

Abstract
The hydrolytic decomposition of four peptides containing S-aroylthiooximes (SATOs) with variable N-arylidene substituents was investigated in 10 aqueous buffer solutions at pH values ranging from 6.0 to 10.9. UV-Vis spectroscopy was employed to study the reaction kinetics, which revealed V-shaped pH-rate profiles for all peptides with a minimum near pH 8, suggesting a change from an acid-catalyzed to a base-activated reaction. Hammett plots showed positive ρ values above pH 8 and negative ρ values below pH 8, providing further evidence for a mechanism change. Based on these data, along with mass spectrometry evidence, we propose a specific acid catalysis under mildly acidic and neutral conditions and multiple base-promoted decomposition reactions under mildly basic conditions.
Graphical Abstract
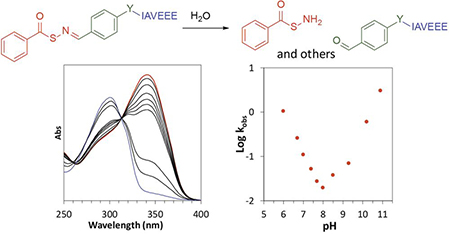
INTRODUCTION
Functional groups containing carbon–nitrogen double bonds, (e.g., imines, hydrazones, and oximes) are easy to prepare and are relatively stable in aqueous media near neutral pH.1 Under acidic conditions, however, these functional groups hydrolyze rapidly.2 As a result of this pH-dependent stability, functional groups containing C=N bonds find use in several applications, including drug delivery,3 and tissue engineering.4 For example, hydrazones are linkers in cancer therapeutics, where active drugs can be released in the acidic tumor microenvironment,5–7 and oximes are used widely in bio-conjugation and in constructing hydrogels as matrices for cell encapsulation.8–10
Despite extensive knowledge of the pH-dependent hydrolyses of functional groups with structures R1R2C=N–X where X = R3, NHR3, or OR3 (imines, hydrazones, and oximes, respectively), little is known about the sulfur-containing analogues (X = SR3) of oximes.11 Our recent investigations of S-aroylthiooximes (SATOs), which are sulfur analogues of oximes, have the general structure R1R2C=NSC(O)Ar.12 SATOs decompose upon reaction with a thiol to release hydrogen sulfide (H2S), a biological signaling gas with many physiological roles and extensive therapeutic potential.13–16 Since our initial report on SATOs in 2014, others have studied synthetic routes to compounds with C=N–S linkages,17,18 and we have employed SATOs as H2S donors in polymers,19 polymer micelles,20 and peptide-based gels.21
SATOs are prepared through a condensation reaction between an S-aroylthiohydroxylamine and an aldehyde or ketone. In our original report on SATOs, we noted that in addition to thiol-triggered decomposition to produce H2S, SATOs can also undergo hydrolysis in a reaction that is the reverse of the condensation reaction through which they form. They are fairly stable near physiological pH, with hydrolysis half-lives ranging from days to weeks for most compounds. Despite the stability of SATOs at near-neutral pH, significant hydrolysis of SATO-containing peptides occurs when purified under basic conditions. In an effort to increase the stability of these H2S-releasing peptides, we quantify the kinetics and probe the mechanism(s) of hydrolytic decomposition at pH 6–10.9 of four water-soluble, SATO-containing peptides.
RESULTS
Design and synthesis of SATO-peptides
To probe how pH affects SATO decomposition, we designed four SATO-peptides for this study. We attached a short peptide chain with the sequence IAVEEE, as we have used previously,21 to promote water solubility. Each SATO-peptide consists of a central aromatic linker, an arylidene, that is attached on one side to IAVEEE and on the other side to SATO (Figure 1A). Only the linker varies among the four compounds; we selected linkers to include both electron-donating (–OCH2C(O)–), and electron-withdrawing groups (–C(O)–, –SO2–), plus a group with extended conjugation (–CH=CHC(O)–) (Figure 1B).
Figure 1.
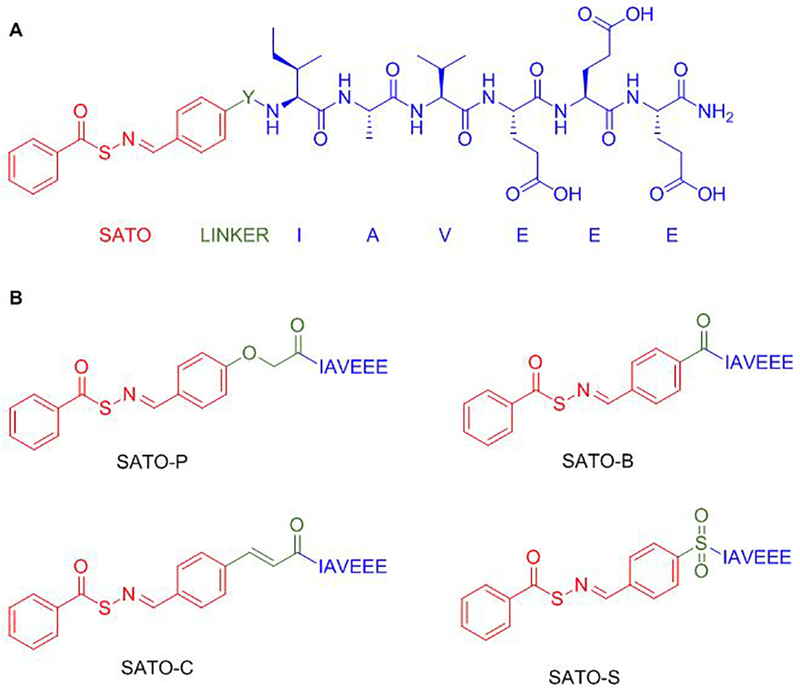
(A) Complete general structure of SATO-peptides with the SATO group in red, the linker (Y) in green, and the peptide chain (IAVEEE) in blue (B) Structures of SATO-containing peptides with the various linkers, Y, shown in green.
Peptides were constructed by using Fmoc-based solid phase peptide synthesis, installing the linker in the final coupling step. After cleavage from the resin and isolation of each functionalized peptide, S-benzoylthiohydroxylamine was added to each peptide aldehyde in a condensation reaction before purification by preparative HPLC. Peptides are named with a two-unit code, where the first unit indicates the thiooxime part of the peptide (SATO in all cases) and the second unit consists of a letter that indicates the type of the linker (P, C, B, or S, as indicated in Figure 1). For example, peptide SATO-C refers to a thiooxime containing peptide with a cinnamoyl linker. As expected, the linkers did not prevent H2S release in the presence of cysteine (Figure S1).
Kinetics hydrolytic decomposition of SATO-containing peptides
Hydrolytic decomposition rate measurements of SATO-peptide solutions were carried out at rt in buffered water at 10 different pH values ranging from 6.0–10.9 (Scheme 1). Reactions were followed via UV–Vis absorption spectroscopy by monitoring the decrease in the absorption maximum near 330 nm. Figure 2 includes representative time-dependent spectra for peptide SATO-C at pH 7.7; data for the other peptides can be found in Supporting Information (Figure S2–S11). We observed a well-anchored isosbestic point near 300 nm for each hydrolysis at each pH value (Figure 2A) except for pH values above 10, in which case the isosbestic point moved slightly, likely due to the formation multiple hydrolytic decomposition species (S10-S11).
Scheme 1.
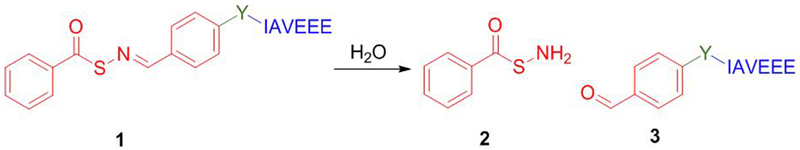
General scheme representing hydrolysis of SATO-peptides (1) to S-benzoylthiohydroylamine (2) and the corresponding peptide aldehyde unit (3).
Figure 2.
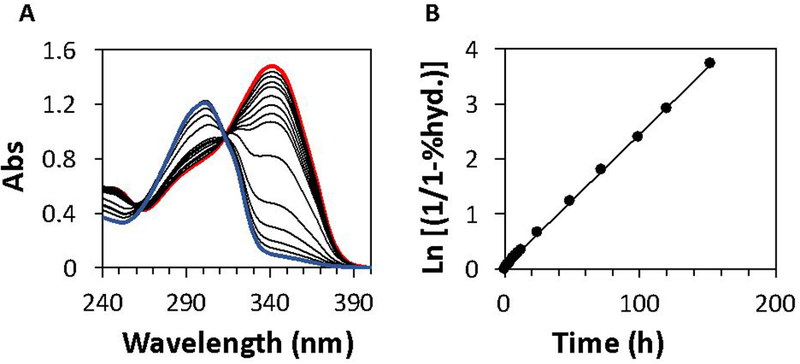
(A) Example UV–Vis absorbance curves for peptide SATO-C at 50 μM in aqueous phosphate buffer solution (10 mM) at pH 7.7. The red and blue traces correspond to the first and final timepoints, respectively, with intermediate timepoints in black. (B) First-order kinetics plot of peptide C3 exhibiting a straight-line fit corresponding to pseudo-first-order kinetics.
First-order kinetics plots based on these spectral data revealed straight lines, and the pseudo-first-order rate constants were calculated from the slope of these lines (Figure 2B, also see calculation section in the SI). Using this method for each peptide at each pH, we obtained 40 pseudo-first-order rate constants (Table 1).
Table 1.
Hydrolytic decomposition rate constants of SATO-peptidesa
| kobs*103 (min–1) | p value | ||||
|---|---|---|---|---|---|
| pH | SATO-P | SATO-C | SATO-B | SATO-S | |
| 6.0 | 27 ± 2 | 18 ± 1 | 17 ± 1 | 13 ± 1 | –0.3 |
| 6.7 | 6.4 ± 0.1 | 4.4 ± 0.1 | 4.3 ± 0.3 | 3.2 ± 0.3 | –0.4 |
| 7.0 | 3.4 ± 0.3 | 2.2 ± 0.1 | 1.8 ± 0.1 | 1.3 ± 0.1 | –0.5 |
| 7.4 | 1.3 ± 0.1 | 0.87 ± 0.04 | 0.87 ± 0.02 | 0.69 ± 0.02 | –0.3 |
| 7.7 | 0.72 ± 0.07 | 0.47 ± 0.06 | 0.42 ± 0.05 | 0.40 ± 0.02 | –0.3 |
| 8.0 | 0.50 ± 0.04 | 0.33 ± 0.03 | 0.33 ± 0.02 | 0.28 ± 0.01 | –0.3 |
| 8.5 | 0.35 ± 0.02 | 0.53 ± 0.08 | 0.63 ± 0.05 | 0.62 ± 0.05 | 0.3 |
| 9.3 | 0.57 ± 0.04 | 0.81 ± 0.04 | 1.5 ± 0.2 | 2.1 ± 0.1 | 0.7 |
| 10.2 | 3.1 ± 0.2 | 6.9 ± 0.8 | 10 ± 1 | 8.9 ± 0.2 | 0.6 |
| 10.9 | 13 ± 0.6 | 34 ± 3 | 49 ± 1 | 37 ± 3 | 0.5 |
Rate constants and standard deviations of measurement were calculated from the average of three replicates. Buffers (all 10 mM) were as follows: pH 6.0–8.0: phosphate buffer; pH 8.5: Bicine buffer; pH 9.3–10.9: carbonate buffer.
Four pH–rate profiles (Figure 3) were constructed to identify the hydrolytic decomposition mechanisms of the SATO-peptides. For each SATO-peptide, a pH–rate profile was generated by graphing Log pseudo-first-order rate constants vs. pH, yielding V-shaped profiles for each peptide with minima in the region of pH 8.0–8.5 (Figure 3). Comparing the nearly identical pH–rate profiles among the 4 SATO-peptides revealed only one notable difference. The minimum Log kobs was at pH 8.5 for SATO-P but was at pH 8.0 for SATO-B, SATO-C, and SATO-S. For all peptides, linear regression of Log kobs in the range of pH 6.0–8.0 gave a slope of approximately −0.9, indicating a linear dependence of rate on [H3O+], which is consistent with an acid-catalyzed reaction. A similar analysis in the range of pH 8.5–10.9 gave a slope in all plots of approximately 0.8, indicative of a linear dependence of the rate on [−OH], which is consistent with a base-activated reaction.
Figure 3.
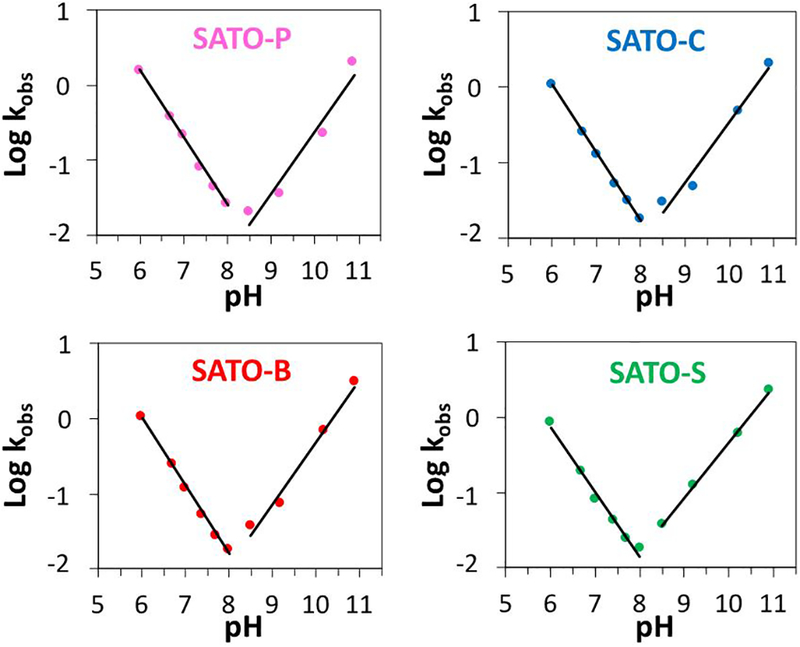
Plots showing pH-rate profiles for all four SATO-containing peptides. Lines are linear regressions of Log kobs at pH 6.0–8.0 and 8.5–10.9 in each profile.
Hammett plots at different pH values
To evaluate how different substituents affect rates of hydrolytic decomposition of SATO-containing peptides, we plotted Log kobs versus Hammett sigma (σ) of the linker at given pH values (Figure 4 and Figure S12). We estimated Hammett σ values for the linkers by first plotting predicted pKa values22 for eight para-substituted benzoic acid derivatives against their known Hammett σ values.22,23 The plot revealed a strong linear correlation for these eight known compounds (Figure S14). We then estimated Hammett σ values for the linkers by applying predicted pKa values for para HOOC-C6H4-Y-NHMe to the fitted line for pKa’s of eight known acids versus their Hammett σ values (see Supporting Information).
Figure 4.
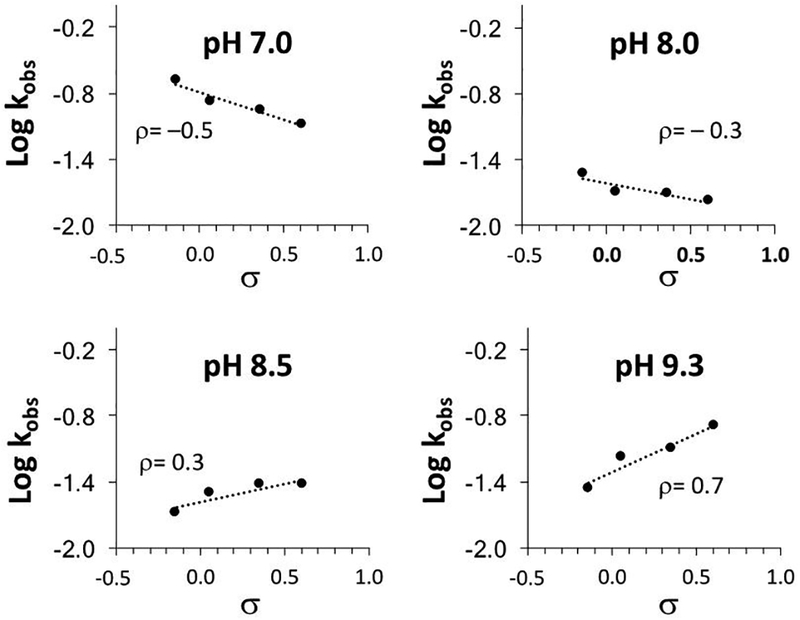
Hammett plots at pH 7.0, 8.0, 8.5, and 9.3, illustrating the change in sign of values of ρ as the pH moves from acidic to basic.
At pH 6.0–8.0, good correlations were observed in the Hammett plots, with ρ values around −0.3 (Table 1). At pH 8.5, Hammett ρ was 0.3; for pH 9.3, 10.0, and 10.9 Hammett ρ was 0.7, 0.6, and 0.5, respectively. This change in the sign of ρ observed for crossover from acidic to basic conditions supported the proposed change in mechanism. Electron-withdrawing and electron-donating substituents affected the acidic and basic reactions in opposite ways.
Identification of products at different pH values
Mass spectrometric analysis of the SATO-peptide fragments formed at various pH values after 5 min provided information on the products of hydrolytic decomposition (see Discussion). A mass spectrum for SATO-B at pH 6.0 (Figure 5A) showed evidence for the formation of peptide aldehyde at m/z = 819.4 (the product of C=N bond hydrolysis) and peptide nitrile at m/z = 816.4 (a decomposition product). At pH 10.9, the same products were observed, along with peptide imine thiol (the product of thioester hydrolysis) at m/z = 850.7 (Figure 5B).
Figure 5.
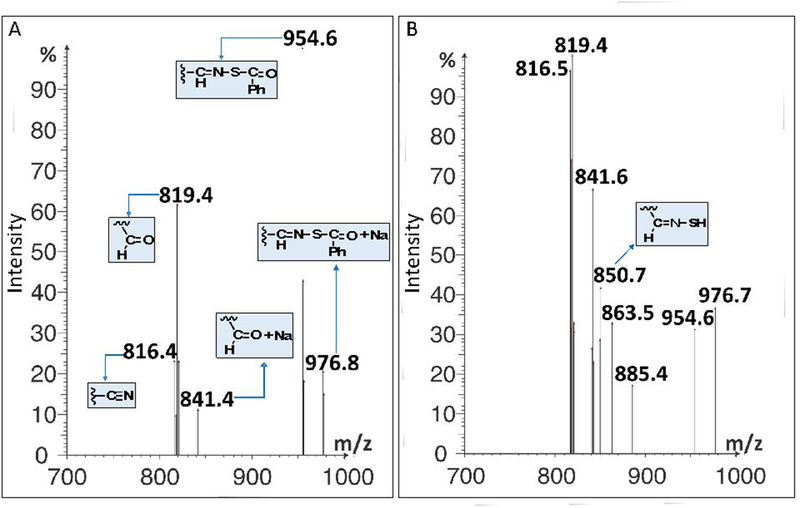
Mass spectra for peptide SATO-B after 5 min depicting reaction progress and products at pH a) 6.0 and B) 10.9 recorded in negative electrospray ionization mode.
DISCUSSION
Possible mechanisms as a function of pH
A V-shape in pH–rate profiles indicates a change in mechanism when transitioning from acidic to basic conditions. The minima in the pH–rate profiles indicate either a minimum in the sum of the rates for the acid-catalyzed and base-activated reactions, or an uncatalyzed reaction, as has been observed for some imines.2,24–26 Based on fittings of the pH–rate profiles to a model involving acidic, basic, and uncatalyzed reactions, we conclude that the uncatalyzed reaction (or buffer catalysis) contributes a small amount to the observed rates under neutral to mildly basic conditions (Figure S13).
In general, three mechanisms are possible for the hydrolysis reactions involving C=N bonds (Scheme 2)24,25 These include (I) equilibrium protonation of –C=N– followed by the addition of water to –C=NH+– to form a protonated carbinolamine adduct (specific acid-catalyzed); (II) addition of water to neutral –C=N– (with concomitant proton transfer from water and proton transfer to the imine N atom) or addition of −OH to cationic –C=NH+– to form a neutral carbinolamine; and (III) addition of −OH to neutral –C=N– to form an anionic carbinolamine adduct (base-activated), which picks up a proton immediately to form neutral carbinolamine (vertical arrow) or forms the neutral carbinolamine in a mechanism of synchronous −OH addition and proton transfer (diagonal arrow). All forms of carbinolamine adducts decompose in subsequent steps to give the hydrolyzed products, an aldehyde and S-benzoylthiohydroxylamine in this case. The formation or decomposition of the carbinolamine adduct can both act as rate-determining steps depending on the reaction conditions and substrate structure.27 The shape of the pH-rate profiles suggests a crossover from acid-catalyzed to base-mediated hydrolytic decomposition.
Scheme 2.

General representation of rate-determining steps possible during the hydrolysis of C=N. The constants k1–k3 represent the respective rate constants of the reactions.
Based on the reactions of O-acylaldoximes,28 three additional decomposition mechanisms might operate in S-acylaldoximes. These include (IV) water addition to the carbonyl of the thioester (with concomitant proton transfer from water and proton transfer to the carbonyl O atom) followed by elimination of thiohydroxylamine (R-C=N-SH); (V) addition of −OH to C=O to form an anionic tetrahedral adduct (base-activated) followed by elimination of thiooximate (R–C=N–S–); and (VI) proton transfer from C—H with synchronous elimination of thiocarboxylate to form a nitrile. For O-acylaldoximes, mechanism IV predominates at pH < 7; mechanisms V and VI operate at a ratio of 3:1 at pH >7.
Hydrolysis in mildly acidic to neutral media (pH 6.0–7.7)
The rate of hydrolysis in this region increases linearly with increasing solution acidity, as evidenced by the slope near −1 in the pH–rate profiles (Figure 3). Under mildly acidic to neutral conditions, the reaction likely proceeds via a fast equilibrium protonation of the –C=N– group followed by addition of water to –C=NH+– to give a protonated carbinolamine adduct, which then decomposes to give the final products (Scheme 2, Mechanism I). Breakdown of the carbinolamine intermediate is likely rate-determining.27,29 The proposed rate law is therefore given equation 1:
| (1) |
Further verification of this mechanism comes from the negative values of ρ obtained from the Hammett plots in this pH range. Negative values of ρ indicate that the transition state is electron deficient compared to the reactant; therefore, addition of water or breakdown of the tetrahedral intermediate must be rate-determining. Electron-withdrawing groups therefore increase the electrophilicity of the carbon atom in –C=N–, decreasing the stability of the cationic intermediate and decelerating the reaction. A more negative value of ρ (closer to −1) would be expected if protonation of the imine were rate determining. This mechanism is consistent with results obtained on the hydrolyses of oximes and hydrazones, where Kalia and Raines concluded that protonated carbinolamine adduct formation is rate-determining under mildly acid and neutral conditions. The appearance of nitrile in the mass spectrum (Figure 5A) suggests an elimination mechanism may be operating as well (Scheme 3, Mechanism VI). The mechanistic details of such an elimination reaction at this pH remain to be explored.
Scheme 3.
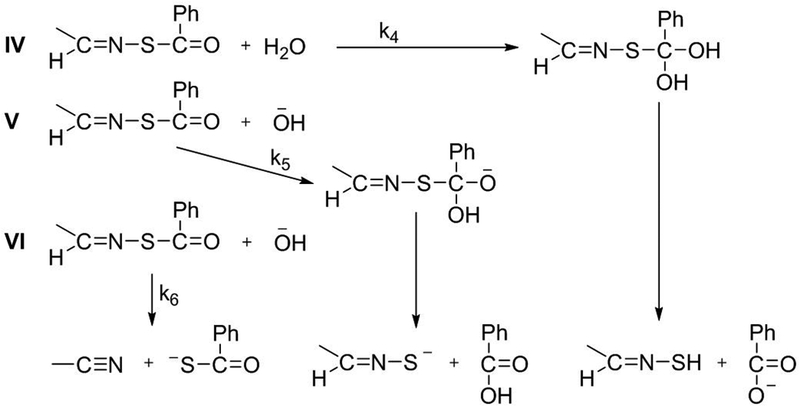
General representation of rate-determining steps possible during the reactions not involving hydrolysis of C=N. The constants k4–k6 represent the respective rate constants of the reactions.
Reactions in basic media (pH 9.3–10.9)
In the range of pH 9.3–10.9, hydrolytic decomposition rates increase with increasing basicity with a slope near 1, which corresponds to the increase in concentration of −OH. Therefore, three mechanisms are possible. (Scheme 2, Mechanism III and Scheme 3, Mechanisms V and VI). The corresponding rate law is 1st order in both −OH and C=N (2nd order overall), as indicated by equation 2:
| (2) |
Values of ρ in this pH region are positive, which is opposite in sign to those in the acidic pH region, indicating build-up of negative charge in the transition state. These data are consistent with addition of −OH to the neutral C=N (Mechanism III), addition of −OH to the neutral thioester (Mechanism V), or proton abstraction from the imine carbon by −OH (Mechanism VI) as the rate determining step. The expected values of ρ would likely be similar for each mechanism.
The pH–rate profiles for the hydrolyses of oximes,30 hydrazones,30 and O-acylaldoximes28 provide precedence for predicting mechanisms. For oximes and hydrazones, pH–rate profiles have negative slopes at pH 5–9. This observation likely rules out Mechanism III, where a positive pH-rate profile under basic conditions would be observed. For O-acylaldoximes, pH–rate profiles have a zero slope at pH 4.8–6.6 and positive slopes starting at pH 7.2–9.5. These measurements support Mechanisms V and VI. Further evidence for Mechanisms V and VI comes from mass spectral product analysis (Figure 5B). The data reveal the formation of three major species: 1) A peptide nitrile (m/z = 816.5), which supports mechanism VI; 2) A peptide thiooxime (m/z = 850.7), which supports mechanism V; and 3) A peptide aldehyde (m/z = 819.4, along with additional Na-adduct peaks at m/z = 841.6, 863.5 and 885.4), which may come from hydrolysis of the thiooximate product in Mechanism V, as we and others have previously suggested.12,31
Reactions in mildly basic media (pH 8.0–8.5)
At pH 8 and 8.5, the reaction proceeds slowly and the rate constants for all four peptides are close. The lack of a plateau region in any of the four pH–rate profiles suggests that this changeover region reflects a sum of rates for acid-catalyzed (I) and base activated (V and VI) mechanisms. Thus, kobs would comprise k1(kH/k–H)[H3O+] + (k5 + k6)[−OH]. However, we cannot exclude mechanisms II and IV. These two mechanisms both generate neutral intermediates, presumably with small values of ρ. The values of ρ flip from negative to positive in this pH range (–0.3 for pH 8.0; 0.3 for pH 8.5). Unfortunately, one cannot distinguish among the mechanisms as an observed ρ is a weighted average of all contributing mechanisms. Because multiple processes likely occur in this pH range, the rate law cannot be explicitly determined.
Hammett ρ values
At pH 6.0–8.0, values of ρ hover near −0.3; at pH >8.5, they increase to approximately 0.6. The crossover point from negative to positive ρ values occurs between pH 8.0 and pH 8.5 (Table 1). The change in sign of ρ agrees with the change from the proposed acid-catalyzed to base-activated mechanisms.
At pH 6.0–8.0, equilibrium protonation of –C=N– precedes rate-determining addition of water to the cationic intermediate. The equilibrium protonation of –C=N– is favored by electron-donating substituents, whereas the attack of water is favored by the electron-withdrawing substituents. As a result of these competing forces, the positive charge build-up in the transition state is small, leading to a small negative value of ρ. The appearance of the nitrile in the mass spectrum at pH 6 (Figure 5A) suggests a contribution from a mechanism that may have a small positive or near zero value of ρ. This contribution means that the observed value would be less negative than if acid-catalyzed hydrolysis of C=N were the only mechanism.
Interestingly, the absolute values of ρ at higher pH are larger than those at lower pH. Above pH 8.5, values of ρ increase in magnitude. Presuming mechanisms V and VI operate at high pH, one would expect the value of ρ would be higher for mechanism VI. Precedence for a large positive value of ρ derives from formation of nitriles in O-pivaloyloximes in acetonitrile.32 Our previous study12 on hydrolysis of arylidenethiooximes at pH 7.4 in water/acetonitrile suggests a very small positive value of ρ for mechanism V. The values in Table 1 and the mass spectrum at pH 10.9 (Figure 5B) suggest that both mechanisms operate.
CONCLUSION
In summary, we have studied in detail hydrolytic decomposition kinetics of a series of SATO-peptides in aqueous buffer to increase our knowledge of the sulfur-containing analogues of imines and oximes. Based on pH-rate profile data, Hammett plots at different pH values, and mass spectral data, we conclude that several mechanisms for hydrolytic decomposition of SATO-peptides operate under acidic and basic conditions. Consistent with C=N functional groups, SATOs undergo acid-catalyzed hydrolysis under mildly acidic to neutral conditions (pH 6.0–7.4), and base-activated decomposition reactions under basic conditions (pH 9.3–10.9). Rates are slowest under mildly basic conditions (pH 7.7–8.5), where crossover occurs. This study provides further insight into the chemistry of sulfur-containing analogues of imines and oximes at physiologically relevant pH. Such insight may be useful in designing new H2S donors and exploiting the reactivity of these compounds for therapeutic purposes.
EXPERIMENTAL SECTION
Materials
Fmoc-protected amino acids, HBTU, and Rink amide MBHA resin were purchased from P3biosystems and used as received. N,N-Diisopropylethylamine (DIEA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were purchased from Chem Impex and trifluoroacetic acid and piperidine were purchased form Oakwood chemicals and BTC chemicals, respectively. All the solvents employed for peptide synthesis were reagent grade. NMR spectra were measured on Agilent 400 MHz or Bruker 500 MHz spectrometers. 1H and 13C NMR chemical shifts are reported in ppm relative to internal solvent resonances. Yields refer to chromatographically and spectroscopically pure compounds unless otherwise stated. The microwave reactor on a Liberty1 microwave-assisted peptide synthesizer (CEM) equipped with an internal temperature probe was used to synthesize the linker ((4-formylphenyl)sulfonyl)-l-isoleucine with a power setting at 50 W. Linkers (2-(4-formylphenoxy)acetic acid and 4-formylbenzoic acid were purchased from Chem Impex and TCI, respectively, and 4-formylcinnamic acid was synthesized according to the literature procedure.33
Methods
General method for peptide synthesis:
Synthesis of the SATO-peptides was accomplished in two steps: 1) The peptides were made by using solid-phase peptide synthesis (SPPS), with installation of the linker on resin; 2) After cleavage of the peptide from the resin and purification by preparative HPLC, S-benzoylthiohydroxylamine was added to form the SATO group in a condensation reaction. Final SATO peptides were then purified again by preparative HPLC. Detailed synthetic conditions are provided below.
For all four SATO-peptides, IAVEEE was synthesized either manually or by using a Liberty1 microwave-assisted peptide synthesizer (CEM) following standard procedures21 for Fmoc-based SPPS. For peptides SATO-P, SATO-C and SATO-B, the linkers (2-(4-formylphenoxy)acetic acid, 4-formyl cinnamic acid, and 4-formylbenzoic acid, respectively) were coupled respectively to the peptide N-terminus on-resin under identical coupling conditions as those for the amino acids. Peptide SATO-S was synthesized by following a different procedure. First, 4-formylsulfonylchloride was coupled to l-isoleucine in water under microwave heating by using a modified literature procedure. This unit was added under standard SPPS conditions to the AVEEE peptide on-resin. After cleavage and isolation, peptides were dissolved in water containing 0.1% NH4OH and filtered through a 0.45 μm PTFE filter before purifying. Purification by preparative-scale reverse phase-high performance liquid chromatography (RP-HPLC) was carried out on an Agilent Technologies 1260 Infinity HPLC system, eluting with a gradient of 2% ACN to 90% ACN in milliQ H2O over 33 min on an Agilent PLRP-S column (100-Å particle size, 25 × 150 mm) and monitoring at 220 nm. To both mobile phases, 0.1% NH4OH was added to aid in solubility. Fractions were analyzed by mass spectrometry (Advion ExpressIon Compact Mass Spectrometer (CMS)), and product-containing fractions were combined, rotovapped to remove ACN, and lyophilized (LabConco).
The lyophilized linker-containing peptides were dissolved in dry DMSO; S-benzoylthiohydroxylamine along with catalytic TFA were added to afford the final SATO-peptides—SATO-P, SATO-C, SATO-B, and SATO-S (Figure 1B). Peptides were dissolved in 10 mM phosphate buffer at pH 7.4 and filtered through a 0.45 μm PTFE filter before injecting onto the HPLC. Purification was carried out by RP-HPLC, eluting with a gradient of 2% ACN to 90% ACN in milliQ H2O without any additives. The protocol for analyzing and recovering the peptides was the same as described above. The final SATO-peptides were dissolved in milliQ water and distributed into aliquots (100 μg each). Aliquots were frozen, lyophilized, and stored at −20 °C.
Synthesis of ((4-formylphenyl)sulfonyl)-l-isoleucine:
Modified amino acid ((4-formylphenyl)sulfonyl)-l-isoleucine was synthesized according to literature procedures.34 Briefly, a microwave reaction tube was charged with 4-formylsulfonyl chloride (0.100 g, 0.5 mmol), l-isoleucine (0.064 g, 0.5 mmol), K2CO3 (0.146 g, 1.0 mmol), and water (2 mL) to give a suspension. The reaction mixture was sonicated for 30–45 min to afford a turbid solution. The reaction mixture was subjected to microwave heating at 75 °C for 10 min to yield a clear, pale yellow solution. Reaction progress was monitored by TLC, eluting with 100 % EtOAc (UV visualization). Once the starting material had been completely consumed, the reaction mixture was acidified by addition of 1N HCl, and the mixture was extracted with EtOAc (2 × 10 mL) in a separatory funnel. The organic layers were then combined and washed with brine (1 × 10 mL). The clear solution was then dried over Na2SO4 and concentrated under vacuum to afford the product as a light orange solid (0.093 g, 63 % yield). The final product was used directly in the next step without further purification. 1H NMR (DMSO-d6): δ 0.78 (m, 6H), 1.13 (m, 1H) δ 1.38 (m, 1H), 1.68 (m, 1H) δ 3.63 (t, 1H), 7.96 (d, 2H) δ 8.06 (d, 2H), 8.34 (d, 1H) δ 10.10 (s, 1H), 12.62 (bs, 1H). 13C NMR (DMSO-d6): δ 11.3, δ 15.8, δ 24.7, δ 37.2, δ 60.7, δ 127.7, δ 130.4, δ 138.7, δ 146.4, δ 172.4, δ 193.1.
pH measurements
Buffer solutions (10 mM) were freshly prepared in milliQ H2O from their respective salts in calibrated volumetric glassware. Phosphate, carbonate, and bicine buffers were used to achieve the required pH range of 6.0–8.0, 9.3–10.9, and 8.5, respectively. The pH of the buffer solutions was checked with a calibrated digital pH-meter equipped with an external glass electrode (Accument Basic [AB15 Plus], Fisher Scientific). Required adjustments were made by adding H3PO4 and NaOH for phosphate buffers or only NaOH for bicine buffer. The pH for the carbonate buffers was not adjusted.
Spectral measurements
Peptides (100 μg lyophilized aliquots) were dissolved in buffer (2 mL) to achieve a final concentration of about 50 μM. Once homogeneous, solutions were transferred into a 10-mm quartz cuvette (Starna Cells) for analysis. UV–Vis absorption spectra were recorded at pre-determined timepoints from 400 to 200 nm on a Varian Cary 100 UV–Vis spectrophotometer with a scanning speed of 600 nm · min–1 with light source changeover set to 400 nm. Spectral data were analyzed and plotted as described in the Supporting Information.
Mass spectrometry
For mass spectrometry analysis, peptides were dissolved at 200 μM in the indicated buffer (10 mM) and vortexed. After 5 min, an aliquot was removed and directly injected into the mass spectrometer (Advion CMS with a resolution of 0.5–0.7 m/z units) in negative electrospray ionization mode.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation (DMR-1454754) and the National Institutes of Health (R01GM123508). We thank Professor Tijana Grove (Virginia Tech) for assistance with instrumentation and Professor Larry Byers (Tulane University) for a critical reading of the manuscript. We thank the reviewers for their thorough reading of the manuscript and several constructive suggestions.
Footnotes
NMR spectra, Hammett plots, H2S-release plots, fittings to experimental rate data, and UV-vis hydrolysis data for all SATO-peptides. See DOI: 10.1039/x0xx00000x
Notes and references
- (1).Jencks WP Mechanism and Catalysis of Simple Carbonyl Group Reaction Prog. Phys. Org. Chem 1964, 2, 63–128. [Google Scholar]
- (2).Cordes EH; Jencks WP On the Mechanism of Schiff Base Formation and Hydrolysis J. Am. Chem. Soc 1961, 84, 832–837. [Google Scholar]
- (3).Matson JB; Stupp SI Drug Release from Hydrazone-Containing Peptide Amphiphiles Chem. Commun 2011, 47, 7962–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Grover GN; Lam J; Nguyen TH; Segura T; Maynard HD Biocompatible Hydrogels by Oxime Click Chemistry Biomacromolecules 2012, 13, 3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baul TS; Basu S; de Vos D; Linden A Amino Acetate Functionalized Schiff Base Organotin(IV) Complexes as Anticancer Drugs: Synthesis, Structural Characterization, and In Vitro Cytotoxicity Studies Invest. New Drugs 2009, 27, 419–431. [DOI] [PubMed] [Google Scholar]
- (6).da Silva CM; Silva MM; Reis FS; Ruiz A; de Carvalho JE; Santos JCC; Figueiredo IM; Alves RB; Modolo LV; de Fatima A Studies on Free Radical Scavenging, Cancer Cell Antiproliferation, and Calf Thymus DNA Interaction of Schiff Bases J. Photochem. Photobiol. B 2017, 172, 129–138. [DOI] [PubMed] [Google Scholar]
- (7).Feng X; Li D; Han J; Zhuang X; Ding J Schiff Base Bond-Linked Polysaccharide-Doxorubicin Conjugate for Upregulated Cancer Therapy Mater. Sci. Eng. C Mater. Biol. Appl 2017, 76, 1121–1128. [DOI] [PubMed] [Google Scholar]
- (8).Rashidian M; Mahmoodi MM; Shah R; Dozier JK; Wagner CR; Distefano MD A Highly Efficient Catalyst for Oxime Ligation and Hydrazone-Oxime Exchange Suitable for Bioconjugation Bioconjug. Chem 2013, 24, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lin F; Yu J; Tang W; Zheng J; Defante A; Guo K; Wesdemiotis C; Becker ML Peptide-Functionalized Oxime Hydrogels with Tunable Mechanical Properties and Gelation Behavior Biomacromolecules 2013, 14, 3749–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mukherjee S; Hill MR; Sumerlin BS Self-healing Hydrogels Containing Reversible Oxime Crosslinks Soft Matter 2015, 11, 6152–6161. [DOI] [PubMed] [Google Scholar]
- (11).Barton DHR; Magnus PD; Pennanen SI Evidence for the Existence of a Thio-oxime J.C.S. Chem. Commun 1974, 1007. [Google Scholar]
- (12).Foster JC; Powell CR; Radzinski SC; Matson JB S-aroylthiooximes: A Facile Route to Hydrogen Sulfide Releasing Compounds with Structure-Dependent Release Kinetics Org. Lett 2014, 16, 1558–1561. [DOI] [PubMed] [Google Scholar]
- (13).Olson KR The Therapeutic Potential of Hydrogen Sulfide: Separating Hype from Hope Am. J. Physiol. Regul. Integr. Comp. Physiol 2011, 301, R297–312. [DOI] [PubMed] [Google Scholar]
- (14).Szabo C Hydrogen Sulphide and its Therapeutic Potential Nat. Rev. Drug Discov 2007, 6, 917–935. [DOI] [PubMed] [Google Scholar]
- (15).Wallace JL; Wang R Hydrogen Sulfide-Based Therapeutics: Exploiting a Unique but Ubiquitous Gasotransmitter Nat. Rev. Drug Discov 2015, 14, 329–345. [DOI] [PubMed] [Google Scholar]
- (16).Wang R Physiological Implications of Hydrogen Sulfide: A Whiff Exploration that Blossomed Physiol. Rev 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- (17).Lee C; Wang X; Jang HY Copper-Catalyzed Oxidative N-S Bond Formation for the Synthesis of N-sulfenylimines Org. Lett 2015, 17, 1130–1133. [DOI] [PubMed] [Google Scholar]
- (18).Long W; Qiu W; Li C; Song L; Bai G; Zhang G; He H Direct Synthesis of N-sulfenylimines through Oxidative Coupling of Amines with Disulfides/Thiols Over Copper Based Metal–Organic Frameworks RSC Adv. 2016, 6, 40945–40952. [Google Scholar]
- (19).Foster JC; Matson JB Functionalization of Methacrylate Polymers with Thiooximes: A Robust Postpolymerization Modification Reaction and a Method for the Preparation of H2S-Releasing Polymers Macromolecules 2014, 47, 5089–5095. [Google Scholar]
- (20).Foster JC; Radzinski SC; Zou X; Finkielstein CV; Matson JB H2S-Releasing Polymer Micelles for Studying Selective Cell Toxicity Mol. Pharm 2017, 14, 1300–1306. [DOI] [PubMed] [Google Scholar]
- (21).Carter JM; Qian Y; Foster JC; Matson JB Peptide-Based Hydrogen Sulphide-Releasing Gels Chem. Commun 2015, 51, 13131–13134. [DOI] [PubMed] [Google Scholar]
- (22).Scifinder Scholar, Chemical Abstracts Service: Columbus, OH (accessed April, 2018); pKa values calculated using Advanced Chemistry Development (ACD/Labs) software, version V11.02; (ACD/Labs 1994–2006) [Google Scholar]
- (23).Hansch C; Leo A; Taft RW A Survey of Hammett Substituent Constants and Resonance and Field Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (24).Dash AC; Dash B; Praharaj S Hydrolysis of Imines: Kinetics and Mechanism of Spontaneous Acid-, Base-, and Metal Ion-Induced Hydrolysis of N-salicylidene-2-aminothiazole J. Chem. Soc., Datlon Trans 1961, 2063–2069. [Google Scholar]
- (25).Dash AC; Dash B; Mahapatra PK Hydrolysis of Imines. Part 2. Kinetics and Mechanism of Hydrolysis of N-Salicylidene-2-aminopyridine in the Presence and Absence of the Copper (I) Ion. A Study of the Catalytic Effects of some Mixed-Ligand Complexes of Copper. J. Chem. Soc., Datlon Trans 1983, 1503–1509. [Google Scholar]
- (26).Reeves RL Schiff Bases. Kinetics of Hydrolysis of p-Trimethylammoniumbenzylidene-p’-hydroxyaniline chloride in Aqueous Solution from pH 1 to 11.5. J. Am. Chem. Soc 1961, 84, 3332–3337. [Google Scholar]
- (27).Rosenberg S; Silver SM; Sayer JM; Jencks WP Evidence for Two Concurrent Mechanisms and a Kinetically Significant Proton Transfer Process in Acid-Catalyzed O-Methyloxime Formation J. Am. Chem. Soc 1974, 96, 7986–7998. [Google Scholar]
- (28).Blanch JH; Onsager OT Stability of N-Heterocyclic Oxime Derivatives. Part I. Decomposition of N-Methylpyridinium 3- and 4-O- Acetylaldoxime Iodides in Aqueous Solution J. Chem. Soc 1965, 0, 3729–3724. [Google Scholar]
- (29).More O’Ferral RA; O’Brien DM; Murphy DG Rate and Equilibrium Constants for Formation and Hydrolysis of 9-Formylfluorene xime: Diffusion- Controlled Trapping of a Protonated Aldehyde by Hydroxylamine Can. J. Chem 2000, 78, 1594–1612. [Google Scholar]
- (30).Kalia J; Raines RT Hydrolytic Stability of Hydrazones and Oximes Angew. Chem. Int. Ed. Engl 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Branchaud BP Studies on the Preparation and Reactions of Tritylsulfenimines J. Org. Chem 1983, 48, 3531–3538. [Google Scholar]
- (32).Cho BR; Cho NS; Lee SK Elimination Reactions of (E)- and (Z)-Benzaldehyde O-Pivaloyloximes. Transition-State Differences for the Syn and Anti Eliminations Forming Nitriles J. Org. Chem 1997, 62, 2230–2233. [DOI] [PubMed] [Google Scholar]
- (33).Rao GK; Kumar A; Singh MP; Kumar A; Biradar AM; Singh AK Influence of Pendent Alkyl Chains on Heck and Sonogashira C–C Coupling Catalyzed with Palladium(II) Complexes of Selenated Schiff bases having Liquid Crystalline Properties J. Organomet. Chem 2014, 753, 42–47. [Google Scholar]
- (34).Milne HB; Peng CH The use of Benzylsulfonyl Chloride in Peptide Synthesis J. Am. Chem. Soc 1956, 79, 639–644. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.