

Abstract
Background:
We have previously described 19 pedigrees with apparent lamin (LMNA)-related dilated cardiomyopathy (DCM) manifesting in affected family members across multiple generations. In 6 of 19 families, at least one individual with idiopathic DCM did not carry the family’s LMNA variant. We hypothesized that additional genetic cause may underlie DCM in these families.
Methods:
Affected family members underwent exome sequencing to identify additional genetic cause of DCM in the 6 families with non-segregating LMNA variants.
Results:
In 5 of 6 pedigrees, we identified at least one additional rare variant in a known DCM gene that could plausibly contribute to disease in the LMNA variant-negative individuals. Bilineal inheritance was clear or presumed to be present in 3 of 5 families and was possible in the remaining two. At least one individual with a LMNA variant also carried a variant in an additional identified DCM gene in each family. Using a multivariate linear mixed model for quantitative traits, we demonstrated that the presence of these additional variants was associated with a more severe phenotype after adjusting for sex, age, and the presence/absence of the family’s non-segregating LMNA variant.
Conclusions:
Our data support DCM as a genetically heterogeneous disease with, at times, multigene causation. Although the frequency of DCM resulting from multigenic cause is uncertain, our data suggest it may be higher than previously anticipated.
Keywords: pedigree lamin type A multifactorial inheritance inheritance patterns cardiomyopathy, dilated genetics exome sequencing
Introduction:
Recent advances in next-generation sequencing have facilitated significant progress in identification of putative causative variants in dilated cardiomyopathy (DCM) genes1. Nevertheless, a comprehensive understanding of the genetic architecture of idiopathic DCM (IDC) remains elusive. It has been previously estimated that 20–35% of DCM can be demonstrated to be familial when clinical screening of at-risk relatives of an IDC proband is conducted2–5. In many of these cases, DCM presents as an adult-onset, autosomal dominant, Mendelian condition with reduced, age-dependent penetrance and variable expressivity. Our hypothesis has been that DCM, regardless of familial occurrence, has a predominantly genetic basis6, 7.
Because most DCM manifests in adulthood, it is not unusual to identify pedigrees with multiple affected individuals who carry the family mutation but who have not yet shown any clinical (phenotypic) evidence of disease. These mutation-positive, phenotype-negative individuals are consistent with the well-known age-dependent penetrance of DCM. In contrast, individuals with IDC who do not carry the family’s pathogenic variant are more atypical. The traditional explanation has been that these individuals represent “phenocopies” – that is, individuals whose DCM stems from non-Mendelian causes such as long-standing hypertension, environmental exposure, or other unknown factors.
Mendelian models of genetic disease are premised on the idea that a single locus is responsible for disease in all affected members of an identified family. In these models, pedigree-based genetic testing aims to identify high-risk variant alleles that clearly segregate with disease and are exceedingly rare or absent from unaffected relatives and control populations. Not all observed segregation patterns conform to Mendelian expectations, however, and recent years have seen the emergence of multigene models for a diverse array of disorders8–11, including genetic cardiovascular disease12–15. Based on these models and unpublished data from our own family studies we have developed the working hypothesis that a proportion of familial DCM may similarly be explained by combined effect of multiple genetic loci. This focus remains novel for DCM genetics.
Sequence variants in the lamin A/C (LMNA) gene represent one of the most common genetic causes of DCM16. As conduction system disease (CSD) typically precedes onset of cardiomyopathy by a median of seven years17, detection of a clinically actionable LMNA variant can guide management towards detection and treatment of arrhythmia, as well as genetic screening of additional family members at risk for CSD and DCM. In 2008, in an effort to determine the frequency of rare LMNA variants among individuals with IDC, we conducted Sanger sequencing of a cohort of 324 unrelated patients with clinically confirmed IDC18, defined as left ventricular enlargement (LVE) with systolic dysfunction occurring in the absence of other clinically detectable causes19. We identified 19 pedigrees carrying 18 unique, protein-altering LMNA variants, representing an overall yield of 5.8% for the cohort. Unexpectedly, in 6 of the 19 families, one or more individuals with IDC did not carry the familial LMNA variant. At the time of publication, we defined these as “non-segregation” pedigrees and speculated that additional, presumably unlinked, genetic cause(s) may contribute to the DCM in these families.
We herein summarize follow-up genetic studies in these families. Our analyses have expanded some of the originally published pedigrees and include a more informative approach to DCM phenotyping. In 5 of 6 pedigrees (Figures 1, 2, 3, 4, 5), additional genetic information now provides evidence for at least one other rare variant contributing to DCM.
Figure 1: Pedigrees with non-segregating LMNA variants, Pedigree L.
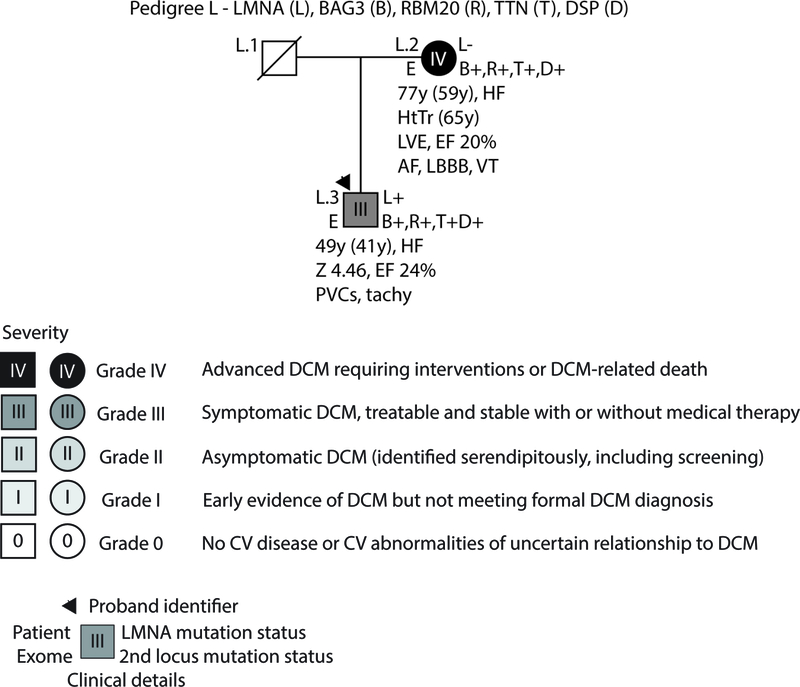
As per convention, the proband for each family is indicated by an arrow. Males are shown as squares and females as circles. Deceased individuals are denoted by a diagonal slash. Individuals for whom exome sequencing was completed are marked on the left side with an E. Genetic testing results are indicated on the right side by either a + (variant present) or a – (variant absent). Gene abbreviations are provided in the title of each pedigree. Obligate carriers are marked in parentheses (+). The upper right symbol reports genetic testing results for LMNA, while the bottom right symbols report testing results for additional identified loci (Table 2). Clinical data is structured as previously described19. First line: current age if the individual is still living or age of death if the individual is deceased, age of onset (in parentheses), major diagnosis or cause of death. Second line: age of transplantation, if applicable. Third line: left ventricular size defined by magnitude of standard deviation (Z-score, z) and function defined by left ventricular ejection fraction (EF, %). Fourth line: arrhythmias or other conduction system disease. Abbreviations: AFib, atrial fibrillation; Asym, asymptomatic; AVB atrioventricular block; 1AVB or 2AVB, first-degree or second-degree AVB, respectively; A/W, alive and well; BiFB, bifascicular block; Br, bradycardia; CAD, coronary artery disease; CHD, congenital heart disease; CM, cardiomyopathy, unknown type; CVD, cardiovascular disease; DCM, dilated cardiomyopathy; EF, ejection fraction; HF, heart failure; HTN, hypertension; HtTr, heart transplant; ICD, implantable cardioverter defibrillator; IDC, idiopathic dilated cardiomyopathy; IBBB, incomplete bundle branch block; IRBBB or ILBBB, right or left IBBB, respectively; IVCD, interventricular conduction delay; LBBB, left bundle branch block; MVP, mitral valve prolapse; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes on ECG; NSVT, non-sustained ventricular tachycardia; PACs, paroxysmal atrial contractions; PM, pacemaker; PSVT, paroxysmal supraventricular tachycardia; PVCs, premature ventricular contractions; RBBB, right bundle branch block; SCD, sudden cardiac death; tachy, sinus tachycardia; SSS, sick sinus syndrome; VSD, ventricular septal defect; VT, ventricular tachycardia; Z, Z-score of left ventricular end-diastolic dimension.
Figure 2: Pedigrees with non-segregating LMNA variants, Pedigree N.
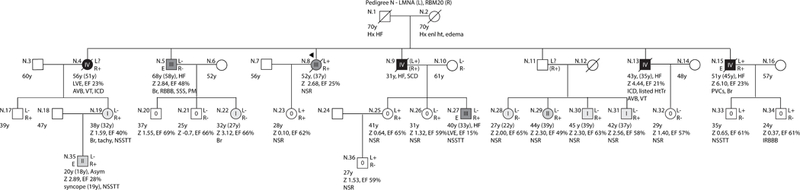
As per convention, the proband for each family is indicated by an arrow. Males are shown as squares and females as circles. Deceased individuals are denoted by a diagonal slash. Individuals for whom exome sequencing was completed are marked on the left side with an E. Genetic testing results are indicated on the right side by either a + (variant present) or a – (variant absent). Gene abbreviations are provided in the title of each pedigree. Obligate carriers are marked in parentheses (+). The upper right symbol reports genetic testing results for LMNA, while the bottom right symbols report testing results for additional identified loci (Table 2). Clinical data is structured as previously described19. First line: current age if the individual is still living or age of death if the individual is deceased, age of onset (in parentheses), major diagnosis or cause of death. Second line: age of transplantation, if applicable. Third line: left ventricular size defined by magnitude of standard deviation (Z-score, z) and function defined by left ventricular ejection fraction (EF, %). Fourth line: arrhythmias or other conduction system disease. Abbreviations: AFib, atrial fibrillation; Asym, asymptomatic; AVB atrioventricular block; 1AVB or 2AVB, first-degree or second-degree AVB, respectively; A/W, alive and well; BiFB, bifascicular block; Br, bradycardia; CAD, coronary artery disease; CHD, congenital heart disease; CM, cardiomyopathy, unknown type; CVD, cardiovascular disease; DCM, dilated cardiomyopathy; EF, ejection fraction; HF, heart failure; HTN, hypertension; HtTr, heart transplant; ICD, implantable cardioverter defibrillator; IDC, idiopathic dilated cardiomyopathy; IBBB, incomplete bundle branch block; IRBBB or ILBBB, right or left IBBB, respectively; IVCD, interventricular conduction delay; LBBB, left bundle branch block; MVP, mitral valve prolapse; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes on ECG; NSVT, non-sustained ventricular tachycardia; PACs, paroxysmal atrial contractions; PM, pacemaker; PSVT, paroxysmal supraventricular tachycardia; PVCs, premature ventricular contractions; RBBB, right bundle branch block; SCD, sudden cardiac death; tachy, sinus tachycardia; SSS, sick sinus syndrome; VSD, ventricular septal defect; VT, ventricular tachycardia; Z, Z-score of left ventricular end-diastolic dimension.
Figure 3: Pedigrees with non-segregating LMNA variants, Pedigree O.
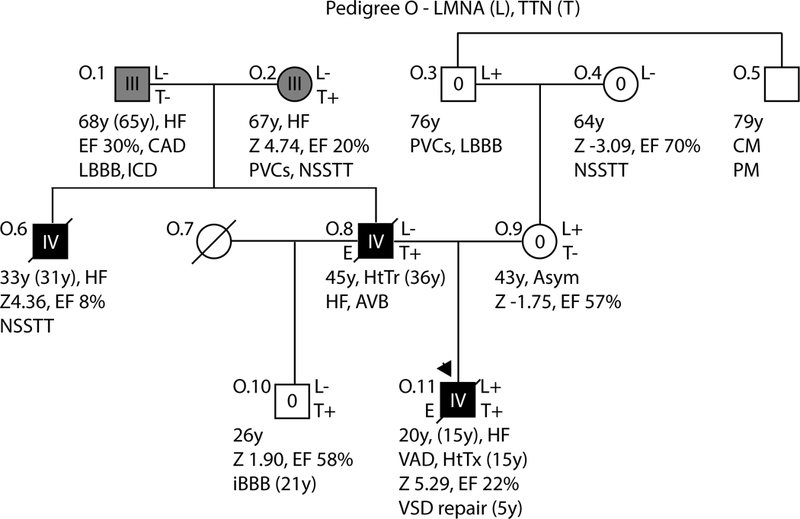
As per convention, the proband for each family is indicated by an arrow. Males are shown as squares and females as circles. Deceased individuals are denoted by a diagonal slash. Individuals for whom exome sequencing was completed are marked on the left side with an E. Genetic testing results are indicated on the right side by either a + (variant present) or a – (variant absent). Gene abbreviations are provided in the title of each pedigree. Obligate carriers are marked in parentheses (+). The upper right symbol reports genetic testing results for LMNA, while the bottom right symbols report testing results for additional identified loci (Table 2). Clinical data is structured as previously described19. First line: current age if the individual is still living or age of death if the individual is deceased, age of onset (in parentheses), major diagnosis or cause of death. Second line: age of transplantation, if applicable. Third line: left ventricular size defined by magnitude of standard deviation (Z-score, z) and function defined by left ventricular ejection fraction (EF, %). Fourth line: arrhythmias or other conduction system disease. Abbreviations: AFib, atrial fibrillation; Asym, asymptomatic; AVB atrioventricular block; 1AVB or 2AVB, first-degree or second-degree AVB, respectively; A/W, alive and well; BiFB, bifascicular block; Br, bradycardia; CAD, coronary artery disease; CHD, congenital heart disease; CM, cardiomyopathy, unknown type; CVD, cardiovascular disease; DCM, dilated cardiomyopathy; EF, ejection fraction; HF, heart failure; HTN, hypertension; HtTr, heart transplant; ICD, implantable cardioverter defibrillator; IDC, idiopathic dilated cardiomyopathy; IBBB, incomplete bundle branch block; IRBBB or ILBBB, right or left IBBB, respectively; IVCD, interventricular conduction delay; LBBB, left bundle branch block; MVP, mitral valve prolapse; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes on ECG; NSVT, non-sustained ventricular tachycardia; PACs, paroxysmal atrial contractions; PM, pacemaker; PSVT, paroxysmal supraventricular tachycardia; PVCs, premature ventricular contractions; RBBB, right bundle branch block; SCD, sudden cardiac death; tachy, sinus tachycardia; SSS, sick sinus syndrome; VSD, ventricular septal defect; VT, ventricular tachycardia; Z, Z-score of left ventricular end-diastolic dimension.
Figure 4: Pedigrees with non-segregating LMNA variants, Pedigree P.
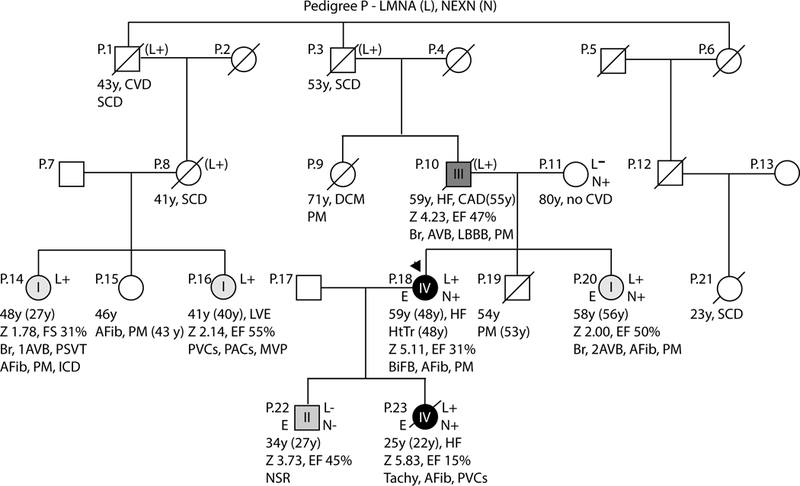
As per convention, the proband for each family is indicated by an arrow. Males are shown as squares and females as circles. Deceased individuals are denoted by a diagonal slash. Individuals for whom exome sequencing was completed are marked on the left side with an E. Genetic testing results are indicated on the right side by either a + (variant present) or a – (variant absent). Gene abbreviations are provided in the title of each pedigree. Obligate carriers are marked in parentheses (+). The upper right symbol reports genetic testing results for LMNA, while the bottom right symbols report testing results for additional identified loci (Table 2). Clinical data is structured as previously described19. First line: current age if the individual is still living or age of death if the individual is deceased, age of onset (in parentheses), major diagnosis or cause of death. Second line: age of transplantation, if applicable. Third line: left ventricular size defined by magnitude of standard deviation (Z-score, z) and function defined by left ventricular ejection fraction (EF, %). Fourth line: arrhythmias or other conduction system disease. Abbreviations: AFib, atrial fibrillation; Asym, asymptomatic; AVB atrioventricular block; 1AVB or 2AVB, first-degree or second-degree AVB, respectively; A/W, alive and well; BiFB, bifascicular block; Br, bradycardia; CAD, coronary artery disease; CHD, congenital heart disease; CM, cardiomyopathy, unknown type; CVD, cardiovascular disease; DCM, dilated cardiomyopathy; EF, ejection fraction; HF, heart failure; HTN, hypertension; HtTr, heart transplant; ICD, implantable cardioverter defibrillator; IDC, idiopathic dilated cardiomyopathy; IBBB, incomplete bundle branch block; IRBBB or ILBBB, right or left IBBB, respectively; IVCD, interventricular conduction delay; LBBB, left bundle branch block; MVP, mitral valve prolapse; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes on ECG; NSVT, non-sustained ventricular tachycardia; PACs, paroxysmal atrial contractions; PM, pacemaker; PSVT, paroxysmal supraventricular tachycardia; PVCs, premature ventricular contractions; RBBB, right bundle branch block; SCD, sudden cardiac death; tachy, sinus tachycardia; SSS, sick sinus syndrome; VSD, ventricular septal defect; VT, ventricular tachycardia; Z, Z-score of left ventricular end-diastolic dimension.
Figure 5: Pedigrees with non-segregating LMNA variants, Pedigree S.
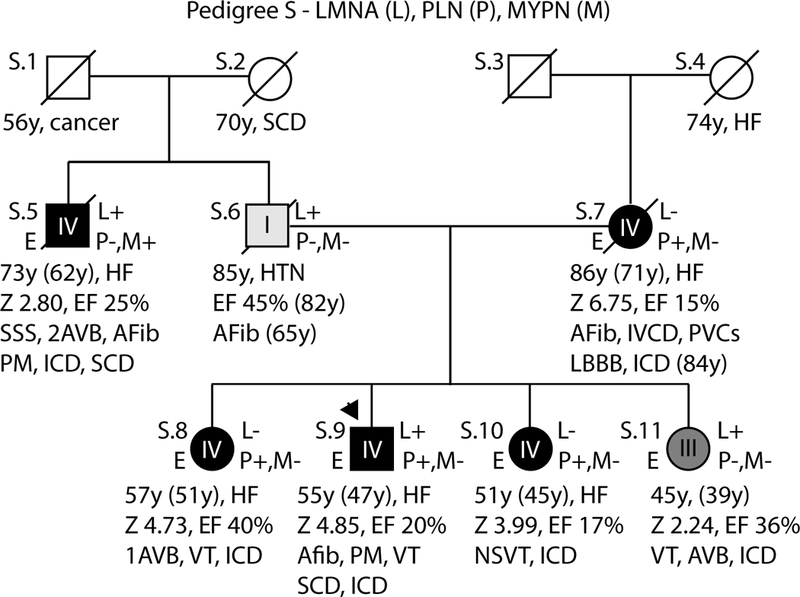
As per convention, the proband for each family is indicated by an arrow. Males are shown as squares and females as circles. Deceased individuals are denoted by a diagonal slash. Individuals for whom exome sequencing was completed are marked on the left side with an E. Genetic testing results are indicated on the right side by either a + (variant present) or a – (variant absent). Gene abbreviations are provided in the title of each pedigree. Obligate carriers are marked in parentheses (+). The upper right symbol reports genetic testing results for LMNA, while the bottom right symbols report testing results for additional identified loci (Table 2). Clinical data is structured as previously described19. First line: current age if the individual is still living or age of death if the individual is deceased, age of onset (in parentheses), major diagnosis or cause of death. Second line: age of transplantation, if applicable. Third line: left ventricular size defined by magnitude of standard deviation (Z-score, z) and function defined by left ventricular ejection fraction (EF, %). Fourth line: arrhythmias or other conduction system disease. Abbreviations: AFib, atrial fibrillation; Asym, asymptomatic; AVB atrioventricular block; 1AVB or 2AVB, first-degree or second-degree AVB, respectively; A/W, alive and well; BiFB, bifascicular block; Br, bradycardia; CAD, coronary artery disease; CHD, congenital heart disease; CM, cardiomyopathy, unknown type; CVD, cardiovascular disease; DCM, dilated cardiomyopathy; EF, ejection fraction; HF, heart failure; HTN, hypertension; HtTr, heart transplant; ICD, implantable cardioverter defibrillator; IDC, idiopathic dilated cardiomyopathy; IBBB, incomplete bundle branch block; IRBBB or ILBBB, right or left IBBB, respectively; IVCD, interventricular conduction delay; LBBB, left bundle branch block; MVP, mitral valve prolapse; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes on ECG; NSVT, non-sustained ventricular tachycardia; PACs, paroxysmal atrial contractions; PM, pacemaker; PSVT, paroxysmal supraventricular tachycardia; PVCs, premature ventricular contractions; RBBB, right bundle branch block; SCD, sudden cardiac death; tachy, sinus tachycardia; SSS, sick sinus syndrome; VSD, ventricular septal defect; VT, ventricular tachycardia; Z, Z-score of left ventricular end-diastolic dimension.
Methods:
All study methods are available as Supplemental Materials. For purposes of reproduction and replicability, the data and programs used for statistical analyses are publicly available at https://github.com/kinnamon-lab/lmna_nonseg. Informed consent was obtained from all study participants following approval by the Oregon Health & Science University, the University of Miami, or the Ohio State University.
Results:
The overall clinical profile of our DCM cohort has already been extensively described19. Prior sequencing of a subset of 312 unrelated probands identified LMNA variants in 19 pedigrees with evidence of familial disease18. In 6 families, the identified LMNA variant did not completely segregate with observed disease phenotypes suggesting existence of additional causative genetic factors. The majority of these pedigrees have been expanded with additional clinical, family, or genetic data since their initial publication1, 18, 20–22 and are presented in updated form in Figures 1–5. For consistency, these pedigrees retain their previously published letter designations18–20. In order to systematically analyze potential genotype/phenotype correlations, affection status for each individual with DCM was categorized using an ordinal grading scale ranging from 0 (no phenotypic evidence of DCM) to 4 (DCM with advanced disease) (Table 1). These grades were then applied to the pedigrees.
Table 1.
Categories for Clinical Grading of DCM Presence and Severity
| Grade | Definition | Comment |
|---|---|---|
| Grade 0 | No cardiovascular disease, or no cardiovascular abnormalities related to DCM | |
| Grade 1 | Left ventricular enlargement (LVE) without systolic dysfunction, or systolic dysfunction [Left ventricular ejection fraction (LVEF) < 50%] without LVE. | Grade 1 is early evidence of DCM but not meeting formal criteria for a DCM diagnosis. |
| Grade 2 | Asymptomatic DCM. | DCM is defined as systolic dysfunction (LVEF < 50%) and LVE with other usual clinically detectable causes of DCM excluded. In most cases asymptomatic DCM is identified serendipitously or via cascade clinical screening of at-risk family members. |
| Grade 3 | Symptomatic DCM with or without medical therapy. | Most commonly DCM presents with heart failure (80%), less commonly with arrhythmia (15–20%). |
| Grade 4 | DCM requiring advanced interventions (heart transplant, ventricular assist device (VAD), implantable cardioverter defibrillator (ICD) for life threatening symptomatic disease), or DCM-related death. | Advanced interventions are used for late-phase, life-threatening DCM. |
Exome sequences were obtained for a total of 20 individuals across all six pedigrees (indicated by an “E”). Only one pedigree (Pedigree B, not shown) was uninformative as genetic cause remained unresolved in 2 affected individuals lacking the familial LMNA mutation. This pedigree is not discussed further. In the remaining 5 families, all of which were Non-Latino Caucasian, sequencing identified at least one other rare genetic variant affecting another plausible disease locus (Table 2). For each pedigree, these additional variants met many of the usual criteria for potential disease-association (rare, occurred in conserved regions of genes with strong biological associations with DCM), but did not meet ACMG criteria for pathogenicity or likely pathogenicity (Suppl. Table 1). Bilineal inheritance could be clearly determined or inferred from 2 pedigrees (O, S). Based on available evidence, bilineal inheritance was presumed in one other family (P), and possible in two others (L, N).
Table 2.
Summary of Genetic Findings
| Pedigree | Gene | RefSeq | UCSC Genomic Position (GRCh37/hg19) |
Nucleotide Change* |
Amino Acid Change | Max ExAC MAF† | Max gnomAD MAF† | dbSNP | Bilineal Inheritance |
|---|---|---|---|---|---|---|---|---|---|
|
L |
LMNA | NM_170707.3 | chr1:156105707 | c.952G>A | p.(Ala318Thr) | 0 | 0.00003984 (NFE) |
rs267607574 | Possible versus de novo |
| BAG3 | NM_004281.3 | chr10:121429393 | c.211C>T | p.(Arg71Trp) | 0.0001348 (EAS) | 0.0001590 (EAS) |
rs387906874 | ||
| RBM20 | NM_001134363.2 | chr10:112541380 | c.1013T>C | p.(Met338Thr) | 0 | 0.00003519 (NFE) |
rs876657970 | ||
| TTN | NM_001267550.2 | chr2:179437290 | c.73568del | p.(Pro24523Hisfs*4) | 0 | 0 | n/a | ||
| DSP | NM_004415.3 | chr6:007585972 | c.8477G>A | p.(Arg2826His) | 0.000100624 (AFR) |
0 | rs548754771 | ||
|
N |
LMNA | NM_170707.3 | chr1:156106010 | c.1163G>A | p.(Arg388His) | 0.00001526 (NFE) |
0.00006498 (SAS) |
rs267607576 | Possible |
| RBM20 | NM_001134363.2 | chr10:112572302 | c.2205G>A | p.(Arg716Gln) | 0.0005057 (SAS) |
0.0003506 (SAS) |
rs375798246 | ||
|
O |
LMNA | NM_170707.3 | chr1:156106042 | c.1195C>T | p.(Arg399Cys) | 0.00001519 (NFE) |
0.00005808 (EAS) |
rs58672172 | Yes |
| TTN | NM_001267550.2 | chr2:179438190 | c.72669del | p.(Asp24224Ilefs*8) | 0 | 0 | rs727504531 | ||
|
P |
LMNA | NM_170707.3 | chr1:156106142- 156106143 |
c.1304_130 7dup |
p.(Ser437Hisfs*2) | 0 | 0.00003251 (SAS) |
rs267607577 | Presumptive |
| NEXN | NM_144573.3 | chr1:78401709 | c.1453G>A | p.(Glu485Lys) | 0.00008684 (AMR) | 0.0001108 (NFE) |
rs368812830 | ||
|
S |
LMNA | NM_170707.3 | chr1:156108540 | c.1960C>T | p.(Arg654*) | 0.000026 (NFE) | 0.00003698 (NFE) |
rs267607544 | Yes |
| PLN | NM_002667.3 | chr6:118880157 | c.73C>T | p.(Arg25Cys) | 0.00001501 (NFE) |
0.00008935 (AMR) | rs761056344 | ||
| MYPN | NM_001256267.1 | chr10:69881503 | c.308A>T | p.(Asp103Val) | 0.000172801 (AMR) |
0.0001489 (AMR) | rs758026056 |
By our application of the ACMG approach, all variants were classified as variants of uncertain significance23
The maximum non-Finnish minor allele frequency (MAF) and associated population for each variant is reported. A MAF of 0 implies absence of the variant from ExAC/gnomAD and sufficient coverage of the genomic position. Reported gnomAD results include both exomes and genomes. Abbreviations: AFR, African; AMR, Latino; EAS, East Asian; NFE, non-Finnish European; SAS, South Asian
Pedigree L:
In addition to a previously reported p.(Ala318Thr) LMNA variant18, exome sequencing identified rare missense variants affecting BAG3, p.(Arg71Trp)1; RBM20, p.(Met338Thr); and DSP, p.(Arg2826His); as well as a truncating, A-band TTN variant (TTNtv), p.(Pro24523Hisfs*4) in the proband (L.3) and his mother (L.2), who did not carry the LMNA variant (Pedigree L, Figure 1). Although variants in BAG3 are well known to cause advanced disease, with less evidence for missense DSP variants, predictive analyses were divided for the BAG3 and DSP variants, with half of the tested algorithms supporting pathogenicity of each allele. The p.(Pro24523Hisfs*4) TTNtv, which was predicted to affect the A-band and severely truncate the full-length protein (24525 vs 35991 amino acids), was carried by both affected family members. While absent from online databases of genetic variation, the RBM20 variant occurred outside of the exon 9 hotspot and was predicted to be benign by almost all tested predictive pathogenicity algorithms (Suppl. Table 2). While both L.2 and L.3 exhibited severe (Grade III/IV) DCM and CSD, onset occurred two decades earlier in the proband than in his mother, suggesting potential for increased disease severity in the presence of the LMNA variant. As is true for all other variants identified in this study (Table 2, Suppl. Table 2), the 5 variants identified in this family are considered to be VUSs under current ACMG guidelines.
Pedigree N:
A rare p.(Arg716Gln) RBM20 variant was identified in 18 members of a large family with DCM and CSD (Pedigree N, Figure 2). This family, which also carries an p.(Arg388His) LMNA variant, has previously been described as pedigree F in Li et al.21 and is presented here in updated form. Both parents (N.1 and N.2) of the affected proband (N.8) survived into their 70s with reported histories of heart failure and cardiomyopathy, suggesting potential for bilineal inheritance. Five of the proband’s 6 siblings (N.4, N.5, N.9, N.13, N.15) developed Grade III/Grade IV DCM by the third to fifth decade. A total of 5 individuals in the second and third generation could be definitively classified as carriers of both RBM20 and LMNA variants, either through direct DNA testing (N.8, N.15, N.23, N.25) or inferred from pedigree structure (N.9). Three double carriers in the second generation exhibited severe (Grade III/IV) disease (N.8, N.9, N.15), while the youngest (N.23, N.25) lacked either clinical or subclinical signs of DCM at time of screening. One RBM20 single variant carrier (N.27) had clinically detectable DCM by 33 years of age, while 7 additional members of the 2 youngest generations exhibited signs of subclinical disease (Grade II)(N.35) or early signs of LVE and/or systolic dysfunction not meeting clinical thresholds for DCM (Grade I) (N.19, N.22, N.28, N.29, N.30, N.31). One other family member in the third generation carried the RBM20 variant but was phenotypically unaffected (N.26). The average age of onset for DCM in this family was 37.6 years, older than the average age of non-penetrant RBM20 variant carriers (23.5 years), supporting possible age-related non-penetrance in affected family members. Three subjects carried only the LMNA variant (N.33, N.34, N.36) and all were similarly unaffected. Two additional family members (N.4, N.13) had unknown LMNA variant status, but exhibited Grade IV disease. While N.4 could be definitively determined to carry the RBM20 variant, the mutation status of N.13 is unknown. On a population level, both the RBM20 and LMNA variants are extremely rare and impact highly conserved residues (Suppl. Table 2). Supporting pathogenicity, nuclear morphology and lamin A subcellular localization were markedly affected by the p.(Arg388His) LMNA variant with most affected protein aberrantly localizing to the cytoplasm20. Collectively these data support potential contribution of both variants to DCM in this family. As a single severely affected individual in the second generation (N.5) as well as two individuals in the youngest generation with early evidence of DCM not meeting formal criteria (N.22, N.28) tested negative for both variants, future genetic testing efforts may help to elucidate whether any additional genetic variation is relevant to this family.
Pedigree O:
A rare p.(Arg399Cys) LMNA variant was identified in the proband (O.11) of Pedigree O (Figure 3), but not in his affected father (O.8). The proband, his mother (O.9), and his maternal grandfather (0.3) all carried the LMNA variant, with evidence of mild LMNA-related cardiac phenotype in O.3 and his brother, the proband’s maternal great uncle (O.5). The proband had no known antecedent CSD or arrhythmias consistent with LMNA-related cardiomyopathy, but rather presented with advanced heart failure with extremely severe DCM requiring rescue ventricular assist device (VAD) placement and transplantation by 15 years of age. Exome sequencing subsequently identified a paternally inherited p.(Asp24224Ilefs*8) TTNtv transmitted to both the proband and his unaffected brother (O.10) through the father (O.8) from the paternal grandmother (O.2) . As with the p.(Pro24523Hisfs*4) TTNtv identified in pedigree L, the p.(Asp24224Ilefs*8) TTNtv affected the A-band and was predicted to severely truncate the full-length TTN protein (24230 vs 35991 amino acids). The proband’s father died at 45 years following transplantation for severe DCM and heart failure that presented at 36 years. Remarkably, the paternal grandparents (O.1, O.2), also had Class III disease, and might have contributed not only the TTNtv (from O.2) but also a second unknown risk allele (from O.1) to cause the early onset of DCM in O.8. It is possible that co-occurrence of the paternal TTNtv and maternal LMNA variant in the proband contributed to his very severe and early onset disease. Supporting this interpretation were damaging predictions from most predictive algorithms for the LMNA variant, although no obvious cellular effect was indicated by nuclear imaging studies20. Notably, both variants affected conserved residues and were extremely rare (LMNA) or absent (TTN) from control populations (Table 2).
Pedigree P:
A C-terminal paternally inherited p.(Ser437Hisfs*2) LMNA variant was identified in 7 individuals (P.1, P.3, P.8, P.10, P.14, P.16, P.20) spanning 3 generations of a large family with multigenerational DCM and arrhythmia (Pedigree P, Figure 4). In 6 of 7 LMNA variant carriers, cardiomyopathy manifested as subclinical symptoms or early signs not meeting formal diagnostic criteria. Nevertheless, this variant was predicted to prematurely truncate the encoded lamin A and C proteins (by 227 and 135 amino acids, respectively) and highly abnormal lamin A aggregation in nuclear imaging studies strongly supported functional effect20. Exome sequencing and follow-up targeted testing subsequently identified a rare p.(Glu485Lys) NEXN variant in 4 family members (P.11, P.18, P.20, P.23). The proband (P.18) and her daughter (P.23) carried both the LMNA and NEXN variants and each exhibited severe (Grade IV) DCM, either requiring transplantation (P.18) or resulting in mortality (P.23). The proband’s sister (P.20), the only other known double variant carrier, was less severely affected. Their mother (P.11), the sole known family member to carry only the variant NEXN allele, showed no evidence of cardiovascular disease. What remains unexplained is the even earlier onset of disease and mortality from advanced heart failure in P.23, who also carried both variants. While we are at this time unable to assign a specific cause, it is possible that P.23 may have had a de novo third modifier, or may have inherited a possible third modifier through P.17, whose personal and family history is unknown. This postulate is suggested by Grade II DCM in P.22, who carried neither the LMNA nor the NEXN variants and whose disease was identified at 27 years by cascade clinical screening following his sister’s diagnosis. Such an early age of onset is highly suggestive of a genetic cause.
Pedigree S:
We identified a rare p.(Arg25Cys) PLN variant, shown to disrupt PLN function,34 in multiple individuals spanning 2 generations of a severely affected family that also carried a paternal p.(Arg654*) LMNA nonsense variant (Pedigree S, Figure 5). The presence of severe disease on both sides of the family as well as in single variant carriers was highly suggestive of bilineal inheritance of 2 pathogenic alleles. The p.(Arg25Cys) PLN variant was identified in the proband (S.9), as well as in 2 of 3 sisters (S.8, S.10) and his affected mother (S.7). All 4 carriers developed arrhythmia requiring ICD placement alongside severe (Grade IV) adult-onset DCM progressing to heart failure. Both the youngest sister (S.11) and her paternal uncle (S.5) were also severely affected but carried only the LMNA variant. An p.(Asp103Val) MYPN variant was additionally identified in S.5, but was not identified in any other family member. Usual criteria for variant adjudication (amino acid conservation, pathogenic potential by predictive algorithms, absence from control populations) supported pathology of both the PLN and LMNA variants (Table 2, Suppl. Table 2) while in vitro lamin A nuclear imaging provided additional evidence for pathologic LMNA variant effect20. A single subject, S.9 (the proband), carried both variants. While degree of DCM was generally comparable between the proband and his siblings (S.8 to S.11), there was an increased burden of arrhythmias, including sudden cardiac death and atrial fibrillation. Disease onset was not significantly earlier than in single variant carriers.
Modeling the Impact of Non-LMNA Variants on Disease Severity:
We used a linear mixed model (see Methods in Suppl. Material) to determine whether the presence of additional potentially relevant variants besides the family’s LMNA variant was associated with DCM-relevant quantitative traits. Table 3 presents descriptive statistics for the individuals in the pedigrees in Figures 1–5, and the mixed model results are presented in Table 4. The variance components point estimates are reasonable and reflect the expected negative correlation between LVEF and LVEDD z-score and phenotypic grade as well as the positive correlation between LVEDD z-score and phenotypic grade. The imprecision in these estimates precludes further inference about the additive contribution of unmeasured loci to phenotypic variance. The mean model intercept parameters estimate the mean trait values for males not carrying any of the family’s variants at the average evaluation age. Trait values did not differ substantially for females, although each increase of 1 standard deviation in the age was associated with a decrease in LVEF and an increase in phenotypic grade. The presence of the familial LMNA variant itself had an equivocal effect on phenotype, likely reflecting the lack of segregation in these families. However, the presence of one or more of the family’s non-LMNA variants was associated with a measurably worse phenotype, including a decrease of 12.6% (95% CI: −23.0 to −2.3) in LVEF and an increase of 1.44 (95% CI: 0.19 to 2.68) in LVEDD z-score. While we cannot formally exclude an effect on phenotypic grade of zero, the increase of 0.8 (95% CI: −0.1 to 1.7) is also concordant with the results for the other traits. Conclusions regarding the effects of LMNA and non-LMNA variants were unchanged in models that (1) included LVEF and LVEDD z-score only and (2) modeled the effect of the number of the family’s non-LMNA variants rather than simply presence/absence (data available on GitHub at https://github.com/kinnamon-lab/lmna_nonseg).
Table 3.
| Variable | Values Available |
N (%) or Mean ± Standard Deviation (Minimum, Maximum) |
|---|---|---|
| Alive | 84 | 51 (61%) |
| Female | 88 | 43 (49%) |
| LMNA variant positive | 50 | 25 (50%) |
| # of relevant variants in other DCM genes | 44 | |
| 0 | 14 (32%) | |
| 1 | 28 (64%) | |
| 4 | 2 (5%) | |
| Phenotypic grade | 48 | 2.1 ± 1.6 (0, 4)* |
| 0 | 13 (27%) | |
| 1 | 9 (19%) | |
| 2 | 3 (6%) | |
| 3 | 8 (17%) | |
| 4 | 15 (31%) | |
| Age at echocardiogram (years) | 45 | 41.0 ± 17.5 (15, 82) |
| LVEF (%) | 44 | 41.7 ± 18.8 (8, 70) |
| LVEDD (z-score) | 40 | 2.64 ± 2.08 (−3.09, 6.75) |
Includes parents of siblings O.3 and O.5 as well as siblings P.1, P.3, and P.6. These 4 parents did not contribute variant or phenotype data and so were not displayed on pedigrees to save space. Some percentages may not sum to exactly 100 because of rounding. DCM indicates dilated cardiomyopathy; LVEDD, left ventricular end-diastolic dimension; and LVEF, left ventricular ejection fraction. *Phenotypic grade is defined as a categorical variable but used as a quantitative trait in analysis.
Table 4.
Multivariate Linear Mixed Model Results
| LVEF (%) | LVEDD (z-score) | Phenotypic grade | |
|---|---|---|---|
|
Mean model parameters Estimate ± Standard Error (95% Confidence Interval)* |
|||
| Intercept | 49.19 ± 6.18 (37.07, 61.30) |
2.24 ± 0.67 (0.93, 3.55) |
1.55 ± 0.46 (0.63, 2.46) |
| Female | 3.47 ± 5.35 (−7.01, 13.95) |
−0.24 ± 0.64 (−1.50, 1.02) |
−0.09 ± 0.46 (−0.99, 0.82) |
| Age at echocardiogram (standardized) | −6.26 ± 2.92 (−11.98, −0.55) |
0.50 ± 0.34 (−0.17, 1.17) |
0.74 ± 0.24 (0.28, 1.21) |
| LMNA variant positive | 3.38 ± 5.57 (−7.54, 14.30) |
−1.19 ± 0.65 (−2.46, 0.08) |
−0.57 ± 0.47 (−1.49, 0.35) |
| At least one relevant non-LMNA variant in other DCM genes | −12.62 ± 5.27 (−22.96, −2.29) |
1.44 ± 0.63 (0.19, 2.68) |
0.82 ± 0.45 (−0.07, 1.71) |
|
Additive genetic covariance components† Estimate ± Standard Error |
|||
| LVEF (%) | 87.81 ± 578.79 | −5.94 ± 13.43 | −4.85 ± 21.78 |
| LVEDD (z-score) | −5.94 ± 13.43 | 0.40 ± 1.64 | 0.33 ± 1.02 |
| Phenotypic grade | −4.85 ± 21.78 | 0.33 ± 1.02 | 0.27 ± 1.01 |
|
Environmental covariance components† Estimate ± Standard Error |
|||
| LVEF (%) | 117.38 ± 105.05 | −10.26 ± 10.33 | −9.62 ± 7.22 |
| LVEDD (z-score) | −10.26 ± 10.33 | 2.28 ± 1.16 | 1.28 ± 0.76 |
| Phenotypic grade | −9.62 ± 7.22 | 1.28 ± 0.76 | 1.20 ± 0.56 |
Wald 95% confidence intervals were calculated from the reported estimate and standard error using the standard normal distribution.
Standard errors for covariance components were calculated using formula (10) of Bauman et al.25, which is reported as STD_ERR_2 by the Mendel software.
We used causal diagrams26–29 to facilitate interpretation of genetic effects in the linear mixed model and determine whether the set of adjustment variables was appropriate (Methods and Suppl. Figure 1). Under the assumed causal model, the genetic effects in the linear mixed model can be interpreted as total causal effects of an increased burden of putatively pathogenic rare variants on the quantitative trait under typical diagnostic and treatment protocols. As such, they are indicative of genetic effects on the quantitative trait not counteracted by typical treatment protocols. Importantly, adjustment for treatment history at the time of data collection in the linear mixed model is inappropriate because it would render the genetic effects biased estimates of both total causal effects and direct causal effects with treatment held constant. On the basis of this result, we did not perform any analysis adjusting for treatment history.
Discussion:
Our observation of variant non-segregation in 6 of 19 families with LMNA-related DCM was originally met with skepticism18. The current work now presents variants putatively relevant for DCM that explain additional phenotypic variation in 5 of these 6 pedigrees. This strengthens our sense that the traditional view of IDC as a strictly monogenic disorder may be incomplete6.
Non-LMNA variants identified in our study affected genes with known DCM associations. RBM20 variants are estimated to occur in approximately 2% of all patients with IDC (Suppl. Table 1). The p.(Arg716Gln) variant detected in Pedigree N was 1 of 6 unique variants previously reported in our wider DCM cohort21 and, like the newly identified p.(Met338Thr) variant detected in pedigree L, affected residues outside of the exon 9 hotspot30. RBM20 encodes an RNA splicing protein that regulates splicing of several other heart-relevant genes, including titin (TTN)31. We detected one TTNtv, p.(Pro24523Hisfs*4), in Pedigree L and another, p.(Asp24224Ilefs*8), in Pedigree O. Both variants were predicted to prematurely truncate the large cardiac N2BA transcript at the highly conserved A-band. Rare variants were additionally identified in 5 other DCM associated genes: BAG31, encoding a Bcl-2-associated molecular chaperone with roles in Z-disc assembly and stress-induced apoptosis; DSP24, encoding a key component of desmosomal junctions in the heart; MYPN32, encoding a z-disc protein with roles in sarcomere assembly and cardiac gene expression, NEXN33, encoding an f-actin binding protein important for maintenance of the sarcomere z-line; and PLN34, encoding a key regulator of calcium homeostasis and myocardial contractility.
Our multivariate linear mixed model demonstrated that the presence of one or more non-LMNA variants was associated with a measurably worse phenotype, including a decrease of 12.6% (95% CI: −23.0 to −2.3) in LVEF and an increase of 1.44 (95% CI: 0.19 to 2.68) in LVEDD z-score, after adjusting for sex, age, and the presence/absence of the family’s non-segregating LMNA variant. The adjusted impact of each family’s LMNA variant on these traits was equivocal in our model, although this result could arise if causal LMNA variants have more subtle effects on these quantitative traits that were not captured with adequate precision in our modest sample. The availability of in vitro evidence supporting pathogenicity for some of these variants20 buttresses this interpretation, but statistical confirmation would require a larger sample of extended pedigrees.
In our cardiovascular genetics practice we are careful to avoid stating that lack of an identified family variant, even one considered pathogenic, relieves family members who do not carry that variant (or variants) of any future disease liability. We rather state that the risk is decreased, as other genetic and/or environmental risk factors35 may be present. While useful as a framework for variant adjudication in classic Mendelian pedigrees, current ACMG guidelines23 do not allow for sufficient flexibility to accurately assess contributions of multiple putative disease variants. This is particularly true when detected variants impact known disease genes that otherwise meet criteria for plausible pathogenicity. For the purposes of this study, we have considered rare variants that may be classified as VUSs to be of similar interest as those classified as pathogenic or likely pathogenic using existing ACMG criteria. Our statistical model suggests that these variants may in fact be relevant and may even explain more of the variation in phenotypic severity than the more established LMNA variants in these pedigrees. Future analyses will benefit greatly from increased knowledge of influencing environmental factors, protective variants, regulatory elements, or other systemic factors. In addition, continued cataloguing of normal human variation in online genetic databases (ExAC, gnomAD) and careful phenotyping of increasing numbers of patients will help to further refine determinations of variant expressivity and penetrance.
Study Limitations:
By definition, exome-based sequencing strategies preclude detection of structural variants and other relevant variants affecting regulatory or functionally significant non-coding domains. Second, while pedigrees have been expanded to incorporate all available clinical and genetic information, small sizes or unavailability of genetically relevant subjects in some families limited our ability to more fully assess possible genotype/phenotype correlations. Third, while we have already emphasized the limited applicability of current ACMG variant adjudication guidelines to multi-variant models, all variants identified in this study must be considered to be VUSs in the absence of additional supportive genetic or functional data. Adjudications may change with access to much larger datasets. Even larger exome datasets may result in future reassessment or provide new, previously unforeseen insights into variant pathogenicity. The multivariate linear mixed model used in our analysis is at best an approximation of a complex biological system, and simplifying assumptions were required to make the model tractable with our modest sample size. Phenotypic grade was modeled as a quantitative trait despite being defined on an ordinal scale. Goodness of fit tests showed no evidence of lack of fit, but these tests have extremely limited power with the number of pedigrees analyzed. Moreover, variance components were estimated rather imprecisely so conclusions about additive genetic variance and covariance from unmeasured loci could be drawn. A significantly larger sample of pedigrees would remedy this issue. Finally, the type of complete-case analysis performed by Mendel is valid if an individual's probability of having missing data is independent of his or her phenotype values given his or her predictors.36, 37 Although this assumption cannot be empirically tested, it is reasonable as long as (1) phenotype values do not directly affect the probability of being a complete case and (2) measured genotypes, sex, and age are the only factors affecting phenotype values that also affect this probability.
Conclusions and Summary:
In summary, we have identified additional putative genetic contributors to DCM in 5 of 6 families with non-segregating LMNA variants, 3 with clear evidence of bilineal inheritance. We suggest that this yield (5 of the 19 families, 26%) places multigene causation as a significant and plausible alternative to the more usual single gene Mendelian DCM model in some cases. Our data suggest that the extent of DCM arising from multigenic cause may be higher than previously anticipated. Much larger ongoing7 and future studies will be required to more precisely determine the genetic architecture of DCM.
Supplementary Material
Acknowledgements:
We thank all probands and family members for their ongoing participation. This work was supported in part by computational infrastructure provided by The Ohio State University Division of Human Genetics Data Management Platform and the Ohio Supercomputer Center.
Funding Sources: NIH RO1 HL58626 (REH), 5 M01 RR000334. Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant HG006493 to Drs. Deborah Nickerson, Michael Bamshad, and Suzanne Leal.
Footnotes
Disclosures: None
References:
- 1.Norton N, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet 2011;88:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grunig E, et al. Frequency and phenotypes of familial dilated cardiomyopathy [see comments]. J Am Coll Cardiol 1998;31:186–94. [DOI] [PubMed] [Google Scholar]
- 3.McKenna C, et al. Idiopathic dilated cardiomyopathy: familial prevalence and HLA distribution. Heart 1997;77:549–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mestroni L, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999;34:181–90. [DOI] [PubMed] [Google Scholar]
- 5.Michels VV, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992;326:77–82. [DOI] [PubMed] [Google Scholar]
- 6.Hershberger RE, et al. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–47. [DOI] [PubMed] [Google Scholar]
- 7.Kinnamon DD, et al. DCM Consortium. Toward Genetics-Driven Early Intervention in Dilated Cardiomyopathy: Design and Implementation of the DCM Precision Medicine Study. Circ Cardiovasc Genet 2017;10: e001826. doi: 10.1161/CIRCGENETICS.117.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoefele J, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol 2007;18:2789–95. [DOI] [PubMed] [Google Scholar]
- 9.Katsanis N The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet 2004;13 Spec No 1:R65–71. [DOI] [PubMed] [Google Scholar]
- 10.Sykiotis GP, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A 2010;107:15140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Blitterswijk M, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:3776–84. [DOI] [PubMed] [Google Scholar]
- 12.Kelly M, et al. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet 2009;2:182–90. [DOI] [PubMed] [Google Scholar]
- 13.Li L, et al. A Potential Oligogenic Etiology of Hypertrophic Cardiomyopathy: A Classic Single-Gene Disorder. Circ Res 2017;120:1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Priest JR, et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet 2016;12:e1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu T, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2010;55:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norton N, et al. Assessment of LMNA copy number variation in 58 probands with dilated cardiomyopathy. Clin Transl Sci 2011;4:351–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brodt C, et al. Temporal relationship of conduction system disease and ventricular dysfunction in LMNA cardiomyopathy. J Card Fail 2013;19:233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parks SB, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008;156:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kushner JD, et al. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Cardiac Failure 2006;12:422–29. [DOI] [PubMed] [Google Scholar]
- 20.Cowan J, et al. Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circ Cardiovasc Genet 2010;3:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li D, et al. Identification of novel mutations In RBM20 in patients with dilated cardiomyopathy. Clin Trans Sci 2010;3:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu GS, et al. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res 2015;107:164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norgett EE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000;9:2761–6. [DOI] [PubMed] [Google Scholar]
- 25.Bauman LE, et al. Fishing for pleiotropic QTLs in a polygenic sea. Ann Hum Genet 2005;69:590–611. [DOI] [PubMed] [Google Scholar]
- 26.Glymour MM, et al. Causal Diagrams. In: Rothman KJ, et al. , eds. Modern Epidemiology Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 27.Greenland S, et al. Causal Diagrams. In: Lovric M, ed. International Encyclopedia of Statistical Science Berlin, NY: Springer-Verlag Berlin Heidelberg; 2011. [Google Scholar]
- 28.Greenland S, et al. Causal diagrams for epidemiologic research. Epidemiology 1999;10:37–48. [PubMed] [Google Scholar]
- 29.Pearl J Causal diagrams for empirical research. Biometrika 1995:669–688.
- 30.Brauch KM, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009;54:930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18:766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duboscq-Bidot L, et al. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res 2008;77:118–25. [DOI] [PubMed] [Google Scholar]
- 33.Hassel D, et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med 2009;15:1281–8. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt JP, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003;299:1410–3. [DOI] [PubMed] [Google Scholar]
- 35.Poller W, et al. Genome-environment interactions in the molecular pathogenesis of dilated cardiomyopathy. J Mol Med (Berl) 2005;83:579–86. [DOI] [PubMed] [Google Scholar]
- 36.White IR, et al. Bias and efficiency of multiple imputation compared with complete-case analysis for missing covariate values. Stat Med 2010;29:2920–31. [DOI] [PubMed] [Google Scholar]
- 37.Little RJA, et al. Statistical analysis with missing data Hoboken: John Wiley & Sons; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.