under the ubiquitous PGK promoter was used. As a negative control, LVs expressing GFP under hepato-(TTR) and myeloid-specific (CD11b) promoters were used. All LVs were used at MOI = 10 (around 106 TU/ml).
Animals and Procedures
Animal studies were approved by the Animal Care and Use Committee of the Università del Piemonte Orientale “A.Avogadro” (Novara, Italy). Hemophilic NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG-HA) mice, generated in our laboratory (Zanolini et al., 2015), at 6–8 weeks of age, were used for cell transplantation studies. Animals received 200 mg/kg MCT (Sigma-Aldrich) in saline intraperitoneally 24 hr prior to intraportal transplantation. To blunt immune responses against GFP transgene reporter, mice were given 30 mg/kg cyclophosphamide (Cytoxan; Bristol-Myers Squibb) in normal saline intraperitoneally twice a week for the duration of the study, starting from the day prior to surgery. Mice were anesthetized with isoflurane. For cell transplantation, 2 × 106 ECs in 0.3 mL serum-free DMEM were injected into the portal vein, as described previously (Follenzi et al., 2008). Controls received serum-free medium.
For bead transplantation, cells were mixed with Cytodex 3 microcarrier beads (Amersham Pharmacia Biotech) in a ratio of 107 cells/mL of rehydrated microcarriers and injected intraperitoneally.
FVIII Activity and FVIII Antigen Assay
Plasma samples of transplanted mice and supernatants of uncorrected and LV-VEC-hBDDFVIII--corrected ECs were analyzed for FVIII activity by aPTT. Standard curves were generated by serial dilution of recombinant human BDD FVIII (ReFacto) starting from 1 U/mL (100%) in hemophilic mouse plasma for aPTT assay. Results were expressed in percentage of correction. FVIII antigen in supernatant of cultured uncorrected and LV-VEC-hBDDFVIII-corrected ECs was quantified by ELISA sandwich using the Matched-Pair Antibody Set for ELISA of human Factor VIII antigen (Affinity Biologicals) following the manufacturer's protocol. Standard curves were generated by serially diluting ReFacto in culture medium and results were expressed as ng/mL. For bleeding assay methods see Supplemental Experimental Procedures.
Statistical Analysis
All data were expressed as mean ± SD. Graphs were generated, and statistical analysis was performed with Prism 5 (Graph Pad). The p values were calculated using Student's t test with two-tailed distribution, assuming equal standard deviation distribution, one-way ANOVA with Bonferroni post-hoc test; p < 0.05 values were considered statistically significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Author Contributions
C.O., M.T., G.R., S.M., A.C., D.C., F.D.S., C.B., G.N.B., M.P., and F.F. performed research and analyzed data; A.L. provided reagents and advice; P.C.S., F.V., and M.M. selected patients; Y.R.-P. and A.R. were instrumental in designing the experiments for iPSC generation, A.F. conceived the study, generated funding, designed the research, and analyzed data. C.O., M.T., and A.F. wrote the manuscript that was revised by all authors.
Acknowledgments
The authors thank V. Bruscaggin,V. Fiorio, C. Firrito, and A. Stevano for technical assistance. Dr G. Walker for critical reading and editing of the manuscript. Dr. L. Piemonti (HSR-Milan) for providing human HIMEC cells. A.F. was supported in part by ERC startup grant no. 261178, AIRC 2012 (project 13166), and in part from Horizon 2020, HemAcure n. 667421. P.C.S., A.R., and A.F. were supported by E-Rare HEMO-iPS 2011. A.F. was a recipient of a CSL-Behring Prof. Heimburger 2010 Award. Additional support for this work came from Instituto de Salud Carlos III-ISCIII/FEDER (Red de Terapia Celular - TerCel RD16/0011/0024), Generalitat de Catalunya - AGAUR (2014-SGR-1460), and CERCA Program/Generalitat de Catalunya to A.R. and from Telethon (TGT18F01) to A.L.
Published: November 8, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.10.012.
Supplemental Information
References
- Barroso-del Jesus A., Lucena-Aguilar G., Menendez P. The miR-302-367 cluster as a potential stemness regulator in ESCs. Cell Cycle. 2009;8:394–398. doi: 10.4161/cc.8.3.7554. [DOI] [PubMed] [Google Scholar]
- Boettler T., Schneider D., Cheng Y., Kadoya K., Brandon E.P., Martinson L., von Herrath M. Pancreatic tissue transplanted in TheraCyte encapsulation devices is protected and prevents hyperglycemia in a mouse model of immune-mediated diabetes. Cell Transplant. 2016;25:609–614. doi: 10.3727/096368915X688939. [DOI] [PubMed] [Google Scholar]
- Bolton-Maggs P.H., Pasi K.J. Haemophilias A and B. Lancet. 2003;361:1801–1809. doi: 10.1016/S0140-6736(03)13405-8. [DOI] [PubMed] [Google Scholar]
- Cantore A., Ranzani M., Bartholomae C.C., Volpin M., Valle P.D., Sanvito F., Sergi L.S., Gallina P., Benedicenti F., Bellinger D. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015;7:277ra228. doi: 10.1126/scitranslmed.aaa1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card D.A., Hebbar P.B., Li L., Trotter K.W., Komatsu Y., Mishina Y., Archer T.K. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Christoforou N., Fattahi A., Herzog R.W., Leong K.W. A robust strategy for negative selection of Cre-loxP recombination-based excision of transgenes in induced pluripotent stem cells. PLoS One. 2013;8:e64342. doi: 10.1371/journal.pone.0064342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Uijl I.E., Mauser Bunschoten E.P., Roosendaal G., Schutgens R.E., Biesma D.H., Grobbee D.E., Fischer K. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 2011;17:849–853. doi: 10.1111/j.1365-2516.2011.02539.x. [DOI] [PubMed] [Google Scholar]
- Eminli S., Foudi A., Stadtfeld M., Maherali N., Ahfeldt T., Mostoslavsky G., Hock H., Hochedlinger K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009;41:968–976. doi: 10.1038/ng.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett L.A., Cleuren A.C., Khoriaty R.N., Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–3705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahs S.A., Hille M.T., Shi Q., Weiler H., Montgomery R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706–3713. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali E.E., Hiralall J.K., van Veen H.A., Stolz D.B., Seppen J. Human liver endothelial cells, but not macrovascular or microvascular endothelial cells, engraft in the mouse liver. Cell Transplant. 2013;22:1801–1811. doi: 10.3727/096368912X657594. [DOI] [PubMed] [Google Scholar]
- Follenzi A., Benten D., Novikoff P., Faulkner L., Raut S., Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J. Clin. Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomin M.E., Zhou Y., Beyer A.I., Publicover J., Baron J.L., Muench M.O. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS One. 2013;8:e77255. doi: 10.1371/journal.pone.0077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhata S., Ando K., Oki M., Aoki K., Ohnishi S., Aoyagi K., Sasaki H., Sakamoto H., Yoshida T., Ohnami S. Gene expression profiles of endothelial progenitor cells by oligonucleotide microarray analysis. Mol. Cell. Biochem. 2007;298:125–138. doi: 10.1007/s11010-006-9359-4. [DOI] [PubMed] [Google Scholar]
- Fusaki N., Ban H., Nishiyama A., Saeki K., Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcon L., Ge J., Manjunath S.H., Mills J.A., Apicella M., Parikh S., Sullivan L.M., Podsakoff G.M., Gadue P., French D.L. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood. 2013;122:912–921. doi: 10.1182/blood-2013-01-478321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graw J., Brackmann H.H., Oldenburg J., Schneppenheim R., Spannagl M., Schwaab R. Haemophilia A: from mutation analysis to new therapies. Nat. Rev. Genet. 2005;6:488–501. doi: 10.1038/nrg1617. [DOI] [PubMed] [Google Scholar]
- Harb R., Xie G., Lutzko C., Guo Y., Wang X., Hill C.K., Kanel G.C., DeLeve L.D. Bone marrow progenitor cells repair rat hepatic sinusoidal endothelial cells after liver injury. Gastroenterology. 2009;137:704–712. doi: 10.1053/j.gastro.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran V., Benten D., Follenzi A., Joseph B., Sarkar R., Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J. Thromb. Haemost. 2005;3:2022–2031. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- Lenting P.J., Denis C.V., Christophe O.D. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130:2463–2468. doi: 10.1182/blood-2017-08-801662. [DOI] [PubMed] [Google Scholar]
- Loh Y.H., Agarwal S., Park I.H., Urbach A., Huo H., Heffner G.C., Kim K., Miller J.D., Ng K., Daley G.Q. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476–5479. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin S., Cannizzo E.S., Borroni E., Bruscaggin V., Schinco P., Tulalamba W., Chuah M.K., Arruda V.R., VandenDriessche T., Prat M. A novel platform for immune tolerance induction in hemophilia A mice. Mol. Ther. 2017;25:1815–1830. doi: 10.1016/j.ymthe.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merling R.K., Sweeney C.L., Choi U., De Ravin S.S., Myers T.G., Otaizo-Carrasquero F., Pan J., Linton G., Chen L., Koontz S. Transgene-free iPSCs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood. 2013;121:e98–e107. doi: 10.1182/blood-2012-03-420273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M., Koyanagi M., Tanabe K., Takahashi K., Ichisaka T., Aoi T., Okita K., Mochiduki Y., Takizawa N., Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani A.C., Davidoff A.M., Tuddenham E.G.D. Advances in gene therapy for hemophilia. Hum. Gene Ther. 2017;28:1004–1012. doi: 10.1089/hum.2017.167. [DOI] [PubMed] [Google Scholar]
- Nelson G.M., Padera T.P., Garkavtsev I., Shioda T., Jain R.K. Differential gene expression of primary cultured lymphatic and blood vascular endothelial cells. Neoplasia. 2007;9:1038–1045. doi: 10.1593/neo.07643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oceguera-Yanez F., Kim S.I., Matsumoto T., Tan G.W., Xiang L., Hatani T., Kondo T., Ikeya M., Yoshida Y., Inoue H. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods. 2016;101:43–55. doi: 10.1016/j.ymeth.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Okada M., Oka M., Yoneda Y. Effective culture conditions for the induction of pluripotent stem cells. Biochim. Biophys. Acta. 2010;1800:956–963. doi: 10.1016/j.bbagen.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Pan J., Dinh T.T., Rajaraman A., Lee M., Scholz A., Czupalla C.J., Kiefel H., Zhu L., Xia L., Morser J. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood. 2016;128:104–109. doi: 10.1182/blood-2015-12-684688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.Y., Kim J., Kweon J., Son J.S., Lee J.S., Yoo J.E., Cho S.R., Kim J.H., Kim J.S., Kim D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. U S A. 2014;111:9253–9258. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.H., Kim D.W., Kim J.S. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y. AAV5-factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- Raya A., Rodriguez-Piza I., Guenechea G., Vassena R., Navarro S., Barrero M.J., Consiglio A., Castella M., Rio P., Sleep E. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiano V., Zhen H.H., Haddad B., Bashkirova E., Melo S.P., Wang P., Leung T.L., Siprashvili Z., Tichy A., Li J. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2014;6:264ra163. doi: 10.1126/scitranslmed.3009540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani T., Covens K., Lavend'homme R., Jazouli N., Sokal E., Peerlinck K., Jacquemin M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014;12:36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- Shima M., Lillicrap D., Kruse-Jarres R. Alternative therapies for the management of inhibitors. Haemophilia. 2016;22(Suppl 5):36–41. doi: 10.1111/hae.13005. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Wallen M.C., Gaj T., Barbas C.F., 3rd Redesigning recombinase specificity for safe harbor sites in the human genome. PLoS One. 2015;10:e0139123. doi: 10.1371/journal.pone.0139123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang X., Wang L., Chiu J.D., van de Ven G., Gaarde W.A., Deleve L.D. Hepatic vascular endothelial growth factor regulates recruitment of rat liver sinusoidal endothelial cell progenitor cells. Gastroenterology. 2012;143:1555–1563.e2. doi: 10.1053/j.gastro.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang X., Xie G., Wang L., Hill C.K., DeLeve L.D. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J. Clin. Invest. 2012;122:1567–1573. doi: 10.1172/JCI58789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zheng C.G., Jiang Y., Zhang J., Chen J., Yao C., Zhao Q., Liu S., Chen K., Du J. Genetic correction of beta-thalassemia patient-specific iPS cells and its use in improving hemoglobin production in irradiated SCID mice. Cell Res. 2012;22:637–648. doi: 10.1038/cr.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D., Alipio Z., Fink L.M., Adcock D.M., Yang J., Ward D.C., Ma Y. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc. Natl. Acad. Sci. U S A. 2009;106:808–813. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav N., Kanjirakkuzhiyil S., Ramakrishnan M., Das T.K., Mukhopadhyay A. Factor VIII can be synthesized in hemophilia A mice liver by bone marrow progenitor cell-derived hepatocytes and sinusoidal endothelial cells. Stem Cells Dev. 2012;21:110–120. doi: 10.1089/scd.2010.0569. [DOI] [PubMed] [Google Scholar]
- Zanolini D., Merlin S., Feola M., Ranaldo G., Amoruso A., Gaidano G., Zaffaroni M., Ferrero A., Brunelleschi S., Valente G. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100:881–892. doi: 10.3324/haematol.2014.123117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.