

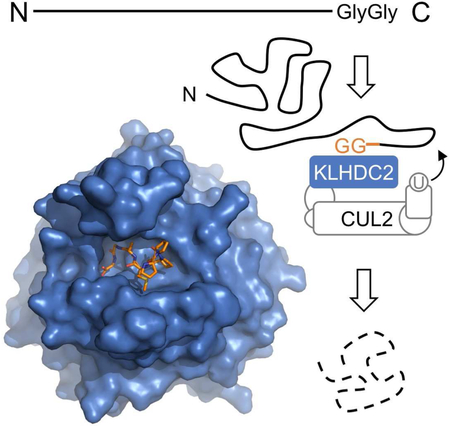
Summary
Aberrant proteins can be deleterious to cells and are cleared by the ubiquitin-proteasome system. A group of C-end degrons has recently been identified in some of these abnormal polypeptides, which are recognized by specific cullin-RING ubiquitin E3 ligases (CRLs). Here we report three crystal structures of a CRL2 substrate receptor, KLHDC2, in complex with the diglycine-ending C-end degrons of two early terminated selenoproteins and the N-terminal proteolytic fragment of USP1. The E3 recognizes the degron peptides in a similarly coiled conformation and cradles their C-terminal diglycine with a deep surface pocket. By hydrogen bonding with multiple backbone carbonyls of the peptides, KLHDC2 further locks in the otherwise degenerate degrons with a compact interface and unexpected high affinities. Our results reveal the structural mechanism by which KLHDC2 recognizes the simplest C-end degron, and suggest a functional necessity of the E3 to tightly maintain the low abundance of its select substrates.
Keywords: C-end degron, DesCEND, KLHDC2, USP1, selenoproteins, cullin, crystal structure, protein degradation, ubiquitin, E3
Graphical Abstract
Introduction
The ubiquitin-proteasome system (UPS) removes aberrant proteins and regulates diverse cellular functions by promoting the turnover of numerous protein substrates (Goldberg, 2003; Hershko and Ciechanover, 1998). The high capacity and selectivity of UPS is conferred by a plethora of ubiquitin E3 ligases, which number in the hundreds for humans (Zheng and Shabek, 2017). Acting at the final step of a three-enzyme cascade, many ubiquitin E3 ligases recognize their cognate substrates through a short linear sequence motif, known as a degron (Guharoy et al., 2016; Lucas and Ciulli, 2017; Mészáros et al., 2017). To achieve high specificity, eukaryotic cells have evolved a variety of mechanism governing degron-E3 interactions. For many degrons, proper forms of post-translational modifications, such as serine/threonine phosphorylation and proline hydroxylation, have been shown to play a key role in enabling E3 binding (Hao et al., 2007; Hao et al., 2005; Min et al., 2002; Orlicky et al., 2003; Wu et al., 2003). On the other hand, an increasing number of degrons have also been identified to function in a modification-independent manner (da Fonseca et al., 2011; Kraft et al., 2005; Padmanabhan et al., 2006; Uljon et al., 2016; Zhuang et al., 2009).
In the classical N-end rule pathway, both aforementioned degrons have been discovered at the extreme N-terminus of proteins (Tasaki et al., 2012; Varshavsky, 2011). Some of these N-degrons are generated by proteolytic cleavage, whereas others are created by N-terminal modifications. While certain resulting N-terminal amino acids can stabilize a protein, other ones, such as arginine, have been shown to substantially reduce the half-life of a substrate. Previous studies have identified a family of UBR-box-containing proteins, classified as N-recognins, responsible for recognizing the N-terminal degrons (Tasaki et al., 2005; Tasaki et al., 2009; Xia et al., 2008). The crystal structure of the UBR-box domain from yeast UBR1 has been determined in complex with the N-degron peptide of the cohesion subunit Scc1 (Choi et al., 2010). In this structure, the E3 recognizes the N-end degron of the proteolytically generated Scc1 C-terminal fragment through its free backbone amino group, the side chain of the leading arginine, and the penultimate residue. A similar recognition interface has also been revealed for the mammalian UBR1 ortholog (Matta-Camacho et al., 2010). By recognizing the N-end degrons exposed by endopeptidase cleavage, the N-end rule not only dictates the half-life of full-length proteins, but also participates in protein quality control by eliminating proteolytic products.
Like the C-terminal polypeptide fragments generated by proteolysis, the N-terminal segments and early terminated protein products could also impose a threat to the normal functions of the cell. Recent studies have unraveled a cohort of ubiquitin ligases from the cullin-RING superfamily, which are implicated in the recognition of specific sequence elements embedded in the extreme C-terminus of these truncated polypeptides (Koren et al., 2018; Lin et al., 2018). This novel protein degradation mechanism, named DesCEND (destruction via C-end degron), relies on select amino acids at key positions within the degrons, which are usually less than ten residues long. Interestingly, one of these C-end degrons is characterized by a strikingly simple diglycine motif at the extreme C-terminus. With a highly degenerate N-terminal region, this diglycine-containing sequence is thought to be recognized by a BC-box protein, KLHDC2, which functions as a substrate receptor of the CRL2 E3 complex. Remarkably, the diglycine C-end degron has been found in multiple classes of DesCEND substrates, including early terminated selenoproteins (SelK and SelS), the N-terminal proteolytic product of a deubiquitinating enzyme (USP1), and a number of full-length proteins (Koren et al., 2018; Lin et al., 2018). Klhdc2 homozygous knockout mice display an embryonic lethal phenotype (Dickinson et al., 2016). KLHDC2 has thus emerged as an essential E3 ligase with a multifaceted role in protein homeostasis.
Similar to other known degrons, the C-end degrons are self-sufficient and can induce proteasomal degradation when fused to otherwise stable proteins. To reveal the mechanism by which a prototypical C-terminal degron is decoded in the DesCEND pathway, we determined the crystal structures of KLHDC2 in complex with three different diglycine C-degron peptides and performed detailed analyses of the interaction interface. Our studies have not only mapped the essential elements dictating the specific interactions between the C-end degron and its cognate E3, but also provided a detailed quantitative understanding of the system and shed light on a unique role of DesCEND that is distinct from the N-end rule pathway.
Results
Mapping key elements in SelK C-end degron
The selenoprotein SelK contains a selenocysteine (Sec), which is encoded by the stop codon UGA (Ambrogelly et al., 2007). With help from a Sec insertion sequence element in the 3’ untranslated region, this terminal signal in the SelK mRNA is translated into Sec, allowing the production of the full-length protein (Driscoll and Copeland, 2003). When selenium is scarce, the lack of Sec-transfer RNA causes premature termination of translation. Recent studies have shown that the resulting early terminated SelK protein bears a C-terminal diglycine degron, which triggers its CRL2KLHDC2-mediated proteasomal degradation (Lin et al., 2015). The BC-box protein KLHDC2 contains a C-terminal region responsible for CUL2-Elongin-B/C binding and an N-terminal kelch repeat β-propeller domain, which is commonly used by CRL substrate receptor subunits to engage substrates (Duda et al., 2011; Mahrour et al., 2008; Zimmerman et al., 2010). To formally establish the direct interaction between SelK C-end degron and KLHDC2, we purified the recombinant KLHDC2 kelch domain (hereafter referred to as KLHDC2) and performed GST pull-down assays using GST-fused SelK C-end degron that are 8 or 12 aa long. KLHDC2 displayed robust interaction with both degron peptides, but not GST alone (Figure 1A). This result indicates that the BC-box protein can directly recognize the SelK C-end degron and that the kelch repeat domain of KLHDC2 is sufficient for this interaction.
Figure 1. KLHDC2 directly recognizes SelK C-end degron with a high affinity.

(A) Validation of direct interactions between purified KLHDC2 kelch repeat domain and an 8 aa and a 12 aa SelK C-end degron fused to GST in a GST-pull down assay.
(B) AlphaScreen competition assay for assessing the affinity of the SelK C-end degron peptides with variable lengths to KLHDC2. The dose response curves of the peptides ranging from 2aa to 12aa are colored in the same scheme as the peptide labels. The IC50 value and the 95% confidence interval for each peptide is listed together with its amino acid sequence at the bottom table. AFU: arbitrary fluorescence units. Data are measured in triplicates and represented as mean ± SEM.
(C) Affinity determination of KLHDC2 binding to the 12 amino acids SelK degron peptide by Octet BioLayer Interferometry. Green lines represent the kinetics of association and dissociation of GST-KLHDC2 to biotinylated SelK peptide, which is immobilized on the probe. Red lines represent global fit of the binding curves with a 1:1 ligand model. The dissociation constant (Kd) was calculated from the kon and kdis values.
(D) AlphaScreen competition assay for assessing the affinity of the 8 aa WT (…GGCOOH), C-terminally amidated (…GGCONH2), and mutant (…GLCOOH) SelK C-end degron peptides to KLHDC2. The IC50 values and the 95% confidence intervals for all three peptides are listed together with their amino acid sequence at the bottom table in (B).
See also Figure S1.
To map the minimal length of the SelK C-end degron for high affinity binding to KLHDC2, we established the degron peptide-E3 interaction in an Amplified Luminescence Proximity Homogenous Assay (AlphaScreen) with a biotinylated 12 aa SelK degron peptide and GST-fused KLHDC2 immobilized on the donor and acceptor beads, respectively (Figure S1A). We then carried out competition experiments with label-free SelK degron peptides of varying lengths ranging from 12 to 2 amino acids. These peptides share the critical extreme C-terminal diglycine motif with variably shortened N-terminal ends. By titrating the concentration of the competing peptides, we were able to derive dose response curves and obtain the IC50 values for each peptide. Due to the extremely low concentration of the immobilized components, the IC50 value determined from our competition assay can be approximated to Kd. To our amazement, the 12 aa SelK degron peptide abolished the AlphasScreen signal with an IC50 of ~3 nM (Figure 1B). Decreasing the length of the degron peptide from 12 to 5 aa caused a gradual and slight decrease in affinity. The 5-residue degron, nevertheless, retained an affinity toward the E3 ligase in the low-nanomolar range. We observed a dramatic 30-fold increase in Kd when the peptide length is reduced to 4 amino acids, indicating the loss of a critical contact between the degron and the E3. Interestingly, even diglycine was able to bind KLHDC2 and compete with the biotinylated 12 aa degron that is immobilized on the AlphaScreen donor beads (Figure 1B). The extreme C-terminal diglycine motif, therefore, contributes a substantial amount of binding energy to the degron-E3 interaction. Together, these results revealed a strong correlation between the length of the SelK degron peptide and its strength in binding KLHDC2, and established the minimal degron length of 5 amino acids for high affinity interaction. The remarkably tight binding between the 12 amino acids SelK degron and KLHDC2 was further confirmed by the dissociation constant of their direct interaction measured with Octet BioLayer Interferometry, which yielded a Kd value of ~4 nM (Figure 1C).
The full-length SelK protein contains three additional amino acids beyond the diglycine motif, which allows the polypeptide to evade DesCEND. Recent studies have revealed a requirement for the digycline motif to terminate a polypeptide in order to give rise to a functional C-end degron (Lin et al., 2015). We hypothesized that the free extreme C-terminal backbone carboxyl group, along with the tandem glycine residues, must play a role in E3 binding. Using our AlphaScreen-based competition assay, we compared the affinity of the 8 aa SelK degron peptide that was C-terminally amidated to that of the unmodified peptide. Consistent with our hypothesis, this simple modification of the C-terminal carboxyl group profoundly decreased the affinity of the degron peptide by nearly a 1000 fold (Figure 1D). Using an 8 aa peptide with the last glycine residue mutated to leucine, we further characterized and validated the importance of the diglycine residues, which has been previously established in cell-based assays (Lin et al., 2018). In contrast to the single digit nM affinity of the wild type SelK degron peptide, the glycine to leucine mutation increased the IC50 value to ~100 μM (Figure 1D).
Crystal structure of KLHDC2 bound to SelK degron peptide
In agreement with the high affinity binding determined from the AlphaScreen assay, KLHDC2 forms a stable complex with the SelK degron peptide as detected by native protein mass spectrometry (Figure S1B). We crystallized and determined the structure of the KLHDC2 kelch repeat domain in complex with the 8 aa SelK C-end degron peptide (Table 1). Akin to most kelch repeats, KLHDC2 adopts a six-bladed β-propeller fold with each blade composed of four antiparallel β-strands (named A to D from inner to outer position) (Figure 2A). Conventionally, the side of a β-propeller fold constructed by the loops linking strands A and D and strands B and C is referred to as the “top” surface (Sprague et al., 2000). These loops are highly variable between different β-propeller proteins and are frequently involved in protein interaction or enzymatic catalysis. In KLHDC2, these loops form a deep binding pocket, which is responsible for recognizing the C-end degron.
Table 1.
Data Collection and Refinement Statistics
| KLHDC2-SelK | KLHDC2-SelS | KLHDC2-USP1 | |
|---|---|---|---|
| PDB: accession number | PDB: 6D03 | PDB: 6D04 | PDB: 6D05 |
| Data collection statistics | |||
| Space group | P 1 21 1 | P 1 21 1 | P 1 21 1 |
| Wavelength (Å) | 1 | 1 | 1 |
| Cell dimensions: a, b, c (Å) | 44.8, 87.8, 88.6 | 44.6, 88.6, 88.8 | 44.5, 87.1, 88.3 |
| Cell dimensions: a, b, g (°) | 90.0, 104.5, 90.0 | 90.0, 104.8, 90.0 | 90.0, 104.6, 90.0 |
| Resolution (Å)a | 50.0 - 2.2 (2.19 - 2.15) | 50.0 - 2.2 (2.24 - 2.20) | 50.0 - 2.2 (2.24 - 2.20) |
| Rmeas | 0.18 (0.68) | 0.16 (0.70) | 0.13 (0.60) |
| Mean I/σ(I) | 15.2 (2.2) | 11.2 (1.6) | 15.3 (1.6) |
| CC1/2 (%) | (77.2) | (66.5) | (71.4) |
| Completeness (%) | 99.9 (99.2) | 99.3 (93.0) | 98.0 (80.3) |
| Redundancy | 6.9 (4.3) | 4.5 (3.3) | 5.9 (3.8) |
| Refinement statistics | |||
| Resolution (Å) | 43.4-2.2 | 39.4-2.2 | 43.1-2.5 |
| Number of reflections | 35101 | 33734 | 22666 |
| R-work/R-free | 0.17/0.22 | 0.17/0.22 | 0.20/0.25 |
| Number of atoms | |||
| Protein | 5136 | 5133 | 5095 |
| Ligand | 0 | 0 | 0 |
| Water | 208 | 230 | 128 |
| Average B-factor | 22.1 | 27.4 | 32.6 |
| RMSD bond length | 0.008 | 0.007 | 0.013 |
| RMSD bond angle | 1.160 | 0.894 | 1.303 |
| Ramachandran plot (%) | |||
| Favored | 96.3 | 97.1 | 96.3 |
| Allowed | 3.7 | 2.9 | 3.5 |
Values in parentheses are for the highest-resolution shell.
Figure 2. Crystal structure of the KLHDC2-SelK C-end degron complex.

(A) Two orthogonal views of KLHDC2 kelch repeat domain in complex with a SelK C-end degron peptide. The β-propeller domain of KLHDC2 is shown in grey ribbon with its six blades labeled “KR1” to “KR6” The four anti-parallel β-strands in repeat 1 are labeled “A” to “D”. The SelK C-end degron peptide is shown in orange sticks and surface representation. The N- and C-termini of the two polypeptides are labeled “N” and “C”.
(B) Conservation surface mapping of the KLHDC2 kelch repeat domain in its top and bottom views. Residues that are strictly (100%) and highly (80-100%) conserved are colored in magenta and light-grey, respectively. The rest of the molecule is colored in dark grey. The C-end degron-binding pocket is annotated for its dimension, two separate chambers, and the middle ridge.
(C) Electrostatic surface potential map (positive in blue and negative in red) of the KLHDC2 kelch repeat domain in the same view as shown in (B). The SelK C-end degron in shown in orange sticks. (D) A close-up stereo view of the interface formed between KLHDC2 and the Selk C-end degron peptide. KLHDC2 is colored in light grey with its SelK-interacting residues shown in sticks. The SelK peptide is shown in orange sticks. Hydrogen bonds and salt bridges are indicated by yellow dashed line. Water molecules are shown as cyan sphere.
(E) Ligplot diagram of the interactions between KLHDC2 and the SelK C-end degron peptide. The SelK peptide is shown in orange and the KLHDC2 residues forming hydrogen bonds with SelK or water molecules are shown in grey. Residues in KLHDC2 involved in van der Waals packing are shown in black with 1/3 circle eyelash shape in maroon. Water molecules are shown as cyan spheres.
KLHDC2 is evolutionarily conserved from amoeba to humans (Figure S2). Surface conservation mapping of its kelch repeat domain reveals a group of solvent-exposed and highly conserved residues clustered at the bottom and one side of the top surface pocket. This special feature of the binding pocket highlights its functional importance (Figure 2B). The KLHDC2 pocket is ~16 Å long and ~12 Å wide. It can be divided into two chambers, which are separated by a low ridge in the middle. Interestingly, one chamber is more conserved than the other. Moreover, electrostatic potential mapping of KLHDC2 unveils a basic patch at the bottom of the pocket, which is co-localized with the highly conserved surface area (Figure 2C).
The SelK C-end degron peptide is anchored to the KLHDC2 top surface pocket in a highly coiled conformation. Its C-terminal end is deeply embedded in the more conserved pocket chamber, hereafter referred to as “chamber C”. By contrast, the N-terminal end of the degron peptide is accommodated by “chamber N” and more exposed to solvent (Figure 2B, and S3A). The complex structure unmasks an intimate and compact interface between the E3 ligase and the degron peptide, which is dominated by inter-molecular hydrogen bond networks and salt bridges. The extreme C-terminal carboxyl group simultaneously forms two hydrogen bonds and two salt bridges with three highly conserved KLHDC2 residues, Ser269, Arg241, and Arg236, which demarcate one side of chamber C (Figure 2B, 2D, and 2E). The tandem glycine resides are buttressed by two KLHDC2 alanine residues, Ala219 and Ala220, from the bottom, and flanked by three KLHDC2 aromatic residues, Tyr163, Trp191, and Trp270, on the sides. The backbone carbonyl group of the penultimate glycine residue is further locked in through a hydrogen bond donated by the side chain of KLHDC2 Trp191 (Figure 2D and 2E). Overall, the strict requirement of a glycine residue at the penultimate position can be explained by two factors. First, the backbone dihedral angles (φ ≅ 90°, ψ ≅ −5°) are limited to glycine. Second, the space between the Cα atom and the surrounding KLHDC2 residues can only accommodate a residue without a side chain. Although glycine is strongly preferred at the last position of the diglycine degron, alanine is also allowed based on previous findings (Lin et al., 2018). The backbone geometry of the terminating residue is less constrained than internal amino acids, but the limited space between its Cα atom and the surrounding KLHDC2 residues would only accommodate a small side chain.
Beyond the C-terminal diglycine motif, the SelK degron makes three additional hydrogen bonds with the E3 ligase, all through its backbone carbonyls. Pointing up from the bottom ridge of the degron-binding pocket, Lys147 of KLHDC2 stabilizes the SelK peptide by hydrogen bonding with the backbone carbonyl groups at the −3 and −5 positions (Figure 2D and 2E). Arg189 of the E3, meanwhile, holds the N-terminal end of the SelK degron in place by interacting with the −6 carbonyl. This hydrogen bond network is further substantiated by four stably bound water molecules, which either bridge the degron and the E3 or stabilize the conformation of the peptide itself. In contrast to these extensive polar interactions, the side chains of the SelK degron peptide only make limited van der Waals packing against KLHDC2. Out of the seven degron residues observed in our structure, only two at −4 and −5 positions use their side chains to make contacts with the boundary of chamber N (SelK-M88 with Y50, L342, L343 in KLHDC2, and SelK-P87 with Y62, H109, and W321 in KLHDC2). Collectively, the KLHDC2-SelK degron interface is predominantly mediated by backbone groups on the degron side and the side chains of amino acids lining the E3 pocket. The physical size of the E3 pocket, which can only accommodate 7 amino acids from the degron, limits the impact of a longer degron sequence on binding affinity. Meanwhile, the close and continuous contacts the pocket makes with the C-terminal 5 amino acids of the degron explain its minimum length requirement for high affinity interactions.
Mutational analysis of the KLHDC2 degron-binding pocket
To dissect the functional roles of the degron-binding residues in KLHDC2, we engineered thirteen single amino acid mutants and tested their activities both in vitro and in vivo. We first purified individual KLHDC2 mutants and assessed their ability to bind GST-fused SelK degron in a pull-down assay (Figure 3A and S3B). In parallel, we generated a Global Protein Stability (GPS) reporter cell line expressing GFP-fused SelK C-end degron with endogenous KLHDC2 knocked down by shRNA. We then individually introduced wild type KLHDC2 or its single point mutants and compared their abilities to promote the degradation of SelK degron (Figure 3B and S3C). In this cell-based system, degradation of the GFP-fused substrate protein can be visualized by a lower steady-state abundance of GFP-substrate to RFP among the entire cell population (peak shift from right to left).
Figure 3. Mutational analysis of degron-binding residues of KLHDC2.

(A) Interactions between purified KLHDC2 mutants and SelK C-end degron fused with GST detected by GST pull-down assay and visualized on SDS-PAGE with Coomassie stain.
(B) GPS experimental design for assessing the effects of wild type (KLHDC2+) and mutant KLHDC2 (KLHDC2*+) on the stability of GFP fused to the SelK or USP1-NTD degron. The GFP/RFP ratio is used to indicate the stability of GFP fused with a C-end degron and was analyzed by flow cytometry and presented in histogram plots. KD: knock down (KLHDC2-).
(C) Stability of GFP-fused SelK or USP1-NTD C-end degron monitored by global protein stability assay with endogenous KLHDC2 knocked down by shRNA and complemented by exogenously expressed KLHDC2 wild type and mutant proteins. Destabilization of the target protein by the wild type (black line) or mutant (red line) KLHDC2 is indicated by a sharp peak at the left side of each panel.
See also Figures S3.
Among the three KLHDC2 residues directly interacting with the C-terminal carboxyl group, alanine mutation of Arg241, but not the other two amino acids, Arg236 and Ser269, abrogated the binding of the SelK degron and failed to induce the degradation of the substrate (Figure 3A and 3C). These results accentuate the importance of the C-terminal carboxyl group in the SelK degron and indicate a central role of Arg241. This strictly conserved and positively charged residue makes a bidentate interaction with the peptide-terminating group via a salt bridge and a hydrogen bond. Intriguingly, when Arg241 is mutated to lysine, KLHDC2 retained its ability to bind and destabilize SelK, highlighting the “hot-spot” nature of the inter-molecular salt bridge. In support of this notion, removal or reversal of the positive charge at this position (R241L and R241E), or neutralization of its positive charge by mutating the nearby R236 residue to a glutamate (R236E), prevented KLHDC2 from binding and degrading the substrate. Noticeably, altering Ser269, which lies underneath the carboxyl group, to a bulkier hydrophobic or negatively charged amino acid also abolished the E3-degron engagement. This result implies that the C-terminal carboxyl group has to be precisely positioned for productive interaction.
Due to their potential roles in stabilizing the kelch repeat fold, we chose not to mutate the three aromatic residues sandwiching the diglycine motif. Although the two nearby alanine residues, A219 and A220, at the bottom of the KLHDC2 pocket are solvent-exposed and become buried by the tandem glycine residues in the complex, mutating either one to leucine altered the solution behavior of KLHDC2 as detected by size exclusion chromatography (data now shown). The integrity of this portion of the pocket, therefore, is important to the proper folding of the β-propeller. Moving to the middle of the pocket, we next mutated the ridge residue, Lys147, which donates hydrogen bonds to the −3 and −5 backbone carbonyl groups. The KLHDC2 K147A mutant has a severely impaired degron-binding activity and loses its function in downregulating SelK. This data echoes the importance of the −5 amino acid of the SelK degron in maintaining high affinity binding, as observed in our AlphaScreen competition assay. In chamber N of the KLHDC2 pocket, Arg189 makes the only direct hydrogen bond interaction with the degron backbone at the −6 position. Mutating this positively charged residue to alanine had little to no effect on degron binding and SelK stability, in accordance with the subtle change in IC50 when the degron peptide length is reduced from 6 to 5 amino acids. As anticipated, degron binding determined by our in vitro pull-down experiments is strongly correlated with protein stability measured by the cell-based GPS assay.
Diglycine C-end degrons from other substrates
In addition to SelK, the extreme C-terminal diglycine degron has been found in a number of proteins, either in their full length, early terminated, or proteolytically processed form (Koren et al., 2018; Lin et al., 2018). These degrons do not share any consensus sequence except the C-terminal diglycine motif. To understand how degenerate N-terminal sequences impact KLHDC2 binding and how they are accommodated by the E3, we chose to focus on the diglycine degrons from two additional proteins, SelS, and USP1-NTD, which have been previously suggested to be KLHDC2 substrates. In our AlphaScreen-based competition assay, the 8 aa SelS degron peptide displayed an IC50 value slightly above SelK, whereas the potency of the USP1-NTD degron peptide is about 7 folds lower (Fig. 4A). The N-terminal sequences of USP1 and SelS degron peptides are distinct from SelK, with the exception of a single proline residue at the −5 position in SelS. Some variation in this portion of the degron, therefore, can have detectable, albeit minor, effects on the degron-KLHDC2 interaction.
Figure 4. Recognition of SelS and USP1 C-end degrons by KLHDC2.

(A) AlphaScreen competition assay for quantifying the affinity of the SelS (green) and USP1 (blue) C-end degron peptides in comparison to the SelK 8 aa degron peptide (orange, dashed line). The IC50 value and the 95% confidence interval for each peptide is listed in a table at the bottom. The dose response curve for the SelK 8 aa degron peptide represented in a dashed line is identical to the data shown in Figure 1B. Data are measured in triplicates and represented as mean ± SEM.
(B) Superposition analysis of the three complexes formed between KLHDC2 and the three C-end degron peptides included in this study. The surface representation of the KLHDC2 pocket is shown in grey. SelK, SelS, and USP1 degrons are colored in orange, green, and blue, respectively. Superposition of the three structures was performed using the entire KLHDC2-peptide complex. The conformation of each individual peptide bound to the E3 is illustrated at the bottom with the amino acid positions from the C-terminal end labeled.
(C) AlphaScreen competition assay for quantifying the affinity of the SelK and USP1 C-end degron peptides towards KLHDC2-R241K mutant in comparison to the wild type E3 protein. The dose response curves of SelK against wild type and mutant KLHDC2 are displayed in orange dashed line and tangerine solid line, respectively. The dose response curves of USP1 against wild type and mutant KLHDC2 are displayed in blue dashed line and cyan solid line, respectively. The orange and blue dashed lines are identical to the dose response curves shown in (A). Data are measured in triplicates and represented as mean ± SEM.
(D) Stability of GFP-fused SelK or USP1-NTD C-end degron monitored by global protein stability assay with endogenous KLHDC2 knocked down by shRNA and complemented by two exogenously expressed KLHDC2 mutant proteins (R236A and R241K).
See also Figures S4.
To compare the binding modes of the two degrons to SelK, we determined their crystal structures in complex with KLHDC2. The 8 amino acids SelS and USP1 degron peptides dock to the KLHDC2 top surface pocket in a nearly identical topology to SelK. Superposition of the three structures reveals a perfectly aligned C-terminal diglycine motif and a small degree of variation between the backbone atoms of the N-terminal residues (Figure 4B). From the distal end of chamber C to the opposite end in chamber N, the KLHDC2 pocket widens, thus harboring side chains of different sizes decorating the N-terminal end of the three degrons. For instance, at the −3 position of the degron, the side chain can extend from a single methyl group to an isobutyl group, while at the −4 position, the KLHDC2 pocket is able to house a degron residue ranging from a serine to a methionine. Among the three degron peptides, SelK has the highest affinity toward KLHDC2 and displays a clear electron density for its residue at the −7 position. These properties could be attributed to the rigidity provided by the three sequential prolines at its N-terminal end. By contrast, USP1 features a glycine at the −5 position, which could destabilize the conformation of the degron and contribute to its weaker binding.
We next subjected the C-end degron of USP1-NTD to the GPS-based stability assay in conjunction with the series of KLHDC2 mutants previously described. As expected, the majority of the KLHDC2 mutations located in the degron-binding pocket, such as R241A, K147A, and R189A, elicited the same effects on USP1-NTD degron stability, as they did to SelK (Figure 3C). To our surprise, though, two KLHDC2 mutants yielded opposite results for the two substrates. While the KLHDC2 R241K mutant maintained its wild type ability to destabilize SelK, the same mutant failed to promote the degradation of USP1-NTD. This differential effect was also observed for the KLHDC2 R236A mutant. Overall, the USP1-KLHDC2 interface, particularly at the position where the C-terminal carboxyl group of the degron is recognized, seems to be less tolerant to perturbation.
Affinity requirement for substrate degradation
The opposite effects of the KLHDC2 arginine mutations on the stability of SelK and USP1 degrons is in stark contrast to the structural similarity of their E3-bound forms. Why would the KLHDC2 R241K mutant degrade one substrate but spare the other? Multiple factors, such as E3 binding, the nature of E2, and the availability of ubiquitin-conjugating sites, could influence the efficiency of substrate ubiquitination. In our case, most of these factors are constant and can be ruled out. Moreover, USP1 can be destabilized by the wild type E3, and the KLHDC2 R241K mutant is able to degrade SelK. The defective degradation of USP1 by the mutated E3, therefore, is not due to loss of function in either the degron or the KLHDC2 mutant. We postulated that the answer to the question above lies in the altered affinity of the two substrates toward the kelch repeat mutant and their cellular concentrations, which dictate their complex formation.
To test our hypothesis, we first sought to quantify the binding of the substrate degrons to the KLHDC2 R241K mutant. In the crystal structures of wild type KLHDC2 bound to SelK and USP1 degron peptides, Arg241 interacts with the extreme C-terminus of both peptides via a salt bridge and a hydrogen bond. The R241K mutation is predicted to weaken the binding by losing the hydrogen bond while retaining the salt bridge. We replaced wild type KLHDC2 with the R241K mutant in the AlphaScreen assay and re-established the degron-E3 binding with the biotinylated 12 aa SelK degron peptide. Next, we individually applied the label-free 8 aa SelK and USP1 degron peptide at different concentrations and derived their IC50 values. The SelK and USP1 peptides showed IC50 of 5.9 μM and 62.6 μM for the KLHDC2 R241K mutant, which represents a 630- and 900-fold increase over the wild type E3, respectively (Fig. 4C). Consistent with a ~7-fold lower affinity of USP1 vs. SelK for the wild type ubiquitin ligase (9.4 nM vs. 69.5 nM), USP1 binds the mutant E3 ~10-fold weaker than SelK. The similar degree of affinity changes for the two degron peptides when binding to the mutant KLHDC2 can be explained by the free energy loss association with the missing hydrogen bond at −3.9 to −4.1kcal/mol.
The diminished affinities of the two degrons and their consistent difference, in and of themselves, cannot unmask the reason for the differential effects of the E3 mutation on the stability of SelK and USP1. However, once the cellular concentrations of the two proteins and the E3 mutant are considered, a possible explanation for the disparity in their degradation arises. Using purified KLHDC2 as standard, we performed quantitative western blot analysis and determined the cellular concentration of the KLHDC2 R241K mutant expressed in our GPS assay to be ~1μM. Through a similar analysis with purified GFP as standard, the concentration of GFP-fused USP1-NTD degron was calculated to be 11.2 μM, 6-fold below the affinity of its degron toward the E3 mutant (Kd ~62.6 μM). These results suggest that only a small fraction of the GFP-USP1-NTD degron fusion protein will be complexed with KLHDC2 at any time. The E3 mutant, therefore, will not be able to degrade USP1 efficiently. By contrast, the cellular concentration of GFP-fused SelK degron was measured to be ~8.8 μM which is above the concentration at which 50% of the substrate is bound to the E3 (Kd ~5.9 μM) and favors complex formation. Thus, GFP-fused SelK degron can be effectively destabilized by the KLHDC2 R241K mutant. In support of this reasoning, when we increased the dosage of the exogenous KLHDC2 mutants to reach a concentration of ~8 μM we observed noticeable enhancement in GFP-SelK degron degradation, but only marginal effect on USP1 (Figure 4D). Besides the inefficiency in complex formation, the low affinity between USP1-NTD and KLHDC2 R241K mutant might also prevent the ubiquitin transfer reaction itself from happening. The fast koff associated with weak binding will particularly impact polyubiquitin chain assembly by impeding the attachment of the first ubiquitin to the CRL substrates (Pierce et al., 2009).
Discussion
Here we elucidate for the first time how a C-end degron is recognized by its cognate E3 ligase. Among the different classes of C-end degradation signals, the diglycine degron is distinguished from others by the simplicity of its consensus sequence that lacks any side chains. The crystal structures of KLHDC2 in complex with three diglycine C-end degrons reveal a surprisingly condensed degron-E3 interface, which is cemented by a network of inter-molecular polar interactions. Upon docking to the E3, the degron adopts a compact conformation, which allows its carboxyl terminus and backbone carbonyl groups to make interactions with a cluster of conserved E3 side chains. This binding mode licenses the overall promiscuous diglycine degron with a minimum of five amino acids to achieve high affinity KLHDC2 binding. Previous studies have shown that the SelK degron, when fused to an otherwise stable protein, has to be seven amino acids long in order to induce proteasomal degradation (Lin et al., 2015). In light of our structural results, this requirement can be explained by the potential steric clash between the globular domain of the substrate and the opening edge of the degron-binding pocket of the kelch repeat protein. Consistent with this notion, the 6 aa C-terminal tail of ubiquitin, which is characterized by its terminating diglycine motif, has to be extended by a linker to become a C-end degron (Lin et al., 2018).
Despite their functional similarity and positional symmetry within a polypeptide, the N-end and C-end degrons have different features, implicating them in distinct regulatory processes. The most striking difference lies in the affinity of the two classes of degrons toward their cognate E3s. The dissociation constants of the complexes formed between the classical Arg/N-end degrons and the UBR box of N-recognins have been documented to be ~4 μM or higher (Choi et al., 2010; Matta-Camacho et al., 2010). Similarly, the recently identified Pro/N-end degron binds to its E3 ligase, GID4, with 2.5 μM affinity (Chen et al., 2017; Dong et al., 2018). By contrast, the three diglycine C-end degron peptides included in this study display affinity toward KLHDC2 in the low nanomolar range. Remarkably, the binding constant of the SelK degron for the E3 was determined to be below 10 nM. Such a strong interaction is reminiscent of the binding of several known CRL substrates, such as Nrf2 and CyclinE, to their E3s (Hao et al., 2007; Suzuki and Yamamoto, 2015). These cellular regulatory proteins are characterized by the tight regulation of their protein abundance in key signaling pathways. The high affinity between the diglycine C-end degron and KLHDC2 suggests that this specific branch of the DesCEND pathway could have evolved to degrade substrates of low abundance and/or eliminate substrates quickly in response to certain cellular events. It is conceivable that a high affinity might also be required to keep substrates at a very low level due to their potential toxicity.
The diglycine degron belongs to a small cohort of C-end degrons characterized by a preferred C-terminal glycine residue (Koren et al., 2018; Lin et al., 2018). The majority of these glycine-terminating C-end degrons are recognized by kelch domain-containing proteins, including KLHDC2, KLHDC3 and KLHDC10. With a common kelch repeat β-propeller fold, KLHDC3 and KLHDC10 are predicted to interact with their specific degrons via a similar top surface pocket as the one revealed in our structure. The particular residues involved in degron binding most likely vary among these kelch domain proteins. Nevertheless, the carboxyl terminus and backbone carbonyls of these glycine-ending degrons are expected to directly interact with interface residues conserved within the KLHDC3 and KLHDC10 orthologs. The compact substrate-binding pocket of KLHDC2 renders the E3 ligase a potential druggable target. In our competition assay, a peptide as small as a diglycine motif with a molecular weight of 132 Da was able to bind to the KLHDC2 pocket with an estimated affinity of ~360 μM. It is conceivable that the KLHDC2 pocket can be exploited by degron-mimicking small molecule compounds to reprogram the E3 ligase for degrading disease-related targets (Raina and Crews, 2017; Zheng and Shabek, 2017).
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ning Zheng (nzheng@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For DNA extraction, E.coli DH5α was grown for 16hr at 37°C. For bacmid production, E.coli DH10Bac was grown for 16hr at 37°C. For baculovirus production and amplification, Sf9 insect cells were grown for 2-3 days at 26°C. For protein expression, both E.coli BL21(DE3) (grown for 4hr at 37°C) and HighFive insect cells (grown for 2-3 at 26°C, 105RPM) were used. LB Broth Miller (Fisher BioReagents) was used for E.coli. Sf9 insect cells were maintained in Grace’s Insect Medium (Gibco) supplemented with 7% FBS (Gibco) and 1% Penicillin-Streptomycin (HyClone) solution. Suspension Hi5 cells were grown in EXPRESS™ FIVE SFM(Gibco) supplemented with 5% L-Glutamine 200mM (HyClone) and 1% Penicillin-Streptomycin (HyClone) solution. HEK293T cells were maintained in DMEM with 10% FBS and antibiotics at 37°C in a 6% CO2 atmosphere. Tissue culture media and supplements were from GIBCO Life Technologies (Carlsbad, CA, USA).
METHOD DETAILS
Molecular biology and protein purification
The kelch repeat domain of human KLHDC2 (amino acid 1–362) was subcloned into the pFastBac vector with an N-terminally fused glutathione-S-transferase (GST) and a TEV-cleavage site. A recombinant baculovirus was produced and amplified three times in Sf9 monolayer cells to produce P4. The P4 virus was used to infect Hi5 suspension insect cell cultures to produce the recombinant GST-KLHDC2 protein. The cells were harvested 2-3 days post-infection, re-suspended and lysed in lysis buffer (20 mM Tris, pH 8.0, 200 mM NaCl, 5 mM DTT) in the presence of protease inhibitors (1μg/ml Leupeptin, 1μg/ml Pepstatin and 100μM PMSF) using a microfluidizer. The GST-KLHDC2 protein was isolated from the soluble cell lysate by Pierce™ Glutathione Agarose (Thermo Scientific). For AlphaScreen competition assays, the GST tagged KLHDC2 was further purified by Q Sepharose High Performance resin (GE Healthcare). The NaCl eluates were subjected to Superdex-200 size exclusion chromatography column (GE Healthcare). For crystallization and GST-pull down assays, the same purification steps were employed, with a GST tag removal step following affinity purification. The samples used for crystallization were concentrated by ultrafiltration to 19–29 mg mL−1. All single amino acids KLHDC2 mutants were purified by glutathione agarose resin, cleaved with TEV, and further purified by ion exchange chromatography following the same procedure as described for the wild type protein. GST-fused SelK degron proteins were overexpressed and purified from BL21 (DE3) E. coli cells. Bacterial cells transformed with the pGEX-based expression plasmid for GST, GST-8 aa and −12 aa SelK degron were grown in LB broth at 37 °C to an OD600 of 0.8 and induced with 0.1 mM IPTG for 4 hrs. Cells were harvested, re-suspended and lysed in lysis buffer. The proteins were isolated from soluble cell lysate by glutathione agarose resin. All protein samples were flash frozen in liquid nitrogen for future use.
Protein Crystallization
The crystals of KLHDC2 in complex with the 8 aa peptides from SelK and USP1-NTD C-end degrons (Bio-Synthesis, Inc) were grown at 25 °C by the hanging-drop vapor diffusion method with 0.150 μL protein complex sample mixed with 0.075 μL volume of reservoir solution containing 0.03 M MgCl2*6H2O, 0.03 M CaCl2*2H2O, 0.05 M imidazole, 0.05 M 2-(N-morpholino)ethanesulfonic acid (MES) monohydrate, 12.5% (v/v) 2-methyl-2,4-pentanediol (MPD), 12.5% (w/v) PEG1000, 12.5% (w/v) PEG3350, pH 6.5. KLHDC2 in complex with the 8 aa SelS C-end degron peptide (Bio-Synthesis, Inc) was crystallized in 0.03 M MgCl2*6H2O, 0.03 M CaCl2*2H2O, 0.05 M Imidazole, 0.05 M MES monohydrate, 13.25% (v/v) MPD, 13.25% (w/v) PEG1000, 13.25% (w/v) PEG3350, pH 6.8. Crystals of maximal sizes were obtained and harvested after a few days. Cryoprotection was provided by the crystallization condition.
Data collection and structure determination
After collecting a native dataset at Advanced Light Source Beamline 8.2.1, X-ray diffraction data were integrated and scaled with HKL2000 package (Otwinowski and Minor, 1997). The structure was solved by molecular replacement with ensembler and phaser from the PHENIX suite of programs. Ensembler was used to superimpose multiple kelch domain containing structures (PDB: 2VPJ, 5A10, 1ZGK, 4CHB, 5CGJ, and 3II7). The output ensembler search model was used for molecular replacement in phaser. Initial structural models were built, refined and rebuilt using COOT (Emsley et al., 2010) and PHENIX (Adams et al., 2002). All structural models were manually built, refined, and rebuilt with PHENIX and COOT. PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC), CHIMERA (Pettersen et al., 2004), and LIGPLOT (Laskowski and Swindells, 2011) were used to generate figures. Protein sequence alignment was performed with CLC Sequence Viewer 7.
AlphaScreen luminescence proximity assay
AlphaScreen assays for determining and measuring protein-protein interactions were performed using EnSpire reader (PerkinElmer). GST-tagged KLHDC2 (WT or R241K) was attached to anti-GST AlphaScreen acceptor beads. Synthetic biotinylated 12 aa SelK degron peptide (Bio-Synthesis, Inc.) was immobilized to streptavidin-coated AlphaScreen donor beads. The donor and acceptor beads were brought into proximity by the interactions between the SelK peptide and KLHDC2. Excitation of the donor beads by a laser beam of 680 nm promotes the formation of singlet oxygen. When an acceptor bead is close proximity, the singlet oxygen reacts with thioxene derivatives in the acceptor beads and causes the emission of 520-620 nm photons, which are detected as the binding signal. If the beads are not in close proximity to each other, the oxygen will return to its ground state and the acceptor beads will not emit light. Competition assays were performed in the presence of numerous peptides which were titrated at various concentrations. Different lengths of the SelK peptides were used (12, 10, 8, 6, 5, 4, 3, and 2 amino acids) in order to determine the minimal length of a high affinity binding degron. In order to dissect the role of the extreme C-terminal diglycine motif, two 8 aa SelK peptides were used for competition assays, one with the C-terminal glycine mutated to leucine and the other with an amidated C-terminus. Lastly, to assess the binding of degrons of different substrate proteins to KLHDC2, we used 8 aa peptides from the extreme C-terminus of early terminated SelS and USP1-NTD.
The experiments were conducted with 0.12 nM of GST-KLHDC2 and 1.7 nM biotinylated 12 aa SelK peptide or with 11 nM GST-KLHDC2-R251K and 11 nM biotinylated 12 aa SelK peptide in the presence of 5 μg/ml donor and acceptor beads in a buffer of 25 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM TCEP, 0.1% Tween-20, and 0.05 mg/ml Bovine Serum Albumin. The concentrations of the peptides used in competition assays ranged from 2 nM to 3 mM. The precise concentrations of the stock peptide were determined by amino acid analysis (TAMU Protein Chemistry Lab, Texas A&M University). The experiments were done in triplicates. IC50 values were determined using non-linear curve fitting of the dose response curves generated with Prism 4 (GraphPad).
Octet BioLayer Interferometry measurement
Binding affinity of biotinylated 12 aa SelK peptide with GST-KLHDC2 was measured using the Octet Red 96 (ForteBio, Pall Life Sciences) following the manufacturer’s procedures in quadruplicates. The optical probes were coated with streptavidin, loaded with 200 nM biotinylated 12 aa SelK peptide as ligand and quenched with 0.1 mM biocytin prior to kinetic binding analysis. The reaction was carried out in black 96 well plates maintained at 30°C. The reaction volume was 200 μL in each well. The binding buffer contained 20 mM Tris-HCl, 200 mM NaCl, 5 mM DTT and 0.1% BSA, pH 8.0. The concentrations of GST-KLHDC2 as the analyte in the binding buffer were 100, 50, 25, 13, and 6.3 nM. There was no binding of the analyte to the unloaded probes. Binding kinetics of the analyte at all five concentrations were measured simultaneously using instrumental defaults. The data was analyzed by the Octet data analysis software. The association and dissociation curves were globally fit with a 1:1 ligand model. The kon and kdis values were used to calculate the dissociation constant, Kd, with kinetic analysis of the direct binding.
Global protein stability assay
The GPS assays using SelK and USP1-NTD reporter cell lines were performed (Emanuele et al., 2011; Lin et al., 2018). The GPS reporter system was based on the co-expression of GFP and RFP from a single transcript enabled by an internal ribosome entry site (IRES). GFP was fused with the C-end degron of SelK or USP1-NTD while RFP served as a non-degradable internal control. The GFP/RFP ratio thus indicated the stability GFP-fused C-end degron and was analyzed by flow cytometry. To testing the activity of KLHDC2 mutants, endogenous KLHDC2 was knocked down by shRNA and exogenous KLHDC2 with indicated single point mutations was introduced in GPS reporter cells. Targeting sequence of KLHDC2 shRNA is CTTGGTGTCTGGGTATATA.
To calculate the volume of a cell, ~10^7 cells were collected and resuspended with PBS buffer with known volume and the final volume was measured. By subtracting the initial volume of PBS from the final volume and dividing the value by number of cells, the volume of a single cell was estimated to be 7950 fL. To obtain the absolute abundance of the GFP-tagged SELK and USP1 proteins, purified Flag-GFP protein was quantified and used as a standard for quantitative western blot analysis.
Protein native mass spectrometry
The KLHDC2-SelK complex was prepared for native mass spectrometry by exchanging the solution prepared for crystallography into aqueous 200 mM ammonium acetate at pH 7 using four cycles of dilution and re-concentration with a 10K MWCO Corning Spin-X UF centrifugal concentrator (14,000 g, 1 hr, 4 °C). The final concentration of the KLHDC2-SelK complex used for experiments was approximately 20 μM. That solution was then ionized using electrokinetic nanoelectrospray ionization. The ionization source used in these experiments consisted of a platinum wire electrode and a borosilicate glass capillary emitter. A micropipette puller (Sutter Instruments Model P-97) was used to fabricate the emitter from a borosilicate capillary.
Approximately 3 uL of sample was added into the tip of the capillary. The platinum wire electrode was placed into the capillary and in direct contact with the sample. Between 0.5 and 1.0 kV was applied to the wire to achieve ionization (Xing et al., 2013). Those ions were then analyzed using a hybrid electrospray/quadrupole/ion-mobility/time-of-flight mass spectrometer (Waters Synapt G2 HDMS) (Allen et al., 2016), in which the original traveling-wave ion mobility cell was replaced by a radio-frequency confining drift cell. Briefly, the native mass spectrum was acquired using a 45 V bias between the sampling and extraction cones in the atmospheric-pressure interface (operated at room temperature) as well as using a 3 V bias between the quadrupole mass filter (operated as an RF-only ion guide) and the trap collision cell (containing ~20 mTorr of argon gas). Activation in the trap cell was performed with the identical conditions used to acquire the native mass spectrum, but the bias between the quadrupole and trap collision cell was increased to 45 V. In-source activation was performed by increasing the bias between the sampling and extraction cones to 120 V. Tandem mass spectrometry was performed using in-source activation and by increasing the bias between the quadrupole mass filter (now used to isolate the released [SelK+H]+ ions) and the trap collision cell to 30 V. Mass spectra were calibrated externally using electrospray generated ions from a 30 mg/ml solution of CsI.
Affinity pull-down assay
Pull-down assay was preformed using ~200 μg of purified GST or GST-peptides as the bait and ~ 250-300 μg of KLHDC2 WT or mutant proteins. Reaction mixtures (140 μl) were incubated with 50 μl GST beads (Thermo Scientific) at 4 °C for 1 hr in the binding buffer with 20 mM Tris-HCl, pH 8, 200 mM NaCl, and 5 mM DTT. After extensive wash with 0.5 ml binding buffer three times and 140 μl binding buffer once, the protein complexes on the beads were eluted by 3 column volumes of 5 mM glutathione. SDS-PAGE loading buffer was added to the samples and proteins were analyzed by Coomassie staining. Inputs samples represent 5-10% of the total reaction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein quantification was done using Bio-rad Protein Assay with comparison against a standard curve constructed using BSA, and by using an A280 extinction coefficient of 97,860 M−1cm−1 for KLHDC2 and 142,210 M−1cm−1 for GST-KLHDC2 on a Nanodrop spectrophotometer (Thermo-Fisher).
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the data reported in this paper are PDB 6DO3, 6DO4, and 6DO5.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFP | Clontech | JL-8 |
| Mouse anti-Flag M2 antibody | Sigma Aldrich | F3165 |
| Bacterial and Virus Strains | ||
| E. coli BL21 (DE3) | NEB | C2527I |
| E. coli DH5α | NEB | C2987I |
| E. coli DH10Bac | Life Technologies | 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pierce™ Glutathione Agarose | Thermo Scientific | 16101 |
| Q Sepharose™ High Performance | GE Healthcare | 17-1014-01 |
| Superdex 200 Increase 10/300 GL | GE Healthcare | 28-9909-44 |
| TEV protease | In house | N/A |
| SelK, SelS, USP1 peptides | Bio-Synthesis, Inc | N/A |
| SelK 3aa peptide | GenScript | N/A |
| AlphaScreen™ Streptavidin-coated donor beads | PerkinElmer | 6760002S |
| AlphaLISA™ anti-GST AlphaScreen acceptor beads | PerkinElmer | AL110C |
| Cesium Iodide | MP Biomedicals | 21004 |
| Ammonium Acetate | Sigma-Aldrich | A7262 |
| ANTI-FLAG M2 Affinity Gel | Sigma-Aldrich | A2220 |
| Streptavidin Biosensors | Pall Life Sciences/ForteBio | 18-5019 |
| Critical Commercial Assays | ||
| Amino acid analysis | TAMU Protein Chemistry Lab | N/A |
| Deposited Data | ||
| KLHDC2-SelK | This paper | 6DO3 |
| KLHDC2-SelS | This paper | 6DO4 |
| KLHDC2-USP1 | This paper | 6DO5 |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda: Sf9 | Life Technologies | B825-01 |
| Trichoplusia ni: HighFive | Life Technologies | B85502 |
| Human: HEK293T | ATCC | CRL-3216 |
| Recombinant DNA | ||
| pFB-GST-KLHDC2(1-362) | This paper | N/A |
| pFB-GST-KLHDC2 K147A | This paper | N/A |
| pFB-GST-KLHDC2 R189A | This paper | N/A |
| pFB-GST-KLHDC2 A219L | This paper | N/A |
| pFB-GST-KLHDC2 A220L | This paper | N/A |
| pFB-GST-KLHDC2 R236A | This paper | N/A |
| pFB-GST-KLHDC2 R236E | This paper | N/A |
| pFB-GST-KLHDC2 R241A | This paper | N/A |
| pFB-GST-KLHDC2 R241E | This paper | N/A |
| pFB-GST-KLHDC2 R241K | This paper | N/A |
| pFB-GST-KLHDC2 R241L | This paper | N/A |
| pFB-GST-KLHDC2 S269A | This paper | N/A |
| pFB-GST-KLHDC2 S269E | This paper | N/A |
| pFB-GST-KLHDC2 S269L | This paper | N/A |
| pGEX SelK 8aa | This paper | N/A |
| pGEX SelK 12aa | This paper | N/A |
| pLenti-GPS | Emanuele et al., 2011 | N/A |
| Software and Algorithms | ||
| Phenix | Adams et al., 2002 | https://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com/ |
| PyMOL | Pymol | https://pymol.org/2/ |
| Prism 4 | GraphPad | N/A |
| FlowJo | FLOWJO | N/A |
| ImageJ | NIH | N/A |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Data Analysis V 9.0 | Pall Life Sciences/ForteBio | N/A |
| MassLynx v. 4.1 | Waters Corporation | https://www.waters.com |
| LigPlot+ | Laskowski and Swindells (2011) | https://www.ebi.ac.uk/thornton-srv/software/LigPlus/ |
| Other | ||
| EnSpire reader | PerkinElmer | N/A |
| Octet Red 96 | Pall Life Sciences/ForteBio | N/A |
| Modified Synapt G2 HDMS Time-of-Flight mass spectrometer | Waters Corporation |
https://www.waters.com
DOI: 10.1039/C5AN02107C |
| Model P-97 Micropipette Puller | Sutter Instrument | https://www.sutter.com |
| Borosilicate capillaries 1.0 mm OD, 0.78 mm ID, 150 mm L | Harvard Apparatus | 30-0035 |
| LSR Fortessa | BD Biosciences | N/A |
Acknowledgements.
We thank the beamline staff of the Advanced Light Source at the University of California at Berkeley and the Advanced Photon Source at Argonne National Laboratory for help with data collection. We also thank members of the Zheng laboratory and Wenqing Xu laboratory for helpful discussion. This work is supported by the Howard Hughes Medical Institute (N. Z.), Career Development Award 101-CDA-L05 from Academia Sinica and MOST grant 106-2321-B-001-045 (H.-C. S. Y0, NIH grant T32GM008268 (D.C.), and NSF grant CHE-1807382 (M.F.B.).
Footnotes
Declaration of Interests.
N.Z. is a scientific advisory board member of Kymera Therapeutic.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Giles K, Gilbert T, and Bush MF (2016). Ion mobility mass spectrometry of peptide, protein, and protein complex ions using a radio-frequency confining drift cell. Analyst 141, 884–891. [DOI] [PubMed] [Google Scholar]
- Ambrogelly A, Palioura S, and Söll D (2007). Natural expansion of the genetic code. Nat Chem Biol 3, 29–35. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Wu X, Wadas B, Oh JH, and Varshavsky A (2017). An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Jeong BC, Joo YJ, Lee MR, Kim J, Eck MJ, and Song HK (2010). Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat Struct Mol Biol 17, 1175–1181. [DOI] [PubMed] [Google Scholar]
- da Fonseca PC, Kong EH, Zhang Z, Schreiber A, Williams MA, Morris EP, and Barford D (2011). Structures of APC/C(Cdh1) with substrates identify Cdh1 and Apc10 as the D-box co-receptor. Nature 470, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, Weninger WJ, Westerberg H, Adissu H, et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Zhang H, Li L, Tempel W, Loppnau P, and Min J (2018). Molecular basis of GID4-mediated recognition of degrons for the Pro/N-end rule pathway. Nat Chem Biol 14, 466–473. [DOI] [PubMed] [Google Scholar]
- Driscoll DM, and Copeland PR (2003). Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr 23, 17–40. [DOI] [PubMed] [Google Scholar]
- Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, and Schulman BA (2011). Structural regulation of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol 21, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, et al. (2011). Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. [DOI] [PubMed] [Google Scholar]
- Guharoy M, Bhowmick P, Sallam M, and Tompa P (2016). Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat Commun 7, 10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, and Pavletich NP (2007). Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26, 131–143. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, and Pavletich NP (2005). Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell 20, 9–19. [DOI] [PubMed] [Google Scholar]
- Hershko A, and Ciechanover A (1998). The ubiquitin system. Annu Rev Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Koren I, Timms RT, Kula T, Xu Q, Li MZ, and Elledge SJ (2018). The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons. Cell 173, 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, and Peters JM (2005). The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell 18, 543–553. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, and Swindells MB (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- Lin HC, Ho SC, Chen YY, Khoo KH, Hsu PH, and Yen HC (2015). SELENOPROTEINS. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding. Science 349, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Yeh CW, Chen YF, Lee TT, Hsieh PY, Rusnac DV, Lin SY, Elledge SJ, Zheng N, and Yen HS (2018). C-Terminal End-Directed Protein Elimination by CRL2 Ubiquitin Ligases. Mol Cell 70, 602–613. e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas X, and Ciulli A (2017). Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr Opin Struct Biol 44, 101–110. [DOI] [PubMed] [Google Scholar]
- Mahrour N, Redwine WB, Florens L, Swanson SK, Martin-Brown S, Bradford WD, Staehling-Hampton K, Washburn MP, Conaway RC, and Conaway JW (2008). Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J Biol Chem 283, 8005–8013. [DOI] [PubMed] [Google Scholar]
- Matta-Camacho E, Kozlov G, Li FF, and Gehring K (2010). Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat Struct Mol Biol 17, 1182–1187. [DOI] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, and Pavletich NP (2002). Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886–1889. [DOI] [PubMed] [Google Scholar]
- Mészáros B, Kumar M, Gibson TJ, Uyar B, and Dosztanyi Z (2017). Degrons in cancer. Sci Signal 10. [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, and Sicheri F (2003). Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112, 243–256. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W, eds. (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode (New York: Academic Press; ). [DOI] [PubMed] [Google Scholar]
- Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, and Yamamoto M (2006). Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21, 689–700. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pierce NW, Kleiger G, Shan SO, and Deshaies RJ (2009). Detection of sequential polyubiquitylation on a millisecond timescale. Nature 462, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K, and Crews CM (2017). Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol 39, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague ER, Redd MJ, Johnson AD, and Wolberger C (2000). Structure of the C-terminal domain of Tup1, a corepressor of transcription in yeast. EMBO J 19, 3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, and Yamamoto M (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88, 93–100. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Mulder LC, Iwamatsu A, Lee MJ, Davydov IV, Varshavsky A, Muesing M, and Kwon YT (2005). A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol 25, 7120–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Sriram SM, Park KS, and Kwon YT (2012). The N-end rule pathway. Annu Rev Biochem 81, 261–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Zakrzewska A, Dudgeon DD, Jiang Y, Lazo JS, and Kwon YT (2009). The substrate recognition domains of the N-end rule pathway. J Biol Chem 284, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uljon S, Xu X, Durzynska I, Stein S, Adelmant G, Marto JA, Pear WS, and Blacklow SC (2016). Structural Basis for Substrate Selectivity of the E3 Ligase COP1. Structure 24, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2011). The N-end rule pathway and regulation by proteolysis. Protein Sci 20, 1298–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, and Pavletich NP (2003). Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell 11, 1445–1456. [DOI] [PubMed] [Google Scholar]
- Xia Z, Webster A, Du F, Piatkov K, Ghislain M, and Varshavsky A (2008). Substratebinding sites of UBR1, the ubiquitin ligase of the N-end rule pathway. J Biol Chem 283, 24011–24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, and Zheng N (2013). SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature 496, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, and Shabek N (2017). Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 86, 129–157. [DOI] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. (2009). Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman ES, Schulman BA, and Zheng N (2010). Structural assembly of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol 20, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.