

Abstract
We present the development of the first small molecule degraders that can induce anaplastic lymphoma kinase (ALK) degradation, including in non-small-cell lung cancer (NSCLC), anaplastic large-cell lymphoma (ALCL), and neuroblastoma (NB) cell lines. These degraders were developed through conjugation of known pyrimidine-based ALK inhibitors, TAE684 or LDK378, and the cereblon ligand pomalidomide. We demonstrate that in some cell types degrader potency is compromised by expression of drug transporter ABCB1. In addition, proteomic profiling demonstrated that these compounds also promote the degradation of additional kinases including PTK2 (FAK), Aurora A, FER, and RPS6KA1 (RSK1).
TABLE OF CONTENTS GRAPHIC
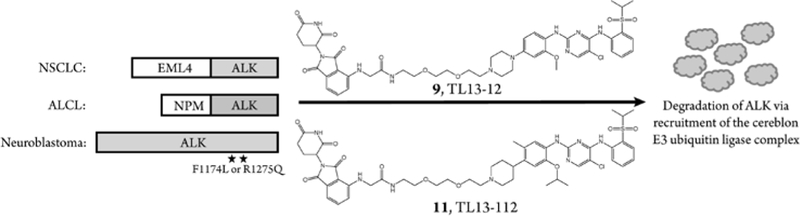
INTRODUCTION
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase that was first identified in a chromosomal translocation associated with anaplastic large cell lymphoma (ALCL), a subtype of T-cell non-Hodgkin’s lymphoma.1 Chromosomal translocations involving the kinase domain of ALK are seen in many cancers. In addition to ALCL, ALK fusion proteins are seen in diffuse large B-cell lymphoma (DLBCL), inflammatory myofibroblastic tumor (IMT), breast cancer, colorectal cancer, esophageal squamous cell cancer (ESCC), renal cell cancer (RCC), and non-small-cell lung cancer (NSCLC).2 ALK fusion partners drive dimerization of the ALK kinase domain, leading to autophosphorylation, which in turn causes the kinase to become constitutively active.3 Oncogenic ALK may also be expressed due to point mutations as is seen in neuroblastoma (NB), where germline mutations in ALK have been documented to drive the majority of hereditary NB cases.4,5 Constitutively active oncogenic ALK signals through multiple pathways, including PI3K/AKT, RAS/ERK, and JAK/STAT3; this signaling leads to enhanced cell proliferation and survival.6
ALK is an attractive target for cancer therapies not only for its prominent role in a number of malignancies, but also for its scant expression in normal adult tissue, which is restricted to a small subset of neural cells, reducing off-target toxicities of ALK-selective agents.2,7 There are currently four FDA approved kinase inhibitors for the treatment of ALK-positive NSCLC: crizotinib, ceritinib (LDK378), alectinib, and, most recently, brigatinib. ALK-positive tumors are highly sensitive to ALK inhibition, indicating that these tumors are addicted to ALK kinase activity. Hwever, despite initial dramatic responses of variable median duration (10.9 months for crizotinib; 16.6 months for ceritinib; 25.7 months for alectinib), resistance to therapy typically develops. 8–12
While next-generation ALK inhibitors such as lorlatinib have been able to successfully target resistant tumors and have shown improvements in potency and overall response rates relative to approved inhibitors, resistance to these inhibitors still consistently arises in patients.13–16 Therapeutic strategies that target ALK with a novel mechanism of action may provide ways to further delay the emergence of resistance mutations. Here we described the development and characterization of bivalent small molecules that are capable of inducing proteasome-mediated degradation of ALK.
We developed small molecule degraders (also called PROTACs or degronimides) which are hetero-bifunctional small molecules that can induce degradation of a protein by bringing it into proximity of an E3 ligase.17 When the ternary complex is formed, the E3 ligase ubiquitinates the target protein, leading to its proteasomal degradation. It has recently been shown that this technology may be used to induce both kinase and kinase fusion protein degradation.18
Here we present two examples of degraders that can induce ALK degradation in NSCLC cells expressing the fusion protein echinoderm microtubule-associated protein-like 4 (EML4)-ALK, ALCL cells expressing the fusion protein nucleophosmin (NPM)-ALK, and NB cells expressing either ALK F1174L or ALK R1275Q.
RESULTS AND DISCUSSION
We designed the degraders 9 and 11 based on known ALK inhibitors TAE684 and LDK378 (ceritinib), respectively, and used the cereblon ligand pomalidomide to recruit the E3 ubiquitin ligase complex (Figure 1A). A 2-polyethylene glycol (PEG) linker was selected for these prototypical ALK degraders due to the success of a previously generated TAE684 based multi-kinase degrader.19 As control compounds to complement the degraders 9 and 11, we designed the analogs 10 and 12 with des-carbonyl pomalidomide groups that exhibit substantially weakened binding to cereblon, as confirmed by a biochemical cereblon binding assay (Figures 1A, S1). Using an ALK activity assay, we validated that both the degraders and their des-carbonyl counterparts are still able to bind ALK, with IC50s comparable to their parental kinase inhibitors (Figure 1B, Table S1).
Figure 1.
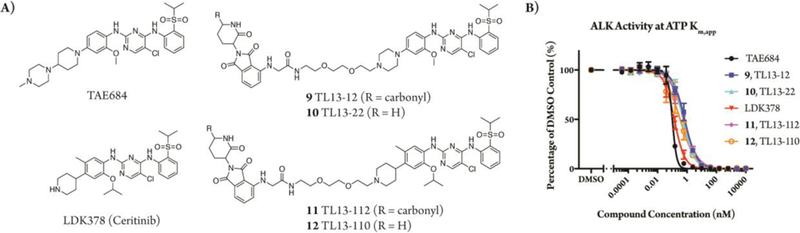
Chemical structures and characterization of ALK degraders. (A) TAE684 is the parental kinase inhibitor of 9 and 10. LDK378 is the parental inhibitor of 11 and 12. 9 and 11 are ALK targeted degraders while 10 and 12 contain des-carbonyl versions of the pomalidomide group, causing them to exhibit substantially weakened binding to cereblon. (B) TR-FRET ALK activity assay, plotted as the mean of three technical replicates ± SD
We examined the effects of the degraders on cell proliferation in ALK-driven NSCLC line H3122, and ALCL lines Karpas 299 and SU-DHL-1. In both NSCLC and ALCL cell lines the degraders and parental kinase inhibitors were about equipotent, while the des-carbonyl compounds were less active than the inhibitors, indicating that the potent anti-proliferative effects seen with the degraders are due in part to their ability to degrade ALK (Figure 2).
Figure 2.
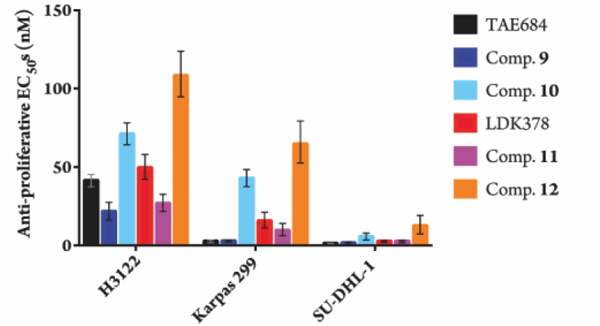
Anti-proliferative best fit EC50 values with 95% CI in NSCLC and ALCL cell lines after 72-hr treatments (three biological replicates; Graphpad Prism 7 software).
We next studied the induced degradation of ALK and a known off-target of TAE684 and LDK378, Aurora A kinase, in H3122 and Karpas 299 cells.20,21 A dose titration of 9 and 11 in each cell line for 16 hours demonstrated that 9 is more selective for degradation of ALK versus Aurora A than 11, which is consistent with LDK378’s higher selectivity for ALK compared to TAE684 (Figure 3A).21 From these dose titrations the DC50s of 9 and 11 against ALK were calculated to be the same in H3122 cells (10 nM, two independent experiments), while 11 induced more potent ALK degradation in Karpas 299 than 9 with DC50s of 40 nM and 180 nM, respectively (two independent experiments). Time course treatments with both degraders showed that some ALK degradation is observed at 4 hours of treatment in H3122 cells and at 8 hours of treatment in Karpas 299 cells, and maximum degradation is achieved at 16 hours in both cases (Figure 3B).
Figure 3.
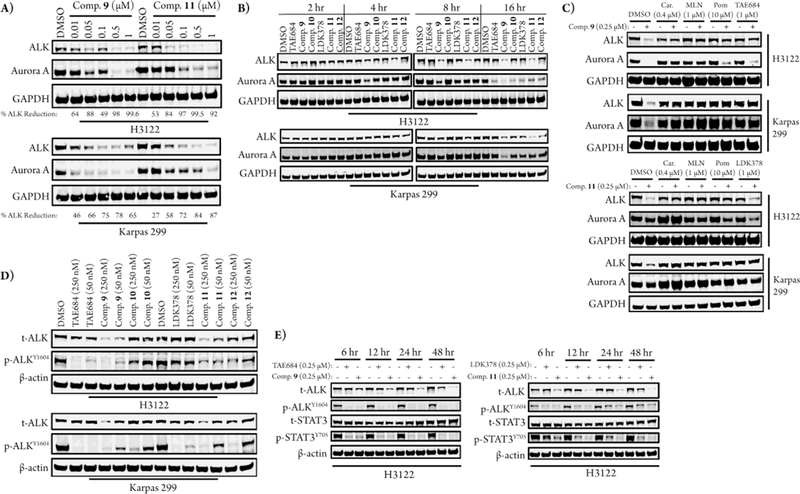
Degrader behavior in H3122 and Karpas 299 cells. (A) Immunoblots after 16 hours of treatment with dose titrations of 9 and 11. (B) Immunoblots after treatment with 250 nM of compound for the indicated amount of time. (C) Immunoblots after 2-hour pre-treatments with DMSO, carfilzomib (Car), MLN4924 (MLN), pomalidomide (Pom), TAE684, or LDK378 followed by 16-hour treatments with 9 or 11. (D) Immunoblots of downstream ALK signaling after 16-hour compound treatments; t-ALK indicates total ALK and p-ALK indicates phosphorylated ALK. (E) Immunoblots of sustained downstream ALK signaling after the indicated treatment times.
Pre-treatment of the cell lines with the proteasome inhibitor carfilzomib prevented degradation of both ALK and Aurora A, validating that the degradation seen is occurring via the proteasome (Figure 3C). MLN4924 indirectly inhibits the cereblon E3 ligase by blocking neddylation of the cullin RING ligase, CUL4, in the protein complex, which is required for ligase activity. Pre-treatment with MLN4924 also prevented degradation of ALK and Aurora A, confirming that the degradation seen is occurring via recruitment of the cereblon E3 ligase complex (Figure 3C). Pre-treatment with pomalidomide or the parental kinase inhibitor also prevented ALK degradation, although pre-treatment with LDK378 only showed partial rescue in H3122 cells. This demonstrates that engagement of both ALK and cereblon is required for the observed degradation.
We compared the effects on downstream signaling between the parental kinase inhibitors, the degraders, and their des-carbonyl counterparts by examining ALK phosphorylation by western blot after 16 hours of treatment; this demonstrated that the extent of downstream signaling inhibition by the degraders is cell line dependent (Figure 3D). In H3122 cells, 9 inhibits downstream signaling to a similar extent as its parental kinase inhibitor, while 11 shows improved downstream inhibition. In Karpas 299 cells the degraders affect downstream signaling to a lesser extent than the kinase inhibitors. In both cell lines the degraders affect downstream signaling more than their des-carbonyl counterparts, indicating that the degrader downstream effects are due to both degradation and inhibition of ALK.
When observing downstream signaling over a 48-hour time course in H3122 cells, 9 sustains inhibition of ALK and STAT3 phosphorylation to a similar extent as TAE684, while 11 shows a distinct improvement on sustained inhibition compared to LDK378 (Figure 3E). This is significant because inhibition of STAT3 signaling leads to significant anti-proliferative effects in EML4-ALK expressing cells and sustained ALK pathway inhibition has been linked to greater anti-tumor efficacy. 22–24
When we tested the anti-proliferative effects of the degraders in NB cell lines we observed an unreported, as of this writing, potential obstacle with the degrader technology related to the ATP-binding cassette sub-family B member 1 (ABCB1) drug transporter. We used Kelly and Lan5 lines, which are ABCB1 low expressing cells, and SH-SY5Y and CHLA20 lines, which are ABCB1 high expressing cells. ABCB1 has been shown to efflux hydrophobic and amphipathic compounds.25 This drug transporter activity can be inhibited by tariquidar.26 In the ABCB1 high expressing cells the degraders had low anti-proliferative activity compared to the parental inhibitors. However, when we co-treated with tariquidar we saw an increase in anti-proliferative activity of the degraders with EC50s comparable to the parental kinase inhibitors (Figure 4). In addition, knockdown of ABCB1 using shRNA also increased degrader-mediated ALK degradation and inhibition of proliferation (Figure S6). Thus, we concluded that degraders can be substrates of the ABCB1 transporter. Future SAR efforts will need to focus on modifications to the degrader molecules that will prevent them from being effluxed by ABCB1 transporters.
Figure 4.
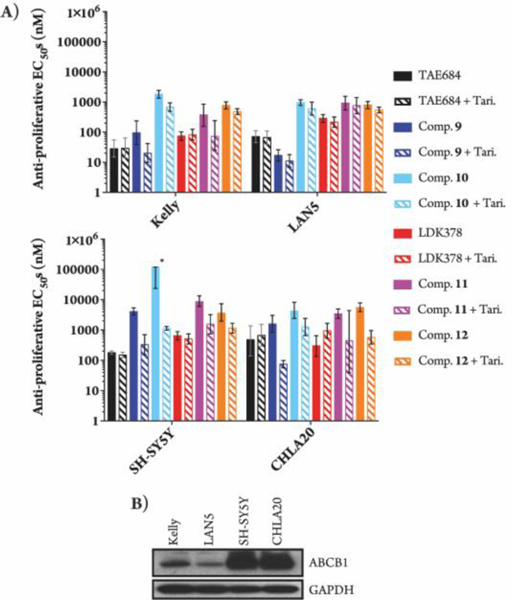
(A) Anti-proliferative best fit EC50 values with 95% CI in NB cell lines after 72-hr treatments (two biological replicates; Graphpad Prism 7 software: * = upper CI not calculated by model). 125 nM of tariquidar (Tari.) was used for the indicated co-treatments. (B) Immunoblot of ABCB1 expression in NB lysates.
We next studied induced ALK degradation by immunoblots using Kelly and CHLA20 cells; CHLA20 cells were always co-treated with 125 nM tariquidar. In Kelly cells 9 and 11 were equipotent ALK degraders, with DC50s of 50 nM each as determined by western blot after 16 hr dose titrations (two independent experiments) (Figure 5A). As in the NSCLC and ALCL cell lines, 9 was less selective for ALK over Aurora A than 11 in both NB lines (Figure 5A). Time courses in the Kelly and CHLA20 cells showed that ALK degradation by the degraders could be observed after 4 hours of treatment in CHLA20 and after 8 hours in Kelly cells (Figure 5B). Interestingly, although pre-treatment with carfilzomib, MLN4924, or pomalidomide in both Kelly and CHLA20 cells did prevent Aurora A degradation (Figure 5C), only pomalidomide and parental inhibitor pre-treatment prevented ALK degradation. This may be due to carfilzomib and MLN4924 causing an upregulation of lysosomal degradation by suppressing the ubiquitin-proteasome system.27
Figure 5.
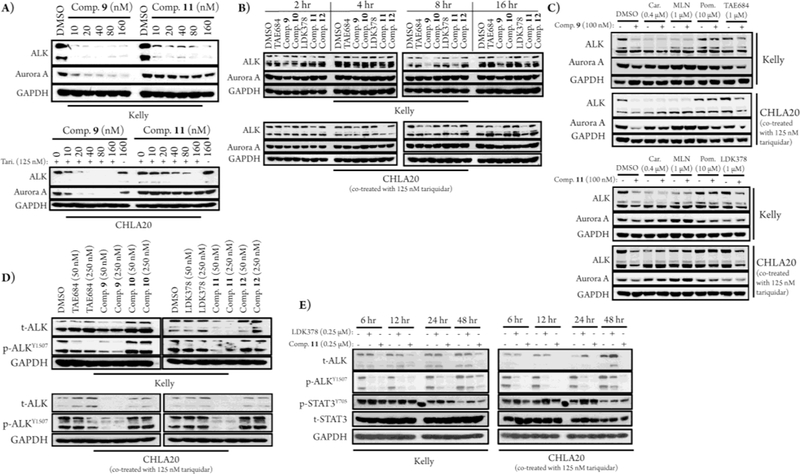
Degrader behavior in Kelly and CHLA20 cells. (A) Immunoblots after 16 hours of treatment with dose titrations of 9 and 11. Tari. indicates tariquidar. (B) Immunoblots after treatment with 100 nM of compound for the indicated amount of time. (C) Immunoblots after 2-hour pre-treatments with DMSO, carfilzomib (Car), MLN4924 (MLN), pomalidomide (Pom), TAE684, or LDK378 followed by 16-hour treatments with 9 and 11. (D) Immunoblots of downstream ALK signaling after 16-hour compound treatments; t-ALK indicates total ALK and p-ALK indicates phosphorylated ALK. (E) Immunoblots of sustained downstream ALK signaling after the indicated treatment times.
In Kelly cells 9 is equipotent to TAE684 at inhibiting downstream ALK signaling, while 11 is more potent than LDK378 at 16 hr (Figure 5D). In CHLA20 cells both degraders are more potent inhibitors of downstream ALK signaling than their parental inhibitors. 11 shows improved sustained inhibition of ALK phosphorylation compared to LDK378 in both Kelly and CHLA20 cells (Figure 5E). In Kelly and CHLA20 cells STAT3 feedback signaling was activated by the parental inhibitors, but not the degraders (Figure 5E). This observation indicates that STAT3 was not a downstream player in the anti-proliferative effects observed in these cells and that kinase degraders may have the advantage of preventing feedback activation of STAT3 that has been previously reported as a major drawback of kinase-targeted therapeutics.28
Expression proteomics was performed after 4 hours of treatment with 9 and 11 in Kelly cells; this early time point was selected in order to minimize secondary effects. However, a decrease in ALK abundance was not measured at a significant level for either compound (Figure S7). The degradation of targets other than ALK, including PTK2, FER, RPS6KA1, and Aurora A, was measured at significant levels and confirmed by western blot (Figures S8, S9); the degraded proteins detected by proteomics were shown during time courses to be more rapidly degraded than ALK, which may have resulted in the differences in detection by proteomics. PTK2, FER, and Aurora A have also been previously demonstrated to be very susceptible to kinase targeting degraders.19 Interestingly, although 9 is a better inhibitor of RPS6KA1 than 11 (Table S2), 11 is a better RPS6KA1 degrader (Figures S7–9). These results highlight the importance of determining degrader selectivity profiles separately from their parental inhibitors. Since the ability to be degraded may vary for different protein targets, degrader selectivity must be carefully assessed. ALK degraders may be improved upon in the future by using more selective inhibitor scaffolds.
The potential ability of degraders to overcome acquired resistance mutations was assessed using Ba/F3 cells expressing EML4-ALK with the resistant mutations L1196M, C1156Y, or G1202R (Figure S10). 9 and 11 both show a drop in anti-proliferative activity similar to the behavior seen with the parental ALK inhibitors when EML4-ALK has a resistant mutation. Since 9 and 11 have demonstrated that ALK is amenable to small molecule induced degradation, it is likely that future ALK targeting degraders based on ALK inhibitors that are able to overcome resistance mutations may have improved pharmacodynamic properties in these resistant mutant cell lines.
While the efficacy of ALK degraders against resistant mutations must be explored further, this work does speak to potential challenges with resistance that may be inherent in the degrader technology since several processes must function together. In addition to the potential resistance by drug transporters demonstrated here, it is possible that factors such as cereblon down-regulation or deubiquitinase upregulation may impact degrader activity.
CONCLUSION
By linking known ALK inhibitors TAE684 and LDK378 to pomalidomide we have provided prototypical examples that ALK degradation may be induced by E3 ligase recruitment in three major ALK-positive disease models. These compounds have displayed the ability to improve upon the pharmacodynamic properties of their parental inhibitors, especially sustained inhibition of downstream ALK signaling, indicating that degraders are a promising new avenue for targeted ALK therapies.
EXPERIMENTAL METHODS
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 500 MHz (Bruker A500), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
tert-butyl 4-(4-((5-chloro-4-((2(isopropylsulfonyl)phenyl) amino)pyrimidin-2yl)amino)-3-methoxyphenyl) piperazine-1-carboxylate (3). Intermediate 1 was prepared according to the literature,29 while tert-butyl 4-(4-amino-3-methoxyphenyl) piperazine-1-carboxylate (2) was commercially available. To 1 (693 mg, 2.0 mmol) and 2 (740 mg, 2.4 mmol) in sec-butanol (4 mL) was added TFA (185 µL, 2.4 mmol) and the mixture was stirred overnight at 80 °C. The mixture was then concentrated and purified by column chromatography (dichloromethane:methanol = 20:1) to yield 925 mg (75%) of 3 as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1H), 8.60 (d, J = 8.4 Hz, 1H), 8.13 (s, 1H), 8.06 (d, J = 7.2 Hz, 1H), 7.91 (d, J 8.0 Hz, 1H), 7.62 (dd, J = 8.8, 8.4 Hz, 1H), 7.33 (s, 1H), 7.25 (dd, J = 8.4, 8.4 Hz, 1H), 6.55 (s, 1H), 6.47 (d, J = 8.8 Hz, 1H), 3.88 (s, 3H), 3.60 (m, 4H), 3.24 (m, 1H), 3.09 (m, 4H), 1.49 (s, 9H), 1.30 (d, J = 7.2 MS (ESI) m/z 617 (M+H)+.
N2-(4-(4-(2-(2-(2-azidoethoxy)ethoxy)ethyl)piperazin-1-yl)-2-methoxyphenyl)-5-chloro-N4-(2-(isopropylsulfonyl)phenyl) pyrimidine-2,4-diamine (5). To 3 (620 mg, 1.0 mmol) in dichloromethane (18 mL) TFA was added (1.8 mL) and the mixture was stirred at room temperature (RT) for 2 h, then was concentrated and dried under vacuum. To the obtained crude intermediate in acetonitrile (5 mL) was added commercially available bromide 4 (300 mg, 1.2 mmol) and potassium carbonate (414 mg, 3.0 mmol). The resulted mixture was stirred under 80 oC overnight, then cooled down to RT and diluted with 50 mL of dichloromethane. The precipitation was filtered, and the filtrate was concentrated and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 524 mg (78%) of 11 as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 8.62 (d, J = 8.4 Hz, 1H), 8.13 (s, 1H), 8.02 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.62 (dd, J = 8.0, 8.0 Hz, 1H), 7.30 (m, 2H), 6.56 (s, 1H), 6.48 (d, J = 8.4 Hz, 1H), 3.88 (s, 3H), 3.70 (m, 10H), 3.25 (m, 1H), 3.41 (t, J = 5.2 Hz, 2H), 3.20 (m, 4H), 2.70 (m, 4H), 1.32 (d, J = 7.2 Hz, 6H). MS (ESI) m/z 674 (M+H)+.
tert-butyl(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycinate (7). Intermediate 6 was prepared according to the literature.30 To 6 (550 mg, 2.0 mmol) and glycine tert-butyl ester (260 mg, 2.0 mmol) in anhydrous DMSO (20 mL) was added N,N-diisopropylethylamine (DIEA) (700 μL, 4.0 mmol). The reaction mixture was stirred under 90 oC for 1 day, then cooled down. The mixture was diluted with ethyl acetate (200 mL), washed with water and brine, dried with Na2SO4, then filtered and concentrated, purified by column chromatography (dichloromethane:ethyl acetate = 2:1) to yield 530 mg (68%) of 7 as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.51 (dd, J = 8.4, 7.2 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 6.76 (d, J = 6.76 Hz, 1H), 4.93 (dd, J = 12.0, 6.4 Hz, 1H), 3.94 (s, 2H), 2.67–2.92 (m, 2H), 2.12 (m, 1H), 1.93 (m, 1H), 1.50 (s, 9H). MS (ESI) m/z 388 (M+H)+.
(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl) glycine (8). To 7 (390 mg, 1.0 mmol) in dichloromethane (18 mL) added TFA (1.8 mL). The mixture was stirred at RT overnight, then was concentrated and dried under vacuum to give 8 as a yellow solid, which was used in next step without purification. MS (ESI) m/z 330 (M-H)-.
N-(2-(2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl) phenyl)amino)pyrimidin-2-yl)amino)-3-methoxyphenyl) piperazin-1-yl)ethoxy)ethoxy)ethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)acetamide (9). Under a nitrogen atmosphere, to 5 (135 mg, 0.2 mmol) in tetrahydrofuran (18 mL) and water (1.8 mL) was added triphenylphosphine (63 mg, 0.24 mmol). The reaction mixture was stirred overnight, then concentrated and dried under vacuum. To the obtained crude oil in anhydrous dichloromethane (3 mL) was added 8 (73 mg, 0.22 mmol) and (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo [4,5-b]pyridinium 3-oxid hexafluorophosphate) (HATU) and DIEA (110 μL, 0.6 mmol). The reaction mixture was stirred for 2 h, then concentrated and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 136 mg (71%) of 9 as a yellow foam. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 9.76 (br, 1H), 9.57 (s, 1H), 8.56 (br, 1H), 8.41 (H, 1H), 8.19 (s, 1H), 8.18 (m, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.60 (m, 1H), 7.57 (dd, J = 8.0, 7.5 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.30 (dd, J = 8.0, 7.5 Hz, 1H), 7.07 (d, J = 7.0 Hz, 1H), 6.95 (m, 1H), 6.85 (d, J = 8.5 Hz, 1H), 6.71 (d, J = 2.5 Hz, 1H), 6.53 (dd, J = 8.5, 2.5 Hz, 1H), 5.07 (dd, J = 13.0, 5.5 Hz, 1H), 3.94 (d, J = 5.0 Hz, 2H), 3.85 (m, 2H), 3.80 (m, 2H), 3.76 (s, 3H), 3.58 (m, 4H), 3.45 (m, 4H), 3.40 (m, 4H), 3.30 (m, 2H), 3.24 (m, 2H), 3.03 (m, 2H), 2.53–2.63 (m, 2H), 1.16 (d, J = 7.0 Hz, 6H). MS (ESI) m/z 961 (M+H)+.
10, 11 and 12 were synthesized with similar procedures as 9.
N-(2-(2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl) phenyl)amino)pyrimidin-2-yl)amino)-3-methoxyphenyl) piper azin-1-yl)ethoxy)ethoxy)ethyl)-2-((1,3-dioxo-2-(2- oxopiperidin-3-yl)isoindolin-4-yl)amino)acetamide (10). 1H NMR (500 MHz, DMSO-d6) δ 9.78 (br, 1H), 9.59 (s, 1H), 8.55 (br, 1H), 8.44 (s, 1H), 8.20 (s, 1H), 8.18 (dd, J = 6.0, 5.5 Hz, 1H), 7.82 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.60 (m, 1H), 7.55 (dd, J =8.0, 7.5 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.31 (dd, J = 7.5, 7.5 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 6.95 (m, 1H), 6.82 (d, J = 8.5 Hz, 1H), 6.71 (d, J = 2.5 Hz, 1H), 6.53 (dd, J = 8.5, 2.5 Hz, 1H), 4.52 (dd, J = 12.0, 6.5 Hz, 1H), 3.93 (d, J = 4.5 Hz, 2H), 3.85 (m, 2H), 3.80 (m, 2H), 3.77 (s, 3H), 3.60 (m, 4H), 3.57 (m, 2H), 3.46 (m, 4H), 3.40 (m, 2H), 3.30 (m, 2H), 3.22 (m, 2H), 3.03 (m, 2H), 2.20 (m, 1H), 1.96 (m, 1H), 1.89 (m, 2H), 1.16 (d, J = 6.5 Hz, 6H). MS (ESI) m/z 947 (M+H)+.
N-(2-(2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl) phenyl)amino)pyrimidin-2-yl)amino)-5-isopropoxy-2- methylphenyl)piperidin-1-yl)ethoxy)ethoxy)ethyl)-2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino) acetamide (11). 1H NMR (500 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.59 (s, 1H), 9.46 (s, 1H), 8.44 (d, J = 8.5 Hz, 1H), 8.38 (br, 1H), 8.25 (s, 1H), 8.18 (dd, J = 6.0, 5.5 Hz, 1H), 8.09 (s, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.60 (dd, J = 8.5, 8.0 Hz, 1H), 7.57 (dd, J = 8.0, 7.5 Hz, 1H), 7.34 (dd, J = 8.5, 8.0 Hz, 1H), 7.06 (d, J = 6.0 Hz, 1H), 6.93 (dd, J = 5.5, 5.5 Hz, 1H), 6.84 (d, J = 9.0 Hz, 1H), 6.75 (s, 1H), 5.06 (dd, J = 13.0, 5.5 Hz, 1H), 4.48 (m, J = 6.0 Hz, 1H), 3.93 (d, J = 5.5 Hz, 2H), 3.78 (m, 2H), 3.59 (m, 4H), 3.56 (m, 2H), 3.44 (m, 4H), 3.28 (m, 2H), 3.13 (m, 4H), 2.83–2.99 (m, 2H), 2.52–2.61 (m, 1H), 2.13 (s, 3H), 1.85–2.04 (m, 4H), 1.22 (d, J = 6.0 Hz, 6H), 1.15 (d, J = 7.0 Hz, 6H). MS (ESI) m/z 1002 (M+H)+.
N-(2-(2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl) phenyl)amino)pyrimidin-2-yl)amino)-5-isopropoxy-2-methylphenyl)piperidin-1-yl)ethoxy)ethoxy)ethyl)-2-((1,3-dioxo-2-(2-oxopiperidin-3-yl)isoindolin-4-yl)amino) acetamide (12). 1H NMR (500 MHz, DMSO-d6) δ 9.46 (s, 1H), 8.46 (d, J = 8.5 Hz, 1H), 8.25 (s, 1H), 8.16 (dd, J = 5.5, 5.0 Hz, 1H), 8.07 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.81 (s, 1H), 7.61 (dd, J = 8.5, 8.0 Hz, 1H), 7.55 (dd, J = 8.0, 7.5 Hz, 1H), 7.52 (s, 1H), 7.35 (dd, J = 8.5, 8.0 Hz, 1H), 7.03 (d, J = 6.0 Hz, 1H), 6.93 (dd, J = 6.0, 5.5 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H), 6.78 (br, 1H), 4.52 (dd, J = 12.0, 6.0 Hz, 1H), 3.92 (d, J = 5.5 Hz, 2H), 3.54 (m, 6H), 3.45 (m, 4H), 3.29 (m, 4H), 3.21 (m, 4H), 2.20 (m, 3H), 2.12 (s, 3H), 1.81–2.00 (m, 6H), 1.22 (d, J = 6.0 Hz, 6H), 1.16 (d, J = 6.5 Hz, 6H). MS (ESI) m/z 988 (M+H)+.
Supplementary Material
Scheme 1.
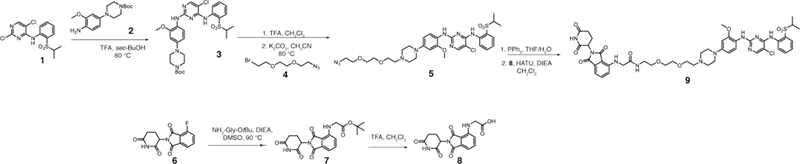
Synthesis of 9
Acknowledgments
Funding Sources
Funding for this work was received from the National Institutes of Health (NIH): Grant R01 CA136851–08 to P.A.J. and N.S.G.; Grant R01 CA148688 to R.E.G.; Grant R01 CA214608–01 to E.S.F. This work was also supported by NIH Grant Numbers 5 T32 GM095450–04 and 2 T32 GM007306–40 (C.E.P.), and a Friends for Life Neuroblastoma Fellowship (Y.G.).
ABBREVIATIONS
- ABCB1
ATP-binding cassette sub-family B member 1
- ALCL
anaplastic large-cell lymphoma
- ALK
anaplastic lymphoma kinase
- EML4
echinoderm microtubule-associated protein-like 4
- NB
neuroblastoma
- NPM
nucleoplasmin
- NSCLC
non-small-cell lung cancer
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental methods for biochemical assays, cell culture, cellular assays, and expression proteomics.
IC50s for biochemical assays and cellular IC50 curves. Dose-response westerns. shRNA studies for ABCB1. Expression proteomics in Kelly cells. Cellular IC50 values for Ba/F3 EML4-ALK cells.
Molecular formula strings
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Chiarle R; Voena C; Ambrogio C; Piva R; Inghirami G The Anaplastic Lymphoma Kinase in the Pathogenesis of Cancer. Nature Reviews Cancer 2008, 8, 11–23. [DOI] [PubMed] [Google Scholar]
- (2).Roskoski R Jr, Anaplastic Lymphoma Kinase (ALK): Structure, Oncogenic Activation, and Pharmacological Inhibition. Pharmacological Research 2013, 68, 68–94. [DOI] [PubMed] [Google Scholar]
- (3).Bayliss R; Choi J; Fennell DA; Fry AM; Richards MW Molecular Mechanisms that Underpin EML4-ALK Driven Cancers and Their Response to Targeted Drugs. Cellular and Molecular Life Sciences 2016, 73, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).George RE; Sanda T; Hanna M; Fröhling S; II WL; Zhang J; Ahn Y; Zhou W; London WB; McGrady P; Xue L; Zozulya S; Gregor VE; Webb TR; Gray NS; Gilliland DG; Diller L; Greulich H; Morris SW; Meyerson M; Look AT Activating Mutations in ALK Provide a Therapeutic Target in Neuroblastoma. Nature 2008, 455, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mossé YP; Laudenslager M; Longo L; Cole KA; Wood A; Attiyeh EF; Laquaglia MJ; Sennett R; Lynch JE; Perri P; Laureys G; Speleman F; Kim C; Hou C; Hakonarson H; Torkamani A; Schork NJ; Brodeur GM; Tonini GP; Rappaport E; Devoto M; Maris JM Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene. Nature 2008, 455, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Palmer RH; Vernersson E; Grabbe C; Hallberg B Anaplastic Lymphoma Kinase: Signalling in Development and Disease. Biochem. J. 2009, 420, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Morris SW; Naeve C; Mathew P; James PL; Kirstein MN; Cui X; Witte DP ALK, the Chromosome 2 Gene Locus Altered by the t(2;5) in Non-Hodgkin’s Lymphoma, Encodes a Novel Neural Receptor Tyrosine Kinase that is Highly Related to Leukocyte Tyrosine Kinase (LTK). Oncogene 1997, 14, 2175–2188. [DOI] [PubMed] [Google Scholar]
- (8).Peters S; Camidge DR; Shaw AT; Gadgeel S; Ahn JS; Kim D-W; Ou S-HI; Pérol M; Dziadziuszko R; Rosell R; Zeaiter A; Mitry E; Golding S; Balas B; Noe J; Morcos PN; Mok T Alectinib Versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. New England Journal of Medicine 2017, 377, 829–838. [DOI] [PubMed] [Google Scholar]
- (9).Soria J-C; Tan DSW; Chiari R; Wu Y-L; Paz-Ares L; Wolf J; Geater SL; Orlov S; Cortinovis D; Yu C-J; Hochmair M; Cortot AB; Tsai C-M; Moro-Sibilot D; Campelo RG; McCulloch T; Sen P; Dugan M; Pantano S; Branle F; Massacesi C; de Castro G Jr, First-Line Ceritinib Versus Platinum-Based Chemotherapy in Advanced ALK-Rearranged Non-Small-Cell Lung Cancer (ASCEND-4): a Randomised, Open-Label, Phase 3 Study. The Lancet 2017, 389, 917–929. [DOI] [PubMed] [Google Scholar]
- (10).Katayama R; Shaw AT; Khan TM; Mino-Kenudson M; Solomon BJ; Halmos B; Jessop NA; Wain JC; Yeo AT; Benes C; Drew L; Saeh JC; Crosby K; Sequist LV; Iafrate AJ; Engelman JA Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Science Translational Medicine 2012, 4, 120ra17–120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cooper MR; Chim H; Chan H; Durand C Ceritinib: A New Tyrosine Kinase Inhibitor for Non-Small-Cell Lung Cancer. Annals of Pharmacotherapy 2014, 49, 107–112. [DOI] [PubMed] [Google Scholar]
- (12).Sullivan I; Planchard D ALK Inhibitors in Non-Small Cell Lung Cancer: the Latest Evidence and Developments. Therapeutic Advances in Medical Oncology 2015, 8, 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mologni L Expanding the Portfolio of Anti-ALK Weapons. Translational Lung Cancer Research 2015, 4, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Katayama R; Friboulet L; Koike S; Lockerman EL; Khan TM; Gainor JF; Iafrate AJ; Takeuchi K; Taiji M; Okuno Y; Fujita N; Engelman JA; Shaw AT Two Novel ALK Mutations Mediate Acquired Resistance to the Next-Generation ALK Inhibitor Alectinib. Clinical Cancer Research 2014, 20, 5686–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Qin A; Gadgeel S The Current Landscape of Anaplastic Lymphoma Kinase (ALK) in Non-Small Cell Lung Cancer: Emerging Treatment Paradigms and Future Directions. Targeted Oncology 2017, 12, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shaw AT; Friboulet L; Leshchiner I; Gainor JF; Bergqvist S; Brooun A; Burke BJ; Deng Y-L; Liu W; Dardaei L; Frias RL; Schultz KR; Logan J; James LP; Smeal T; Timofeevski S; Katayama R; Iafrate AJ; Le L; McTigue M; Getz G; Johnson TW; Engelman JA Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. New England Journal of Medicine 2016, 374, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angewandte Chemie 2015, 128, 818–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Huang H-T; Dobrovolsky D; Paulk J; Yang G; Weisberg EL; Doctor ZM; Buckley DL; Cho J-H; Ko E; Jang J; Shi K; Choi HG; Griffin JD; Li Y; Treon SP; Fischer ES; Bradner JE; Tan L; Gray NS A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chemical Biology 2017, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Davis MI; Hunt JP; Herrgard S; Pietro Ciceri; Wodicka LM.; Pallares G.; Hocker M.; Treiber DK; Zarrinkar, PP. Comprehensive Analysis of Kinase Inhibitor Selectivity. Nature Biotechnology 2011, 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- (21).Marsilje TH; Pei W; Chen B; Lu W; Uno T; Jin Y; Jiang T; Kim S; Li N; Warmuth M; Sarkisova Y; Sun F; Steffy A; Pferdekamper AC; Li AG; Joseph SB; Kim Y; Liu B; Tuntland T; Cui X; Gray NS; Steensma R; Wan Y; Jiang J; Chopiuk G; Li J; Gordon WP; Richmond W; Johnson K; Chang J; Groessl T; He Y-Q; Phimister A; Aycinena A; Lee CC; Bursulaya B; Karanewsky DS; Seidel HM; Harris JL; Michellys P-Y Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro- N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)- N4-(2-(isopropylsulfonyl)phenyl) pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [DOI] [PubMed] [Google Scholar]
- (22).Takezawa K; Okamoto I; Nishio K; Janne PA; Nakagawa K Role of ERK-BIM and STAT3-Survivin Signaling Pathways in ALK Inhibitor-Induced Apoptosis in EML4-ALK-Positive Lung Cancer. Clinical Cancer Research 2011, 17, 2140–2148. [DOI] [PubMed] [Google Scholar]
- (23).Ott GR; Tripathy R; Cheng M; McHugh R; Anzalone AV; Underiner TL; Curry MA; Quail MR; Lu L; Wan W; Angeles TS; Albom MS; Aimone LD; Ator MA; Ruggeri BA; Dorsey BD Discovery of a Potent Inhibitor of Anaplastic Lymphoma Kinase with in Vivo Antitumor Activity. ACS Medicinal Chemistry Letters 2010, 1, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang S; Anjum R; Squillace R; Nadworny S; Zhou T; Keats J; Ning Y; Wardwell SD; Miller D; Song Y; Eichinger L; Moran L; Huang W-S; Liu S; Zou D; Wang Y; Mohemmad Q; Jang HG; Ye E; Narasimhan N; Wang F; Miret J; Zhu X; Clackson T; Dalgarno D; Shakespeare WC; Rivera VM The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clinical Cancer Research 2016, 22, 5527–5538. [DOI] [PubMed] [Google Scholar]
- (25).Yu DMT; Huynh T; Truong AM; Haber M; Norris MD ABC Transporters and Neuroblastoma; 1st ed Elsevier Inc: Cambridge, MA, 2015; Vol. 125, pp. 139–170. [DOI] [PubMed] [Google Scholar]
- (26).Roe M; Folkes A; Ashworth P; Brumwell J; Chima L; Hunjan S; Pretswell I; Dangerfield W; Ryder H; Charlton P Reversal of P-Glycoprotein Mediated Multidrug Resistance by Novel Anthranilamide Derivatives. Bioorganic & Medicinal Chemistry Letters 1999, 595–600. [DOI] [PubMed] [Google Scholar]
- (27).Korolchuk VI; Menzies FM; Rubinsztein DC Mechanisms of Cross-Talk Between the Ubiquitin-Proteasome and Autophagy-Lysosome Systems. FEBS Letters 2010, 584, 1393–1398. [DOI] [PubMed] [Google Scholar]
- (28).Lee H-J; Zhuang G; Cao Y; Du P; Kim H-J; Settleman J Drug Resistance via Feedback Activation of Stat3 in Oncogene-Addicted Cancer Cells. Cancer Cell 2014, 26, 207–221. [DOI] [PubMed] [Google Scholar]
- (29).Galkin AV; Melnick JS; Kim S; Hood TL; Li N; Li L; Xia G; Steensma R; Chopiuk G; Jiang J; Wan Y; Ding P; Liu Y; Sun F; Schultz PG; Gray NS; Warmuth M Identification of NVP-TAE684, a Potent, Selective, and Efficacious Inhibitor of NPM-ALK. Proceedings of the National Academy of Sciences 2006, 104, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).An J; Ponthier CM; Sack R; Seebacher J; Stadler MB; Donovan KA; Fischer ES pSILAC Mass Spectrometry Reveals ZFP91 as IMiD-Dependent Substrate of the CRL4CRBN Ubiquitin Ligase. Nature Communications 2017, 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem. 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Team, R. C. R: A Language and Environment for Statistical Computing http://www.R-project.org/ (accessed Nov. 1, 2017).
- (34).Ritchie ME; Phipson B; Wu D; Hu Y; Law CW; Shi W; Smyth GK Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Research 2015, 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chipumuro E; Marco E; Christensen CL; Kwiatkowski N; Zhang T; Hatheway CM; Abraham BJ; Sharma B; Yeung C; Altabef A; Perez-Atayde A; Wong K-K; Yuan G-C; Gray NS; Young RA; George RE CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.