

Summary
Background
There is still no reliable, specific biomarker for precision diagnosis and clinical monitoring of systemic lupus erythematosus. The aim of this study was to investigate the importance of the determination of immunofenotypic profiles (T, B lymphocytes and NK cells) and serum cytokine concentrations (IL-17 and IFN-alpha) as potential biomarkers for this disease.
Methods
The study included 55 patients with SLE and 25 healthy controls. The proportion of T, B, NK cells were assessed in peripheral blood using flow cytometric assays while the serum cytokine concentration (IL-17 and IFNalpha) was determined by ELISA test.
Results
ROC curve analysis showed good accuracy to distinguish between patients and healthy individuals for activated T cells (AUC=0.798; p<0.001), Treg (AUC= 0.651; p=0.036), and memory B cells (AUC=0.285; p=0.002). We found statistically significant difference (p=0.036) in the levels of serum IL-17 between patients with SLE (IL-17=49.27 pg/mL) and controls (IL-17= 28.64 pg/mL).
Conclusions
Significant increase in the relative number of Treg lymphocytes, and decrease in memory B cells, as well as decrease level of IL-17, in SLE patients may be implicated in the pathogenesis of the disease. These parameters, as biomarkers, could distinguish SLE patients and no-SLE patients. Monitoring subpopulations of immune cells in peripheral blood using flow cytometry provides insight into abnormal T and B cell function in SLE. Progress in understanding the immunity at SLE, results in concrete benefits for the SLE patients, which include new clinical management and therapeutic strategies.
Keywords: SLE, biomarkers, T and B lymphocytes, cytokines, flow cytometry
Kratak sadržaj
Uvod
Još uvijek ne postoji pouzdan, specifičan biomarker za precizno dijagnosticiranje i kliničko praćenje sistemskog lupus eritematodesa. Cilj ovog istraživanja je da se ispita značaj određivanja imunofenotipskog profila (udio T, B limfocita i NK ćelija) i serumske koncentracije citokina (IL-17 i IFN-alfa) kao potencialnih biiomarkera za ovo oboljenje.
Metode
U studiju je uključeno 55 pacijenata oboljelih od SLE i 25 zdravih osoba. Za određivanje imunofenotipskog profila (T, B limfociti i NK ćelije), iz periferne krvi ispitanika, korištena je metoda protočne citometrije, dok je koncentracija citokina u serumu (IL-17 i IFN-alfa) određena ELISA metodom.
Rezultati
Analizom ROC krive uočena je statistički značajna razlika za udio aktiviranih T limfocita (AUC=0,798; p< 0,001), T regulatornih limfocita (AUC=0,651; p=0,036), i memorijskih B ćelija (AUC=0,285; p=0,002) između pacijenata sa SLE i zdravih individua. Pronašli smo značajno veći nivo serumskog IL-17 (p=0,036) kod pacijenata sa SLE (IL-17=49,27 pg/mL) u odnosu na kontrolnu grupu (IL-17=28,64 pg/mL).
Zaključak
Značajno povećanje relativnog broja Treg limfocita i smanjenje memorijskih B ćelija, kao i povećanje nivoa IL-17 kod pacijenata s SLE može biti povezano sa patogenezom bolesti. Ovi parametri, kao biomarkeri, zbog njihove odlične specifičnosti, mogu razlikovati osobe sa i bez SLE-a. Monitoring subpopulacija imunih ćelija u perifernoj krvi pomoću protočne citometrije omogućava uvid u abnormal-nu funkciju T i B limfocita u SLE. Napredak u razumijevanju imunološke osnove kod SLE-a rezultira konkretnom koristi za pacijente sa ovim oboljenjem, što podrazumjeva nove načine kliničkog praćenja pacijenta i nove terapijske strategije.
Ključne reči: SLE, biomarkeri, T i B limfociti, citokini, protočna citometrija
Introduction
Systemic lupus erythematosus (SLE) is clinically and serologically diverse autoimmune disease of unknown aetiology. It is characterized by the presence of autoantibodies, primarily to the nuclear material of the cell, and by the deposition of immune complexes in various tissues (1, 2). The specific class of autoantibodies are anti-nuclear antibodies (ANA). These antibodies are capable of binding and destroying certain structures within the nucleus of cells. There are two main groups of ANA: autoantibodies to DNA (single and double stranded) and autoantibodies to extractable nuclear antigens (ENA): SSA/Ro, SSB/La, Sm, Sm/RNP, Scl-70, Jo-1 (3).
Patients with SLE typically suffer exacerbations of the disease, with periods of remission between these. It predominantly affects younger women, but can occur in up to 20% of patients aged 50 years or older. The disease affects almost every system in the body, with varying degrees of severity. It can occasionally be refractory to therapy, and is associated with a higher mortality rate than that of the general population. In a certain proportion of patients SLE gradually worsens over time, so the resulting organ damage can be life threatening (4). Despite improvements in survival of patients with SLE, the standardized mortality ratios (SMR) are still 3-fold higher than for the general population. Infections, cardiovascular disease, and end-organ damage remain the major causes of death (5). SLE can affect several organs, and has an unpredictable disease course (6, 7).
As in all autoimmune diseases, loss of tolerance, in both T and B cells, plays a significant role in the pathogenesis of SLE. Recent studies focusing on the immuno-pathogenesis of the disease suggest that some patients have a genetic predisposition towards loss of tolerance to autoantigens (damaged immunoregulation of T cells), causing overproduction of autoantibodies responsible for the resultant tissue and organ destruction (autoreactive B lymphocytes) (8, 9). T cells of patient with SLE display numerous phenotypic and numerological abnormalities, thereby contributing to SLE pathogenesis and serving as a potentially rich source of SLE biomarkers (10). Significant T cells in SLE include: T-helper cell type 1 (Th1), T-helper cell type 2 (Th2), Th17 cells, follicular T cells (Tfh) and T-regulatory (Treg) cells. T cells from SLE patients are more resistant to induction of apoptosis by thymic stromal cells (11). The TCR complex on the surface of T lymphocyte, consists of a T-cell receptor (TCR), CD3 and (zeta) chain. According to their function, T cells are separated into T helper cells (CD4+) and their cytotoxic T lymphocytes (CD8+). The activation of T lymphocytes is characterized by the expression of number of antigens, including HLA-DR (12).
The diagnosis of SLE is based on criteria set by the American College of Rheumatology (ACR), which were developed in 1971, revised in 1982 and in 1997. These criteria were newly revised and validated by Systemic Lupus International Collaborating Clinics (SLICC) group in 2012. According to SLICC, a SLE positive patient must satisfy at least 4 from 17 criteria, including at least 1 of 11 clinical criteria and 1 of 6 laboratory criteria (13). The activity and severity of disease are measured using several indexes such as the SLEDAI – Systemic Lupus Erythematosus Disease Activity Index (14).
Improved understanding of the immunological basis and pathological mechanisms of SLE has resulted in tangible benefits to patients, such as the introduction of targeted therapy. However, there remains a lack of reliable biomarkers to recognize the disease, predict response to therapy, and monitor this response (15). The aim of this study was to examine the role of several biomarkers (T and B lymphocytes, IL-17, IFN-α) as potential biomarkers for the diagnosis of SLE, and as markers of disease severity and progression.
Material and Methods
Fifty five (N=55) SLE patients were included in the study (4 male and 51 female), with an average age of 33 (25.5–40.0) years.
Inclusion criteria: All SLE patients fulfilled the revised ACR classification criteria: malar and discoid rash, photosensitivity, oral ulcers, arthritis, serositis, renal, neurological, hematologic and immunologic disorder, and positive antinuclear antibody. The presence of 4 out of 11 criteria confirmed the diagnosis of SLE (16).
Exclusion criteria: age less than 18 years; patients with confirmed HIV infection or any other immune deficient; patients undergoing biologic therapy; patients with metastatic disease and acute infectious disease.
SLE group: Most patients (N=45, 81.8%) included in this study were under routine follow-up in the Rheumatology Department at the Clinical Center in Sarajevo from January 2014 to October 2015. Five (9.1%) patients were being treated at the Department of Nephrology, and 5 (9.1%) patients were hospitalized at the Department of Dermatology at Clinical Center in Sarajevo during the same period. At every inspection, patients were clinically evaluated for the presence of signs and symptoms of SLE, and they underwent biochemical laboratory tests and specific immunology tests, and immunological profile (T, B, NK, cytokines). The immunological research of this study was carried out at the Department of Clinical Immunology at Clinical Center of Sarajevo.
Control group: 25 healthy individuals were matched for age and gender with the SLE group. Subjects for the healthy control group were randomly selected from the pool of representative blood samples of healthy adults. This group was not subjected to additional diagnostic procedures.
The blood samples for immune analysis were taken into two tubes: (i) a tube with a gel for determination of IFN-α and IL-17 by ELISA (Enzyme-linked immunosorbent assay), (ii) and the second with an anticoagulant EDTA (ethylene-diamine tetra-acetate) to determine the expression of T and B cells by flow cytometry. Levels of IFN-α from the serum of SLE patients was determined by standardized VeriKine Human Interferon Alpha Multi-Subtype Serum ELISA kit (PBL Assay Science, New York, USA), and for the detection of IL-17 levels, a R&D SystemsTM Human IL-17 kit (R&D SystemsTM, Inc. Minneapolis, USA) was used.
Immunophenotyping of lymphocytes
In order to evaluate the immune status, we analysed peripheral blood lymphocytes using flow cytometry (diagnostic technique), which was performed on BD FACS Canto II (Becton Dickinson, New York, USA) instrument. After acquisition, data were analysed with Diva Software, version 6.1.3 (DB Bioscience). Immunophenotyping of cells was carried out with standard method of sample preparation. Human lymphocytes, after lysis of erythrocytes, were stained with monoclonal antibodies (Abs) conjugated to: FITC (fluorescein isothiocyanate), PE (phycoeryrhrin), PerCP (peridinin-chlorophyll protein complex) and APC (allophycocyanin). All Abs volumes were 20 μL, exept for APC, which was 5 μL. Previously pipetted antibodies were added to a 100 μL whole blood sample and then incubated for 15–30 min. FACS Lysing Solution (2 mL) was added to the sample and incubated for 10 min. The cells were washed with 2–3 mL CellWash, twice, and then pelleted and resuspended for flow cytometric analysis. Every time, between procedure steps, the cells were incubated at room temperature (25–30 °C) in the dark place. All Abs and solutions were provided by BD Bioscience (San Jose, CA, USA) and are summarized in Table I. Furthermore, Treg (CD4+CD25++FoxP3+CD127-), Breg (CD19+ CD24+CD38+), memory B (CD19+CD24+38-), mature (CD19+CD24+CD38inter) were also evaluated. cyFox P3 was prepared according to principles of intracellular staining.
Table I.
Combinations of cells surface markers for the fluorescently labelled monoclonal antibodies.
| FITC | PE | PerCP | APC | |
|---|---|---|---|---|
| T1 T1 | CD3 | CD8 | CD45 | CD4 |
| T2 T2 | CD3 | CD16+56 | CD45 | CD19 |
| T3 T3 | CD3 | HLA-DR | CD45 | – |
| T4 T4 | CD3 | cyFoxP3 | CD4 | CD25 |
| T5 T5 | CD38 | CD24 | CD45 | CD19 |
FITC, Fluorescein Isothiocyanate; PE, Phycoerythrin; APC, Allophycocyanin; PerCP, Peridin hlorophill protein complex; CD3, T lymphocytes; CD4, T helper lym; CD8, T cytotoxic lym; CD19, B cells; CD16+56, Natural killer cells; HLA-DR, marker for activated cells; CD45, leukocytes, FoxP3, forkhead box P3; CD25 and CD38, activation lymphocytes markers.
Ethics
The Institutional Ethics Committee reviewed and approved the study design. Patients were informed and agreed with publishing of their data.
Statistical analysis
Statistical analysis was performed using SPSS 21.0 software for Windows (SPSS, Chicago, IL, USA). The Shapiro-Wilk test was used to test normality and variance homogeneity of data. Data were presented as median and interquartile ranges for skewed variables. Categorical variables were shown as frequencies and analysed using Pearson’s chi-square test. The difference in normally distributed data was tested with independent t-test. The differences in parameters values showing skewed distribution were assessed by Mann-Whitney U test. To determinate the accuracy and respective best cut-off values of biomarkers differentiating SLE patients from healthy controls, Receiver Operating Characteristic (ROC) curves were designed. The accuracy of diagnosing measures was calculated with 95% confidence interval (95% CI). A p value <0.05 was considered statistically significant for all comparisons.
Results
Demographics
The study included a total of 80 subjects; 25 healthy controls and 55 SLE patients with disease at various stages based on the SLEDAI score. All patients were diagnosed according to the ACR criteria (at least 4 out of 11 criteria). Active vs. non-active stage of disease were discriminated according to ACR classification criteria, clinical evaluation, SLEDAI score >4 and compulsory positive antinuclear antibodies for active SLE. There were no statistically significant differences in age or in gender between the patient and the control group (Table II).
Table II.
Baseline characteristics of SLE patients and patients in the control group.
| Variables | SLE group (n=55) | Control group (n=25) | p-value |
|---|---|---|---|
| Age (years) | 33.0 (25.5–40) | 33.0 (27–46) | 0.614* (NS) |
| Sex female male | 51 (92.7%) | 23 (92.0%) | 0.909** (NS) |
| 4 (7.3%) | 2 (8.0%) | ||
| SLEDAI female male | 18.9 (5.0–32.0) | / | |
| 6.9 (2.9–15) | / |
*Data are presented as median (25th and 75th percentiles); NS-not significant compared between SLE and control group; p-probability; *Fisher’s Exact Test; **Pearson Chi-Square Test
Immunophenotyping profile (CD3, CD4, CD8, CD19, NK and Ratio)
Table III summarizes the immuno-phenotype profile of T, B, NK cells. The relative values of activated T cells (CD3HLA-DR+), B lymphocytes (CD19+), regulatory T cells (Treg), and values of serum IL-17 were significantly higher in the SLE group than in the control group. Meanwhile average value of memory B cells was statistically lower for SLE patients compared to the control group. For other parameters, Student’s t-test for independent samples did not demonstrate any statistical significance when comparing the average values of analysed groups. Moreover, the percentage of Treg cells was significantly decreased in patients with active compared to non-active stage of disease (Table IV). IL-17 was much higher in active SLE (54.84 pg/mL) compared to non-active (31.27 pg/mL), but the difference was not statisically significant (p=0.106). Other parameteres did not show significant difference between stages of SLE.
Table III.
Immunophenotype profile: relative values of T, B, NK cells in SLE patients vs. control group.
| Groups | t | p | ||
|---|---|---|---|---|
| SLE group (n=55) | Control group (n=25) | |||
| Mean±SD | Mean±SD | |||
| CD3 (%) | 76.76±9.31 | 76.28±6.91 | 0.23 | 0.819 |
| CD4 (%) | 45.09±11.74 | 46.35±6.87 | -0.60 | 0.551 |
| CD8 (%) | 28.58±11.42 | 25.03±7.27 | 1.68 | 0.098 |
| CD3HLA-DR (%) | 15.51±8.35 | 8.01±3.83 | <0.001 | |
| CD19 (%) | 10.43±6.23 | 8.15±3.63 | 2.06 | 0.043 |
| NK (%) | 11.71±7.39 | 13.13±5.96 | -0.85 | 0.401 |
| Ratio | 1.95±1.17 | 2.02±0.68 | -0.92 | 0.363 |
| Treg (%) | 9.79±5.05 | 7.35±1.64 | -2.309 | 0.024 |
| Breg (%) | 10.02±7.17 | 8.86±4.14 | -0.750 | 0.456 |
| Memory B (%) | 25.11±19.30 | 33.71±11.73 | 2.048 | 0.044 |
| Mature B (%) | 55.13±18.96 | 51.96±11.16 | -0.773 | 0.442 |
| IL-17 (pg/mL) | 49.27±45.88 | 28.64±21.70 | -2.136 | 0.036 |
| IFN-α (pg/mL) | 0.47±1.63 | 0.24±0.44 | -0.691 | 0.491 |
*95% CI Mean-95% confidence interval; n-sample size, t-independent t-test, SD-standard deviation, p-probability rejection of the null hypothesis
Table IV.
Immunophenotype profile: relative values of T, B, NK cells in SLE patients with active vs. non-active disease.
| SLE | P | ||
|---|---|---|---|
| Active (n=14) | Non-active (n=41) | ||
| CD3 (%) | 76.42±9.04 | 76.88±9.50 | 0.526 |
| CD4 (%) | 43.52±14.00 | 45.63±11.00 | 0.217 |
| CD8 (%) | 29.61±13.42 | 28.23±10.81 | 0.700 |
| CD19 (%) | 11.09±6.12 | 10.20±6.32 | 0.280 |
| NK (%) | 11.61±8.06 | 11.74±7.25 | 0.977 |
| Ratio | 1.85±1.03 | 2.0±1.21 | 0.672 |
| Treg (%) | 4.78±1.08 | 11.50±4.71 | <0.001 |
| Breg (%) | 7.22±6.38 | 10.88±7.25 | 0.123 |
| Memory B (%) | 24.86±23.95 | 25.20±17.85 | 0.215 |
| Mature B (%) | 59.72±25.04 | 53.93±16.81 | 0.701 |
| IFN-α (pg/mL) | 0.49±1.81 | 0.40±0.91 | 0.870 |
| IL-17 (pg/mL) | 53.84±48.20 | 31.27±32.79 | 0.106 |
Values of biomarkers for SLE prediction
The ROC curve for accurate prediction of SLE showed that proportion of activated T cells (CD3HLA-DR+) in the SLE patients vs. healthy controls, proportion of regulatory T lymphocytes and proportion of memory B lymphocytes demonstrated a significant area under the curve, shown in Figure 1. ROC curve
Figure 1.
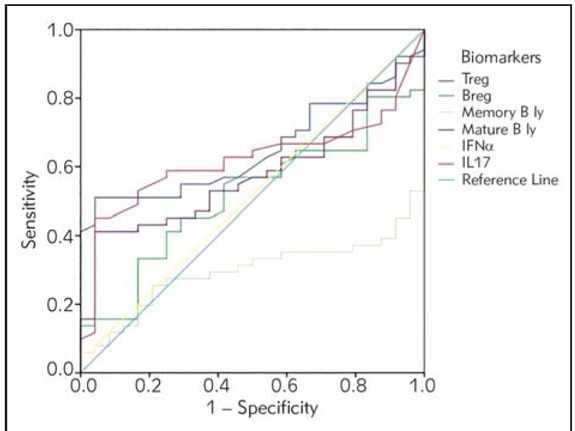
Receiver operating characteristic (ROC) curve of potential biomarkers for differentiation between SLE group and healthy controls. The smallest cut-off value was the minimum observed test value minus 1, and the largest cutoff value was the maximum observed test value plus 1. All the other cut-off values were the average of two consecutively ordered observed test values.
analysis suggested that cut-off for the HLA-DR+ T cells, Treg cells and memory B cells showed good specificity and positive predictive values for diagnosis of SLE (Table V).
Table V.
Optimal cut-off values, area under the curve with 95% confidence interval (AUC, 95% CI), sensitivity, specificity, positive and negative predictive value for biomarkers in differentiating between SLE group and healthy controls.
| Variable and cut-off values | Diagnosing measures | p-value of SLE prediction | ||||
|---|---|---|---|---|---|---|
| AUC(95% CI) | SEN | SPE | PPV | NPV | ||
| CD3HLA-DR+ (≥13.5%) | 0.798 (0.700–0.890) | 49.0% | 96.0% | 96.0% | 46.0% | <0.001 |
| CD3 (78.25%) | 0.546 (0.414–0.678) | 54.5% | 64.0% | 76.9% | 39.0% | 0.513 |
| CD4 (44.45%) | 0.504 (0.375–0.632) | 60.0% | 52.0% | 73.3% | 37.1% | 0.959 |
| CD8 (25.2%) | 0.575 (0.450–0.700) | 56.4% | 72.0% | 81.6% | 42.9% | 0.285 |
| CD19 (7.55%) | 0.603 (0.478–0.727) | 60.0% | 64.0% | 78.6% | 42.1% | 0.143 |
| NK (10.8%) | 0.409 (0.282–0.537) | 45.5% | 44.0% | 64.1% | 26.8% | 0.196 |
| Ratio (2.35) | 0.442 (0.315–0.569) | 30.9% | 72.0% | 70.8% | 32.1% | 0.409 |
| Treg (9.35%) | 0.651 (0.514–0.753) | 49.1% | 95.8% | 96.4% | 45.1% | 0.036 |
| Breg (9.65%) | 0.515 (0.385–0.645) | 45.1% | 72.0% | 71.9% | 36.4% | 0.834 |
| Memory B (34.2%) | 0.285 (0.173–0.398) | 26.9% | 72.0% | 67.9% | 33.3% | 0.002 |
| Nature B (63.3%) | 0.587 (0.461–0.713) | 41.2% | 96.0% | 95.5% | 44.4% | 0.220 |
| I IFN–α (0.34 pg/mL) | 0.517 (0.382–0.653) | 7.3% | 96.0% | 80.0% | 32.0% | 0.807 |
| I IL–17 (33.95 pg/mL) | 0.616 (0.494–0.738) | 58.2% | 76.0% | 84.2% | 45.2% | 0.098 |
*AUC–Area under the curve; CI–Confidence Interval; SLE–Systemic Lupus Erythematosus; SEN–sensitivity; SPE–specificity; PPV–positive predictive value; NPV–negative predictive; p–probability
Discussion
Although the aetiopathogenesis of SLE is incompletely understood, altered immune processes play a significant role, an idea that was supported by the demonstration of autoantibodies against various tissues, primarily to the cells nuclear material (1, 2, 3). Scientific contribution of current study is to estimate the value of specific cellular immunity and the level of cytokines as markers, on the basis of which it would be possible to identify patients with SLE in immunopathogenesis of the disease. Progress in understanding the immunity of SLE could improve insight into the disease mechanism and result in concrete benefits for patients.
For a more accurate diagnosis it is necessary to screen combination of biomarkers for SLE, such as lymphocyte population. Because of high specificity, Treg could potentially refer to real SLE patients. In this way it would be possible to provide an optimal treatment for patients who have a direct benefit from the specific anti-cytokine therapies. We figured out that SLE subjects had higher average value (M=9.79%) of Treg cells compared to control group (M=7.35%) (p=0.024). But when we examined patients with active SLE, the percent of Treg was lower compared to non-active disease (4.78±1.08; 11.50±4.71; p<0.001). This value was also statistically lower compared to control group (p<0.001). The values of Breg cells did not demonstrate statistically significant difference between the SLE and control group. According to the results ROC curve analysis showed that Treg could represent good predictor for the classification of patients with SLE, while Breg cells is not a good classifier. The cut-off value of Treg was 9.35%, according to this criterion the sensitivity was 49,1 % and specificity 95.8%. In relation to these results, we believe that the value of Treg could be a significant marker for the patients with (non-active) SLE evaluation, since this cut-off proved to be extremely specific. However, some authors described different results regarding Treg cells. Crispin (17) examined 30 untreated patients of whom 10 had active disease, 10 inactive, and 10 were healthy volunteers. Treg cells were significantly reduced in patients with active disease compared to controls and patients with inactive disease (p<0.001), showing the same results for active SLE from our study. Previous studies have also examined CD4+CD25hi regulatory T cell frequency and function in patients with SLE with mixed results. Suppressive Treg cells are only those with the highest CD25 expression levels (CD25hi), with the majority of Treg cells non-suppressive (18). The results have been mixed in SLE, with different studies finding a reduced proportion. Possible results diversity can be explained due to remission (non-active) phase of SLE patients in our study, so the values of Treg were higher. Yates et al. (19) investigated 21 patients with SLE and 6 with inactive disease. They found no reduction in the CD25hi subset, although active disease was associated with an increased proportion of Treg cells, but with significant difference when comparing controls and patients with non-active lupus. This indicates and confirms that the apparent increase in Treg cells in patients with active disease is accounted by an increase in the proportion of CD25int cells. Although there was no apparent increase in the proportion of CD4+CD25hi T cells, despite an increase in total Treg cells in patients with active disease, it remained possible that in lupus patients, in vivo activation may have resulted in enhanced expression of CD25 in the CD25int cells. It is possible that in patients with acute onset of disease, CD4+CD25hi T cells may be reduced, that is why active lupus is associated with an increase in total Treg cells by increase of CD25int cells (19).
In part, the reason for discrepancy between studies could also be technical difficulties in the phenotypic characterization of Treg cells by flow cytometry, or characterizing patients with SLE. Crispin (17) included only small number of patients with active SLE, while we included 14 patients with active and 41 patients with non-active SLE. Moreover, Suarez (20) went even further and characterize and quantify cell populations Treg as CD4+CD25low and CD25high in controls and patients with SLE. Quantitatively measured percentage of CD25high cells in 110 SLE patients was significantly higher compared to the controls, what correlates with our results. Accordingly, our analysis of Treg cells was well placed and had no bias to the results. Yan et al. (21) found that the number of CD4+CD25highFoxP3 cells in inactive SLE patients is actually higher than in normal controls, and in the active SLE CD4+CD25high cells show impaired suppressive function. Venigalla et al. (22) reported that the number of CD4+CD25highFoxP3 cells in active SLE is higher than in inactive SLE. However, Valencia et al. (23) in addition to reduced number of CD4+CD25high Treg in the peripheral blood of patients with active SLE, demonstrated that CD4+CD25high for active SLE reduce levels of FoxP3, and were weaker suppressors of cytokine secretion and proliferation of effector T cells in vitro.
As T cells also B cells are hyperactive in SLE. They produce variety of autoantibodies, which result in the formation of immune complexes, and play a key role in the effector phase of the disease. In addition, it also became clear that in SLE T cells are involved in the attack on target cells and tissues through the overproduction of proinflammatory cytokines and increasing adhesion cell-to-cell, which ultimately leads to apoptosis of target cells (23). One possibility to explain the phenomenon of autoimmunity diseases such as SLE may refer to lower Treg function. The lack of function in Treg may result in increased activity of Th cells or directly in the increased activity of B cells, which are both regulated by the Treg in healthy people (17). There are evidences of significance for IL-17, produced by the various T cells, in SLE. Yang et al. (24) demonstrated connection with T cells, which produce IL-17 (Th17 cells), and clinical features of the disease activity score (SLEDAI). Serum IL-17 and the number of Th17 cells were significantly increased in SLE patients compared to controls. Elevated levels of IL-17 frequently significantly correlated with SLEDAI (25). The pathogenesis of SLE is not well understood but it is known that IL-17 and Th17 cells play an important role. IL-17A, a family member of IL-17 enhances the immune response by inducing local production of cytokines, recruiting neutrophils and monocytes, increasing antibody production and exacerbates inflammation and organ damage in SLE (26). In our study, we found statistically significant difference (p=0.036) in the levels of serum IL-17 between patients with SLE (IL-17=49.27 pg/mL) and controls (IL-17=28.64 pg/ml), and the levels were much higher in patients with active SLE compared to nonactive, but serum levels of IL-17 did not show significant accuracy in diagnosing SLE (AUC=0.696; p=0.098). While the levels of IL-17 were significantly higher in patients with SLE, correlation between IFN-α and IL-17 was not found, as expected. IFN-α, as a cytokine with pleiotropic effects, may generate an anti-inflammatory environment or inhibit specific inflammatory T cells such as Th1 and Th17 cells. However, it is not known whether IFN-α inhibits the pro-inflammatory T cells or induce anti-inflammatory properties in T cells. IFN-α does not inhibit IL-17 in SLE patients, because it promotes regulatory Th1 response through the secretion of IL-10 (27). In vitro stimulated CD3+ cells from active SLE patients have significantly higher levels of IL-17 compared to healthy controls. Exceptional only T cells isolated from patients with active disease produce higher levels of IL-17. These results are consistent with previous research by Yang et al. in which they tested 50 patients (24). Furthermore, Wong and colleagues (25) have also shown increased concentration of serum IL-17 in SLE patients compared to healthy controls. Lee et al. (26) proved that the values of IFN-α were statistically higher in the group of SLE patients than in the control group. Similar was obtained in our report, but the difference was not statistically significant, and prediction values, despite excellent specificity, did not show significance for SLE (Table IV). As observed in our study also Dolff et al. (27) discovered that patients with active disease had significantly elevated levels of IL-17 expressing T-cells in comparison to healthy controls (1.46±0.58% vs. 0.93±0.30%, p=0.007), and active patients had also increased levels of IL17+ T-cells as compared to inactive patients (1.46±0.58% vs. 0.88±0.5%, p=0.002). The results suggest that IL-17 producing cells play a pivotal role in the pathogenesis of SLE. Dolff et al. performed in vitro stimulation of CD3+ cells from active SLE patients which only produced significantly higher levels of IL-17 compared to healthy controls. These findings are in accordance with a large previous study by Yang et al. (24) in which 50 patients were enrolled. In addition, Wong et al. (25) demonstrated increased circulating plasma concentrations of IL-17 in SLE patients as compared to healthy controls.
Flow cytometry might provide useful insights into the immune status and distribution of lymphocyte subpopulations. Our study also highlighted the importance of B cells in the immunopathogenesis of SLE, demonstrating that CD19+ (B lymphocytes) were significantly higher in the SLE group than in control group (p<0.043), but could not predict SLE (p=0.143). SLE was traditionally classified as a »B-cell disease« (28). Subgroups of B lymphocytes play a key role and have strong association with the disease. Particularly abnormal activation of human B cells is associated with the phenotypic markers that correlate with disease activity. For example, increased peripheral CD27-IgD-CD19+ memory B cells in SLE patients were associated with SLE disease activity, renal disease and autoantibodies (29). Also, for the number of peripheral CD19+CD27high B cells a positive correlation with serological abnormalities in SLE patients was found (16). To understand whether Treg inhibit these subgroups of B cells in patients with SLE, the Treg were cultivated with autologous CD19+CD27-IgD-B cells or autologous CD19+CD27high B cells. Authors found that Treg induced apoptosis of both B cells subgroups, and Treg inhibited both subgroups after B cells stimulation. Treg lymphocytes reduce B cells, including those, which are associated with active disease and the production of autoantibodies. Quantitative deficit of Treg may be primarily responsible for the impossibility of reducing the B cells. In other words, less functional Treg attempt to counteract with the development of autoimmunity with B cells in SLE, but they were overwhelmed with unbalanced immune homeostasis, which is being developed together with the progression of disease (29). Our results showed that group of patients with SLE had a higher percentage of B lymphocytes (10.43%) compared to controls (8.15%) (p=0.043), while the levels were lower in active form of SLE (9.92%), so we can assume, that in the SLE group, patients had inactive form of disease (12.07% of CD19+ cells), which was largely unaffected by the Treg, and consequently no decrease in B cells was observed.
We found that the percentage of activated T lymphocytes (CD3HLA-DR+) in the patient group was higher than in the control group (p<0.0005). A number of studies have demonstrated that T cells isolated from patients with SLE are abnormal, with regard to their phenotypes and functions compared to healthy subjects (30, 31). Other studies in this area have compared activated T cells in the peripheral blood of SLE patients with healthy controls. Viallard et al. (32) found that activated subpopulation of T lymphocytes (cytotoxic T lymphocytes CD8+HLA-DR+) was increased in SLE patients compared to healthy controls, as was also observed in our analysis.
The phenotype of T cells isolated from patients with SLE is abnormal: SLE T cells partially resemble activated cells and partially behave like anergic (unresponsive) cells. Their response to stimulation through the T-cell receptor (TCR) is exaggerated, and their gene expression profile is altered in comparison with cells obtained from healthy individuals (33). We also analysed sensitivity and specificity of CD3HLA-DR+ for the detection of SLE using ROC curves and defined the optimal cut-off for activated T lymphocytes, which statistically distinguished between patients and healthy individuals. The optimal cut-off was set at 13.5% of CD3HLA-DR+ of total T lymphocytes. CD3HLA-DR+ may differentiate SLE patients from healthy controls, with specificity of 96% and sensitivity of 46%. Despite low sensitivity the CD3HLA-DR+ lymphocytes could represent potential biomarker for the diagnosis of SLE, due to excellent specificity and negative predictive value (NPV=96%) we could evaluate an algorithm that would exclude patients with SLE from further evaluation and lower the time and cost of diagnostics. Different data indicate that in the complex pathogenesis of SLE activated T cells have an important role (34). Despite this, there is a lack of reliable biomarkers for the diagnosis of SLE, monitoring and prediction of response to the therapy. The existing SLE biomarkers are often used to diagnose or monitor disease activity, but the challenge is to discover biomarkers that will be able to predict the beginning of SLE in susceptible individuals and predict disease worsening (development of flares). New generation of SLE biomarkers should provide monitoring of the treatment and evaluate the success of therapy (15).
The reason for the divergent results in terms of the number of immune cells could be sought in the context of several elements. In fact, there are differences in the phenotypic characterization of immune cells with flow cytometry, the method of quantifying cells by flow cytometry and more accurate definition of SLE disease as active or inactive. We believe, if we had a larger cohort of patients with SLE, we would be able to find better statistical differences between the parameters. Therefore, it is necessary to develop larger studies, also be focused on the multicentre analyses, to assemble more patients that would have given wider image, because the results in the literature are divergent. Potential algorithm for SLE patients could be determined with elevated levels of HLA-DR+CD3+ (active T-cells), CD19+ B-cells and drop in relative values of memory B-cell. Differentiation of patients with active disease, according to our results, can solely be established with levels of low Treg and high Th17 cells.
Conclusions
Our results suggested that significant increase in the relative number of activated T cells, Treg lymphocytes, and decrease in memory B cells, as well as increase level of IL-17 in SLE patients compared to healthy controls may be implicated in the pathogenesis of the disease. These parameters, as biomarkers, could distinguish patients with no SLE and SLE patients. Moreover, stratification of patients according to the diseasestage could also be determined with the relative value of Treg and Th17 lymphocytes. Monitoring subpopulations of immune cells in peripheral blood using flow cytometry provides insight into abnormal T and B cell function in SLE. Further investigations are required to explain the role and clinical significance of lymphocytes in the pathogenesis of SLE. Progress in understanding the immunity at SLE, results in concrete benefits for the SLE patients, which include new clinical management and therapeutic strategies.
List of abbreviations
- ACR,
American College of Rheumatology
- APC,
allophycocyanin
- AUC,
Area under the curve
- Breg,
B regulatory cells
- CI,
Confidence Interval
- ENA,c
extractable nuclear antigens
- FITC,
fluorescein isothiocyanate
- IFN,
interferon
- IL,
interleukin
- NPV,
negative predictive value
- PE,
phycoeryrhrin
- PerCP,
peridinin-chlorophyll protein complex
- p,
probability
- PPV,
positive predictive value
- ROC,
Receiver operating characteristic
- SEN,
sensitivity
- SLEDAI,
Systemic Lupus Erythe matosus Disease Activity Index
- SLICC,
Systemic Lupus International Collaborating Clinics
- SPE,
specificity
- TCR,
T-cell receptor
- Treg,
T regulatory cells
Footnotes
Conflict of interest statement The authors stated that they have no conflicts of interest regarding the publication of this article.
References
- 1.D’Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet. 2007;369(9561):587. doi: 10.1016/S0140-6736(07)60279-7. –. [DOI] [PubMed] [Google Scholar]
- 2.Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus: an update. Curr Opin Immunol. 2012;24:651. doi: 10.1016/j.coi.2012.10.004. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castro C, Gourley M. Diagnostic testing and interpretation of tests for autoimmunity. J Allergy Clin Immunol. 2010;125:238. doi: 10.1016/j.jaci.2009.09.041. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomioka R, Tani K, Sato K. Observations on the occurrence of exacerbations in clinical course of systemic lupus erythematosus. J Med Invest. 2008;55:112. doi: 10.2152/jmi.55.112. et al. –. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Shakra M, Novack V. Mortality and Multiple Causes of Death in Systemic Lupus Erythematosus − Role of the Death Certificate. J Rheumatol. 2012;39:458. doi: 10.3899/jrheum.111556. –. [DOI] [PubMed] [Google Scholar]
- 6.Crispin J, Liossis S, Kis-Toth K. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med. 2011;16:47. doi: 10.1016/j.molmed.2009.12.005. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahman A, Isenberg DA. Systemic Lupus Erythematosus. N Engl J Med. 2008;358:929. doi: 10.1056/NEJMra071297. –. [DOI] [PubMed] [Google Scholar]
- 8.Sestak AL, Fürnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis. 2011;70:37. doi: 10.1136/ard.2010.138057. –. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y, Tsao BP. Advances in lupus genetics and epigenetics. Curr Opin Rheumatol. 2014;26:482. doi: 10.1097/BOR.0000000000000086. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman R. T cells in the pathogenesis of systemic lupus erythematosus. Clin Immunol. 2004;113:4. doi: 10.1016/j.clim.2004.05.001. –. [DOI] [PubMed] [Google Scholar]
- 11.Lloyd P, Doaty S, Hahn BH. Gordon C, Isenberg D. Systemic Lupus Erythematosus. Oxford University Press; 2016. Aetiopathogenesis of systemic lupus erythematosus; pp. 7–23. –. [Google Scholar]
- 12.Abbas A, Lichtman A, Pillai S. Abbas AK. Cellular and Molecular Immunology. Philadelphia Saunders: 2014. Activation of T lymphocytes; pp. 199–212. –. [Google Scholar]
- 13.Petri M, Orbai AM, Alarc n GS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677. doi: 10.1002/art.34473. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Formiga F, Moga I, Pac M, Mitjavila F, Rivera A, Rujol R. Mild presentation of systemic lupus erythematosus in elderly patients assessed by SLEDAI. SLE Disease Activity Index. Lupus. 1999;8:462. doi: 10.1177/096120339900800609. –. [DOI] [PubMed] [Google Scholar]
- 15.Liu CC, Kao A, Manzi S, Ahearn J. Biomarkers in systemic lupus erythematosus: challenges and prospects for the future. Ther Adv Musculoskelet Dis. 2013;5:210. doi: 10.1177/1759720X13485503. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 17.Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21:273. doi: 10.1016/s0896-8411(03)00121-5. –. [DOI] [PubMed] [Google Scholar]
- 18.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245. doi: 10.4049/jimmunol.167.3.1245. –. [DOI] [PubMed] [Google Scholar]
- 19.Yates J, Whittington A, Mitchell P, Lechler RI, Lightstone L, Lombardi G. Natural regulatory T cells: number and function are normal in the majoriy of patients with lupus nephritis. Clin Exp Immunol. 2008;153:44. doi: 10.1111/j.1365-2249.2008.03665.x. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+ CD25high T cell population in patinets with systhemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis. 2006;65:1512. doi: 10.1136/ard.2005.049924. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dys-functional CD4+,CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon-alpha-producing antigenpresenting cells. Arthritis Rheum. 2008;58:801. doi: 10.1002/art.23268. –. [DOI] [PubMed] [Google Scholar]
- 22.Venigalla RK, Tretter T, Krienke S.. Reduced CD4+,CD25-T cell sensitivity to the suppressive function of CD4+, CD25high, CD127-/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. 2008;58:2120. doi: 10.1002/art.23556. et al. –. [DOI] [PubMed] [Google Scholar]
- 23.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007;178:2579. doi: 10.4049/jimmunol.178.4.2579. –. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Chu Y, Yang X. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1472. doi: 10.1002/art.24499. et al. –. [DOI] [PubMed] [Google Scholar]
- 25.Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol. 2008;127:385. doi: 10.1016/j.clim.2008.01.019. –. [DOI] [PubMed] [Google Scholar]
- 26.Lee JY, Park JK, Lee EY, Lee EB, Song YW. Circulating exosomes from patients with systemic lupus erythematosus induce an proinflammatory immune response. Arthritis Res Ther. 2016;18:264. doi: 10.1186/s13075-016-1159-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolff S, Quandt D, Wilde B, Feldkamp T, Hua F, Cai X. Increased expression of costimulatory markers CD134 and CD80 on interleukin-17 producing T cells in patients with systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R150. doi: 10.1186/ar3100. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mak A, Kow NY. The Pathology of T Cells in Systemic Lupus Erythematosus. J Immunol Res. 2014;2014:419029. doi: 10.1155/2014/419029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iikuni N, Lourenço EV, Hahn BH, La Cava A. Cutting edge: Regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol. 2009;183:1518. doi: 10.4049/jimmunol.0901163. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collinson P. Laboratory medicine is faced with the evolution of medical practice. J Med Biochem. 2017;36:211. doi: 10.1515/jomb-2017-0032. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comte D, Karampetsou PM, Tsokos CG. T cells as therapeutic target in SLE. Lupus. 2015;24:351. doi: 10.1177/0961203314556139. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viallard JF, Bloch-Michel C, Neau-Crasac M. HLADR expression on lymphocyte subsets as a marker of disease activity in patients with systemic lupus erythematosus. Clin Exp Immunol. 2001;125:485. doi: 10.1046/j.1365-2249.2001.01623.x. et al. –. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cripsin JC, Tsokos GC. Hahn BH, Wallace DJ. Dubois’ lupus erythematosus and related syndromes. Philidelphia Saunders: 2013. T Cells; pp. 96–104. –. [Google Scholar]
- 34.Madhok R, Wu O. Systemic lupus erythematosus. American Family Physician. 2007;76:1351. –. [PubMed] [Google Scholar]