

Abstract
Post-translational modification is a common mechanism to affect conformational change in proteins, which in turn, regulates function. Herein, this principle is expanded to instruct the formation of supramolecular assemblies by controlling the conformational bias of self-assembling peptides. Biophysical and mechanical studies show that an engineered phosphorylation/dephosphorylation couple can affectively modulate the folding of amphiphilic peptides into a conformation necessary for the formation of well-defined fibrillar networks. Negative design principles based on the incompatibility of hosting residue side-chain point charge within hydrophobic environments proved key to inhibiting the peptide’s ability to adopt its low energy fold in the assembled state. Dephosphorylation relieves this restriction, lowers the energy barrier between unfolded and folded peptide, and allows the formation of self-assembled fibrils that contain the folded conformer, ultimately constituting the formation of cytocompatible hydrogel material.
Keywords: Enzyme, dephosphorylation, conformational change, self-assemble, supramolecular hydrogel
Summary
Post-translational modification is a common mechanism to control protein conformational change and regulate function. Herein, we expand this principle to instruct the formation of supramolecular assemblies by controlling the conformational bias of self-assembling peptides.
Graphical Abstract
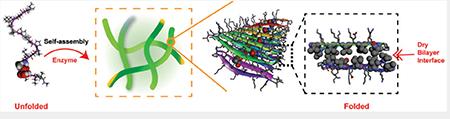
Phosphorylation/dephosphorylation to control protein conformation is a common mechanism to modulate the biological activity of proteins.[1] For example, the K-homology splicing regulator protein (KSRP) is regulated by phosphorylation of a serine in its N-terminal β-sheet domain. Introduction of the phosphoryl group results in the destabilization of the N-terminal sheet and local unfolding that ultimately impairs its ability to foster RNA degradation.[2] In this and other examples,[1] the local environment provided by the protein fold is not conducive to the inclusion of the negatively charged phosphorylated side chain either sterically and/or electronically and thus, the unfolded state is favored. Importantly, in many cases, unfolding is reversible and dephosphorylation results in the restoration of the native fold. Thus, modification of a single residue via this post translational modification can provide exquisite control over conformational equilibria. Herein, we extend this concept beyond globular proteins to control the conformational equilibrium of peptides to allow the formation of supramolecular assemblies.
Our lab had previously developed a class of amphiphilic peptides that self-assemble into a physically crosslinked network of fibrils resulting in the formation of hydrogel material.[3] Solid state NMR shows that these designed peptides fold into a well-defined hairpin conformation in their self-assembled state and that hairpins are arranged within each fibril to form a bilayer cross-β structure.[4] Bilayer formation is driven by the burial and association of hydrophobic amino acid side chains within the core of the fibril forming an extremely dry interface void of water. Importantly, formation of this dry core represents the thermodynamic driving force for peptide assembly and is essential for stabilizing the folded conformation of the β-hairpin in its self-assembled state.[5] In contrast, this dry environment would be exceedingly inhospitable to the inclusion of a negatively charged phosphate side chain as there is no water to solvate its formal point charge. Thus, this represents a design opportunity to control the conformational equilibrium of a peptide en-route to its lowest energy self-assembled folded state. Further, this allows one to exploit the enzymatic action of phosphatase to trigger the formation of fibrillar hydrogel, which depends on the ability of the peptide to access its thermodynamic minimum on the energy folding landscape. Although there are elegant reports of using dephosphorylation or phosphorylation events to study amyloid formation[6], silk modification,[7] and to trigger the hydrogelation of small linear peptides,[8] these employ phosphorylated residues to endear water solubility to the peptide; when removed, aqueous buffer becomes a poor solvent and the peptides assemble in response. Herein, we exploit phosphorylation to control the folded conformation of peptides, which is energetically coupled to their ability to form supramolecular assemblies.
This premise was tested with the design of a 20-residue peptide named PP1, Figure 1. When dephosphorylated, PP1 is designed to adopt an amphiphilic β-hairpin conformation in its self-assembled fibrillar state. PP1’s sequence contains two β-strands connected by a four-residue type II’ β-turn. The N- and C-terminal β-stands contain an (AB)n repeat of hydrophilic and hydrophobic residues endearing facial amphiphilicity to its folded conformation. When folded and assembled, dephosphorylated PP1 is designed to place its hydrophobic valine side chains into the core of its resultant fibrils. Importantly, the peptide contains a threonine residue at position 14, a hydrophobic B-position located directly in the middle of the folded hairpin. When PP1 is assembled, multiple copies of the threonine side chain line the entire dry interface of the bilayer as it traverses down the long axis of a given fibril. Phosphorylation of this residue would place an extended array of point charges in this dry environment, an energetically unfavorable proposition. Thus, phosphorylated PP1 should disfavor the folded and assembled state, but favour an unstructured state not competent to self-assemble.
Figure 1.

Enzymatic dephosphorylation shifts the conformational equilibrium favouring folded hairpin in the self-assembled state. The sequences of phosphorylated peptides are also shown.
Circular dichroism (CD) spectroscopy coupled with absorbance sedimentation measurements show that phosphorylated PP1 is, in fact, unfolded and of low molecular weight under solution conditions that normally favour self-assembly, Figure S1. However, the unfolded peptide is a substrate of lambda protein phosphatase (LPP) and is nearly quantitatively dephosphorylated within hours by a relatively low amount of enzyme (0.092 U/μL), Figure 2a. Although the reaction is efficient, around 10% of the peptide substrate remains. This is most likely due to a restriction of either the substrate and/or the enzyme within the evolving fibril network as the gel forms. Interestingly, the peptide is a selective substrate for LLP as its not cleaved by alkaline phosphatase at high enzyme concentration (25 U/mL) known to readily cleave peptide substrates,[9] Figure S2. As predicted, dephosphorylation leads to a distinct conformational change from random coil to β-sheet over a similar time course as evidenced by the time-dependent CD experiment shown in Figure 2b. Here, the negative band at 195 nm, indicative of random coil (green data) increases to positive values of mean residue ellipticity as PP1 is dephosphorylated along with a concomitant formation of a new band at 216 nm, indicative of β-sheet secondary structure. This data is in agreement with that obtained for a dephosphorylated control peptide that undergoes immediate self-assembly in a phosphatase-independent manner to form β-sheet rich fibrils, Figure S3. It should be noted that the enzyme contributed negligibly to the CD spectrum at the concentration used in this experiment (0.575 U/μL), Figure S4. In these experiments, CD spectroscopy reports on both the folded hairpin conformation of the peptide and the extended β-sheet structure of fibrils resulting from self-assembly. Thus, it is difficult to define the folded conformation of PP1 using CD alone.
Figure 2.

(a) Time-dependent dephosphorylation of PP1 (0.5 wt%) in the presence of 0.092 U/μL lambda protein phosphatase (LPP). (b) Time dependent CD spectra of 0.5 wt% PP1 upon the addition of 0.092 U/μL LPP reveals the evolution of β-sheet structure. (c) TEM images of fibrils from the dephosphorylation of 0.5 wt% PP1 employing 0.575 U/μL LPP for 48 h, inset are distribution of nanofibers width. (d) Mean residue ellipticity of 0.5 wt% PP1 at 216 nm as a function of time and enzyme concentration. All experiments were performed in pH 7.4 BTP buffer (50mM BTP, 150 mM NaCl) containing 1mM MnCl2 at 37oC.
Transmission electron microscopy (TEM) was used to further investigate PP1 conformation. Figure 2c shows that dephosphorylated PP1 assembles into well-defined monomorphic fibrils consistent with the model depicted in Figure 1. Importantly, the measured width of the fibrils (~4 nm) is very close to the width of a folded PPI hairpin (~3 nm). As expected, no fibrils were observed by TEM of unreacted phosphorylated PP1, Figure S5. Thus, as PP1 is dephosphorylated by LPP, the conformational equilibrium shifts from unfolded peptide to assembled and folded peptide with the free threonine side-chain accommodated within the dry fibril core. Further support that dephosphorylation is responsible for executing the conformational change is provided in Figure 2d. Here, the rate of β-sheet evolution monitored by CD increases with increasing concentration of phosphatase. Panel d also shows that in the absence of enzyme (black data), phosphorylated PP1 remains unstructured and unable to assemble over an extended period of time.
The enzyme-dependent rate of hydrogelation was investigated by oscillatory rheology, which was used to measure the storage modulus (G’, a measure of material rigidity) as a function of time. Figure 3a shows time-sweep measurements where solutions of PP1 and enzyme are co-incubated in the rheometer. In this experiment, enzymatic dephosphorylation produces peptide capable of self-assembling into a network of fibrils, converting the solution to gel. The fibril network evolves with time further stiffening the gel. These data show that the rate of hydrogelation as well as material stiffness increases as a function of increased enzyme concentration (compare green and blue data). Further, hydrogel rigidity is also dependent on enzyme substrate concentration. When the concentration of PP1 is increased from 0.5 to 1.0 wt% (compare blue and red data), the plateau modulus increases as a result of more peptide being available for self-assembly after dephosphorylation. The CD and rheology data, taken together, indicate that the enzymatic rate of dephosphorylation is coupled to the rate of β-sheet secondary structure formation, which in turn, is coupled to the rate of material formation. Thus, modulating the conformational equilibrium of PP1 via enzymatic action directly affects supramolecular assembly and bulk mechanical properties of the resultant material.
Figure 3.

(a) Rheological dynamic time sweeps of PP1 monitoring the storage modulus (G’) as a function of time, peptide, and enzyme concentration. (b) Shear thin-recovery of 1.0 wt% dephosphorylated PP1 hydrogel. Time sweep (6.0 rad/s and 0.2 % strain) for 1.0 h shows initial formation of gel, followed by an applied 1000% strain for 30 s to thin the gel, with a final time sweep (6.0 rad/s and 0.2 % strain) for 1.0 h to show rapid recovery of hydrogel stiffness.
The rheological properties were further investigated in Figure 3b, which shows that gels formed by dephosphorylated PP1 exhibit shear-thin/recovery mechanical properties. Here, dephosphorylated peptide, obtained non-enzymatically from chemical synthesis, is allowed to self-assemble, forming a gel within minutes that further stiffens with time. At 60 minutes, a large strain is applied to the material that results in thinning, converting the solid-like gel into a viscous material that flows. Under these high-strain conditions, the fibril network is disrupted resulting in the loss of mechanical integrity. After 30 seconds, the strain is reduced. The data clearly shows that material recovery is rapid with a quantitative recovery of storage modulus. This recovery behavior is due, in part, by the fact that the original fibril network is not covalently crosslinked, but is defined by physical crosslinks formed via self-assembly. Thus, although compromised by strain, the crosslinks within this gel can reform on cessation of strain.
We next examined the positional dependence of the phosphorylated threonine on its ability to shift the conformational equilibria towards the unfolded state and its role as enzyme substrate. We also assessed the ability of the resulting dephosphorylated peptide to adopt β-sheet structure and assemble into fibrillar hydrogel. As stated earlier, PP1 contains a phosphothreonine at a central position (residue 14) within the folded β-hairpin, which proved to be destabilizing towards the formation of the hydrophobic dry bilayer interface, an essential component of fibril formation. A second peptide was prepared that incorporates the phosphothreonine near the C-terminus at position 18, Figure 1. Similar to PP1, CD spectroscopy coupled with absorbance sedimentation measurements show that phosphorylated PP2 is unfolded and of low molecular weight under solution conditions that normally favour self-assembly, Figure S6. This indicates that the side chain of residue 18 must be contained in a relatively hydrophobic environment when the peptide is folded and assembled within a given fibril. Compared to PP1, the rate of PP2 dephosphorylation measured by HPLC (Figure S7) is nearly identical. This is expected as both peptide substrates are unstructured and it is reasonable to expect that each peptide’s phosphoryl group is of similar accessibility towards the phosphatase. Unexpectedly, we found that rate of β-sheet evolution measured by CD was significantly slower for PP2 when incubated with the same concentration of enzyme, Figure S8. Thus, the intrinsic propensity of dephosphorylated PP2 to self-assemble is lower than PP1. Although speculative, it may be more difficult to dehydrate a side chain near the termini of the peptide during self-assembly en-route to fibrillization. This assertion is consistent with the observation that the rate of hydrogelation monitored by rheology is also slower, Figure S9. Lastly, PP3 was designed to incorporate phosphothreonine at the i+3 position of the type II’ β-turn within the folded hairpin. Interestingly, phosphorylated PP3 readily self assembles, forming β-sheet structure resulting in the formation of hydrogel without the aid of phosphatase, Figure S10A, B. This indicates that this residue position is at least partially solvent exposed when the peptide is folded and assembled in the fibrillar state. The model in Figure 1 suggests this to be a reasonable expectation. Although exposed to solvent, the phosphoryl group is unable to be hydrolyzed by phosphatase, suggesting that the folded and assembled hairpin has limited accessibility to the enzyme, Figure S11. Overall, these positional studies indicate that the location of the phosphorylated residue is important in defining its ability to govern peptide conformation with respect to assembly formation, with hydrophobic, solvent inaccessible locations offering optimal control. Meanwhile, we also test whether phosphorylation could disassemble the hydrogels of dephosphorylated PP1 (or PP2). As shown in Figure S12 and S13, the addition of protein kinase A fails to trigger phosphorylation of dephosphorylated PP1 (or PP2). This is most likely due to the enzyme substrate being inaccessible to enzyme in its self-assembled state. All the peptides used in this study were prepared by Fmoc solid-phase peptide synthesis and purified to homogeneity, Figures S15–19.
Finally, we investigated the cytocompatibility of a hydrogel resulting from dephosphorylation to help define future use of these materials in biomedical applications. Figure 4 shows live-dead cytocompatibility assays for a PP1 derived gel formed by enzymatic dephosphorylation. Human dermal fibroblasts (HDFs) were incubated on top of the resulting 1.0 wt% hydrogel and cell viability monitored and compared to a control tissue-cultured treated polystyrene surface. In this assay, live cells fluoresce green and dead cells fluoresce red. Panels a and c show that cells are equally viable on the surface of dephosphorylated PP1 gel as compared to the control surface. Next, the morphology of the cells was examined in panels b and d for the control and PP1 material surfaces. F-actin staining with Alexa fluor phalloidin shows that cells adopt spread morphologies characteristic of adherent cells that are capable of forming well-defined stress fibers defining their cytoskeleton. Like the dephosphorylated hydrogel PP1, almost all cells on dephosphorylated PP2 hydrogel are alive and exhibit well-defined F-actin fibers (Figure S14). We also examined the behaviour of HDF cells on the surface of the phosphorylated PP3 gel and found little change in cell behaviour as compared to the dephosphorylated materials. Which was almost identical with dephosphorylated PP1 gel, indicating that phosphorylation in the contexts of these peptide-based materials has little influence on cell viability and morphology, similar to that observed for other phosphorylated surfaces.[10] Further, the behaviour of cells on the gels reported herein are similar to that observed for other β-hairpin based gels.[11]
Figure 4.

Live/dead assay and F-actin staining of human dermal fibroblasts 24h after being seeded onto (a,b) control tissue-culture polystyrene surface, (c,d) 1.0 wt% dephosphorylated PP1 hydrogel and (e,f) 1.0 wt% PP3 hydrogel.
In sum, the folded conformation of peptides can influence the mechanism of formation and resultant architecture of supramolecular assemblies. For example, the α-helix is a ubiquitous fold not only important to protein structure, but also in the formation of supramolecular helical assemblies[12] and filamentous gels[13]. In these examples, the ability of the peptide to adopt its native conformation is absolutely required for the formation of the final self-assembled state. Thus, having control over the conformational bias of peptides offers governance over material construction. Post-translational modifications, especially those that are enzyme-instructed, are convenient effectors of conformational change. Herein, we showed that enzymatic dephosphorylation events can instruct the formation β-hairpin structure, which is essential for the formation of supramolecular hydrogels. Although dephosphorylation was exemplified in this study, one can imagine that other modifications such as glycosylation, farnesylation, or amidation, to name a few, could also be employed to control the conformational landscape of peptides important to the construction of supramolecular assemblies.
Supplementary Material
Acknowledgements
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. We thank Dr. Ziqiu Wang in Electron microscopy core facility at ATFR for assistance. Dr. J. Shi thanks Dr. Stephen Miller for proofreading the manuscript.
Contributor Information
Dr. Junfeng. Shi, Chemical Biological Laboratory, National Cancer Institute. National Institutes of Health 376 Boyles Street, Frederick MD, 21702, US
Dr. Galit. Fichman, Chemical Biological Laboratory, National Cancer Institute. National Institutes of Health 376 Boyles Street, Frederick MD, 21702, US
Dr. Joel.P. Schneider, Chemical Biological Laboratory, National Cancer Institute. National Institutes of Health 376 Boyles Street, Frederick MD, 21702, US:Joel.Schneider@nih.gov
References
- [1].Johnson LN, Lewis RJ, Chem. Rev 2001, 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- [2].Diaz-Moreno I, Hollingworth D, Frenkiel TA, Kelly G, Martin S, Howell S, Garcia-Mayoral M, Gherzi R, Briata P, Ramos A, Nat. Struct. Mol. Biol 2009, 16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Nagy-Smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP, ACS Central Science 2017, 3, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J, J. Am. Chem. Soc 2002, 124, 15030–15037 [DOI] [PubMed] [Google Scholar]; c) Smith DJ, Brat GA, Medina SH, Tong D, Huang Y, Grahammer J, Furtmüller GJ, Oh BC, Nagy-Smith KJ, Walczak P, Brandacher G, Schneider JP, Nat. Nanotechnol 2015, 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nagy-Smith K, Moore E, Schneider J, Tycko R, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9816–9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Sathaye S, Zhang H, Sonmez C, Schneider JP, MacDermaid CM, Von Bargen CD, Saven JG, Pochan DJ, Biomacromolecules 2014, 15, 3891–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Micklitsch CM, Medina SH, Yucel T, Nagy-Smith KJ, Pochan DJ, Schneider JP, Macromolecules 2015, 48, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Broncel M, Wagner SC, Hackenberger CPR, Koksch B, Chem. Commun 2010, 46, 3080–3082 [DOI] [PubMed] [Google Scholar]; b) Valette NM, Radford SE, Harris SA, Warriner SL, Chembiochem. 2012, 13, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Volkov V, Cavaco-Paulo A, Appl Microbiol Biot 2016, 100, 4337–4345. [DOI] [PubMed] [Google Scholar]
- [8].a) Zhou J, Du X, Xu B, Angew. Chem. Int. Ed 2016, 55, 5770–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, Xu B, Adv Mater 2004, 16, 1440–1444 [Google Scholar]; c) Gao J, Wang H, Wang L, Wang J, Kong D, Yang Z, J. Am. Chem. Soc 2009, 131, 11286–11287 [DOI] [PubMed] [Google Scholar]; d) Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I, J. Am. Chem. Soc 2015, 137, 576–579 [DOI] [PubMed] [Google Scholar]; e) Sahoo JK, Pappas CG, Sasselli IR, Abul‐Haija YM, Ulijn RV, Angew. Chem. Int. Ed 2017, 56, 6828–6832 [DOI] [PubMed] [Google Scholar]; f) Akkarachaneeyakorn K, Li M, Davis SA, Mann S, Langmuir 2016, 32, 2912–2919 [DOI] [PubMed] [Google Scholar]; g) Shi J, Yuan D, Haburcak R, Zhang Q, Zhao C, Zhang X, Xu B, Chemistry – A European Journal 2015, 21, 18047–18051 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Jie Zhou XD, Wang Jiaqing, Yamagata Natsuko, Xu Bing, Front. Chem. Sci. Eng 2017, 11, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi J, Du X, Yuan D, Zhou J, Zhou N, Huang Y, Xu B, Biomacromolecules 2014, 15, 3559–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zelzer M, McNamara LE, Scurr DJ, Alexander MR, Dalby MJ, Ulijn RV, J. Mater. Chem 2012, 22, 12229–12237. [Google Scholar]
- [11].Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Egelman EH, Xu C, DiMaio F, Magnotti E, Modlin C, Yu X, Wright E, Baker D, Conticello VP, Structure, 23, 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu C, Liu R, Mehta AK, Guerrero-Ferreira RC, Wright ER, Dunin-Horkawicz S, Morris K, Serpell LC, Zuo X, Wall JS, Conticello VP, J. Am. Chem. Soc 2013, 135, 15565–15578 [DOI] [PubMed] [Google Scholar]; c) Fletcher JM, Harniman RL, Barnes FRH, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P, Woolfson DN, Science 2013, 340, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN, Nat. Mater 2009, 8, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.