

Abstract
Systems biology provides an effective approach to decipher, predict, and ultimately manipulate the complex and inter-connected networks that regulate the immune system. Advances in high-throughput, multiplexed experimental techniques have increased the availability of proteomic and transcriptomic immunological datasets, and as a result, have also accelerated the development of new data-driven computational algorithms to extract biological insight from these data. This review highlights how data-driven statistical models have been used to characterize immune cell subsets and their functions, to map the signaling and intercellular networks that regulate immune responses, and to connect immune cell states to disease outcomes to generate hypotheses for novel therapeutic strategies. We focus on recent advances in evaluating immune cell responses following viral infection and in the tumor microenvironment, which hold promise for improving vaccines, antiviral and cancer immunotherapy.
Graphical abstract
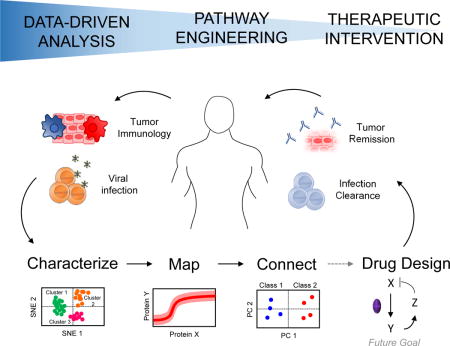
Multiplexed experimental technologies are accelerating data-driven systems immunology
Systems biology seeks to understand how elements of a protein or cellular network interact to produce diverse biological outcomes. Systems approaches that combine high-resolution multiplexed experimental measurements and computational analyses are especially advantageous when studying the immune system, a dynamic network of heterogeneous cell types and non-linear, interconnected signaling pathways [1]. To comprehensively characterize complex cellular behaviors, systems biology relies on multiplexed measurements of signaling and transcription in cell populations or single cells over time and in response to perturbations. Because phenotypic heterogeneity between immune cell types and within immune cell subsets is critical for immune function, the rapid increase in single-cell data collection in particular has catalyzed the growth of systems immunology [2–4].
Experimental techniques include RNA-sequencing, bead-based immunoassays, microwell assays [5,6], flow cytometry, and mass cytometry [7], and each approach has advantages and disadvantages. For example, mass cytometry (or cytometry by time-of-flight, CyTOF) enables multiplexed measurements of more than 40 proteins in thousands of single cell but requires pre-determined antibody panels [7]. In contrast, single-cell RNA-sequencing (scRNA-seq) measures thousands of transcripts for a more comprehensive and unbiased approach, but the measurements are more susceptible to experimental noise, particularly for low-abundance transcripts [2]. These differences must be considered when developing methods to generate biological insights from high-dimensional datasets.
Data-driven statistical algorithms for extracting information from large datasets (Table 1) have paralleled recent advances in multiplexed measurements, especially in single cells. Data-driven modeling uses mathematical analyses and computational algorithms to infer biological relationships. Computational data-driven approaches can be implemented even when knowledge of the underlying immune response mechanism is incomplete, as is often the case. The biological insights gained from data-driven modeling depend on the types of measurements available and the biological relationships being tested [8]. For example, data-driven models can be used to characterize immune cells by their associated phenotypes and functions, map signaling network influences intracellular and extracellularly, or connect biological states with outcomes such as disease status (Figure 1). This review highlights recent examples of data-driven systems modeling that characterize, map, or connect components of the immune system, with a focus on applications in virus-host interactions and cancer immunotherapy.
Table 1.
Data-driven algorithms for high-dimensional analysis discussed in this review.
| Method | Description | Ref. |
|---|---|---|
| PCA | A linear dimensionality reduction method that maps data into a new coordinate system that captures the covariation in the data | [52] |
| PLS-DA | A dimensionality reduction method that linearly links covarying signals to associated outcomes; can be used for classification and prediction | [53] |
| t-SNE | t-distributed stochastic neighbor embedding is a nonlinear dimensionality reduction method used for visualizing high-dimensional data in 2D or 3D | [10,11] |
| Self-organizing maps | A dimensionality reduction and unsupervised clustering method to visualize discrete populations on a map. FlowSOM is modified for flow- and mass- cytometry data. | [43,54] |
| SPADE | A clustering method that hierarchically orders changes in marker expression to depict groups in a minimally spanning tree | [12] |
| Scaffold Maps | A clustering method that visualizes cellular landscapes with pre-defined landmark populations | [17] |
| PhenoGraph | An unsupervised clustering method based on Louvain modularity often used to partition single-cell data into subsets | [25] |
| CellCnn | A clustering method based on convolutional neural networks that has been applied to detect rare cell populations associated with disease | [44] |
| ISOMAP | A trajectory visualization method that can be used to model cellular phenotypic progression based on the geodesic distances between cells | [16] |
| DREVI, DREMI | An inference method that applies conditional density estimation to visualize pairwise interactions and then calculates mutual information to score the strength of the interactions | [21] |
| SLIDE | An inference method that calculates differences between nearest neighbor cells to identify remodeled signaling pathways | [35] |
| Gaussian Graphical Modeling | A network inference method that quantifies partial correlations to define the dependence between variables. | [55] |
| DBScan | A clustering method that uses a density-based algorithm to discover clusters in high-dimensional space. | [56] |
| SC3 | A clustering method that combined multiple clustering solutions to reach a consensus clustering of single-cell RNA-seq data | [57] |
Figure 1.
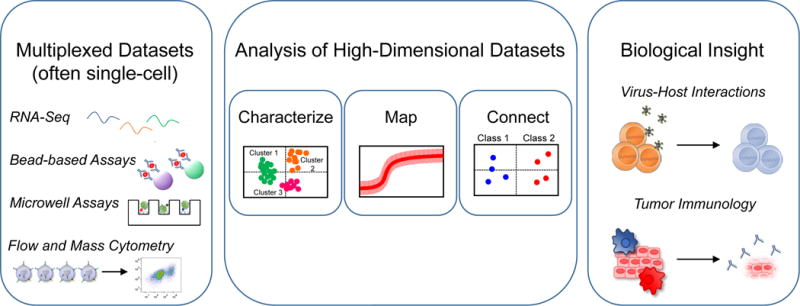
Quantitative analyses shift the interpretation of large-scale systems datasets towards higher-order biological and mechanistic insights. The collection of high-dimensional datasets (left) can be computationally analyzed to characterize immune cells by their associated phenotypes and functions, map intracellular and extracellular signaling network influences, and connect biological states with outcomes (for example, disease status) (middle). These findings can be translated to identify new therapeutic targets to prime the immune system for antiviral and anti-tumor responses.
Characterization of immune cell subsets and their functions
To characterize immune cell heterogeneity across functional subsets of the immune system, many researchers have interpreted high-dimensional protein measurements using dimensionality reduction techniques. Newell et al. measured surface markers and combinatorial cytokine expression of CD8+ T cells–critical effector cells of the adaptive immune system–by mass cytometry. They then used principal component analysis (PCA), a linear transformation of the data that maps cells onto lower dimensional axes of maximum covariance, to separate naïve, central memory, and effector memory CD8+ T cells and identified new functional features associated with these subtypes [9]. Amir et al. robustly separated distinct immune cell types in leukemic bone marrow by visualizing mass cytometry data with t-distributed stochastic neighbor embedding (t-SNE) [10], a non-linear dimensionality reduction algorithm that preserves local structure when mapping the high-dimensional space in two or three dimensions [11].
Functional heterogeneity also exists within a single immune cell type, as seen in natural killer (NK) cells and B cells, two populations critical for immune defense. To understand how both genetic and environmental components contribute to NK cell diversity, NK cells were isolated from monozygotic twins, characterized by 37-dimensional mass cytometry, and clustered using spanning-tree progression analysis of density-normalized events (SPADE) [12]. SPADE was a useful way to visualize how genetics and environment shape NK cell diversity because the algorithm emphasizes rare subpopulations, groups cells with similar phenotypes, and links clusters with weighted edges to identify relationships [13]. In a similar example, clustering of mass cytometry data tracking B-cell differentiation during rotavirus infection identified diverse mucosal trafficking phenotypes associated with humoral B-cell memory to the pathogen [14].
Given the growing number of dimensionality reduction and clustering techniques available to visualize and separate immune cell subsets, it is reasonable to ask how they perform relative to each other. Wong et al. characterized surface and functional markers of CD4+ T helper (TH) cells, a subset of T cells that promote adaptive immunity by activating B cells and cytotoxic CD8+ T cells [15]. They found that t-SNE outperformed PCA for separating known TH cell subsets; but isometric feature mapping (ISOMAP) [16], a method to visualize the relative similarities between subsets by mapping non-linear distances, was better at capturing how TH cells progressed phenotypically over time [15].
A challenge in systems immunology is comparing newly discovered features of immune cell populations that were characterized in studies using different measurements and data-driven statistical methods. The robustness of these discoveries depends on the experimental method used and the number of dimensions measured: as more parameters are measured, clustering and low-dimensionality projections sometimes become less stable due to increased data noise, and thus approaches for accurate, stable comparisons across high-dimensional studies are needed. To address this, Spitzer et al. developed a data-driven reference map, termed Scaffold map, by clustering single-cell data from murine bone marrow using force-directed graphs around “landmark nodes” of commonly recognized immune subsets. They then identified changes in immune composition across studies by comparing this reference map to Scaffold maps of different tissues and species across mass cytometry and archived flow cytometry data [17]. Such frameworks are essential for establishing a reference point to examine changes in immune composition associated with disease [18].
Some studies combine dimensionality reduction and data visualization with clustering to characterize distinct immune cell types within healthy and diseased microenvironments. For example, Tirosh et al. profiled malignant, immune, stromal and endothelial cells in metastatic melanoma patients using scRNA-seq, and used an algorithm called DBScan, which analyzes the density rather than the distance between data points to identify clusters of arbitrary shapes. They first used DBscan to cluster the data after dimensionality reduction using t-SNE, and then used PCA to identify subgroups within these clusters. Together, these methods characterized heterogeneous immune phenotypes and identified exhausted tumor-infiltrating T cells in melanoma tumors [19]. Zheng et al. combined t-SNE visualization with an unsupervised clustering algorithm called SC3, which returns robust and stable clusters by combining multiple clustering solutions to reach a consensus clustered dataset – an approach designed to address the instability of high-dimensional measurements. This analysis identified an enrichment of exhausted CD8+ T cells and inhibitory regulatory T cells in liver tumors relative to healthy liver, as well as potential regulatory genes enriched in these immune subsets [20]. These types of systems-level classifications will aid the design of novel cancer immunotherapies.
Mapping signaling network influences and network connectivity
Data-driven modeling can also map intracellular signaling and transcriptional networks or extracellular cell-cell interaction networks that regulate immune cell responses. Importantly, multiplexed measurements in single cells provide the thousands of observations typically required to mathematically extract network connectivity. To generate hypotheses for how dependencies between proteins change under different conditions, Krishnaswamy et al. developed conditional-density rescaled visualization (DREVI), a technique that emphasizes rare events (e.g., rare cells) by normalizing events based on conditional density, and then further analyzes mutual information between variables (e.g., proteins) in the rescaled data (DREMI) [21]. They applied these techniques to single-cell mass cytometry measurements of signaling network activation in naïve and antigen-exposed CD4+ T cells to show that DREVI effectively reconstructed signal response functions between T-cell signaling proteins, and DREMI quantified how signaling dependencies change in naïve versus antigen-exposed T cells [21]. In type 1 diabetic mice, DREVI analysis of T cell signaling revealed that small impairments in signaling activation proximal to the T cell receptor are amplified to produce larger defects at downstream signaling nodes [22].
A promising approach to map out immune signaling networks relies on taking high-dimensional measurements following targeted perturbations and then using data-driven approaches to identify network-level changes. For example, Martins et al. measured high-dimensional gene expression variation in monocytes after stimulation with cytokine perturbations representing different environmental contexts. By performing multiplexed qPCR on single cells and small cell groups, they were able to overcome “drop-out noise” for transcripts with lower abundance and calculate the propagation of cell-to-cell variation across multiple genes to uncover novel regulatory gene networks in macrophages that modulate noise to adapt to distinct environments [23]. Another study combined single-cell RNA-seq with CRISPR-pooled screens to measure how genetic perturbations affect transcriptome-wide regulatory circuits in single cells [24]. They identified unique cell phenotypes using dimensionality reduction and clustering with Phenograph [25], an unsupervised clustering algorithm that identifies nearest neighbors in high-dimensional space, and linked these phenotypes to their respective genotypes from the CRISPR screen. Similarly, Xue et al. used RNA-seq to measure macrophage transcriptomes after treatment with diverse stimuli, and used a combination of co-expression analysis, hierarchical clustering and self-organizing maps (SOM)–an unsupervised dimensionality reduction algorithm that projects and clusters data as a discrete lower-dimensional map–to identify stimulus-specific gene network programs [26].
To analyze how extracellular signaling networks regulated immune responses, our lab used Gaussian graphical modeling (GGM) to calculate partial correlations between proteins secreted by isolated macrophages, and reconstructed autocrine and paracrine signaling networks that drive the inflammatory stimulus response [27]. Another study characterized communication between the innate and adaptive immune systems by co-culturing monocytes and T cells and implementing self-organizing maps to uncover cytokines regulated by intercellular communication [28]. To dissect cell-to-cell communication across the entire immune system, Rieckmann et al. characterized human hematopoietic populations with mass spectrometry, and computed interaction network maps of “sender” and “receiver” cells by connecting the corresponding receptor-ligand expression levels between different cell populations [29]. Thus, data-driven modeling can identify networks and pathway connectivity in broad immunological settings.
Connecting immune cell states and composition with outcomes
Data-driven modeling can also be used to connect biological states to clinically important outcomes, such as disease progression or therapeutic response, as illustrated below with examples drawn from virus-host interactions and cancer immunotherapy.
Connecting immune cell states to various aspects of infection
Connecting systems-level changes in host cell signaling or gene expression patterns with the state or severity of viral infection can increase understanding of viral pathogenesis. Data-driven models have been used to connect alterations in neutrophil, NK cell, macrophage, and T cell function with increased susceptibility to viral infection [30–34]. Sen et al. developed a distance-based algorithm called SLIDE that quantifies the similarity between a virally-infected cell and its most closely-related uninfected cell in multi-dimensional space [35]. Cavrois et al. used SLIDE to analyze differential surface marker levels between infected and uninfected memory T cells measured by mass cytometry and defined signatures of HIV-susceptible cells, termed ‘predicted precursors’ of infected cells [36].
Data-driven algorithms can also identify molecular or cellular signatures associated with virus manipulation of the host system. Using t-SNE visualization of cytokine and cell surface marker data measured by mass cytometry, Hamlin et al. showed that dengue virus alters the dynamics of cytokine production and primes the immune response to promote viremia rather than clearance [37]. Clustering analysis performed on whole proteome mass spectrometry profiles showed that HIV-infected patient CD4+ T cells on anti-retroviral therapy exhibited decreases in toll-like receptor 4 and type 1 interferon signaling relative to paired uninfected CD4+ T cell data [38]. To classify HIV+, T-cell depleted, and healthy PBMC samples, partial least squares discriminant analysis (PLS-DA)– which identifies linear combinations of variables to maximally separate known biological states –identified classification signatures based on measurements of 16 cytokines and chemokines, and then the underlying molecular changes could be associated with each group [39]. In a study from our lab, we measured phospho-signaling differences in uninfected and latent HIV-infected T cells and used PLS-DA to define signatures that predicted infection status and then identified changes in stress kinase signaling as a novel target for preferential infected-cell killing [40]. Comparing total immune cell composition in healthy and infected donors, SPADE and t-SNE analyses performed on 26-parameter mass cytometry data demonstrated that infected patients had a higher fraction of highly differentiated effector memory cells following HIV infection and ART treatment in comparison to healthy donors [41]. Overall, systems immunology studies of virus-host immune interactions have generated new hypotheses about mechanisms of viral pathogenesis and potential strategies to combat viral infection.
Connecting immune cell states to cancer
Data-driven approaches can also be used to generate immunological signatures from patient data that are predictive of clinical outcomes, a key goal in cancer immunology. Chevrier et al. used mass cytometry to profile tumors from renal carcinoma patients and found Phenograph-defined clusters of T cells and macrophages that correlated with progression-free survival [42]. A recent study by Krieg et al. used FlowSOM [43], a self-organizing map algorithm, and CellCnn [44], an algorithm that uses neural networks to detect rare cell populations associated with disease, to identify unique monocyte populations predictive of a clinical response to anti-PD-1 immunotherapy [45]. Together, these studies show that immune cells or system states can be connected in a predictive manner to biological and clinical outcomes.
Another goal is to identify immune populations correlated with disease progression that may become novel targets for immunotherapy. For example, patient-derived immune cells in tumor, non-involved lung tissue, and blood compartments were profiled using mass cytometry, and immune composition was defined using Phenograph clustering, which highlighted changes associated early lung adenocarcinoma progression and identified tumor-infiltrating myeloid cells as potential therapeutic targets [46]. Spitzer et al. used mass cytometry and Scaffold maps to characterize immune composition across successful and failed immunotherapy treatments and connect peripheral CD4+ T cells to systemic immune responses required for tumor rejection [18]. Together, these systems-level studies uncovered key immune populations and potential therapeutic targets associated with disease.
Systems-level approaches also provide insights into mechanisms underlying cancer immunotherapy efficacy, which remain largely unknown. Infiltrating T cells from human and mouse melanomas were profiled by mass cytometry following treatment with immune checkpoint inhibitors anti-CTLA-4 and anti-PD-1, and condensed via clustering and PCA to reveal conserved and unique mechanisms for both immunotherapies [47]. In an example from our lab, we used microwell assays to characterize functional “secretome” changes in tumor-associated monocytes and macrophages (TAMs) following combination immunotherapy in melanoma [48]. Phenograph clustering of the secretome identified changes in TAM subsets, including the emergence of a novel TAM subset that secretes inflammatory cytokines with anti-tumor activity. Taken together, these studies demonstrate how systems-level profiling is uncovering new strategies to redirect the immune system for therapeutic benefit.
Conclusion
Systems immunology is poised for rapid growth. As the field matures, common analytical methods such as t-SNE have risen as consensus algorithms for non-linear single-cell data visualization, while new methods are being introduced to address data challenges that still limit biological interpretation. For example, new computational methods are being developed to address “drop-out noise” in single-cell RNA-seq data [49], to identify underlying data structures that are continuous and non-linear [50], and to speed up computation time [51]. Other challenges still to be addressed include integrating across different types of data sets: incorporating single-cell data with measurements collected in cell populations; and incorporating prior biological knowledge, such as time series and dose responses, to extract more biological insight from the same data set. In the next few years, we expect to see systems immunology studies move towards mechanistic discovery by combining data-driven approaches with hypothesis-driven modeling. By using data-driven modeling to generate a global view of the immune network and hypothesis-driven modeling to test specific mechanisms within the network, systems immunology will uncover new strategies to engineer the immune system and combat disease.
Highlights (target for each: 85 characters with spaces).
New experimental techniques provide high-content data of immune system function.
Data-driven computational approaches enable new insights from immune cell data.
Recent applications characterize immune cell subsets and map network influences.
Connecting immune states to disease states will uncover new strategies for therapy.
Acknowledgments
We would like to thank all the members of the Miller-Jensen lab for helpful comments and discussion. This work was supported by the National Institutes of Health (R01 GM123011-01 and R21 AI132013-01A1 to K.M.-J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wong HS, Germain RN. Robust control of the adaptive immune system. Semin Immunol. 2017 doi: 10.1016/j.smim.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2017;18:35–45. doi: 10.1038/nri.2017.76. [DOI] [PubMed] [Google Scholar]
- 3.Di Palma S, Bodenmiller B. Unraveling cell populations in tumors by single-cell mass cytometry. Curr Opin Biotechnol. 2015;31:122–129. doi: 10.1016/j.copbio.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay PK, Gierahn TM, Roederer M, Love JC. Single-cell technologies for monitoring immune systems. Nat Immunol. 2014;15:128–135. doi: 10.1038/ni.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varadarajan N, Julg B, Yamanaka YJ, Chen H, Ogunniyi AO, McAndrew E, Porter LC, Piechocka-Trocha A, Hill BJ, Douek DC, et al. A high-throughput single-cell analysis of human CD8+ T cell functions reveals discordance for cytokine secretion and cytolysis. J Clin Invest. 2011;121:4322–4331. doi: 10.1172/JCI58653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, Chen JJ, Mu L, Xue Q, Wu Y, Wu P-H, Li J, Vortmeyer AO, Miller-Jensen K, Wirtz D, et al. High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal Chem. 2013;85:2548–56. doi: 10.1021/ac400082e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 8.Benedict KF, Lauffenburger DA. Insights into Proteomic Immune Cell Signaling and Communication via Data-Driven Modeling. In: Katze M, editor. Systems Biology Current Topics in Microbiology and Immunology. Springer; Berlin Heidelberg: pp. 2012pp. 201–233. [DOI] [PubMed] [Google Scholar]
- 9.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by Time-of-Flight Shows Combinatorial Cytokine Expression and Virus-Specific Cell Niches within a Continuum of CD8 + T Cell Phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Der Maaten LJP, Hinton GE. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 12.Qiu P, Simonds EF, Bendall SC, Gibbs KD, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–893. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Dekker CL, Mackey S, Maecker H, Swan GE, et al. Genetic and Environmental Determinants of Human NK Cell Diversity Revealed by Mass Cytometry. Sci Transl Med. 2013;5:208ra145–208ra145. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair N, Newell EW, Vollmers C, Quake SR, Morton JM, Davis MM, He XS, Greenberg HB. High-dimensional immune profiling of total and rotavirus VP6-specific intestinal and circulating B cells by mass cytometry. Mucosal Immunol. 2016;9:68–82. doi: 10.1038/mi.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Wong MT, Chen J, Narayanan S, Lin W, Anicete R, Kiaang HTK, De Lafaille MAC, Poidinger M, Newell EW. Mapping the Diversity of Follicular Helper T Cells in Human Blood and Tonsils Using High-Dimensional Mass Cytometry Analysis. Cell Rep. 2015;11:1822–1833. doi: 10.1016/j.celrep.2015.05.022. This study compares the performance of dimensionality reduction and automated clustering methods (e.g., t-SNE, PCA and ISOMAP), for analyzing T helper cell developmental progression from mass cytometry measurements. [DOI] [PubMed] [Google Scholar]
- 16.Tenenbaum JB, de Silva V, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–23. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- 17••.Spitzer MH, Gherardini PF, Fragiadakis GK, Bhattacharya N, Yuan RT, Hotson AN, Finck R, Carmi Y, Zunder ER, Fantl WJ, et al. An interactive reference framework for modeling a dynamic immune system. Science. 2015;349:1259425–1259425. doi: 10.1126/science.1259425. This study created and applied Scaffold maps, a cluster-mapping approach that established a reference map for immune compositions and enables comparisons across different samples. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502.e15. doi: 10.1016/j.cell.2016.12.022. In this study, the authors applied Scaffold maps to compare the immune composition of untreated and immunotherapy-treated tumors and identified systemic immunity as an important factor required for succesful cancer immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 21.Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D, Nolan GP. Conditional density-based analysis of T cell signaling in single-cell data. Science. 2014;346:1250689–1250689. doi: 10.1126/science.1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Mingueneau M, Krishnaswamy S, Spitzer MH, Bendall SC, Stone EL, Hedrick SM, Pe’er D, Mathis D, Nolan GP, Benoist C. Single-cell mass cytometry of TCR signaling: Amplification of small initial differences results in low ERK activation in NOD mice. Proc Natl Acad Sci. 2014;111:16466–16471. doi: 10.1073/pnas.1419337111. This study used DREVI and DREMI analysis to analyze TCR signaling dynamics measured by mass cytometry in control C57BL/6 and non-obese diabetic mice to show how diabetes results in dysregulated propagation of TCR-mediated signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martins AJ, Narayanan M, Prüstel T, Fixsen B, Park K, Gottschalk RA, Lu Y, Andrews-Pfannkoch C, Lau WW, Wendelsdorf KV, et al. Environment Tunes Propagation of Cell-to-Cell Variation in the Human Macrophage Gene Network. Cell Syst. 2017;4:379–392.e12. doi: 10.1016/j.cels.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 2016;167:1883–1896. doi: 10.1016/j.cell.2016.11.039. e15. [DOI] [PubMed] [Google Scholar]
- 25.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Q, Lu Y, Eisele MR, Sulistijo ES, Khan N, Fan R, Miller-Jensen K. Analysis of single-cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci Signal. 2015;8:ra59–ra59. doi: 10.1126/scisignal.aaa2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrier SB, Hill AS, Plana D, Lauffenburger DA. Synergistic Communication between CD4+ T Cells and Monocytes Impacts the Cytokine Environment. Sci Rep. 2016;6:34942. doi: 10.1038/srep34942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, Sallusto F, Shen-Orr SS, Lanzavecchia A, Mann M, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. 2017;18:583–593. doi: 10.1038/ni.3693. This study used high-resolution mass-spectrometry to build interaction networks between different immune cell types. They found communication patterns between cells and identified specialized “sender” and “receiver” cells. [DOI] [PubMed] [Google Scholar]
- 30.Whiting CC, Siebert J, Newman AM, Du H, Alizadeh AA, Goronzy J, Weyand CM, Krishnan E, Fathman CG, Maecker HT. Large-Scale and Comprehensive Immune Profiling and Functional Analysis of Normal Human Aging. PLoS One. 2015;10:e0133627. doi: 10.1371/journal.pone.0133627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao Y, Montgomery RR. Role of Immune Aging in Susceptibility to West Nile Virus. In: Colpitts TM, editor. West Nile Virus Methods in Molecular Biology. Springer; New York: 2016. pp. 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao Y, Strauss-Albee DM, Zhou JQ, Malawista A, Garcia MN, Murray KO, Blish CA, Montgomery RR. The natural killer cell response to West Nile virus in young and old individuals with or without a prior history of infection. PLoS One. 2017;12:e0172625. doi: 10.1371/journal.pone.0172625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, Kinuthia J, Montgomery RR, John-Stewart G, Holmes S, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med. 2015;7:297ra115–297ra115. doi: 10.1126/scitranslmed.aac5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustafson CE, Qi Q, Hutter-Saunders J, Gupta S, Jadhav R, Newell E, Maecker H, Weyand CM, Goronzy JJ. Immune Checkpoint Function of CD85j in CD8 T Cell Differentiation and Aging. Front Immunol. 2017;8:1–12. doi: 10.3389/fimmu.2017.00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sen N, Mukherjee G, Sen A, Bendall SC, Sung P, Nolan GP, Arvin AM. Single-Cell Mass Cytometry Analysis of Human Tonsil T Cell Remodeling by Varicella Zoster Virus. Cell Rep. 2014;8:633–645. doi: 10.1016/j.celrep.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.Cavrois M, Banerjee T, Mukherjee G, Raman N, Hussien R, Rodriguez BA, Vasquez J, Spitzer MH, Lazarus NH, Jones JJ, et al. Mass Cytometric Analysis of HIV Entry, Replication, and Remodeling in Tissue CD4+ T Cells. Cell Rep. 2017;20:984–998. doi: 10.1016/j.celrep.2017.06.087. This study implements SLIDE, a distance algorithm which identifies nearest neighbors in an uninfected control, to mass cytometry data to systematically investigate susceptibility to HIV infection. They identify surface markers that increase HIV susceptibility and that are modulated by the virus upon infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37•.Hamlin RE, Rahman A, Pak TR, Maringer K, Mena I, Bernal-Rubio D, Potla U, Maestre AM, Fredericks AC, Amir ED, et al. High-dimensional CyTOF analysis of dengue virus–infected human DCs reveals distinct viral signatures. JCI Insight. 2017;2:e92424. doi: 10.1172/jci.insight.92424. This study is applies t-SNE visualization of 18 phenotypic markers collected by mass cytometry after dengue virus infection and demonstrates that cytokine signaling can prime the immune response to promote viremia rather than clearance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemeth J, Vongrad V, Metzner KJ, Strouvelle VP, Weber R, Pedrioli P, Aebersold R, Günthard HF, Collins BC. In Vivo and in Vitro Proteome Analysis of Human Immunodeficiency Virus (HIV)-1-infected, Human CD4 + T Cells. Mol Cell Proteomics. 2017;16:S108–S123. doi: 10.1074/mcp.M116.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Arnold KB, Szeto GL, Alter G, Irvine DJ, Lauffenburger DA. CD4+ T cell-dependent and CD4+ T cell-independent cytokine-chemokine network changes in the immune responses of HIV-infected individuals. Sci Signal. 2015;8:ra104–ra104. doi: 10.1126/scisignal.aab0808. This study used PLS-DA of 16 secreted signals to discriminate between the effects of T-cell depletion in health versus HIV subsets and found an impairment in NK cell cytokine secretion, thereby identifying a dysregulated immune network that should be taken into consideration during treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Fong LE, Sulistijo ES, Miller-Jensen K. Systems analysis of latent HIV reversal reveals altered stress kinase signaling and increased cell death in infected T cells. Sci Rep. 2017;7:16179. doi: 10.1038/s41598-017-15532-0. This study used PLS-DA to demonstrate that multiplexed phospho-signaling signatures are sufficient to predict the infection status of cells. This study further used PLSR to link dysregulated stress kinase signaling with cell death in HIV-infected T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corneau A, Cosma A, Even S, Katlama C, Le Grand R, Frachet V, Blanc C, Autran B. Comprehensive Mass Cytometry Analysis of Cell Cycle, Activation, and Coinhibitory Receptors Expression in CD4 T Cells from Healthy and HIV-Infected Individuals. Cytom Part B Clin Cytom. 2017;92:21–32. doi: 10.1002/cyto.b.21502. [DOI] [PubMed] [Google Scholar]
- 42.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749. doi: 10.1016/j.cell.2017.04.016. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom Part A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 44.Arvaniti E, Claassen M. Sensitive detection of rare disease-Associated cell subsets via representation learning. Nat Commun. 2017;8:1–10. doi: 10.1038/ncomms14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45••.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, Becher B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24:144–153. doi: 10.1038/nm.4466. The authors used a combination of clustering and machine-learning techniques to identify and characterize an immune biomarker that predicts a patient’s response to cancer immunotherapy with anti-PD-1. This work highlights the direct utility of systems-level approaches to uncover clinically relevant biological insights. [DOI] [PubMed] [Google Scholar]
- 46.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169:750–765. doi: 10.1016/j.cell.2017.04.014. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170:1120–1133. doi: 10.1016/j.cell.2017.07.024. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Perry CJ, Muñoz-Rojas AR, Meeth KM, Kellman LN, Amezquita RA, Thakral D, Du VY, Wang JX, Damsky W, Kuhlmann AL, et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. J Exp Med. 2018;215 doi: 10.1084/jem.20171435. This is the first study to characterize multiplexed single-cell secretion from TAMs in murine melanoma tumors before and after immunotherapy, and then used Phenograph to identify unique functional cell subsets that are induced by immunotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MAGIC. A diffusion-based imputation method reveals gene-gene interactions in single-cell RNA-sequencing data. bioRxiv. 2017 doi: 10.1101/111591. [DOI] [Google Scholar]
- 50.Moon KR. Visualizing Transitions and Structure for High Dimensional Data Exploration. bioRxiv. 2017 doi: 10.1101/120378. [DOI] [Google Scholar]
- 51.Amodio M, Srinivasan K. Exploring Single-Cell Data with Multitasking Deep Neural Networks. bioRxiv. 2017 doi: 10.1101/237065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clark NR, Ma’ayan A. Introduction to Statistical Methods to Analyze Large Data Sets: Principal Components Analysis. Sci Signal. 2011;4:tr3–tr3. doi: 10.1126/scisignal.2001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat Rev Mol Cell Biol. 2006;7:820–8. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 54.Saeys Y, Van Gassen S, Lambrecht BN. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat Rev Immunol. 2016;16:449–462. doi: 10.1038/nri.2016.56. [DOI] [PubMed] [Google Scholar]
- 55.Opgen-Rhein R, Strimmer K. From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst Biol. 2007;1:37. doi: 10.1186/1752-0509-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ester M, Kriegel H-P, Sander J, Xu X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise Martin. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining. 1996:226–231. [Google Scholar]
- 57.Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR, et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat Methods. 2017;14:483–486. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]