

Summary:
High-risk Human Papilloma Viruses (HPV) cause cervical, anal and oropharyngeal cancers, unlike the low-risk HPVs, which cause benign lesions. E6 oncoproteins from the high-risk strains are essential for cell proliferation and transformation in HPV induced cancers. We report that a cellular deubiquitinase, USP46 is selectively recruited by the E6 of high-risk, but not low-risk, HPV to deubiqutinate and stabilize Cdt2/DTL. Stabilization of Cdt2, a component of the CRL4Cdt2 E3 ubiquitin ligase, limits the level of Set8, an epigenetic writer, and promotes cell-proliferation. USP46 is essential for the proliferation of HPV-transformed cells, but not of cells without HPV. Cdt2 is elevated in human cervical cancers and knockdown of USP46 inhibits HPV-transformed tumor growth in xenografts. Recruitment of a cellular deubiquitinase to stabilize key cellular proteins is an important activity of oncogenic E6, and the importance of E6-USP46Cdt2-Set8 pathway in HPV-induced cancers makes USP46 a target for the therapy of such cancers.
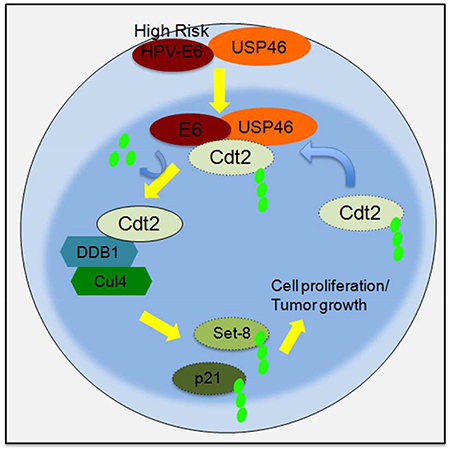
Graphic Abstract
INTRODUCTION
99.7 percent of cervical cancers in women are attributable to human papilloma virus infections (Walboomers et al., 1999), with 500,000 new patients annually worldwide. 84% of cases are in developing countries that will not soon have access to preventive steps like vaccines against HPV and/or Pap smears and so there is still a major need of therapeutic drugs for this type of cancer. The global mortality rate from cervical cancers is about 54%. HPV is also the leading cause of oropharyngeal cancers (>130,000 new cases worldwide) and cancers of the anus and external genitalia (incidence of 1 per 100,000 per year).
HPVs are non-enveloped DNA viruses with a double-stranded circular genome, which infect cutaneous squamous epithelium to cause hyper proliferation (Howley and Livingston, 2009; zur Hausen, 2002). More than 120 types of HPVs have been identified with conserved genomic structures encoding 8–10 ORFs (Bernard et al., 2010; Munger et al., 1989). Based on their ability to cause cancers, HPV types 16, 18, 31, 33, 45 and 52 belonging to alpha-7 and alpha-9 species are considered high-risk HPV. HPV16 is responsible for 50% of all cervical cancers (Lowy et al., 2008). E6 and E7 proteins of only the high risk HPVs are responsible for oncogenic transformations of HPV infected cells. The E6 protein of high risk HPV causes ubiquitin-mediated degradation of p53 by interacting with the ubiquitin ligase E6AP, while the E7 protein of high risk HPV binds to hypophosphorylated Rb to inhibit pRb-E2F complex formation and the suppression of the cell-cycle by Rb (Munger et al., 1989; Scheffner et al., 1990). The N terminal portions of high risk HPV E6 proteins interact with p53 to promote p53 degradation, while the C-terminal portion interacts with PDZ-containing proteins and promotes their degradation also in an E6AP dependent manner (Handa et al., 2007). Cellular proteins containing PDZ domain that are degraded by HPV E6 to promote cellular transformation include TIP-2/GIPC, MAGl1, DLG1, DLG4, MUPP1, Human scribble etc. (Pim et al., 2012).
CRL4Cdt2, is an important E3 ubiquitin ligase required for cell-cycle progression and genomic instability (Abbas and Dutta, 2011; Havens and Walter, 2011). Cdt2 acts as a substrate recognition adaptor protein for CRL4cdt2 E3 ubiquitin ligase complex and is responsible for regulation of the S phase of the cell cycle by degradation of p21, Set8 and Cdt1 (Abbas and Dutta, 2011; Abbas et al., 2010; Abbas et al., 2008). Many cancers e.g. hepatocellular carcinomas, melanomas, breast cancer and gastric cancers maintain high levels of Cdt2 expression compared to their normal tissue counterparts. (Li et al., 2009; Pan et al.,2006; Ueki et al., 2008). Cdt2 itself is targeted for proteasomal degradation by CRL1FBXO11 ubiquitin ligase (Abbas et al., 2013; Rossi et al., 2013).
The ubiquitination of proteins is reversed by deubiquitinases (DUBs), which have an important role in maintaining protein stability in the cells. There are about 90 DUBs in the human genome, classified into seven families (Hutchins et al., 2013; Kwasna et al., 2018).
In the present work we report that E6 recruits a cellular DUB, USP46, to Cdt2 leading to the stabilization of Cdt2. Cells infected with high risk HPVs are dependent on E6 and USP46 for cell proliferation. Only high risk but not low risk HPV-E6s can recruit USP46 for Cdt2 stabilization. Biochemically, the amino terminal 1–43 amino acids of high risk E6 protein are sufficient for Cdt2 stabilization. To our knowledge, this is the first report that HPV E6 recruits a deubiquitinase to a cellular protein for enhancing the latter’s stability. USP46 is essential for the proliferation only of those cancer cells that contain HPV, and USP46 knockdown inhibits HPV-positive cancer xenograft growth in mice.
RESULTS
Viral E6 oncoprotein and cellular deubiquitinase USP46 maintain Cdt2 protein levels in HPV-transformed cancer cell lines.
While studying CRL4Cdt2 (Abbas and Dutta, 2011), we discovered that knockdown of the viral E6 oncoprotein in HPV18-transformed HeLa cervical cancer cells decreased Cdt2. (Fig. 1a). p53 is concurrently elevated because knockdown of E6 prevents the recruitment of the ubiquitin ligase E6AP to p53. However, the elevation of p53 alone does not decrease Cdt2, because knockdown of E6AP elevated p53 but did not decrease Cdt2 (Fig. 1a). Different siRNAs against 18E6 decreased Cdt2 diminishing the possibility of this being an off-target activity of one siRNA (Fig. 1b). The decrease of Cdt2 was countered by the proteasome inhibitor, MG132 (Fig. 1c). Knockdown of E6, by stabilizing p53 increased the level of the p21 mRNA, but did not decrease the Cdt2 mRNA (Fig. 1d). The decrease of Cdt2 protein upon E6 knockdown was seen even if p53 induction was blocked by transfection of a siRNA to p53 (Fig. 1e). These results suggest that E6 increases Cdt2 protein levels by a post-transcriptional mechanism that is independent of E6AP or of p53 degradation.
Figure 1: Silencing USP46 or E6 decreases Cdt2 in HPV E6 transformed cells.

(A) U2OS and HeLa cells were transfected with GL2 (control siRNA) or siRNAs against E6 or E6AP and harvested after 60 hrs. The cell lysates were immunoblotted as indicated. (B) Different siRNAs towards 18E6 (lane 2, 4 and 6) were transfected into HeLa cells and immunoblotted. (C) HeLa cells were transfected with GL2 (control) or 18E6 siRNAs and immunoblotted. Lanes 3 and 4: Cells exposed to 10 μM MG132 for 8 hrs before harvest. (D) Cdt2 and p21 mRNA levels measured by RT-qPCR and normalized to actin mRNA after transfection of indicated siRNA into HeLa cells. Mean ± standard deviation of three replicates (E) HeLa cells were transfected with control siRNA (GL2) or with siRNAs against 18E6 or p53 for 60 hrs and lysates probed with indicated antibodies. (F) Indicated cell lines were transfected with GL2 (control) or USP46 siRNA, harvested after 60 hrs and immunoblotted. Actin was used as loading control. (G) Indicated cell lines were transfected with GL2 (control) or E6 siRNAs, harvested after 60 hrs and immunoblotted. Actin was used as loading control. (H) Indicated cell lines were transfected with GL2 (control) or USP46 siRNA, harvested after 60 hrs and immunoblotted. Actin was used as loading control. (I) Lysates of primary human keratinocytes (PHK) ± HPV18 or U2OS ± plasmid expressing HPV18 E6 were immunoblotted as indicated. (J) 293T cells transfected with indicated siRNAs were transfected with empty vector (V) or plasmid expressing HPV16-E6 and 72hrs after siRNA transfection, cell lysates were immunoblotted for indicated antibodies. (K) U2OS cells, transfected with empty vector or HPV-16 E6 expressing plasmid were enriched in S phase by serum starvation for 48 hours and then released into serum for 8 hr. The cell lysates were then probed with indicated antibodies. Indicated siRNAs were transfected 18 hours before collection.
Since proteasome inhibition stabilized the Cdt2 after knockdown of E6 (Fig. 1c) we screened for deubiquitinases involved in maintaining Cdt2 levels in HeLa cells, by knocking down USP22, USP37 and USP46 deubiquitinases, reported in proteomic screens to interact with Cdt2 or DDB1 (Sowa et al., 2009). Knockdown of USP22 and USP46 decreased Cdt2 (Fig.S1-a). However knockdown of USP22 also decreased FBXO11, of the CRL1-FBXO11 E3 ligase that ubiquitylates Cdt2, so that the decrease of Cdt2 is unlikely to be due to excessive polyubiquitination of Cdt2. USP46 was therefore pursued as a potential deubiquitinase of Cdt2. Later we also show that knockdown of USP46 decreased Cdt2 protein levels in HeLa cells without affecting the levels of another regulator of Cdt2 polyubiquitination, 14–3-3 proteins (Fig. 3b, lane 3) (Abbas et al., 2013; Dar et al., 2014). USP46 was required to sustain Cdt2 protein level only in cell lines transformed by high risk HPV viruses (HeLa and SiHa) but not in cell lines without HPV: C33A cervical cancer cells, 293T embryonic kidney, HCT116 colon cancer, U2OS osteosarcoma and A549 lung cancer cells (Fig. 1f). The requirement of E6 and USP46 for maintaining Cdt2 protein levels was confirmed in four other HPV-transformed cells, C4-II, CaSki, MS751 and ME180 (Fig. 1g,h). Cdt2 decrease by siUSP46 was rescued by overexpression of siRNA-resistant USP46, ruling out off-target action of the siUSP46 as the cause of Cdt2 decrease (Fig. 2a). These results suggest that in E6 transformed cells the USP46 deubiquitinase becomes important for maintaining Cdt2 protein levels.
Figure 3: E6 cooperates with USP46 to stabilize Cdt2 via deubiquitination.

(A) Half-life measurement of Cdt2 in HeLa cells upon 18E6 or USP46 silencing: HeLa cell lysates after incubation with cycloheximide for indicated hrs were immunoblotted. Because si18E6 or siUSP46 decrease the Cdt2 protein, a longer exposure was used for Cdt2 immunoblot after si18E6 or siUSP46 so as to equalize the Cdt2 signal at 0 hr to that after siGL2. p53 induction shows si18E6 knocked down E6 effectively. (B) Co-silencing FbxO11E3 ligase stabilizes Cdt2 in HeLa cells where E6 or US46 have been knocked down. HeLa cells were transfected with indicated siRNA and lysates immunoblotted. (C) Silencing USP46 or E6 in HeLa cells increases polyubiquitination of Cdt2. HeLa cells stably expressing Flag-Cdt2 were transfected with indicated siRNAs. Cells were transfected with HA-Ubiquitin plasmid at 8hr and harvested at 48 hr. Cells treated with MG132 for 6 hrs before harvesting. Flag-Cdt2 immunoprecipitates were immunoblotted with anti-HA and anti-Flag (top) or cell lysates with indicated antibodies (bottom). (D) Knocking down E6 or USP46 increases ubiquitination of endogenous Cdt2 in HeLa cells. Same as in (C), except that endogenous Cdt2 and ubiquitin were monitored without any overexpression. (E) E6 decreases polyubiquitination of Cdt2 but this requires USP46. 293T cells transfected with indicated plasmids and siRNAs were treated with 10μM of MG132 for 8 hrs before harvest at 48 hr of plasmid transfection. Flag-Cdt2 immunoprecipitates immunoblotted with anti-HA and anti-Flag (Top 2 panels) or cell lysates immunoblotted with indicated antibodies (Bottom 3 panels). (F) as in (C) (top) and overexpression of transfected proteins detected by immunoblotting cell lysates (bottom). USP46 (C44S): catalytically dead USP46. (G) Ubiquitinated Cdt2 purified by anti-Flag antibody was incubated with myc-16E6 or IgG immunoprecipitates in presence or absence of recombinant USP46 protein and deubiquitination activity checked by western blotting with HA antibody recognizing HA-ubiquitin. (H) Separate experiment similar to Fig. 3G except that the reaction mixes were immunoblotted with anti-Cdt2 antibody and exposed to see the smear above unubiqutinated Cdt2, which represents poly-ubiquitinated Cdt2.
Figure 2: HPV E6 promotes Cdt2 and USP46 interaction.

(A) Rescue of Cdt2 stabilization after siUSP46 using siRNA-resistant USP46 overexpression. HeLa cells were transfected with indicated plasmids and siRNAs as indicated and immunoblotted. Lane 3: siRNA-resistant HisUSP46. (B) HeLa or U2OS cell lysates immuno-precipitated with control IgG or anti-Cdt2 antibodies, and immunoblotted. (C) E6-USP46-Cdt2 interaction reconstituted in vitro. HisUSP46 from 293T cell lysates bound to nickel-NTA beads in the presence of bacterially purified GST or GST-E6. Immunoblot of bound proteins shows cellular Cdt2 associating with column along with GST-E6. Right hand panel shows 10% input cell lysate or GST protein. (D) Bacterially purified GST or GST-E6 incubated with bacterially purified His-USP46, loaded onto a nickel-NTA column, washed, eluted and immunoblotted with indicated antibodies. (E) Bacterially purified E6 interacts with USP46 purified from mammalian cells. Left: Glutathione beads coated with GST or GST-16E6 were incubated with purified His-USP46 (lanes 1 and 3). Nickel beads coated with nothing or His-USP46 were incubated with GST-16E6 (lanes 2 and 4). Beads were washed and boiled in Laemmli buffer for immunoblotting. Right, Lanes 1 and 2: input (10%) purified GST-E6 or His-USP46. Lane 3: 293T cell lysate from which the HisUSP46 was purified to show that cellular Cdt2 has been removed from the purified His-USP46 in lane 2. (F) E6 promotes interaction between Cdt2 and USP46 but USP46 is essential for the interaction between E6 and Cdt2. HEK293T cells transiently transfected with plasmids or siRNAs indicated at top. 48 hr later, cell lysates were immunoprecipitated with anti-flag antibody and immunoblotted. Input: 15% of input lysates.
Even in primary human keratinocytes Cdt2 levels were elevated upon HPV18 infection (PHK, left panel, Fig 1i). Finally, since the siRNA to E6 could knockdown both E6 and E7 oncoproteins, we tested whether E6 alone can induce Cdt2. HPV-negative U2OS cells transfected with the HPV18 E6 gene alone induced Cdt2 protein without any E7 gene (right panel, Fig. 1i and lanes 1, 2, Fig. 1j). This E6 mediated stabilization of Cdt2 is dependent on USP46 as knockdown of USP46 abolished the effect (Fig. 1j). Additionally we found that stabilized Cdt2 functions as a part of CRL4Cdt2 complex as E6 transfected U2OS cells decreased the levels of CRL4Cdt2 substrates, Set8 and p21 (Fig. 1k, lanes 1,2). Knockdown of Cdt2 in E6 expressing cells reversed the destabilization of Set8 and p21, indicating that their destabilization was indeed due to stabilization of Cdt2 (Fig. 1k, lanes 3,4). This experiment was done in cells enriched in S phase by re-addition of serum to serum-starved cells because CRL4Cdt2 recognizes Set8 and p21 primarily when the latter interact with chromatin-loaded PCNA in S phase (Abbas et al., 2010; Abbas et al., 2008).
E6 protein promotes the interaction of USP46 with Cdt2.
If the USP46 deubiquitinase stabilizes Cdt2 by deubiquitylating the latter, we expected the two proteins to physically interact with each other. Immunoprecipitation of endogenous cellular Cdt2 coimmunoprecipitated cellular USP46 and E6 only from HeLa cell lysates (that contain HPV E6) but not from U2OS cell lysates (that do not contain E6) (Fig 2b; S1b), suggesting that E6 is necessary to promote the interaction of Cdt2 with USP46. To test the dependence of the interaction on E6 in vitro, His6-epitope tagged USP46 from 293T cells (without E6) was purified by immobilization on nickel-NTA resin and incubated in the presence of GST (negative control) with cellular lysate containing Cdt2 (Fig. 2c, lanes 1, 2). The His-USP46 did not bind the cellular Cdt2 under those conditions. However, bacterially produced GST-16E6 (from the high risk HPV16) was retained on the His-USP46 column and promoted the retention of the cellular Cdt2 (Fig. 2c, lanes 3, 4). Thus in vitro, E6 interacts with USP46 and promotes the latter’s interaction with Cdt2.
To test whether E6 and USP46 can interact with each other independent of other cellular proteins, a column with bacterial GST-16E6 was incubated with bacterially produced His-USP46. His-USP46 was retained on the column suggesting that E6 and USP46 can directly interact with one another (Fig. 2d, lane 2). Thus E6 and USP46 interacted with each other directly. A column with GST-16E6 also bound His-USP46 purified from mammalian cells and vice versa (Fig. 2e).
In contrast, Flag-Cdt2 alone from cells without E6 did not co-immunoprecipitate endogenous USP46 (Fig. 2f, lane 1), suggesting that Cdt2 and USP46 do not interact with each other in the absence of E6. Co-expression of Flag-Cdt2 and Myc-E6 led to the coimmunoprecipitation of Cdt2 with USP46 and E6 (Fig. 2f, lane 2). However, the interaction of Cdt2 with E6 was lost after depletion of USP46 with siUSP46 (Fig. 2f, lane 3) suggesting that the E6-USP46 complex is required for the interaction of E6 with Cdt2 and that E6 alone cannot interact with Cdt2. Thus, HPV E6 binds directly to USP46, and this facilitates the interaction of USP46 with its potential substrate Cdt2.
USP46 deubiquitinates and stabilizes Cdt2 only in the presence of E6 protein from high-risk HPV strains.
We next tested whether E6 or USP46 promote the stability of Cdt2 protein. The half-life of Cdt2 after cycloheximide addition in HeLa cells was >2 hours but this was decreased significantly when E6 or USP46 were knocked down (Fig. 3a,S1e).
If USP46 stabilizes Cdt2, we expect this to be through the reversal of polyubiquitination of Cdt2. CRL1FbxO11 poly-ubiquitinates Cdt2. Indeed, the decrease of Cdt2 seen upon knockdown of USP46 was reversed by co-knockdown of FbxO11 (Fig. 3b, lane 3 vs 4). Since E6 stabilizes Cdt2 through USP46, the decrease of Cdt2 upon knockdown of E6 was also mitigated by co-knockdown of FbxO11 (Fig. 3b, lane 5 vs 6). Polyubiquitination of Cdt2 in vivo was studied by immunoprecipitating Cdt2 and immunoblotting the precipitates with antibodies that recognize ubiquitin. The polyubiquitin chains on Flag-Cdt2 in MG132 treated HeLa cells is increased by knockdown of E6 or of USP46 (Fig. 3c, lanes 3 and 4 vs 2;). To ensure that this result was not an artifact from overexpressing Flag-Cdt2, we also immunoprecipitated endogenous Cdt2 and probed for endogenous ubiquitin in the Cdt2 immunoprecipitate to confirm that knockdown of E6 or USP46 increased the ubiquitin chains on Cdt2 in HeLa cells (Fig. 3d, lanes 2 and 3 vs 1).
We next tested whether E6 was essential for USP46 to deubiquitinate Cdt2. Flag-Cdt2 recovered from 293T cells (that normally do not contain E6) was extensively polyubiquitinated (Fig. 3e, lane 2). Overexpression of 18E6 in the 293T cells led to deubiquitination of Cdt2 (Fig. 3e, lane 4 vs. 2), but this required endogenous USP46, because the ubiquitin chains on Cdt2 are restored by knocking down the cellular USP46 (Fig. 3e, lane 5 vs. 4). On the other hand, the simple overexpression of USP46 alone in 293T cells did not significantly decrease the polyubiquitination of Cdt2 in vivo (Fig. 3f, lane 2; S1d), but co-expression of the USP46 with the E6 from the high risk HPV18 virus deubiquitinates Cdt2 (lane 4). This deubiquitination is not seen when the HPV18 E6 is co-expressed with the catalytically dead USP46 (C44S mutation in the catalytic site) (lane 5 vs. 4) proving that the catalytic activity of USP46 is required for the deubiquitination of Cdt2. Interestingly E6 protein from a low risk HPV strain, HPV6, did not induce the deubiquitylation of Cdt2 when co-expressed with USP46 (lane 3), a result that will be explored later. Next we checked whether E6-USP46 deubiquitinates Cdt2 in vitro. Ubiqutinated-Cdt2 was incubated in vitro with E6 immunoprecipitates in presence or absence of purified USP46 and the ubiqutinated state of Cdt2 examined by immunoblotting for HA ubiquitin (Fig. 3g & quantitated from five replicate experiments in S2a). While IgG or E6 immunoprecipitate alone (lanes 4 vs 3 & 8 vs 7) or IgG immunoprecipitates with USP46 (lanes 5, 6 vs 3) did not deubiqutinate Cdt2, E6 immunoprecipitates with USP46 deubiquitinated Cdt2 in vitro (lanes 9 and 10 vs 7). In a separate experiment the ubiquitination of Cdt2 was followed by blotting with anti-Cdt2 antibody alone (Fig. 3h). The smear represents polyubiquitinated Cdt2 and the results are identical to that in Fig. 3g. Therefore cellular E6 cooperates with USP46 to deubiqutinate Cdt2 in vitro.
Only high risk but not low risk E6 proteins could promote Cdt2 interaction with USP46 and stabilize Cdt2.
If the co-opting of USP46 by E6 is important for cancer formation, a prediction is that the E6 oncoprotein from high risk HPV (HPV16, 18 and 31) will be better at the co-opting than the E6 from low-risk HPV (HPV6, 11 and 42). Indeed, transient co-transfection experiments by only high-risk E6s, but not low-risk E6s, increased the Flag-Cdt2 protein in 293T cells (Fig. 4a). As published (Jha et al., 2010) both low- and high-risk E6s promoted the degradation of TIP60 in this experiment (Fig. 4a), establishing that the low risk E6 proteins were functional. Consistent with this differential stabilization of Cdt2 by the two types of E6 proteins, His-tagged USP46 co-immunoprecipitated with Cdt2 and E6 when the high risk E6 proteins were co-expresssed, but not when the low risk E6 proteins were expressed (Fig. 4b). In Fig. 3f we have shown that high risk E6 from HPV18, but not low-risk E6 from HPV6 can deubiquitinate Cdt2 in vivo.
Figure 4: E6 oncoproteins from high risk HPVs translocate cytoplasmic USP46 into nucleus and stabilizes Cdt2.

(A) 293T cells transfected with Flag-Cdt2 and Myc-Tip60 plasmid in combination with plasmids expressing low risk myc-E6s (From HPV 6, 11, 42) or plasmids expressing high risk myc-E6s (HPV 16,18, 31) were immunoblotted. (B) 293T cells transfected with indicated plasmids were immunoprecipitated with anti-His antibody and immunoblotted (top), or the 10% input cell lysates were immunoblotted (bottom). (C) Confocal immunofluorescence images of HeLa or 293T cells transfected with myc-USP46 plasmid and stained with anti-myc antibody, and DAPI (for nuclei). (D-E) Immunofluorescence images with antibodies indicated on top of 293T cells transfected with His-USP46 plasmid along with myc-E6 plasmids indicated on left. Scale bar indicates 10μm. Quantitation in Fig. S1c. (F) 293T cells transfected with Flag-Cdt2 and indicated E6 variants were immunoblotted as indicated. Table summarizes types of E6 mutations studied and there effect on Cdt2 stabilization with respect to their known p53 degradation activity. (G) 293T cells transfected with variants of E6, Flag-Cdt2 and myc-USP46 were immunoprecipitated with anti-Flag antibody and immunoblotted (top) or 10% input cell lysates directly immunoblotted (bottom). (H) Bacterially expressed GST, GST-16E6F2V+P5R or GST-16E6Y79N (as positive control) bound to glutathione agarose in columns were loaded with purified His-USP46 (lanes 1, 3, 5). Nickel-NTA beads coated with nothing or with HisUSP46 (lanes 2, 4) in columns were loaded with GST-16E6F2VP5R. Bound proteins were eluted and immunoblotted.
Although USP46 has been implicated in the deubiquitination of histones, the protein has also been reported to localize in the cytoplasm (Lehoux et al., 2014). By confocal microscopy, it is evident that myc-USP46 is excluded from the nucleus in HPV-negative 293T cells, but is highly enriched in the nucleus and the peri-nuclear space in HPV-transformed HeLa cell (Fig. 4c). Endogenous USP46 protein is cytoplasmic in HPV-negative U2OS cells and nuclear in HPV-positive HeLa cells (Fig. S2b). Upon transient co-transfection of 293T cells USP46 is imported to the nucleus by E6 from the three high-risk (Fig. 4d) but not the three low-risk (Fig. 4e) strains (Fig. S1c quantitates the subcellular localization of USP46).
Collectively, these results suggest that the high risk E6 proteins, but not the low risk E6 proteins, selectively interact with USP46 in cells in situ.
Amino acids conserved in high risk but not low risk HPVs are critical for Cdt2 stabilization.
To address the differential ability of high risk versus low risk E6s to stabilize Cdt2, we checked whether amino acids conserved in only high risk HPVs are critical for Cdt2 stabilization. Mutation of several amino acids conserved in high risk HPV E6 proteins (F2, P5, R8, L12, P112) ablated Cdt2 stabilization. Amino acids E18, F47, and P112 are at the interaction interface with p53 and essential to degrade p53 (Martinez-Zapien et al., 2016). Mutations E18K and P112S also failed to stabilize Cdt2. For F47 the mutation F47R fails to degrade p53 while mutation F47Y degrades p53 efficiently(Crook et al., 1991). The F47Y mutation also stabilized Cdt2 effectively. Other E6 mutations which fail to degrade p53, also failed to stabilize Cdt2 (D4G, R8Q, L12S, A46V, L50A, I52T). Conversely, mutations, which permit p53 degradation, also stabilize Cdt2 (R10A/K11A, Y32G, K34A, F47Y, E113K, D120K). The N-terminal 43 amino acid of E6 protein E6*I, which corresponds to a splice isoform of E6 in HPV infected keratinocytes, is sufficient to stabilize Cdt2 (Fig. 4f, lane 13). However, Y79 of E6 required to interact with E6AP for p53 degradation was dispensable for stabilization of Cdt2. Thus E6 seems to utilize similar structural features to interact with and degrade p53 as to stabilize Cdt2, except for the requirement of interaction with E6AP for p53 degradation. This overlap of structural requirement between the two processes explains why high risk but not low risk E6s stabilize Cdt2 and degrade p53.
If the interaction of USP46 with Cdt2 is essential for the stabilization of Cdt2, we would expect that the residues of E6 required to stabilize Cdt2 are also essential to promote the interaction of USP46 with Cdt2. Immunoprecipitation of Cdt2 and immunoblotting of the precipitate for USP46 showed that high risk 16E6 and the 1–43 fragment of 16E6, but not low risk 6E6 or 16E6-F2V/P5R, promoted the interaction of USP46 and Cdt2 (Fig. 4g), demonstrating the correlation expected above. Similarly, when complex formation was examined in vitro, immobilized GST-16E6 bound His-USP46 in vitro (Fig. 2e), as did the Y79N mutant E6, but not the F2V/P5R mutant (Fig. 4h, lanes 5 and 3). In the opposite direction, immobilized His6-USP46 bound GST-16E6 (Fig. 2e) but not GST-16E6-F2V/P5R (Fig. 4h, lane 4). Thus there was a perfect correlation between an E6 variant’s ability to bind USP46 and promote the interaction of USP46 with Cdt2, and the ability of the variant to stabilize Cdt2.
E6 associates specifically with USP46+Cdt2 independent of E6AP, p53 and USP46 accessory proteins.
In Hela cells, E6 is associates with E6AP and p53, and now we find that the E6 protein also associates with USP46 and Cdt2. To determine whether the two complexes are mutually exclusive, Cdt2 immunoprecipitates from HeLa cells were immunoblotted for E6AP and p53. Cdt2 immunoprecipitates contained USP46 but not E6AP or p53 (Fig. 5a). Conversly, E6AP co-immunoprecipitated p53 but not USP46 (Fig. 5b). Thus the E6-E6AP-p53 complex is independent from the E6-USP46-Cdt2 complex.
Figure 5: The E6-Cdt2-USP46 complex is independent of E6AP, p53, UAF1 or USP12.

(A) Negative control IgG or anti-Cdt2 immunoprecipitates from HeLa cells immunoblotted with indicated antibodies. (B) Same as (A), except with E6AP immunoprecipitates. (C) Same as (A), except with UAF1 (WDR48) immunoprecipitates. (D) Same as (A), except with mycUSP46 immunoprecipitates. (E) HeLa cells transfected with siRNAs indicated at top were immunoblotted as indicated. (F) In vivo ubiquitination of Cdt2 studied by immunoprecipitation of Flag-Cdt2 and immunoblotting, after transfection of HEK-293T cells with plasmids expressing proteins indicated at the top. (G) Lysates from 293T cells transfected with FlagCdt2, Myc-USP46 and Myc-E6 were passed through Superose 6–10/300 column and fractions collected. Even numbered fractions were immunoblotted with indicated antibodies. Peaks of elution of MW markers are indicated at the top. (H) Indicated fractions from gel-filtration chromatography were pooled and immunoprecipitated with either Rabbit IgG or with Cdt2 antibody and probed with Flag or Myc antibodies.
USP46 is normally associated with its accessory proteins UAF1 and WDR20. USP46 also shares substrates and the accessory factor UAF1 with a closely related deubiquitinase USP12. On the other hand, Cdt2 is associated with DDB1 in the CRL4 complex. We tested whether the E6-USP46-Cdt2 complex contains these additional partners. An UAF1 immunoprecipitate from HeLa cells pulled down USP46, USP12 and WDR20, but did not coimmunoprecipitate Cdt2 (Fig. 5c). Immunoprecipitates of myc-USP46 contained Cdt2 and DDB1 indicating that USP46 associates with CRL4-Cdt2 complex (Fig 5d). Finally, knock down of USP12, unlike the knockdown of USP46, did not decrease Cdt2 protein levels in HeLa cells (Fig. 5e). Also an USP46 mutant, USP46-E186K, that cannot bind to UAF1 could still deubiqutinate Cdt2 in the presence of E6 (Fig. 5f, lane 6). These results indicate that USP46 does not associate with its accessory factor UAF1 or WDR20 when interacting with or deubiquitinating Cdt2 in high risk E6 transformed cells. They also show the specificity of the interaction of E6 with USP46, because a closely related deubiquitinase, USP12, fails to enter into the E6-Cdt2 complex and is not responsible for stabilizing Cdt2.
Overexpressed Cdt2, USP46 and E6 co-eluted on a gel filtration column in fractions corresponding to 150 to 500 kDa (Fig. 5g, fractions 20–23). While E6 and USP46 co-elute in fractions 18 to 29, Cdt2 is present in fractions 14–23. Cdt2 immunoprecipitates from the overlapping fractions (20–23) contained both USP46 and E6 (Fig. 5h). From this we conclude that Cdt2 (90 kDa), USP46 (42 kDa) and E6 (16.5 kDa) could be present in a heterotrimeric complex of 150 kDa, but is most likely associated with other cellular proteins. The elution profile also suggests that the Cdt2 associates with E6-USP46, as a low-affinity substoichiometric substrate rather than as a core member of the E6-USP46 complex.
USP46 is essential for proliferation of high risk HPV infected cancer cell lines.
The specific recruitment of USP46 to Cdt2 by high-risk, but not low-risk, HPV E6 proteins suggests that USP46 could be important for malignant transformation by HPV. Indeed, knockdown of USP46 decreases proliferation of HPV-transformed cells (HeLa and SiHa cervical cancer), but not cells without HPV (C33A cervical cancer and U2OS) (Fig. 6a). The decrease in the cell proliferation and increase in Set8 levels could be reversed with si-resistant USP46 (Fig. 6b, 6c) thereby proving a specific role of USP46 in proliferation of HPV transformed cells. Since cell proliferation is essential for malignancies, this result supports the hypothesis that the recruitment of USP46 by high risk HPV E6 is important for cancer formation by this oncoprotein.
Figure 6: USP46 is necessary for viability of E6 positive cervical cancer cells.

(A) Growth rates of HPV E6 positive cervical cancer cells (HeLa, SiHa) or HPV negative cervical cancer cells (C33A) or HPV negative U2OS were transfected with indicated siRNA at day 0 and day 1. Mean ± standard deviation of 3 replicates. (B) Empty vector (V) or plasmid expressing si-USP46 resistant USP46 cDNA were transfected in HeLa cells after USP46 knockdown and lysates were subjected to immunoblotting with indicated antibodies. (C) Cells as in (B) were allowed to proliferate and counted at indicated time points. (D) Growth rates of HeLa cells expressing Flag or Flag-Cdt2. Rest as in (A). (E) Same cells as in (D) immunoblotted with indicated antibodies. (F) % of HeLa cells in different cell-cycle phases, as measured by FACS, following treatment with indicated siRNAs. (G) HeLa cells engineered to induced shUSP46 with doxycycline were grown ±doxycycline and immunoblotted with indicated antibodies. (H) Growth rates of the shUSP46-HeLa cells after transfection with indicated siRNAs at day 1 and day 5. + Dox (1 ug/ml doxycycline added from day 1). – Dox is no doxycycline. Mean ± standard deviation of 3 replicates.
USP46 is recruited by the E6 protein to stabilize Cdt2. To test whether Cdt2 is the critical substrate of USP46 to support proliferation of HPV transformed cells we overexpressed Cdt2 in HeLa cells. Indeed, the decrease in proliferation of HeLa cells upon knockdown of USP46 was rescued by ectopic expression of Flag-Cdt2 (Fig. 6d, e). Thus stabilization of Cdt2 is a critical function of E6+USP46 for the proliferation of HPV-transformed cells. However, the incomplete rescue by exogenous Cdt2 indicates that there are additional substrates of E6USP46 complex, which contribute to loss of cell proliferation upon USP46 depletion.
We next determined what substrates of Cdt2 needed to be degraded for the proliferation of HeLa cells. The decrease of Cdt2 upon USP46 knockdown was accompanied by an increase in the levels of CRL4Cdt2 substrates Set8 and p21 (Fig. 6e, lane 2), and these increases were ameliorated by exogenous Flag-Cdt2 (lane 4). siUSP46 also increased the G2 population of cells and decreased the G1 population, suggesting a block to progression from G2 to G1 (Fig. 6f). Of the known substrates of Cdt2, an increase of the H4K20 methyltransferase Set8 is known to block progression of cells from G2 to G1 (Abbas et al., 2010). Consistent with this, the G2-G1 block seen upon knockdown of USP46 in HeLa cells was relieved by co-knockdown of Set8 but not p21 (Fig. 6f). To confirm whether the increase of Set8 was responsible for the inhibition of HeLa cell proliferation when USP46 was removed, HeLa cells stably transfected with shUSP46 under control of a doxycycline inducible promoter were prepared. Induction of shUSP46 in these cells by doxycycline decreased cell proliferation (Fig. 6g, h). This decrease of cell proliferation was partially rescued by co-knockdown of Set8 (purple line in Fig. 6h). Thus, USP46 recruitment by E6 stabilizes Cdt2, which restrains the level of Set8 through the action of CRL4Cdt2 and thus allows cell proliferation in HPV-transformed cancers.
Cdt2 protein is elevated in HPV-transformed human cancers and USP46 is essential for cervical cancer growth, in vivo.
Knockdown of USP46 in HPV-negative cells does not decrease Cdt2 levels or cell proliferation (U2OS cells in Fig. 7a, b). However, expression of HPV-16E6 in these cells increases Cdt2 levels, which is now decreased upon knockdown of USP46 (Fig. 7a). Strikingly, cell proliferation of U2OS cells expressing E6 is now decreased upon knockdown of USP46 (Fig. 7b). On the other hand, in U2OS cells expressing 16E6F2VP5R, which does not recruit USP46 or stabilize Cdt2, knockdown of USP46 does not inhibit cell proliferation (Supp Fig. S2d). These results indicate that high risk E6 rewires the cellular circuitry so that the Cdt2 and cell proliferation become much more dependent on USP46.
Figure 7:

(A) USP46 was knocked down with two different siRNAs in U2OS vector control cells or those expressing HPV-E6 and the lysates subjected to immunoblotting with indicated antibodies. (B) Proliferation rates of U2OS cells as in (A), with or without transfected E6, after transfection of siGL2 (blue line) or the siUSP46 (red and green lines). Mean and s.d. of 3 replicates. (C) Summary of staining scores in paired normal and cancerous areas of cervical cancer sections. DCC 1–10 indicate normal and cancer pairs from 10 patients. (D) Representative immunohistochemistry staining of Cdt2 in cervical normal and tumor sections. (E) Image of HeLa xenografts injected intratumorally with either non-target or USP46 siRNA. (F) Image showing difference in size of HeLa xenografts tumors injected with non-target or USP46 siRNA. (G) Lysates from Hela xenograft tumors injected with siControl or siUSP46, subjected to immunoblotting with indicated antibodies. (H) Plot showing tumor growth of HeLa xenografts injected with non-target or USP46 siRNAs monitored for 8 weeks after siRNA injections. Each data point represents median tumor volume and error bars represent median absolute deviation (MAD), black and red fonts for “n” denote number of xenografts tumors in study at those time points. Mice were sacrificed once the tumor size grew to 1500 mm3. *,p<0.05; **p<0.005 (I) USP46 interacts with high risk HPVE6 protein to promote its interaction with and de-ubiquitination of Cdt2. The stabilized Cdt2 then cooperates with CRL4 to ubiquitinate and degrade Set8 and p21, and thus promote cell proliferation.
To test whether the E6-USP46 mediated stabilization of Cdt2 is relevant in cervical carcinomas we did immunohistochemistry with Cdt2 and USP46 antibodies on FFPE (Formalin fixed paraffin embedded) sections from cervical carcinomas. Cdt2 immunohistochemistry revealed a significant increase in the level of Cdt2 protein in the cancers compared to adjacent normal areas (Fig. 7c, d). This result is consistent with the stabilization of Cdt2 by E6 from high-risk HPV strains. We could not determine whether the E6 translocated USP46 to the nucleus of the cancer cells, because contrary to the well-known cytoplasmic localization of USP46 in cell lines in culture, IHC with the USP46 antibody revealed that USP46 is nuclear in both normal and malignant areas of cervical squamous epithelium (Fig. S2c).
To determine whether USP46 is a good drug target for inhibiting HPV-transformed cancers we implanted HeLa xenografts in immune compromised CB17SC female mice. Knockdown of USP46 in these tumors using siRNAs with Invivofectamine significantly decreased the growth rate of these tumors (Fig. 7e, f, g and h). In an alternative experiment CaSki cells (also cervical cancer cells transformed with HPV16) engineered to stably express Doxycycline-induced shRNA against USP46, were allowed to form tumors in mice. Knockdown of USP46 in these tumors by gavaging mouse with doxycycline resulted in slower growth of tumors compared to the mice gavaged with normal saline (Fig. S3-a). These experiments show the importance of USP46 in growth of HPV-transformed cervical cancers.
DISCUSSION
High-risk subtypes of Human Papilloma Viruses are associated with cervical cancers, oropharyngeal cancers and anogenital cancers. Mechanistically, E6 oncoproteins from highrisk papilloma viruses act as a scaffold for redirecting a cellular E3 ubiquitin ligase, E6AP to p53 tumor suppressor for polyubiquitination and subsequent proteasomal degradation (Scheffner et al., 1993). Degradation of p53 by E6 prevents the host cell from undergoing cell cycle arrest and apoptosis in response to genotoxicity caused by viral infection. Here we extend the function of high risk E6 by discovering that it also recruits a deubiquitinase, USP46, to stabilize important cellular proteins.
An important regulator of the cell cycle, CRL4Cdt2, degrades several substrate proteins like p21, Cdt1, TDG and Set8 for the smooth progression of G1/S and S-phase into M-Phase (Abbas and Dutta, 2011; Havens and Walter, 2011; Shibata et al., 2014). Elevated Set8 is unique among the substrates in also causing a G2 arrest. In growing cells, Cdt2 itself is subjected to ubiquitin-mediated degradation by another E3 ligase, FbxO11 as part of the CRL1FbxO11 complex (Abbas et al., 2013). We have discovered that in high risk HPV immortalized cervical cancer cells E6 oncoprotein is indispensable for the Cdt2 protein stability. In contrast to promoting p53 degradation, E6 maintains the Cdt2 protein levels by protecting it from proteasomal degradation. This maintenance of Cdt2 by E6 is independent of the conventional E6-E6AP E3 ligase complex that degrades p53. Contrary to the high risk E6, low risk E6 proteins are not involved in maintaining levels of Cdt2 in cells, thus establishing Cdt2 as an important target of high risk E6 oncoproteins. Also, Primary Human Keratinocytes (the natural, and untransformed host of HPV) infected with HPV18 virus have increased level of Cdt2 suggesting that this is not something uniquely occurring only in long-established laboratory cell lines. We discovered that USP46 DUB is essential for Cdt2 stability exclusively in cells transformed with high risk HPV E6 oncoproteins. Ablating or silencing USP46 from high-risk E6 transformed cells decreased the level of Cdt2 protein. Consistent with the suggestion that E6 increases the level of Cdt2, we see an increase in Cdt2 protein in cervical squamous cell carcinoma sections from patients. Our data suggest that although E6 and USP46 can interact with each other in the absence of Cdt2, E6 and Cdt2 or Cdt2 and USP46 cannot interact without the presence of the third factor. We propose that the E6-USP46 interaction promotes the tripartite E6-USP46-Cdt2 complex either due to allosteric changes in USP46, or because Cdt2 interacts simultaneously with sites on E6 and USP46 (Fig. 6i). The formation of a tripartite complex between high-risk E6-USP46-Cdt2 and the failure of a similar complex formation with low-risk E6 proteins explains how high-risk E6 stabilizes Cdt2.
Previous studies have shown that USP46 has very low intrinsic catalytic activity in vivo indicating that this DUB needs additional factors or modulators to stimulate its function. In C. elegans WDR20 and UAF1 form a complex with USP46 to stimulate its catalytic activity (Dahlberg and Juo, 2014). Similarly, USP46 or USP12 in complex with UAF1 has deubiquitination activity towards histones H2A and H2B in Hela and Xenopus extracts (Joo et al., 2011). Our results show E6 and USP46 form an exclusive complex that excludes UAF1 or WDR20 and is recruited to Cdt2 to deubiquitinate the latter. The catalytic activity of USP46 is needed for the stabilization of Cdt2 and in vitro E6 immunoprecipitate cooperates with recombinant USP46 to deubiqutinate Cdt2. Therefore the E6 oncoprotein (perhaps with other yet unidentified cellular proteins) is an important activator of USP46.
Inactivation of Cdt2 in cells results in the accumulation of CRL4Cdt2 substrates, Set8 (Histone H4K20 methyl transferase) or p21 (Cdk inhibitor), both of which play a role in cell cycle blockade. An interesting question here is why do high-risk HPV E6 oncoproteins selectively recruit USP46 for Cdt2 stability, when cells without HPV can maintain enough Cdt2 for cell proliferation in the absence of USP46. A possible explanation would be that high risk E6 oncoproteins suppress a yet unknown canonical DUB for Cdt2 and so have to recruit a different DUB to maintain Cdt2 levels to support cell-cycle progression. Alternatively, some other function of the E6 or E7 oncoproteins may decrease Cdt2 levels, e.g. by activating an E3 ligase or decreasing Cdt2 RNA level, so that it becomes imperative for the virus to use USP46 for maintaining Cdt2 at a level sufficient for S phase progression. Interference with Cdt2USP46-E6 interaction in HPV transformed cells destabilizes Cdt2 (Fig. 1 e-h & 4 f, g), and we know that decrease of Cdt2 impairs cell proliferation (Fig. S3 b, c, d, e).
Just as the recruitment of E6AP E3 ubiquitin ligase by the E6 oncoprotein leads to the ubiquitylation of many substrates besides p53, we anticipate that Cdt2 is the first of many cellular proteins that will be discovered to be stabilized by E6 through its recruitment of USP46 deubiquitinase. Loss of these other substrates of USP46 could contribute to the cell proliferation defect when USP46 is knocked down in HeLa cells, because the defect was not completely rescued by overexpressing Cdt2 (Fig. 6d).
USP46 also interacts with the HPV replicative protein E1 to promote the replicative life-cycle of the virus (Lehoux et al., 2014). The transformation of cervical epithelial cells is seen only when the HPV genome has integrated into the cellular genome, almost invariably due to a break in the E1/E2 genes of the virus (Ziegert et al., 2003). The E1 and E2 are not expressed in transformed cells, and there is no viral replication in the transformed cells. Thus the results reported here cannot be attributed to a role of USP46 in promoting the replicative cycle of the virus.
There are other examples of deubiquitinases involved in (a) viral life cycles or (b) cell transformation. Examples include the interaction of HAUSP (USP7) with herpesvirus ICP0 (Boutell et al., 2005; Everett et al., 1997) and the deubiquitinase activity of the BPLF1 protein of Epstein Barr Virus (Saito et al., 2013; Whitehurst et al., 2015). USP15 stabilizes HPV E6 oncoprotein (Vos et al., 2009). Deubiquitinases have also been implicated in cancer progression (Gelsi-Boyer et al., 2008). This, however, is the first demonstration that oncogenic E6 proteins, but not the non-oncogenic E6 proteins, selectively recruit a deubiquitinase to cellular substrates and that USP46 deubiquitinase is critical for the proliferation of HPV transformed cancer cells and for growth of HPV-transformed cancers in vivo.
In conclusion, this paper identifies a pathway from high risk HPV E6 through USP46 that stabilizes Cdt2 and promotes the degradation of Set8 to allow the cancer cells to proliferate and tumors to grow (Fig. 7i). Since deubiquitinases are proteases that have been successfully targeted by small chemical inhibitors, the dependence of HPV-transformed cells on USP46 offers an opportunity to target cancers caused by HPV. The screening of inhibitors against USP46 enzymatic activity would lead to identification of therapeutic molecules to arrest growth of HPV-positive cancers.
STAR Methods Text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anindya Dutta (ad8q@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HeLa, SiHa, C33A, A549, CaSki and MS751 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). HCT116 and ME180 cells were maintained in McCoy’s 5A medium supplemented with 10% FBS. 293T and U2OS cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS). C-4 II cells were cultured in Waymouth’s MB 752/1 medium containing 10% fetal bovine serum. 1% penicillin/streptomycin (Gibco) was added to all culture media. Primary human keratinocytes (PHKs) were cultured in Keratinocytes serum free media. All cell lines were grown in humidified incubators with 5% CO2 at 370C. All the cell lines were authenticated at Biosythesis (www.biosyn.com) by analyzing Short Tandem Repeats (STR analysis) at 15 genetic loci and the amelogenin gene.
Mouse model
Mouse experiments were done in accordance with institutional guidelines of University of Virginia. Eight week old, female, immunodeficient CB17sc mice were purchased from Taconic. Animals were held in university core animal facility and maintained as per institutional guidelines.
METHOD DETAILS
Co-immunoprecipitation
Cells were lysed on ice for 30 min in a lysis buffer (50 mM Tris-Cl [pH 8.0], 10% glycerol, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 1 mM dithiothreitol [DTT], 50 mM NaF, 1 mM Na3VO4, 1 mM glycol phosphate, protease inhibitor cocktail). It was followed by a brief sonication and centrifugation at 15,000 rpm for 15 min. The supernatant recovered was incubated with control IgG or target antibody overnight at 4°C. The immune complex was then pulled down by adding Protein G Dynabeads (Invitrogen, 10003D) for 1 hour to the immune complex. Immune complex bound to the beads were then separated from rest of the lysate on magnetic rack. Beads were then washed for five times with the lysis buffer to remove nonspecific interactions. 2X Lammeli buffer was then added to the beads and heated to release the bound protein complex. The tubes were then spinned for a minute and supernatant were subjected to SDS-polyacrylamide gel electrophoresis followed by western blotting. Light chain specific secondary antibodies or trueblot (Rockland antibodies-88–7788-31) secondry antibodies were used for western blotting in immunoprecipitation experiments.
Reconstituted GST-pull down assay
GST tagged E6 protein was expressed in bacterial E. Coli BL21 cells at 16° C for 20 hrs (after induction with 0.8 mM IPTG). Cells were lysed in lysis buffer (lysozyme 1mg/ml, 20 mM Tris pH 7.5, 300 mM NaCl, 1mM EDTA, 0.1% Triton X100, 5 mM BME + cocktail protease inhibitor [Roche]) on ice for 1 hr. Lysed mixture was then sonicated (20 amp, 4 cycles of 1 min each on ice) and ultracentrifuged at 25000 rpm to clear the lysate. The clear lysate was then mixed with glutathione sepharose beads (Invitrogen) in binding buffer (lysis buffer devoid of lysozyme) and incubated for 45 mins at 4° C. The beads were then washed with 10X binding buffer and finally eluted in desired volume of elution buffer (binding buffer containing 10mM reduced glutathione). His-USP46 was expressed in HEK293T cells for 60 hrs. Cells were lysed in lysis buffer (20 mM Tris pH 7.5, 300 mM NaCl, 1mM EDTA, 0.05% NP-40, 5 mM BME + protease inhibitor cocktail), sonicated and cleared as described above. Soluble lysates from 293T cells overexpressing His-USP46 were bound to Ni-NTA beads in the presence or absence of purified GST-E6 in above mentioned binding buffer. The Ni-NTA beads were then washed with 10X binding buffer and boiled in 2X SDS buffer followed by western blotting with respective antibodies.
Immunofluorescence
Cells growing on cover slips were transfected with plasmids expressing His6-USP46 without or with myc tagged E6s (high or low risk) for 48 hrs. Cells were then washed with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature, followed by wash with PBS (2 times). This was followed by permeabilization with 0.2% TritonX-100 for 10 min at room temperature, followed by 3 washes with PBS and blocking with 1% BSA (in PBS + 0.1 % Tween 20) for 30 minutes. After this coverslips were incubated with primary antibody in 1% BSA for 2 hours followed by 3 washes with PBS. This was followed by incubation with fluorescently labeled secondary antibodies (Alexa Fluor 555 anti-rabbit (A21429; Life Technologies) or Alexa Fluor 488 anti-mouse (A11029; Life Technologies)) in dark at room temperature for 1 hr. Nuclei were stained with DAPI (Vector Laboratories, Inc). The cover glasses were mounted on glass slides followed by microscopy. The stained cells were imaged by Zeiss AXIO observer A-1 equipped with Zeiss EC Plan-Apochromax 63X/1.4 oil and Zeiss AXIOCAM MRC. Axiovision software was used to analyze the images.
Immunohistochemistry on FFPE sections
Tissue sections were cut from each block at 4μm thick intervals. Antigen retrieval and deparaffinization were performed in PT Link (Dako, Glostrup, Denmark) using high pH EnVison FLEX Rettreival solution (Dako) for 20 minutes at 970C. Immunohistochemistry was performed on a robotic platform (Autostainer, Dako). Endogenous peroxidases were blocked with peroxidase and alkaline Phosphatase blocking reagent (Dako) before incubating the sections with respective antibodies. Antibody for USP46 was purchased from Sigma (HPA007288) and antibody for Cdt2 was homemade. Cdt2 antibody was used at 1:100 dilution and USP46 antibody was used at 1:200 dilution for 30 minutes at room temperature. Antigen-antibody complex was detected using DAKO Envision, anti rabbit polymer followed by incubation with 3,3’-diaminobenzidine tetrahydrochloride (DAB+) chromagen (Dako). All the slides were counterstained with hematoxylin subsequently; they were dehydrated, cleared and mounted for the assessment.
Deubiquitination assay
For in vivo deubiquitination assay, the plasmid- transfected 293T cells were treated with MG132 (10μM) for 6 h before harvesting. Cells were harvested in denaturing ubiquitination buffer (50 mM Tris-Cl [pH 8.0], 5 mM DTT, and 1% SDS) and immediately boiled for 10 min at 95°C, followed by cooling on ice for 10 min. The lysate was sonicated, and supernatant recovered after centrifugation at 15,000 rpm for 20 min. The supernatant was diluted with 9 volumes of buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5% glycerol, 0.4% NP40, and protease inhibitors and subjected to immunoprecipitation followed by western blotting (Dar et al., 2013). For in vitro deubiqutination assay, ubiqutinated Cdt2 substrate was prepared by transfecting 293T cells with plasmids expressing Flag-Cdt2, HA-Ubiquitin and FBXO11 (CRL4 adaptor for ubiquitinating Cdt2). 48 hours after transfection cells were treated with MG132 and lysate prepared by hot lysis method as described above. Ubiqutinated Cdt2 was prepared by immunoprecipitating lysate with Flag antibody and eluted with 100 mM Glycine pH 2.5 followed by neutralization with 1M Tris-HCl pH 8.8. Myc-E6 immunoprecipitate was prepared by transfecting Myc-E6 plasmid in 293T cells and immunoprecipitation with Myc antibody. Ubiquitnated-Cdt2 (from 1/5th of a 10 cm plate) and myc-E6 immunoprecipitates (on dynabeads) were then incubated in 60μl deubiqutination buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM EDTA, 5%Glycerol, 1%BSA, 10 mM DTT) in the presence of Myc peptide (100 μg/ml, to elute the myc-E6 immunoprecipitates), with or without recombinant USP46 (48 or 192 Pico moles, Boston Biochem) for 14h at 37°C. The reaction mix was then heated in Laemmli buffer for western blot analysis with anti-HA and anti-Cdt2 antibody.
Half-life measurements
HeLa cells were treated with freshly prepared cycloheximide (5 mg/ml) and harvested at different time points, followed by western blotting of the whole-cell lysates.
Cell cycle analysis
Cells were harvested by trypsinization followed by 2 washes with PBS. The washed cells were fixed with 70% ethanol in PBS at -20°C overnight. Fixed cells were washed with PBS, f ollowed by staining with a solution containing 50 g/ml propidium iodide plus 50 μg/ml RNase-A in PBS. The stained cells were analyzed in FACSCalibur flow cytometer (Becton Dickinson).
Real-time q-RT-PCR
Total RNA was isolated by using TRIzol reagent (Invitrogen), following the manufacturer’s instructions, and was used for cDNA synthesis with SuperScript III (Invitrogen). The cDNAs were used as the templates for real-time quantitative PCR (q-PCR) using SYBR green PCR master mix (Applied Biosystems).
Cell viability
Cell viability and count of different cell lines was monitored by the trypan blue exclusion method using a 0.4% trypan blue solution (Invitrogen) and counted on automated cell counter (Invitrogen).
In vivo studies
Mouse experiments were done in accordance with institutional guidelines of University of Virginia. Eight week old, female, immunodeficient CB17sc mice were purchased from Taconic. 1 X 107 HeLa cells were injected in both flanks of mice and tumors were allowed to attain a size of at least a 50 mm3. The mice were then divided in two groups such that the mean tumor volume was 50 mm3 in each group. The two groups received either 20 ug of siGl2 or siUSP46 complexed with Invivofectamine-3 intra-tumorally for each tumor, twice a week for two weeks. siRNA complexes were prepared as per manufacturer’s protocol. 100 ul Ambion In Vivo negative control siRNA or siUSP46 (2400 ng/ml) was diluted with 100 ul of the complexation buffer and then mixed with 200 ul of invivofectamine3 followed by incubation at 50° C for 30 minutes. The complex was then diluted with PBS to a volume of 2.4 ml and then concentrated to a volume of 240 ul by passing thorough Amicon Ultra-15 Ultracel-50kDa. 20 ul of the complex was injected intra-tumorally in each tumor. The tumors were then allowed to grow for 8-weeks and the tumor volumes were measured twice every week.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was done on at least three replicates (indicated in figure lgends). Student’s t-test was used to arrive at the significance of results .* and *** stands for p < 0.05 and p < 0.005, respectively.
DATA AND SOFTWARE AVAILABILITY
The raw data files for images are available at Mendeley.
Supplementary Material
Highlights:
E6 forms a complex with USP46 to deubiqutinate and stabilize Cdt2
E6-USP46 complex is different from E6-E6AP or USP46-UAF1 complex
Only high risk E6s interact with USP46 to deubiqutinate Cdt2
USP46 is essential for survival of HPV positive cancers
Acknowledgements
This work is supported by R01 GM084465 to AD. We thank members of the Dutta lab for discussions and S. Van de Pol for plasmids expressing HPV E6s or their mutant versions. We also acknowledge T. Gao for providing myc-USP6 expressing plasmid. We acknowledge the MAPS core facility and CCSG grant number 2P30CA044579–26 at University of Virginia for animal experiments.
Footnotes
Declaration of Interests
The authors declare no competing interests.
The oncogenes E6 and E7 from high Risk HPVs induce many cancers. Kiran et al. show that E6 protein from oncogenic HPV associates with USP46 and directs USP46 to deubiquitinate and stabilize cellular proteins like Cdt2 to promote cell proliferation and tumor growth, making USP46 an attractive target for therapy.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas T, and Dutta A (2011). CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 10, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Mueller AC, Shibata E, Keaton M, Rossi M, and Dutta A (2013). CRL1-FBXO11 promotes Cdt2 ubiquitylation and degradation and regulates Pr-Set7/Set8-mediated cellular migration. Mol Cell 49, 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Shibata E, Park J, Jha S, Karnani N, and Dutta A (2010). CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 40, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, and Dutta A (2008). PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev 22, 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, and de Villiers EM (2010). Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Canning M, Orr A, and Everett RD (2005). Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. Journal of virology 79, 12342–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook T, Tidy JA, and Vousden KH (1991). Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell 67, 547–556. [DOI] [PubMed] [Google Scholar]
- Dahlberg CL, and Juo P (2014). The WD40-repeat proteins WDR-20 and WDR-48 bind and activate the deubiquitinating enzyme USP-46 to promote the abundance of the glutamate receptor GLR-1 in the ventral nerve cord of Caenorhabditis elegans. J Biol Chem 289, 3444–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A, Shibata E, and Dutta A (2013). Deubiquitination of Tip60 by USP7 determines the activity of the p53dependent apoptotic pathway. Molecular and cellular biology 33, 3309–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A, Wu D, Lee N, Shibata E, and Dutta A (2014). 14–3-3 proteins play a role in the cell cycle by shielding cdt2 from ubiquitin-mediated degradation. Molecular and cellular biology 34, 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Meredith M, Orr A, Cross A, Kathoria M, and Parkinson J (1997). A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. The EMBO journal 16, 1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Trouplin V, Adelaide J, Aceto N, Remy V, Pinson S, Houdayer C, Arnoulet C, Sainty D, Bentires-Alj M, et al. (2008). Genome profiling of chronic myelomonocytic leukemia: frequent alterations of RAS and RUNX1 genes. BMC Cancer 8, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa K, Yugawa T, Narisawa-Saito M, Ohno S, Fujita M, and Kiyono T (2007). E6AP-dependent degradation of DLG4/PSD95 by high-risk human papillomavirus type 18 E6 protein. Journal of virology 81, 13791389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens CG, and Walter JC (2011). Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev 25, 1568–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, and Livingston DM (2009). Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins AP, Liu S, Diez D, and Miranda-Saavedra D (2013). The repertoires of ubiquitinating and deubiquitinating enzymes in eukaryotic genomes. Molecular biology and evolution 30, 1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, and Dutta A (2010). Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol Cell 38, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo HY, Jones A, Yang C, Zhai L, Smith A.D.t., Zhang Z, Chandrasekharan MB, Sun ZW, Renfrow MB, Wang Y, et al. (2011). Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J Biol Chem 286, 7190–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasna D, Abdul Rehman SA, Natarajan J, Matthews S, Madden R, De Cesare V, Weidlich S, Virdee S, Ahel I, Gibbs-Seymour I, et al. (2018). Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol Cell 70, 150–164.e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux M, Gagnon D, and Archambault J (2014). E1-mediated recruitment of a UAF1-USP deubiquitinase complex facilitates human papillomavirus DNA replication. Journal of virology 88, 8545–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ng EK, Ng YP, Wong CY, Yu J, Jin H, Cheng VY, Go MY, Cheung PK, Ebert MP, et al. (2009). Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. British journal of cancer 101, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy DR, Solomon D, Hildesheim A, Schiller JT, and Schiffman M (2008). Human papillomavirus infection and the primary and secondary prevention of cervical cancer. Cancer 113, 1980–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, Cousido-Siah A, Masson M, Vande Pol S, Podjarny A, et al. (2016). Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, and Howley PM (1989). Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. The EMBO journal 8, 4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, and Hsu HC (2006). Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle 5, 2676–2687. [DOI] [PubMed] [Google Scholar]
- Pim D, Bergant M, Boon SS, Ganti K, Kranjec C, Massimi P, Subbaiah VK, Thomas M, Tomaic V, and Banks L (2012). Human papillomaviruses and the specificity of PDZ domain targeting. The FEBS journal 279, 3530–3537. [DOI] [PubMed] [Google Scholar]
- Rossi M, Duan S, Jeong YT, Horn M, Saraf A, Florens L, Washburn MP, Antebi A, and Pagano M (2013). Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCF(Fbxo11) ubiquitin ligase. Mol Cell 49, 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, Kawashima D, and Tsurumi T (2013). Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kappaB signaling during productive replication. Journal of virology 87, 4060–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, and Howley PM (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, and Howley PM (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Shibata E, Dar A, and Dutta A (2014). CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J Biol Chem 289, 23056–23064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, and Harper JW (2009). Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki T, Nishidate T, Park JH, Lin ML, Shimo A, Hirata K, Nakamura Y, and Katagiri T (2008). Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene 27, 5672–5683. [DOI] [PubMed] [Google Scholar]
- Vos RM, Altreuter J, White EA, and Howley PM (2009). The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. Journal of virology 83, 8885–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, and Munoz N (1999). Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. The Journal of pathology 189, 12–19. [DOI] [PubMed] [Google Scholar]
- Whitehurst CB, Li G, Montgomery SA, Montgomery ND, Su L, and Pagano JS (2015). Knockout of Epstein-Barr virus BPLF1 retards B-cell transformation and lymphoma formation in humanized mice. MBio 6, e01574–01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegert C, Wentzensen N, Vinokurova S, Kisseljov F, Einenkel J, Hoeckel M, and von Knebel Doeberitz M (2003). A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genomebased amplification techniques. Oncogene 22, 3977–3984. [DOI] [PubMed] [Google Scholar]
- zur Hausen H (2002). Papillomaviruses and cancer: from basic studies to clinical application. Nature reviews. Cancer 2, 342–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.