

Abstract
Krüppel-like factor 5 (KLF5) both suppresses and promotes tumor growth depending on cellular context. The mechanisms underlying tumor promotion could be targetable for therapy. Although a number of transcriptional targets of KLF5 have been identified and implicated in KLF5-mediated tumor growth, how KLF5 regulates these genes remains to be addressed. Here we performed co-immunoprecipitation (co-IP) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) in the TSU-Pr1 bladder cancer cell line, in which KLF5 is shown to promote tumor growth, to identify KLF5-interacting nuclear proteins that are necessary for KLF5’s tumor promoting function. LC-MS/MS revealed 122 potential KLF5 binding proteins in the nuclear proteins precipitated by the KLF5 antibody, and the top nine candidates included AHNAK, TFAM, HSDL2, HNRNPC, CINP, IST1, FBL, PABPC1, and SNRNP40. SRB assays of these 9 proteins indicated that silencing CINP had the most potent inhibitory effect on cell growth in KLF5-expressing cells but did not affect parental TSU-Pr1 cells. Further analyses not only confirmed the physical interaction between KLF5 and CINP, also demonstrated that knockdown of CINP attenuated the effects of KLF5 on cell cycle progression, apoptosis, and tumorigenesis. Silencing CINP also attenuated the effect of KLF5 on the expression of a number of genes and signaling pathways, including cell cycle regulator Cyclin D1 and apoptosis-related Caspase 7. These results suggest that CINP is a cofactor of KLF5 that is crucial for the promotion of tumor growth, and that the KLF5-CINP interaction could be a novel therapeutic target for inhibiting KLF5-promoted tumor growth.
Keywords: Krüppel-like factor 5 (KLF5), cyclin-dependent kinase 2-interacting protein (CINP), cell proliferation, cell survival, tumorigenesis
INTRODUCTION
Deregulated cell proliferation and evasion of cell death are two hallmarks of tumor development that are caused by activated oncogenic signaling and loss of tumor suppressive signaling 1. The KLF5 transcription factor, a member of the Krüppel-like factor (KLF) family 2, 3, regulates a variety of biological processes including cell proliferation, apoptosis, angiogenesis, stemness, and the epithelial-mesenchymal transition (EMT) in cancer cells. KLF5 is involved in several oncogenic signaling pathways including RAS/ERK, PI3K/AKT, p53, and TGF-β, and is a context-dependent regulator of cancer cell behavior 4–6. In the regulation of cell proliferation and tumorigenesis, KLF5 has demonstrated seemingly opposing effects depending on cell context including the status of TGF-β and likely other signaling activities. For example, in prostate cancer cells, KLF5 is suppressive in the presence of TGF-β, which induces the acetylation of KLF5, but becomes tumor promoting when the acetylation of KLF5 is interrupted 7, 8.
The tumor promoting function of KLF5 involves increased cell proliferation and/or attenuated apoptosis, which have been demonstrated in different types of cells to involve the coordination between oncogenic signaling pathways 4, 5. KLF5 has transforming activity in NIH3T3 fibroblasts 9, and is one of the genes associated with aberrant activation of cell proliferation in the ApcMin mouse model of colorectal cancer 10. KLF5 also acts as an oncogene in intestinal epithelial cells, based on the findings that downregulation of KLF5 leads to reduced cell proliferation and transformation 11, and deletion of Klf5 prevents intestinal tumorigenesis induced by ApcMin and KRASV12 mutations in mice 12. In bladder and breast cancer cell lines, KLF5 has also been shown to promote proliferation, survival, and tumor growth 13–15. Attenuation of apoptosis by increased KLF5 has also been reported in different types of cells including leukemia cells 16, and depletion of KLF5 enhances DNA damage-induced apoptosis by reducing BAD phosphorylation and PIM1 protein expression 17. Recently, Klf5 was demonstrated to cooperate with Sox4 to prevent apoptosis associated with lethal EMT in pancreatic cancer 18. A recent study indicates that somatic coding and noncoding genomic alterations activate KLF5 expression, and increased KLF5 expression is a mechanism for enhanced cell proliferation and tumorigenesis 19. Therefore, targeting the tumor promoting function of KLF5 is a potential approach for the treatment of multiple types of cancer.
A number of genes involved in cell proliferation and tumorigenesis have been identified as transcriptional targets of KLF5 9. For example, KLF5 activates aberrant expression of proliferation-associated genes CCND1 and MYC in colorectal cancer cells 10, and upregulates a number of genes including HBP17, ITGA6, and RAIG1 to promote tumorigenesis in bladder cancer cells 13. KLF5 also interacts with a number of transcription factors to regulate gene transcription. For example, KLF5 interacts with c-Jun to suppress p21 expression in vascular smooth muscle cells 20; and a number of other factors also interact with KLF5, including TBP 21, CBP 22, 23, ERα and ERβ 24, 25, p5316, C/EBPb/d 26, SREBP-127, PARP-128 and TEAD429. Related to its suppression of cell proliferation in the context of TGF-β signaling, KLF5 interacts with SMADs, MYC and p300 to regulate the transcription of p15 and MYC 7, 30, 31. KLF5 forms different transcriptional complexes under different cellular contexts to exert different functions.
At present, the factors that interact with KLF5 to promote cell proliferation and tumorigenesis are largely unknown 7, 30, yet this information is critical for developing novel therapeutics for cancer treatment 32. In this study, we used a previously established model of KLF5-mediated tumor promotion, the TSU-Pr1 bladder cancer cell line in which ectopic expression of KLF5 promotes cell proliferation and tumor growth 13, to identify and characterize proteins that partner with KLF5 to promote tumor growth. Using immunoprecipitation combined with mass spectrometry, a number of KLF5-interacting nuclear proteins were identified. Functional screening revealed that one such protein, the Cdk2-interacting protein CINP, is required for KLF5’s tumor promoting function. CINP was established as a cofactor of KLF5 crucial for tumor growth. The KLF5-CINP interaction provides a therapeutic opportunity for targeting KLF5-mediated tumorigenesis.
MATERIALS & METHODS
Cell culture
The TSU-Pr1 cell line is a derivative of the T24 bladder carcinoma cell line 33. K12 is a clone of TSU-Pr1 cells stably expressing KLF5, and V4 is a vector control clone, both of which were established in a previous study and propagated as previously described 13. Briefly, cells were grown at 37°C in a humidified 5% CO2 incubator in RPMI-1640 medium (Gibco, Gaithersburg, MD) supplemented with 8% fetal bovine serum (FBS) (Sigma-Aldrich, St Louis, MO). All human cell lines, including the embryonic kidney cell line 293T, immortalized epidermal cell line HaCaT and mammary epithelial cell line MCF 10A, and all cancer cell lines (HeLa, cervical cancer; TSU-Pr1, bladder cancer; MCF-7, MDA-MB-231 and T47D, breast cancer; PC-3 and DU-145, prostate cancer), were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and propagated according to manufacturer’s instructions.
Plasmids, siRNAs and other materials
The Myc-CINP plasmid and its vector pCI-Myc were previously described and kindly provided by Dr. Zhenghong Yuan of Fudan University 34. The Flag-KLF5 plasmid has been reported in our previous study 35. Sequences of siRNAs used in this study are listed in Table S8. MG-132 was purchased from Selleck Chemicals (Houston, TX).
Preparation of nuclear extracts and co-immunoprecipitation (IP)
One billion K12 cells were harvested and swollen in ice-cold hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 1 mM freshly added DTT), and 10% IGEPAL®CA-630 solution (Sigma-Aldrich) was added to a final concentration of 0.6%. Nuclei pellets were isolated by centrifugation, and lysed in high salt extraction buffer (20 mM HEPES, pH 7.9, 25% glycerol v/v, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM freshly added DTT). A protease inhibitor cocktail (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail, Roche, Indianapolis, IN) and a phosphatase inhibitor solution (10 mM NaF, 1 mM Na3VO4, 1 mM sodium pyrophosphate with 25 mM sodium β-glycerophosphate) were added to both the hypotonic lysis buffer and the high salt extraction buffer.
Sixty mg mouse monoclonal antibody against KLF5, which was generated and purified using peptide PPVPIIPEHKKYRR at Abmart (Abmart, Shanghai), and mouse normal IgG were coupled with 7.5 mg magnetic Dynabeads M-270 Epoxy according to instructions from the Dynabeads® Co-Immunoprecipitation Kit (Invitrogen Life Technologies). Briefly, antibody-coupled beads were added to nuclear lysate, followed by overnight incubation at 4°C, and washing and elution in citric acid buffer. Eluted sample was concentrated using an Amicon Ultra ultrafiltration tube (Millipore, Billerica, MA), and then separated on NuPAGE® Novex 4-12% Bis-Tris gels (Invitrogen). Gels were stained with Coomassie brilliant blue (Beyotime Institute Biotech, Nanjing). Both the light and heavy IgG bands were excised from the two IP lanes (KLF5 and IgG), and the gel from each lane was then subjected to LC-MS/MS proteomics analysis. Gel areas used for MS are identified by red dotted lines in Figure 1B.
FIGURE 1. Isolation and preparation of proteins for mass spectrometric analysis.
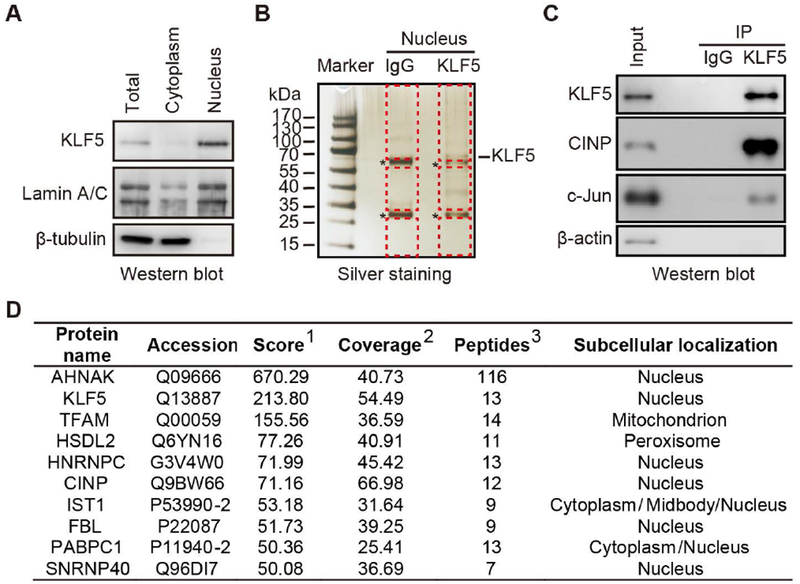
(A) Validation of nuclear extraction from TSU-Pr1 cells stably expressing KLF5 (K12 clone). The lamin A/C nuclear marker and β-tubulin cytoplasmic marker were detected by western blotting. (B) Silver staining gel analysis of KLF5 interacting proteins pulled down using KLF5 antibody by co-IP (about 1/10 of the elution from co-IP) in K12 cells. Proteins pulled down using purified IgG served as a negative control. Bands for the light and heavy chains of IgG are indicated by asterisks. For mass spectrometry, four fifths of the co-IP elution for each KLF5 or the IgG negative control were concentrated, separated in another gel and stained with Coomassie blue, and both the light and heavy chain bands of IgG were excised and discarded to reduce nonspecific readings. The remaining gel of the entire lane, as indicated dotted red lines, was collected and subjected to mass spectrometry. (C) Confirmation of the presence of KLF5, c-Jun, and CINP in the co-IP products by western blotting. c-Jun, a known KLF5 interacting protein, was used as a positive control. β-actin was used as a negative control. (D) Top candidates of KLF5 interacting proteins identified by co-IP and mass spectrometry analysis. 1 Score: Protein scores, calculated by Proteome Discoverer application from a list of peptides identified for a particular protein, indicate the relevance of a protein. 2Coverage: Coverage of identified high-confidence peptides match the protein. 3Peptides: Number of high-confidence peptides which match the protein.
For co-IP validation, 293T cells were co-transfected with pcDNA3.0-Flag or Flag-KLF5 in the same vector with Myc-CINP plasmid using the Lipofectamine 2000 Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. In addition, 293T cells were co-transfected with pCI-Myc vector or Myc-CINP plasmid with Flag-KLF5 plasmid. Co-IP was performed following the standard protocol using anti-FLAG M2 affinity gel (Sigma-Aldrich) or Myc-tag antibody (Cell Signaling Technology) with Protein G Sepharose (GE Healthcare Biosciences, Pittsburgh, PA).
Protein separation and identification by LC-MS/MS analysis
Sample preparation from excised gels and LC-MS/MS were performed by the Center of Biomedical Analysis, Tsinghua University, Beijing, China. Briefly, proteins in gel were reduced by dithiothreitol, alkylated by iodoacetamide, and then digested with trypsin. Peptides were extracted with 50% acetonitrile containing 0.1% formic acid, dried in a speed-vac, and reconstituted in formic acid prepared in water for nano-LC-MS/MS analysis. Peptides were separated by a C18 column (75 µm inner-diameter, 150mm length; Upchurch, Oak Harbor, USA) with a flow rate of 300nL/min. Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The Q-Exactive Orbitrap mass spectrometer was operated in the data-dependent acquisition mode using Thermo Scientific Xcalibur 3.0 software. A full-scan followed by 20 data-dependent MS/MS scans was acquired with collision induced dissociation with normalized collision energy of 35%. The MS/MS spectra from each LC-MS/MS run were queried against the protein database using Proteome Discovery searching algorithm.
Candidate KLF5 interactants were identified from LC-MS/MS analysis by the following parameters: the number of KLF5 associated unique peptides > 2, and that of IgG associated unique peptides = 0, which led to the exclusion of IgG-bound proteins from the KLF5 group. The cellular component function of gene ontology (GO) analysis (http://www.uniprot.org/) was performed to limit KLF5 interactants to nuclear proteins.
Cell proliferation assay
Cells were transfected with siRNAs using Lipofectamine RNAiMax reagent (Invitrogen) according to the manufacturer’s protocol. Twenty-four hours after transfection, cells were seeded into 5 24-well plates. One, 2, 3, 4 and 5 days after seeding, 4 wells of cells for each data point (e.g., siRNA or control) were fixed in 10% TCA (Sigma-Aldrich) culture, stained with 0.4% sulforhodamine-B solution (SRB) (Sigma-Aldrich), and washed with 1% acetic acid. Absorbance was recorded with a spectrometer at 490 nm.
Real-time qPCR
Total RNA was extracted using the Trizol reagent (Invitrogen). Two μg of total RNA from each sample were reverse transcribed to single-stranded cDNA using oligo dT and random primers. One μl of cDNA was used as template for the Real-time RT-PCR, which was performed with the SYBR Green MasterMix reagent (Takara, Tokyo, Japan) in a Mastercycler Realplex thermal cycler (Eppendorf, Shanghai, China). The 2(−△△Ct) method was used to calculate relative fold change with GAPDH as the internal control. The assay was conducted in duplicate or triplicate for each gene. Gene names and primers used for real-time PCR are listed in Table S9.
Tumorigenesis assay
For the tumorigenesis assay, 3-4 week old male BABL/C nude mice were used. For each mouse, a total of 1×106 cells transfected with siCtrl or siCINP, mixed with 0.5 volume of Matrigel, were injected subcutaneously on both sides. Five mice were used for each group. Tumor volumes were measured twice a week. Four weeks later, mice were euthanized; and tumors were surgically dissected, immediately weighed and fixed in 10% formalin for standard histopathological evaluation. These experiments were repeated twice. All of the mice were maintained and handled at an Emory University Division of Animal Resources facility according to the policies of the Institutional Animal Care and Use Committee.
Immunohistochemistry
Immunohistochemistry (IHC) staining was performed to detect protein expression of Ki67, cleaved-caspase3, cyclin D1 and caspase7 in tumor xenografts. Formalin-fixed paraffin-embedded tissues were sectioned at 5 µm, deparaffinized in xylene, rehydrated in graded ethanol, subjected to antigen retrieval by boiling the slides in a pressure cooker for 3 min in a citrate buffer (10 mM trisodium citrate, pH 6.0), and permeabilized with 0.5% (vol/vol) Triton X-100. After 10 min treatment with 3% H2O2, tissue sections were blocked with 5% normal goat serum, incubated first with primary antibodies at 4℃ overnight and then with EnVision Polymer-HRP secondary antibodies (Dako, Glostrup, Denmark) at room temperature for 1 hour. After the application of DAB-chromogen, tissue sections were stained with hematoxylin, dehydrated, and mounted. Antibodies included the following: Ki67 (1:300, Thermo Fisher), cleaved-caspase3 (1:200, Cell Signaling Technology), cyclin D1 (1:250, Abcam), and Caspase 7 (1:250, Abcam).
Cell cycle analysis and apoptosis assay
For cell cycle analysis, cells were collected and fixed in 70% ice-cold ethanol overnight. After washing, cells were resuspended in PBS and incubated with DAPI for 15 min in the dark. Cell cycle analysis was carried out on a Flowsight (EMD Millipore-Amnis, Seattle, WA) instrument. Data was analyzed using the FlowJo software (Treestar Software, San Carlos, CA).
For apoptosis assay, cells were collected, washed with cold PBS, stained with Annexin V-FITC/PI, and analyzed using a Flowsight flow cytometer as previously described 36. Data was analyzed using the Amnis IDEAS software following the manual.
RNA-Seq and bioinformatic analyses
RNA was isolated 48 hours after transfection with siCtrl or siCINP in K12 cells. RNA-Seq analysis was performed using the BGISEQ-500 at the BGI (ShenZhen, China). Briefly, total RNA was extracted, purified and used to construct SE50 RNA-Seq libraries. For each sample, 20M reads were mapped to human HG19 genome using the HISA and Bowtie2 programs. Expression level for a gene was established by the number of fragments per kilobase of exon per million fragments mapped (FPKM) reads using the RSEM tool. Differentially expressed genes were identified using the position distribution method. RNA-Seq data have been deposited to NCBI’s Gene Expression Omnibus database (GSE98930, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98930).
Genes with significantly different expression between siCINP and siCtrl in K12 cells were uploaded to the web-based Meta Core program (http://thomsonreuters.com/metacore), and different algorithms were run with Meta Core’s databases to identify functional processes, molecular pathways, and transcription factor networks. Details of the Meta Core platform and its applications have been described previously 37.
Western blotting
Total protein was collected 72 hours after transfection. The rabbit polyclonal antibody for KLF5 (1:1000 dilution) was generated and described in our previous study 38. Other antibodies used in this study included CINP (1:1000 dilution, Abcam, Cambridge, MA, catalog #ab183519), c-JUN (1:1000 dilution; Cell Signaling Technology, catalog #9155), β-actin (1:5000 dilution, Sigma-Aldrich, catalog #A1978), β-tubulin (1:5000 dilution, Cell Signaling Technology, catalog #2128), cyclin D1 (1:10000 dilution, Abcam, catalog #ab134175), and caspase-7 (1:1000 dilution, Abcam, catalog #ab32522). Western blotting was carried out following an established protocol using the WesternBright ECL (Advansta, Menlo Park, CA), and blots were photographed with the Image Quant LAS 4000 luminescent image analyzer (General Electric, Fairfield, CT).
Statistical Analysis
All experiments were performed at least in duplicate and repeated at least once, and all numerical results are expressed as the mean ± SD. Groups of means among different treatments were compared using the Student’s t test. A value of P < 0.05 was considered statistically significant. Statistical analyses were run using the GraphPad Prism 6 software (Graph Pad Software, San Diego CA).
RESULTS
Discovery of KLF5-interacting proteins in the nucleus of TSU-Pr1
To identify proteins that interact with KLF5 to promote tumorigenesis, we performed a co-immunoprecipitation (co-IP) with anti-KLF5 antibody using nuclear lysates from the previously established K12 cell line, a clone of the TSU-Pr1 bladder cancer cell line that stably expresses KLF5 and has high propensity for cell proliferation and tumor growth 13. Expression levels of the lamin A/C nuclear marker and the β-tubulin cytoplasmic marker were analyzed by western blotting in both the nuclear and cytoplasmic fractions to confirm successful separation of nuclei (Fig. 1A). As expected, KLF5 protein expression was primarily detected in the nuclear fraction (Fig. 1A). A silver staining assay revealed a band corresponding to the size of KLF5 and several other bands of proteins that were not visible in the IgG control group (Fig. 1B). The IgG light and heavy chain bands are indicated by asterisks in Figure 1B. Western blotting confirmed the presence of KLF5 in the precipitate (Fig. 1C). These results indicate the existence of potential KLF5-interacting nuclear proteins in K12 cells.
The same precipitates resulting from treatment with the KLF5 antibody and the IgG negative control were concentrated using an Amicon ultrafiltration tube to reduce their volumes, separated on another gel, and stained with the Coomassie blue dye to visualize proteins. In both KLF5 and IgG lanes, bands for both the light and heavy chains of IgG were cut and discarded to reduce nonspecific proteins, and the remaining gel from the entire lane was collected (Fig. 1B) and subjected to mass spectrometry (MS) to identify potential KLF5-interacting proteins. As a known KLF5-interacting transcription factor 20, 39, c-Jun was used as reference protein for the success of IP and MS analysis. Western blotting clearly detected c-Jun in the KLF5 precipitates, although the band was not as strong as expected (Fig. 1C). Meanwhile, 2 unique peptides of c-Jun were detected in the MS analysis. Therefore, the cutoff value for potential KLF5-interacting proteins in the LC-MS/MS analysis was set to 2. A total of 122 proteins had at least 2 unique peptides in the MS analysis, and were thus considered potential KLF5-interacting proteins (Table S1). The ranking scores for the 122 proteins ranged from 0 to 670 (median 10.7). Ten of the proteins had a score of at least 50, and KLF5 was ranked number 2 among them, with a score of 213.8 and 13 unique peptides (Figure 1D). Interestingly, the protein ranked first on the list was AHNAK, a very large scaffold nuclear protein with a wide range of functions including a tumor suppressor function 40, 41 with a score of 670 and 116 unique peptides (Figure 1D). The other 8 of the top 10 proteins included TFAM, HSDL2, HNRNPC, CINP, IST1, FBL, PABPC1 and SNRNP40 (Figure 1D). Based on the current cellular compartment information in the Gene Ontology database, at least 64 of the 122 molecules were primarily localized in the nucleus (Table S1); and a literature search of the PubMed database suggests that 35 of the 64 proteins are associated with cell cycle and/or apoptosis (Table S2).
Functional identification of CINP as a strong candidate KLF5 partner in cell proliferation
To identify KLF5-interacting molecules that are required for KLF5-promoted cell proliferation and survival, we performed SRB assays for the top 9 proteins (Figure 1D). Both K12 and parental TSU-Pr1 cells were transfected with a mixture of 2 siRNAs targeting each of the 9 genes. Relative mRNA levels of the 9 genes after knockdown were confirmed by real-time qPCR, along with the control siRNA for each gene (Fig. 2A). As indicated by images of stained cells (Fig. 2B) and optical densities of stained cells (Fig. 2C), KLF5-expressing K12 cells indeed grew more rapidly than the parental TSU-Pr1 cells, which is consistent with the original report 13. After knockdown of AHNAK, FBL, SNRNP40 and TFAM, the promoting effect of KLF5 was still detected, suggesting that these genes contribute to the proliferation of TSU-Pr1 cells but are not specifically required for KLF5 to promote cell proliferation. These genes were thus excluded for further analysis. Knockdown of HSDL2, IST1 and PABPC1 abolished the promoting effect of KLF5 on cell proliferation, and knockdown of CINP and HNRNPC even reversed KLF5’s function from promoting to suppressing, suggesting that HSDL2, IST1, PABPC1, HNRNPC, and CINP are partners of KLF5 in the promotion of proliferation and/or survival in TSU-Pr1 cells. Among these 5 molecules, knockdown of CINP showed the greatest effect, reducing the number of cells by more than 50% (Fig. 2C). In addition, CINP was also one of the 11 nuclear localized proteins that has been demonstrated to regulate cell cycle progression (Table S2). CINP was thus selected for additional analyses.
FIGURE 2. Functional screening for KLF5 partners required for KLF5 to promote cell growth.
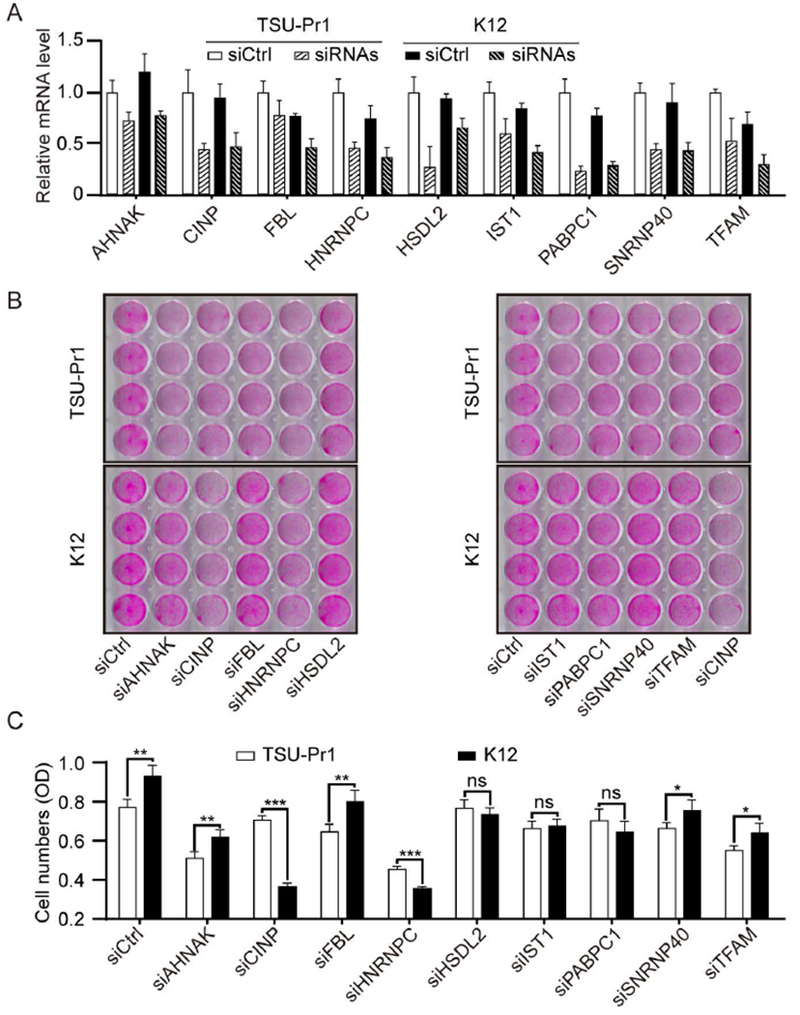
(A) Validation of knockdown efficiencies for siRNAs against different genes by real-time qPCR. TSU-Pr1 and K12 cells were transfected with a mixture of two siRNAs for each indicated gene. (B, C) Effects of gene silencing by RNAi on cell growth, as measured by the SRB assay, for different candidate KLF5 partners. Shown are staining images (B) and optical densities (C) of cells with siRNA transfection for 3 days. *, P<0.5; **, P<0.01; ***, P<0.001.
Validation of KLF5-CINP interaction
We next validated the interaction between KLF5 and CINP proteins. Among the proteins precipitated by the KLF5 antibody in K12 cells, western blotting showed that CINP was much more abundant than c-JUN, a known KLF5-interacting protein (Fig. 1C). We also performed IP and western blotting in different cell lines with or without KLF5 expression. Compared to K12 cells in which KLF5 is ectopically expressed, IP and western blotting showed that in the V4 vector control clone of the TSU-Pr1 cells, which have little KLF5, the KLF5 antibody precipitates did not contain detectable CINP (Fig. 3A). Consistently, when the CINP antibody was used for IP in K12 cells, KLF5 protein was detected in the CINP-containing precipitates (Fig. 3B). We also generated tagged expression constructs Flag-KLF5 and Myc-CINP, transfected them into 293T cells, and performed IP with anti-Flag antibody for Flag-KLF5 or anti-Myc antibody for Myc-CINP. Again, CINP was detected in the Flag-KLF5 precipitate (Fig. 3C) and KLF5 detected in the Myc-CINP precipitates (Fig. 3D). To further determine the binding domains of KLF5 and CINP, we performed similar experiments with truncated KLF5 proteins, and found that the fragment of KLF5 containing residues 201 to 453 bound to CINP (Fig. 3E).
FIGURE 3. Confirmation of the KLF5-CINP interaction by co-IP and western blotting in different cell lines.
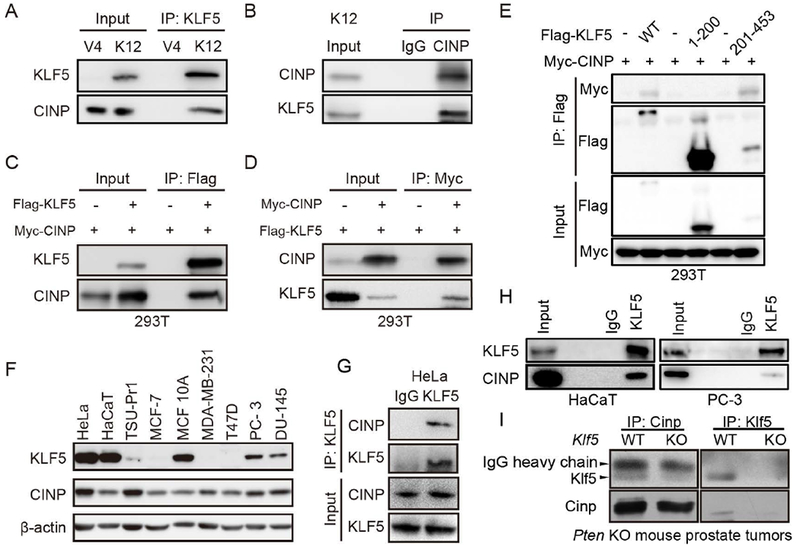
(A, B) Co-IP with KLF5 antibody (A) or CINP antibody (B) and blotting with the other antibody in KLF5-expressing K12 cells and KLF5-low V4 cells. Inputs were from cell lysates before IP. IgG was used as a negative control for co-IP. (C, D) 293T cells were transiently transfected with expression plasmids of FLAG-tagged KLF5 and/or myc-tagged CINP, and then subjected to co-IP and western blotting with anti-FLAG or anti-Myc antibody. (E) Mapping of interacting domains of KLF5 and CINP by co-IP with FLAG antibody and western blotting with anti-FLAG or anti-Myc antibody. (F) Detection of KLF5 and CINP by western blotting in different epithelial cell lines. Except for HaCaT and MCF10A, which are noncancerous epithelial cell lines, all others are cancer cell lines. β-actin was the loading control. (G-I) Co-IP and western blotting were applied to cell lines HeLa (G), HaCaT (H) and PC-3 (H) and Pten-null mouse prostate tumors (I) with KLF5 and CINP antibodies.
Expression of both KLF5 and CINP was ubiquitous in multiple types of cancers, as indicated by analysis of the TCGA database (Fig. S1 and S2). We thus tested whether the endogenous CINP interacts with the endogenous KLF5. We first compared multiple cancerous and non-cancerous cell lines for the expression of KLF5 and CINP using western blotting, and found that HeLa, HaCaT and PC-3 cell lines had higher levels of both KLF5 and CINP (Fig. 3F). We then pulled down the protein complexes by KLF5 antibody in HeLa, HaCaT and PC-3 cells, and were able to detect CINP in KLF5 precipitates from each of the cell line by western blotting (Fig. 3G and 3H). These results further indicate that KLF5 and CINP indeed associate with each other. Furthermore, we detected Klf5-Cinp interaction in Pten-null mouse prostate tumors using co-IP and western blotting assays (Fig. 3I).
We also tested whether the interaction with CINP is necessary for KLF5’s protein stability. We found that knockdown or overexpression of CINP in HeLa cells did not affect the protein level of KLF5 (Fig) lity of KLF5, as indicated by CHX assays. is essential for KLF5’ein stability of these two proteins. Fig. S3A-B). Neither did they affect protein stability of KLF5, as indicated by the CHX assay (Fig. S3C-D).
Knockdown of CINP attenuates the promoting effect of KLF5 on tumor growth
To determine whether the KLF5-CINP interaction is necessary for KLF5 to promote tumor growth, we silenced CINP expression in both parental TSU-Pr1 cells and K12 KLF5-expressing cells, and first performed cell growth assays (Fig. 4A, 4B). Consistent with functional screening experiments (Fig. 2), silencing CINP attenuated the ability of KLF5 to promote cell proliferation, as detected in K12 cells, but did not reduce cell numbers in the KLF5-low parental TSU-Pr1 cells (Fig. 4A, 4B), indicating that the promoting effect of KLF5 on cell growth depends on the presence of CINP. In HeLa, K12 and HaCaT cells, all of which have higher expression levels of KLF5 and CINP (Fig. 3F) and KLF5-CINP interaction (Fig. 3A, 3B, 3G and 3H), silencing either CINP or KLF5 resulted in an inhibition of cell growth, but the effect of KLF5 silencing was significantly attenuated by CINP silencing (Fig. 4C, 4D, S4, S5A and S5B), further indicating the requirement of CINP for KLF5 to promote cell growth.
FIGURE 4. CINP is indispensable for KLF5 to promote cell and tumor growth in TSU-Pr1 cells.
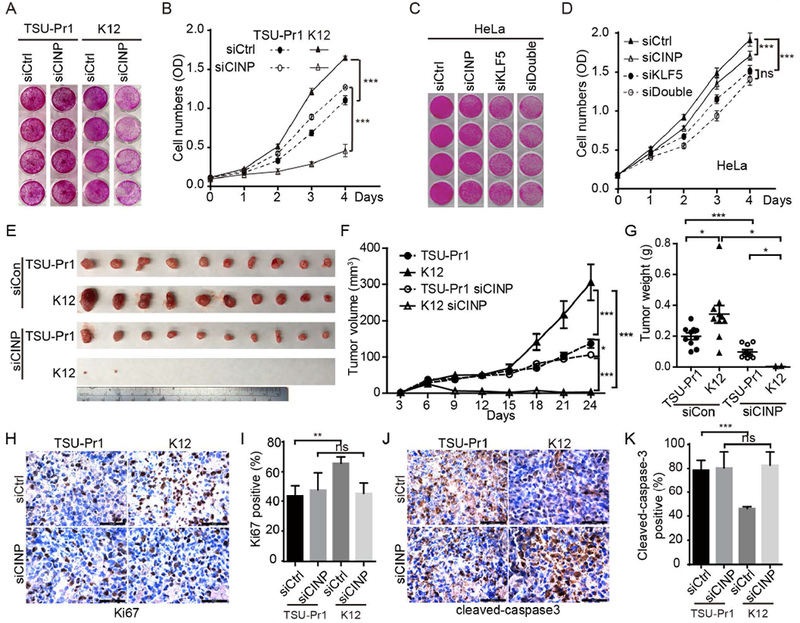
(A, B) CINP silencing by RNAi inhibited KLF5-promoted cell growth, as demonstrated by images (A) and optical densities (B) of stained cells in the SRB assay. siCtrl, control siRNA; siCINP, CINP siRNA. (C, D) Knockdown of CINP and/or KLF5 inhibited cell proliferation in HeLa cells, as determined by the SRB assay. siKLF5, KLF5 siRNA; siDouble, mixture of siRNAs for both KLF5 and CINP. (E-G) Knockdown of CINP attenuated KLF5-promoted tumor growth in TSU-Pr1 cells, as indicated by tumor images (E), tumor volumes at different times (F), and tumor weights at excision (G). (H-K) Cell proliferation and apoptosis in tumor xenografts were evaluated by measuring the expression of Ki67 and cleaved caspase 3, respectively, using IHC staining. *, p<0.05; **, P<0.01; ***, P<0.001.
Parental TSU-Pr1 cells and K12 cells with CINP silencing were inoculated into immunosuppressed 3-4 week old male BABL/C nude mice for tumorigenesis assays. Tumor volumes and weights were measured (Fig. 4E-4G). Consistent with in vitro experiments, silencing CINP significantly attenuated the promoting effect of KLF5 on tumor growth, as indicated by both tumor volumes and weights (Fig. 4E-4G). Without silencing CINP, KLF5 promoted tumor growth as previously reported 13. These results indicate that the interaction with CINP is necessary for KLF5 to promote tumor growth in TSU-Pr1 cells. We also determined cell proliferation and apoptotic indices based on the expression of Ki67 and cleaved caspase 3 in tumor xenografts. Consistent with tumor growth rates, KLF5-expressing K12 tumors had significantly more Ki67-positive cells and fewer cleaved caspase 3-positive cells compared to parental TSU-Pr1 tumors. However, silencing CINP eliminated the effects of KLF5 on cell proliferation and apoptosis (Fig. 4H-4K).
Loss of CINP slows cell cycle progression and causes apoptosis in KLF5-expressing cells
To determine the cellular process responsible for the attenuation of tumor growth by CINP silencing, we examined cell cycle distribution and apoptosis by flow cytometry in TSU-Pr1 and K12 cells with or without CINP knockdown (Fig. 5). Consistent with the previous report 13, KLF5 overexpressing K12 cells had significantly fewer cells in the G1 phase but more cells in the G2/M phase when compared to KLF5-low TSU-Pr1 cells 13, indicating that KLF5 promotes cell proliferation. Silencing CINP in K12 cells significantly slowed cell cycle progression, as more cells accumulated in the S phase (Fig. 5A, 5B), indicating that CINP is needed by KLF5 to promote cell cycle progression. In HeLa cells, knockdown of KLF5 or CINP alone significantly increased the percentage of cells in the G1 population and decreased that in the G2/M population (Fig. 5C, 5D). Interestingly, CINP knockdown diminished the increase in G1 population and the decrease in G2/M population caused by KLF5 silencing, although silencing both genes showed an additive effect (Fig. 5C, 5D). Therefore, KLF5 requires CINP at least in part to accelerate the cell cycle.
FIGURE 5. Loss of CINP compromises KLF5-accelerated cell cycle progression and induces apoptosis.
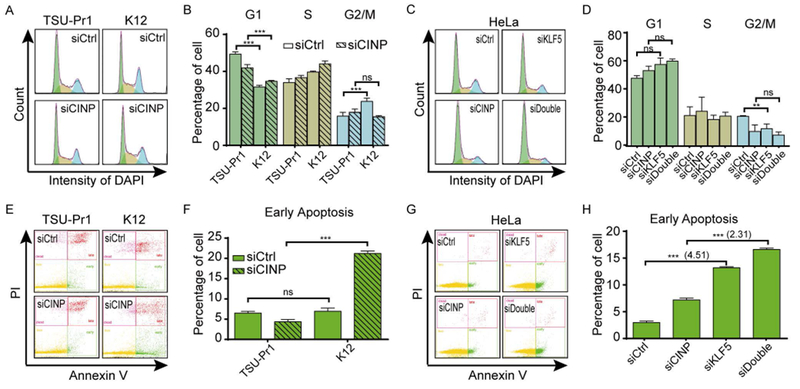
(A, B) Silencing CINP eliminated the promoting effect of KLF5 on cell cycle progression in TSU-Pr1 cells. (C, D) Knockdown of CINP partially blocked the reduction of cells in the G2/M phase caused by silencing KLF5 in HeLa cells. The distribution of cells in different phases of cell cycle (A-D) was measured by cytometry analysis. (E, F) Silencing CINP caused apoptosis in KLF5-expressing K12 cells but not in parental TSU-Pr1 cells. (G, H) Knockdown of CINP diminished the apoptosis caused by KLF5 silencing in HeLa cells. Statistical analysis for early apoptosis (E-H) was conducted using Amnis IDEAS software. n.s., not significant; *, P<0.5; **, P<0.01; ***, P<0.001.
We evaluated whether CINP is also necessary for KLF5 to regulate cell survival by flow cytometry using cells stained with Annexin V-FITC/PI. CINP knockdown dramatically increased cell death in KLF5-expressing K12 cells but not in TSU-Pr1 parental cells (Fig. 5E, 5F), which explains why silencing CINP reduced cell numbers much more in KLF5 overexpressing cells than in parental TSU-Pr1 cells (Fig. 4B). Therefore, CINP is required for the anti-apoptotic function of KLF5. Consistent effects were detected in HeLa cells and HaCaT cells, in which knockdown of KLF5 or CINP alone enhanced early apoptosis and knockdown of CINP compromised the apoptosis caused by KLF5 silencing (Fig. 5G, 5H, S5C and S5D). These results indicate that the anti-apoptosis effect of KLF5 is attributed to CINP at least partially.
CINP is required for the function of KLF5 in gene regulation
KLF5 regulates cellular processes including cell proliferation and survival by regulating gene expression, so we tested the effect of silencing CINP on the expression of genes regulated by KLF5 in K12 cells. RNA-Seq analysis of K12 cells with or without CINP knockdown demonstrated that silencing CINP altered the expression of 760 genes, including 521 that were upregulated and 239 downregulated (Table S3). The RNA-Seq data was validated by Real-time PCR (Fig. S6). Of these genes, knockdown of CINP downregulated the CCND1 oncogene by 2.35-fold, and upregulated the CASP7 apoptosis related gene by 2.24-fold, which is consistent with suppressed cell cycle and enhanced apoptosis by CINP silencing. By comparing CINP-regulated genes to previously reported KLF5-regulated genes 13, we found that 3 of 21 KLF5-upregulated genes in TSU-Pr1 cells, including CCND1, TIMP2 and PDGFA, were downregulated by CINP silencing in K12 cells (Table S4). Similarly, one of the 5 KLF5-downregulated genes in TSU-Pr1 cells from the previous study 13, i.e., CD24, was upregulated by silencing CINP (Table S4). The remainder had no detectable changes in their expression after CINP was knocked down. Therefore, the KLF5-CINP interaction regulates a unique set of genes, including some that are involved in cell cycle progression, apoptosis, and tumorigenesis.
After confirming the expression status of KLF5 and CINP in both TSU-Pr1 and K12 cells, realtime PCR was then performed to validate expression changes caused by CINP silencing (Fig. 6A, 6C). In addition to confirming reduced CINP mRNA by RNAi, we confirmed that silencing CINP significantly compromised the effect of KLF5 on the induction of the CCND1 cell cycle regulator, the down regulation of the CASP7 apoptosis gene, and the expression of other KLF5 regulated genes such as PDGFA, TIMP2, and CD24 (Fig. 6A). We also evaluated whether CINP knockdown affects KLF5 regulated genes at the protein level. As expected, KLF5 up regulated cyclin D1, as indicated by Western blotting (Fig. 6C). In the HeLa cells, KLF5 silencing down regulated CCND1 and up regulated CASP7 at both the RNA and protein level, and the effect was compromised after CINP knockdown (Fig. 6B, 6D). Consistent to the proliferation index and apoptosis index as measured by Ki67 and cleaved caspase3 (Fig.4H-K), CINP knockdown abolished cyclin D1 upregulation in KLF5 overexpressing K12 cells, and compromised the downregulation of caspase 7 caused by KLF5 overexpression in a tumor xenograft model (Fig. 6E, 6F).
FIGURE 6. Necessity of CINP for KLF5-mediated gene regulation and a model for the dependency of KLF5 on CINP to promote tumor growth.
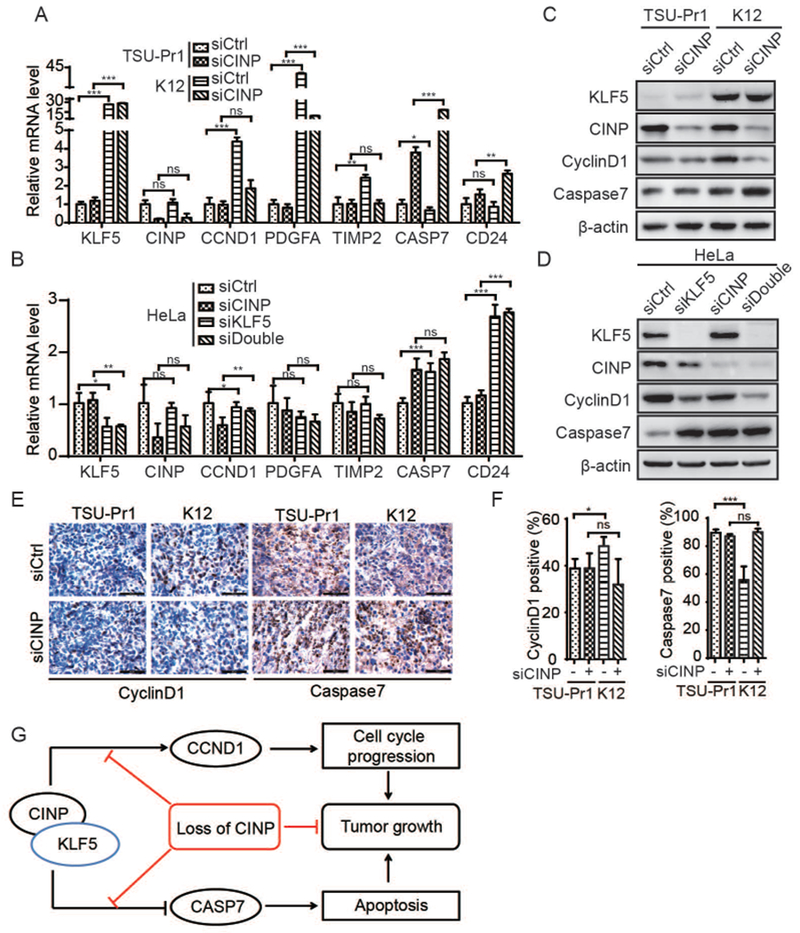
(A-D) Silencing CINP attenuated the function of KLF5 in gene regulation, as detected by both real-time qPCR (A, B) and western blotting (C, D) in both TSU-Pr1 cells (A, C) and HeLa cells (B, D). (E, F) Expression of cyclin D1 and caspase 7 was measured by IHC in TSU-Pr1 xenografts. (G) A model for KLF5-CINP interaction in KLF5-promoted tumor growth. n.s., not significant; *, P<0.5; **, P<0.01; ***, P<0.001.
KLF5 depends on CINP to regulate multiple signaling pathways in tumorigenesis
To further understand the molecular mechanisms underlying the necessity of CINP for KLF5 to promote tumorigenesis, we uploaded differentially expressed genes between K12 cells with and without silencing CINP to the Meta Core platform to identify signaling pathways and transcription factor networks underlying different cellular processes. Knockdown of CINP in K12 cells affected genes involved in cytoskeleton, inflammation, cell adhesion, proteolysis, proliferation, and development, including the enrichment of genes that negatively regulate cell proliferation (Table S5); pathways regulating cytoskeleton remodeling, development, transcription, cell adhesion, protein folding and maturation, immune response, developmental epithelial-to-mesenchymal transition (EMT), and stimulation of TGF-β signaling (Table S6); and 5 key transcription factors involved in tumor development, including CREB1, SP1, c-Myc, p53, and androgen receptor (AR) (Table S7). Interestingly, CREB1, c-Myc, p53, and AR were also enriched as key transcription factors in our previous study8 of DU 145 prostate cancer cells expressing deacetylated KLF5, which also promotes tumorigenesis. These findings further indicate the necessity of CINP for KLF5 to modulate genes and signaling pathways involved in multiple cellular processes during tumorigenesis.
DISCUSSION
Transcription factors interact with other factors to regulate gene transcription and cellular processes. In this study, we focused on the KLF5 transcriptional complex that is responsible for KLF5’s tumor promotion function in the TSU-Pr1 bladder cancer cell line, which was established in a previous study 13. We applied a proteomics approach, isolating KLF5 protein complexes by co-IP from the nucleus and identifying individual proteins by mass spectrometry. The success of IP and MS analysis is indicated by the detection of a known KLF5-interacting protein, c-Jun, and the detection of KLF5 as the second most abundant protein in the MS analysis (Fig. 1). While 122 different proteins were identified, 9 were much more abundant than others and are thus more likely to be true interacting factors involved in KLF5 functions. These 9 proteins included AHNAK, TFAM, HSDL2, HNRNPC, CINP, IST1, FBL, PABPC1, and SNRNP40. Four of the 9 proteins, including HSDL2, IST1, PABPC1, and CINP, could be involved in KLF5-mediated cell proliferation and/or survival, because silencing each of them by RNAi abolished the promoting effect of KLF5 (Fig. 2). Among the 4 proteins, CINP was further characterized in this study because it showed the most potent effect on KLF5 (Fig. 2), but HSDL2, IST1, and PABPC1 could also play important roles in KLF5-mediated tumor growth, which remains to be clarified. For example, silencing the HSDL2 orphan short-chain dehydrogenase suppresses cell proliferation in glioma 42, and the polyadenylate binding protein PABPC1 interacts with and stabilizes a lncRNA that appears to promote tumor growth and metastasis of human gallbladder cancer 43. KLF5 regulates a number of cellular processes 4, so many potential KLF5-interacting nuclear proteins provide candidates for further characterization for their interactions with KLF5 to regulate different cellular processes.
In the context of TSU-Pr1 cells, however, the CDK2-interacting protein CINP 44 of the 9 proteins appears to be the most crucial KLF5-interacting protein involved in KLF5-mediated tumor growth (Fig. 6G). For example, silencing CINP by RNAi dramatically reduced cell proliferation and survival in KLF5-expressing K12 cells but had no effect in KLF5-low TSU-Pr1 cells (Fig. 2B, 2C); and CINP’s protein association with KLF5 was validated across multiple cell lines and mouse tumor samples (Fig. 3). Additional experiments confirmed the necessity of CINP for KLF5 to promote cell proliferation, survival, and tumorigenesis in different cell lines such as K12, HeLa and HaCaT, because silencing CINP eliminated the promoting effect of KLF5 on tumorigenesis by modulating the cell cycle and apoptosis (Fig. 4, 5). CINP also showed a crucial role in the expression of genes regulated by KLF5, as CINP silencing rescued gene expression changes caused by KLF5 overexpression (Table S4, Fig. 6, S6). Genes regulated by KLF5 and CINP include the cyclin D1 cell cycle promoter and the CASP7 apoptosis regulator (Fig. 6), which are likely part of the mechanisms responsible for KLF5-CINP-mediated tumorigenesis in TSU-Pr1 cells (Fig. 6G). These findings establish CINP as a novel cofactor of KLF5 in the promotion of tumor growth, and the KLF5-CINP interaction as a crucial tumor promoter for therapeutic intervention.
The KLF5-CINP interaction likely involves multiple transcriptional regulators, and bioinformatics analysis suggests that CREB1, SP1, and Myc could form complexes with KLF5-CINP to regulate a series of genes (Fig. 6 and Table S7). In addition, the KLF5-CINP interaction appears to regulate multiple cellular processes and signaling pathways, as suggested by MetaCore analyses of RNA-Seq data from K12 cells with and without CINP silencing (Table S5-S7).
KLF5 plays context-dependent roles in tumorigenesis, being both suppressive and oncogenic depending on molecular contexts 4–6, 8, 45. For example, an oncogenic function of KLF5 has been reported in several types of tumors including intestinal and breast tumors 19. In prostate cancer, however, loss of Klf5 clearly promotes tumorigenesis induced by the loss of Pten 45, TGF-β-induced acetylation of KLF5 at lysine 369 renders KLF5 a suppressor of cell proliferation and tumor growth, and deacetylation converts KLF5 from a tumor suppressor to a tumor promoter 7, 8, 30, 31. The TSU-Pr1 cell line was derived from the T24 bladder cancer cell line, which has an activating mutation in the RAS oncogene 33. However, it is largely unknown which contexts render KLF5 oncogenic or tumor suppressive. Findings in the current study suggest that KLF5 could bind with different nuclear factors to promote or suppress tumorigenesis. While CINP could be an essential cofactor of KLF5 for tumor promotion, other KLF5-interacting proteins could be responsible for KLF5’s tumor suppressor function.
Although it is also unknown whether the tumor promoting function of the KLF5-CINP interaction occurs in contexts other than that represented by TSU-Pr1 cells, bioinformatics analyses suggest some similarities between KLF5-CINP interaction and deacetylated KLF5 in terms of signaling pathways and networks. Previously we profiled genes and pathways regulated by the deacetylation of KLF5 in a prostate cancer cell line, in which deacetylated KLF5 promotes tumor growth 8. MetaCore analysis suggests that the gene changes caused by CINP were associated with those caused by deacetylated KLF5, as shown by the involvement of the enriched signaling pathways and the molecular networks in different datasets. According to the enrichment analysis by MetaCore, 3 of the top 10 enriched pathways caused by CINP silencing were also enriched by deacetylated KLF5 (Table S6). In addition, 4 of the top 5 key transcription factors associated with deacetylated KLF5, including CREB1, c-Myc, p53, and AR, were also enriched in by CINP silencing in K12 cells (Table S7). Notably, the identified KLF5 binding protein c-JUN (Fig. 1C) was also a seed node in the Meta Core analysis of key transcriptional networks for deacetylated KLF5 8.
Silencing CINP suppressed cell proliferation (Fig. 4A, 4B) and tumor growth (Fig. 4E-G) much more significantly in KLF5-expressing K12 cells than in TSU-Pr1 cells, suggesting that CINP also requires KLF5 to promote cell proliferation and tumor growth. In addition, CINP knockdown suppressed proliferation of HeLa cells significantly, but this suppression was not significant after KLF5 knockdown (Fig. 4C, 4D), further supporting the idea that CINP may require KLF5 to maintain cell growth. In addition, in the situation of high KLF5 expression, CINP knockdown decreased the proportion of cells in G2/M phase in both K12 and HeLa cells (Fig. 5B, 5D). However, CINP knockdown failed to do so in the setting of low KLF5 expression, i.e in TSU-Pr1 parental cells or HeLa cells with KLF5 knockdown (Fig. 5B, 5D). Consistent with our data, a previous study demonstrated that CINP functions in genome maintenance, and silencing CINP in the U2OS cell line leads to a slight increase in cells of the G2-phase 46. According to the Cancer Cell Line Encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle/home) and the Human Protein Atlas (http://www.proteinatlas.org/), U2OS cells also express little KLF5. Collectively, these findings suggest that the decrease in G2/M phase cells caused by CINP knockdown requires the presence of KLF5. “
The KLF5-CINP interaction provides a potential opportunity to inhibit KLF5’s tumor promoting function for the treatment of cancer. It is typically challenging to develop effective small molecule inhibitors of transcription factors. Targeting protein-protein interactions, on the other hand, is a valid approach for developing chemical inhibitors. Identification of the KLF5-CINP interaction and demonstration of CINP’s necessity for KLF5 to promote tumor growth thus make it possible to identify and/or develop chemicals that inhibit KLF5 function by interrupting the KLF5-CINP interaction.
In summary, we identified and established CINP as a novel KLF5-interacting protein that is an essential cofactor of KLF5 in the regulation of gene expression and the promotion of cell proliferation, survival, and tumorigenesis in an experimental system. The KLF5-CINP interaction regulates a unique set of genes and molecular pathways involved in tumorigenesis, thus providing a novel opportunity for developing chemical inhibitors that can inhibit KLF5’s tumor promoting function by interrupting the KLF5-CINP interaction.
Supplementary Material
WHAT’S NEW?
KLF5 is established in promoting cell proliferation and tumorigenesis, though KLF5 cofactors that facilitate tumor promotion remain unknown. Here, CINP was identified as a KLF5 cofactor by MS and RNAi-based functional screening. In addition to validating the KLF5-CINP interaction, CINP knockdown attenuated the effects of KLF5 on cell and tumor growth by decelerating cell cycle progression and inducing apoptosis in KLF5-expressing cells. Downstream networks of KLF5 regulated by CINP were also identified by RNA-Seq.
ACKNOWLEDGEMENTS
We thank Dr. Zhenghong Yuan of Fudan University for kindly providing various CINP plasmids for the study; Dr. Xiaomeng Shi of Tsinghua University for advice on the LC-MS/MS analysis; Dr. Dan Zhao, Dr. Ang Luo and Shiying Zhang of Nankai University for advice and help throughout the study; and Dr. Anthea Hammond for copy editing the manuscript. This work was supported in part by grant 81130044 from the National Natural Science Foundation of China and grant R01CA171189 from the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001;411: 342–8. [DOI] [PubMed] [Google Scholar]
- 2.Sogawa K, Imataka H, Yamasaki Y, Kusume H, Abe H, Fujii-Kuriyama Y. cDNA cloning and transcriptional properties of a novel GC box-binding protein, BTEB2. Nucleic Acids Res 1993;21: 1527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi H, Zhang Z, Wang X, Liu S, Teng CT. Isolation and characterization of a gene encoding human Kruppel-like factor 5 (IKLF): binding to the CAAT/GT box of the mouse lactoferrin gene promoter. Nucleic Acids Res 1999;27: 4807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci 2009;66: 2691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diakiw SM, D’Andrea RJ, Brown AL. The double life of KLF5: Opposing roles in regulation of gene-expression, cellular function, and transformation. IUBMB Life 2013;65: 999–1011. [DOI] [PubMed] [Google Scholar]
- 6.Ci X, Xing C, Zhang B, Zhang Z, Ni JJ, Zhou W, Dong JT. KLF5 inhibits angiogenesis in PTEN-deficient prostate cancer by attenuating AKT activation and subsequent HIF1α accumulation. Molecular cancer 2015;14: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo P, Dong XY, Zhang X, Zhao KW, Sun X, Li Q, Dong JT. Pro-proliferative factor KLF5 becomes anti-proliferative in epithelial homeostasis upon signaling-mediated modification. J Biol Chem 2009;284: 6071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Zhang B, Wu Q, Ci X, Zhao R, Zhang Z, Xia S, Su D, Chen J, Ma G, Fu L, Dong JT. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int J Cancer 2015;136: 536–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun R, Chen X, Yang VW. Intestinal-enriched Kruppel-like factor (Kruppel-like factor 5) is a positive regulator of cellular proliferation. J Biol Chem 2001;276: 6897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mcconnell BB, Bialkowska AB, Nandan MO, Ghaleb AM, Gordon FJ, Yang VW. Haploinsufficiency of Krüppel-Like Factor 5 Rescues the Tumor-Initiating Effect of the ApcMin Mutation in the Intestine. Cancer Research 2009;69: 4125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nandan MO, McConnell BB, Ghaleb AM, Bialkowska AB, Sheng H, Shao J, Babbin BA, Robine S, Yang VW. Kruppel-like factor 5 mediates cellular transformation during oncogenic KRAS-induced intestinal tumorigenesis. Gastroenterology 2008;134: 120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandan MO, Ghaleb AM, McConnell BB, Patel NV, Robine S, Yang VW. Kruppel-like factor 5 is a crucial mediator of intestinal tumorigenesis in mice harboring combined ApcMin and KRASV12 mutations. Mol Cancer 2010;9: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C, Benjamin MS, Sun X, Otto KB, Guo P, Dong XY, Bao Y, Zhou Z, Cheng X, Simons JW, Dong JT. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. Int J Cancer 2006;118: 1346–55. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Zheng HQ, Zhou Z, Dong JT, Chen C. KLF5 promotes breast cell survival partially through fibroblast growth factor-binding protein 1-pERK-mediated dual specificity MKP-1 protein phosphorylation and stabilization. J Biol Chem 2009;284: 16791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng HQ, Zhou Z, Huang J, Chaudhury L, Dong JT, Chen C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene 2009;28: 3702–13. [DOI] [PubMed] [Google Scholar]
- 16.Zhu N, Gu L, Findley HW, Chen C, Dong JT, Yang L, Zhou M. KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. J Biol Chem 2006;281: 14711–8. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y, Hamza MS, Leong HS, Lim CB, Pan YF, Cheung E, Soo KC, Iyer NG. Kruppel-like factor 5 modulates p53-independent apoptosis through Pim1 survival kinase in cancer cells. Oncogene 2008;27: 1–8. [DOI] [PubMed] [Google Scholar]
- 18.David CJ, Huang Y- H, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massagué J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016;164: 1015–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Choi PS, Francis JM, Gao GF, Campbell JD, Ramachandran A, Mitsuishi Y, Ha G, Shih J, Vazquez F, Tsherniak A, Taylor AM, et al. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov 2018;8: 108–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He M, Han M, Zheng B, Shu YN, Wen JK. Angiotensin II stimulates KLF5 phosphorylation and its interaction with c-Jun leading to suppression of p21 expression in vascular smooth muscle cells. J Biochem 2009;146: 683–91. [DOI] [PubMed] [Google Scholar]
- 21.Kojima S, Kobayashi A, Gotoh O, Ohkuma Y, Fujii-Kuriyama Y, Sogawa K. Transcriptional activation domain of human BTEB2, a GC box-binding factor. J Biochem 1997;121: 389–96. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Teng CT. Phosphorylation of Kruppel-like factor 5 (KLF5/IKLF) at the CBP interaction region enhances its transactivation function. Nucleic Acids Res 2003;31: 2196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oishi Y, Manabe I, Tobe K, Ohsugi M, Kubota T, Fujiu K, Maemura K, Kubota N, Kadowaki T, Nagai R. SUMOylation of Kruppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR-delta. Nat Med 2008;14: 656–66. [DOI] [PubMed] [Google Scholar]
- 24.Guo P, Dong XY, Zhao KW, Sun X, Li Q, Dong JT. Estrogen-induced interaction between KLF5 and estrogen receptor (ER) suppresses the function of ER in ER-positive breast cancer cells. Int J Cancer 2010;126: 81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakajima Y, Akaogi K, Suzuki T, Osakabe A, Yamaguchi C, Sunahara N, Ishida J, Kako K, Ogawa S, Fujimura T, Homma Y, Fukamizu A, et al. Estrogen regulates tumor growth through a nonclassical pathway that includes the transcription factors ERbeta and KLF5. Sci Signal 2011;4: ra22. [DOI] [PubMed] [Google Scholar]
- 26.Oishi Y, Manabe I, Tobe K, Tsushima K, Shindo T, Fujiu K, Nishimura G, Maemura K, Yamauchi T, Kubota N, Suzuki R, Kitamura T, et al. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab 2005;1: 27–39. [DOI] [PubMed] [Google Scholar]
- 27.Lee MY, Moon JS, Park SW, Koh YK, Ahn YH, Kim KS. KLF5 enhances SREBP-1 action in androgen-dependent induction of fatty acid synthase in prostate cancer cells. Biochem J 2009;417: 313–22. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, Nishi T, Nagino T, Sasaki K, Aizawa K, Kada N, Sawaki D, Munemasa Y, Matsumura T, Muto S, Sata M, Miyagawa K, et al. Functional interaction between the transcription factor Kruppel-like factor 5 and poly(ADP-ribose) polymerase-1 in cardiovascular apoptosis. J Biol Chem 2007;282: 9895–901. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, Qin J, Ma Y, Chen L, Li S. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription ofp27Kip1. Oncotarget 2015;6: 17685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo P, Zhao KW, Dong XY, Sun X, Dong JT. Acetylation of KLF5 alters the assembly of p15 transcription factors in transforming growth factor-beta-mediated induction in epithelial cells. J Biol Chem 2009;284: 18184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo P, Dong XY, Zhao K, Sun X, Li Q, Dong JT. Opposing effects of KLF5 on the transcription of MYC in epithelial proliferation in the context of transforming growth factor beta. J Biol Chem 2009;284: 28243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrugia MK, Vanderbilt DB, Salkeni MA, Ruppert JM. Kruppel-like Pluripotency Factors as Modulators of Cancer Cell Therapeutic Responses. Cancer Res 2016;76: 1677–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van BA, Varellagarcia M, Korch C, Miller GJ. TSU-Pr1 and JCA-1 cells are derivatives of T24 bladder carcinoma cells and are not of prostatic origin. Cancer Research 2001;61: 6340–4. [PubMed] [Google Scholar]
- 34.Wang YH, Wang YC, Xu Y, Tong WY, Pan TT, Li JH, Sun SH, Shao JJ, Ding HP, Toyoda T, Yuan ZH. Hepatitis C Virus NS5B Protein Delays S Phase Progression in Human Hepatocyte-derived Cells by Relocalizing Cyclin-dependent Kinase 2-interacting Protein (CINP). J Biol Chem 2011;286: 26603–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CS, Sun XD, Guo P, Dong XY, Sethi P, Cheng XH, Zhou J, Ling JX, Simons JW, Lingrel JB, Dong JT. Human Kruppel-like factor 5 is a target of the E3 ubiquitin ligase WWP1 for proteolysis in epithelial cells. J Biol Chem 2005;280: 41553–61. [DOI] [PubMed] [Google Scholar]
- 36.Qi L, Zhang B, Zhang S, Ci X, Wu Q, Ma G, Luo A, Fu L, King JL, Nahta R, Dong JT. ERRF sensitizes ERBB2-positive breast cancer cells to lapatinib treatment likely by attenuating MCL1 and ERBB2 expression. Oncotarget 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ekins S, Nikolsky Y, Bugrim A, Kirillov E, Nikolskaya T. Pathway mapping tools for analysis of high content data. Methods Mol Biol 2007;356: 319–50. [DOI] [PubMed] [Google Scholar]
- 38.Chen C, Sun X, Ran Q, Wilkinson KD, Murphy TJ, Simons JW, Dong JT. Ubiquitin-proteasome degradation of KLF5 transcription factor in cancer and untransformed epithelial cells. Oncogene 2005;24: 3319–27. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Wen J, Dong L, Zheng B, Han M. Krüppel-like factor (KLF) 5 mediates cyclin D1 expression and cell proliferation via interaction with c-Jun in Ang II-induced VSMCs. Acta Pharmacologica Sinica 2010;31: 10–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis TA, Loos B, Engelbrecht AM. AHNAK: the giant jack of all trades. Cell Signal 2014;26: 2683–93. [DOI] [PubMed] [Google Scholar]
- 41.Lee IH, Sohn M, Lim HJ, Yoon S, Oh H, Shin S, Shin JH, Oh SH, Kim J, Lee DK, Noh DY, Bae DS, et al. Ahnak functions as a tumor suppressor via modulation of TGFbeta/Smad signaling pathway. Oncogene 2014;33: 4675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruokun C, Yake X, Fengdong Y, Xinting W, Laijun S, Xianzhi L. Lentivirus-mediated silencing of HSDL2 suppresses cell proliferation in human gliomas. Tumour Biol 2016;37: 15065–77. [DOI] [PubMed] [Google Scholar]
- 43.Wu XS, Wang F, Li HF, Hu YP, Jiang L, Zhang F, Li ML, Wang XA, Jin YP, Zhang YJ, Lu W, Wu WG, et al. LncRNA-PAGBC acts as a microRNA sponge and promotes gallbladder tumorigenesis. EMBO Rep 2017;18: 1837–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grishina I, Lattes B. A novel Cdk2 interactor is phosphorylated by Cdc7 and associates with components of the replication complexes. Cell cycle (Georgetown, Tex) 2005;4: 1120–6. [PubMed] [Google Scholar]
- 45.Xing C, Ci X, Sun X, Fu X, Zhang Z, Dong EN, Hao ZZ, Dong JT. Klf5 deletion promotes Pten deletion-initiated luminal-type mouse prostate tumors through multiple oncogenic signaling pathways. Neoplasia 2014;16: 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao RX, Ye F, Sirbu BM, Titus LC, Shyr Y, Cortez D. Functional genomic screens identify CINP as a genome maintenance protein. P Natl Acad Sci USA 2009;106: 19304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.