

Abstract
Cystic fibrosis (CF) is a monogenic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR dysfunction is characterized by abnormal mucociliary transport due to a dehydrated airway surface liquid (ASL) and hyperviscous mucus, among other pathologies of host defense. ASL depletion is caused by the absence of CFTR medicated chloride secretion along with continued activity of the epithelial sodium channel (ENaC) activity, which can also be affected by CFTR mediated anion conductance. Therefore, ENaC has been proposed as a therapeutic target to ameliorate ASL dehydration and improve mucus transport. Inhibition of ENaC has been shown to restore ASL hydration and enhance mucociliary transport in induced models of CF lung disease. To date, no therapy inhibiting ENaC has successfully translated to clinical efficacy, in part due to concerns regarding off-target effects, systemic exposure, durability of effect, and adverse effects. Recent efforts have been made to develop novel, rationally designed therapeutics to produce specific, long-lasting inhibition of ENaC activity in the airways while simultaneously minimizing off target fluid transport effects, systemic exposure and side effects. Such approaches comprise next-generation small molecule direct inhibitors, indirect channel-activating protease inhibitors, synthetic peptide analogs, and oligonucleotide-based therapies. These novel therapeutics represent an exciting step forward in the development of ENaC-directed therapies for CF.
Keywords: ion channels, lung disease, airway surface liquid, mucociliary clearance
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive, monogenic disease arising from mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which affects roughly 70,000 patients worldwide. Mutations in the CFTR gene cause dysfunctional or non-functional CFTR protein and dysregulated epithelial anion secretion [1]. CF presents with multi-organ pathologies in the lung, pancreas, and vas deferens (in male patients). Pulmonary manifestations of defective anion secretion are characterized by dehydrated airway surface liquid (ASL) and highly viscous mucus resulting in failure of the mucociliary escalator [2,3]. In concert with other defects in host defense, most prominently airway acidification and defective bacterial killing [4], consequently patients with CF become susceptible to chronic infection, inflammation, and progressive obstructive lung disease [1].
While CFTR plays a crucial role in anion secretion and regulation of epithelial potential difference, it also has absorptive capacity, where it works both in concert and in opposition to the epithelial sodium channel (ENaC) [5]. ENaC is a heterotrimeric protein channel expressed on the apical surface of airway epithelial cells [6] . ENaC is typically composed of three homologous subunits (α-, β-, and γ-subunits), although a δ subunit has identified more recently [6,7], which may alter the biophysical characteristics of the channel when incorporated into the assembled protein. Once assembled, ENaC is activated by proteolytic cleavage of the extracellular loops of the α- and γ -subunits. NaC primarily facilitates sodium and water resorption from the luminal surface into the epithelial cell. The balance of CFTR-mediated secretion and ENaC-mediation absorption plays a key role in maintaining ASL homeostasis, although other ion channels such as the calcium-activated chloride channel (CaCC) are also known to contribute to ASL regulation [8]. Presently, the relationship between CFTR and ENaC remains controversial [9]. Strong evidence suggests that ENaC activity is likely both abnormal in CF, and dependent on CFTR activity [10–17], whereas other studies do not support this [18–21], indicating tonic ENaC-mediated fluid resorption is present in CF and normal subjects. Other studies have shown that while ASL depth contributes to mucus transport, rheological abnormalities of CF mucus independent of dehydration defects can be a dominant factor in delaying MCC [2,3]. The controversy regarding the role of ENaC in CF pathogenesis, combined with the failure of several ENaC-targeted therapeutics in CF despite promising preclinical data, has led some to question whether ENaC can be an effective target in CF [22]. Therefore, the need to validate ENaC as a therapeutic target in CF persists.
In support of continued development of ENaC therapeutics in CF, empirical evidence demonstrates that aberrant ENaC activity contributes to either hyper- or hypo-hydrated ASL, with corresponding effects on mucociliary clearance (MCC) rate [23–26]. Indeed, mutations in the SCNN1A and B genes, which encode the α- and β-subunits of the ENaC protein, respectively, have been associated with CF-like lung disease in the absence of disease-causing CFTR mutations [6].For example, a patient possessing a gain-of-function mutation [27] in the SCNN1B gene (V348M) was identified as having CF-like symptoms in the absence of CFTR mutations [28]. This phenomenon has been recapitulated in a murine model overexpressing the β-subunit of the ENaC protein [23,26]. Conversely, mutations causing hypoactivity of the ENaC channel have been shown to cause pseudohypoaldosteronism [6,24,29], which expands the ASL depth and accelerates the MC [29]. Further, recent evidence has emerged demonstrating that CF patients who possess rare mutations in the SCNN1D gene, which encodes the δ subunit of ENaC, have hypomorphic ENaC channel activity [17]. These patients possessing rare SCNN1D mutations also demonstrated a long-term nonprogressive phenotype in lung function in which their lung function over a period of more than 20 years experienced minimal changes lending further support that ENaC is a plausible therapeutic target [17]. Thus, ENaC has been proposed as a putative target to partially ameliorate the mucus dehydration which is commonly observed in the CF population [30–33]. This therapeutic strategy is particularly enticing because it operates independent of CFTR function and CFTR mutation class [34,35]. Therefore the ENaC therapeutic strategy has the theoretical potential to benefit all CF patients, particularly those possessing rare mutations who do not currently have approved modulator therapy [36]. This review will briefly summarize past attempts at ENaC inhibition as a therapeutic target in CF, and focus on recent (since 2015) development of ENaC-targeted therapeutics.
PHARMACOLOGICAL ATTEMPTS AT ENaC INHIBITION
Previous reviews have discussed in detail, prior unsuccessful attempts at inhibiting ENaC [30–33], although they continue to inform current efforts; Table 1 summarizes these previous ENaC studies. Briefly, the development of the small molecule ENaC inhibitor amiloride marked the first attempt at the therapeutic use of ENaC inhibition in CF [37–40]. Amiloride is a pyrazinoyl guanidine compound which directly inhibits ENaC [41,42]. Despite being an effective inhibitor of ENaC, amiloride failed as a viable therapeutic option in CF, thought to be primarily due to its instability (short half-life) in the ASL and rapid clearance from the lungs (epithelial permeability) [43,44]. This rapid clearance has been observed in subsequently developed amiloride derivatives, including benzamil and phenamil [45]. Curiously, amiloride also obfuscated the beneficial effects of hypertonic saline inhalation [46]. This may have been due to non-selective inhibition of the water channel AQP, depleting water transport into the lumen through the osmotic effects of hypertonic saline, although other reports have demonstrated that amiloride may not affect water permeability [47]. Additionally, clinical development of a promising third generation derivative of amiloride, GS-9411 (formerly P-680, a collaborative effort between Gilead and Parion), was terminated after phase-1 trial participants had transient clinically significant hyperkalemia [48].
Table 1.
Unsuccessful attempts at ENaC inhibition.
| Drug | Type | Mechanism | Reason for Failure |
|---|---|---|---|
| Amiloride | Small molecule | Direct inhibition | Rapid clearance-short half-life and epithelial permeability in lungs; limited therapeutic efficacy [39][43] |
| Benzamil/ Phenamil (2nd Generation Amiloride derivatives) | Small molecule | Direct inhibition | Rapid clearance-short half-life and epithelial permeability in lungs [45] |
| GS-9411 (Parion; formerly P-680; 3rd generation Amiloride derivative) | Small molecule | Direct inhibition | Serious adverse effects in Phaselinical Trial- acute hyperkalemia [48] |
| Camostat (QAU 145) | Small molecule | Channel-Activating Protease (Prostasin) inhibitor | Adverse effects/tolerability issues in Phase II Clinical Trial [50] |
An alternative approach to direct ENaC inhibition is the use of indirect inhibitors, which do not directly act on ENaC but instead indirectly attenuate its function through modulation of other factors which contribute to regulation of ENaC activity. Indirect ENaC inhibition has primarily been achieved by Channel-Activating Protease (CAP) inhibitors. CAP inhibitors prevent the cleavage of extracellular loops on ENaC by proteases including prostasin, matriptase and furin. Targeting trypsin-like serine proteases (such as prostasin) and other channel activating proteins has been of interest since the late 1990s when Vallet et al. demonstrated that aprotinin (a trypsin-like serine protease) inhibited ENaC activity in the kidney [49]. Camostat (also known as QAU 145, developed by Novartis) is a potent P inhibitor that substantially reduces ENaC activity [50]. In phase 2 clinical trial, CF patients receiving camostat as a nasal spray showed reduced sodium transport across the nasal epithelium on transepithelial nasal potential difference (NPD) assays. However, some patients experienced adverse side effects raising concerns regarding the tolerance and safety of camostat in non-optimized formulations as an inhaled treatment for CF [50].
While many previously tested compounds have demonstrated effective inhibition of ENaC, significant challenges persist regarding time-course of efficacy, side-effects of systemic exposure, and unexpected adverse events including epistaxis, hematuria, and condition aggravation. Systemic exposure to potent potassium-sparing ENaC inhibitors such as amiloride may contribute to hyperkalemia. Thus, with these types of molecules, a significant therapeutic effect in the airways must be balanced with minimal off-target effects. Other approaches, which allow for airway-specific targeting of ENaC inhibition may also provide a more directed approach to therapeutic ENaC inhibition.
CHALLENGES OF ENaC-TARGETED THERAPEUTICS
Figure 1 describes the mechanisms of ENaC inhibition which have been attempted, or are currently in development. The success of therapeutic ENaC inhibition in CF will be dependent on their ability to overcome a multitude of challenges associated with ENaC-targeted therapeutics. The most significant challenges to ENaC-targeted therapeutics are: 1) the need to maximize treatment effects in the lung while minimizing systemic exposure and off-target effects, 2) prolonging the duration of effect, particularly with small-molecule inhibitors, which can often be rapidly cleared, and 3) understanding the interaction between ENaC and other airway epithelial ion channels involved in ASL regulation, particularly CFTR, and their ability to complement one another versus the potential risk that CFTR-dependent bicarbonate secretional pH resolution represent independent effects that are not addressed by fluid transport alone. An additional challenge in an effective ENaC-targeted CF therapy is the determination of the “optimal” time for initiating treatment in relation to the onset of lung disease [51]. Our ability to employ rational drug design to maximize specificity to the airway epithelium may be crucial to effective ENaC-targeted therapeutics for CF, as specificity to the respiratory epithelium has been a key issue that has plagued development so far. For example, selectivity could be optimized through a structure-based rational drug design approach of manipulating ligand affinity by leveraging subtle or substantial differences of candidate compounds in the binding determinants of ligand shape, electrostatic properties, hydration, conformational selection/flexibility, hydration, and allostery [52]. Other approaches to limit systemic absorption could be alternatives to achieve this goal.
Figure 1.
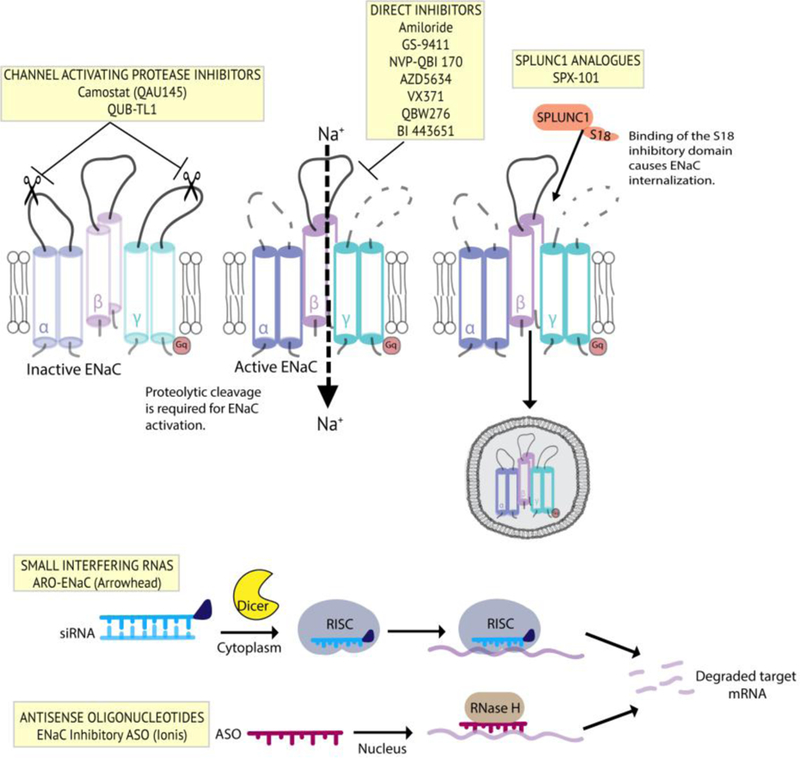
Summary of ENaC inhibitor therapies for CF. Clockwise from top left: channel activating protease inhibitors which prevent proteolytic cleavage of the extracellular loops of a- and g-ENaC; direct inhibitors which decrease channel open probability through direct interaction with the channel; peptide analogues which mimic the regulatory effects of the SPLUNC1 secreted protein; antisense oligonucleotide therapies which degrade ENaC mRNA transcripts through the RNase H mechanism; siRNA therapies which degrate ENaC mRNA transcripts though the RISC mechanism.
ENaC is highly expressed in the cortical collecting duct of the kidney, thus the localization of ENaC inhibitor therapies to the airway epithelial tissue is a highly desirable attribute of any ENaC-targeted therapeutic for CF. Blockade of ENaC activity in the kidney by potassium-sparing diuretic, ENaC inhibitors such as amiloride may lead to hyperkalemia and consequent arrhythmias in severe cases. Therefore, any ENaC-blocking therapy for CF must maximize effects in the lung while limiting ENaC inhibition in the kidney. This may be dose-limiting for ENaC inhibiting drugs, mainly traditional small-molecule global ENaC blockers. However, newer approaches which either exploit innate regulatory mechanisms specific to the airway epithelial milieu [53], or tissue-specific targeting may enhance efficacy in the lung while simultaneously reducing systemic effects [54,55], thus minimizing the risk of hyperkalemia and other off-target drug effects. Furthermore, ongoing development of new small molecule direct ENaC inhibitors which do not cause hyperkalemia are promising (AZD5634; clinicaltrials.gov: NCT02950805).
While minimizing off-target effects and systemic exposure is important for ENaC inhibitor therapies, concurrently maximizing the durability of effect in the lung is also essential. One of the challenges with early small molecule ENaC inhibitors is that they could be rapidly cleared from the lung, thus reducing the therapeutic window following administration of the drug. Subsequent development of molecules with substantially longer dwell time in the lung has somewhat mitigated this concern, but the most promising approaches may be oligonucleotide-based therapeutics, which potentially have a therapeutic window of days- to weeks, thus substantially reducing the frequency of dosing needed for therapeutic inhibition of ENaC in the lungs.
Finally, understanding the interactions between ENaC and other airway epithelial ion channels which regulate ASL homeostasis is an important, and perhaps somewhat understudied consideration. While evidence suggests that ENaC is downregulated by normal CFTR [51], this remains controversial [9,31]; in contrast, little is known regarding whether ENaC, in turn affects CFTR function [12]. A co-expression study performed in Xenopus laevis oocytes demonstrated that CFTR activity is upregulated by ENaC [56]. It has been suggested that this interaction could be facilitated by the Na+/H+ exchanger regulatory factor, isoform 1 (NHERF1), although further investigation is necessary to understand these interactions [12,57]. ENaC activity may also be modulated by the potential difference across the apical cell membrane [58]. The electrophysiological gradient across the membrane and in particular the intracellular Cl-concentration modulate channel activity. To date, whether ENaC influences CFTR function is still unknown, thus the effects of pharmacological inhibition of ENaC on CFTR function are not well described. In particular, it is presently unknown whether ENaC inhibitors and CFTR modulators could interact directly, as suggested by past literature [56,59]. Regardless and independent of this mechanism, the electrophysiological effects of increased anion secretion and decreased cation absorption suggest that a synergistic effect could be expected when ENaC inhibitors and FTR modulators are combined as the potential difference of the membrane is normalized by either modality, providing favorable electrical driving forces for the converse ion. Said another way, if ENaC is inhibited, electrical drive favoring CFTR secretion would be enhanced; in parallel, as CFTR function is augmented, ENaC resorption should be diminished, as observed for ivacaftor in G551D patients [60,61]. Nevertheless, if ENaC does indeed activate CFTR directly, potentially through protein-protein interactions, it is possible that inhibition of ENaC could depress CFTR function, thus blunting the effects of modulator therapy. It is possible that restoration of ASL hydration and consequent improvements in MCC by ENaC inhibition may be enhanced by uncoupling interactions between ENaC and CFTR, if ENaC does indeed activate CFTR.
Future studies elucidating the relationship between ENaC and CFTR will aid in understanding the development and optimization of ENaC-targeted therapeutics for CF. Given the rapid development of next-generation CFTR modulator therapies and triple-combination therapies with the potential to treat broader segment of the CF population, further understanding the relationship between ENaC and CFTR will become particularly important as ENaC inhibitors are combined with CFTR modulators as potentially complementary means to restore ASL volume, or as CFTR modulators achieve more broad utilization. It is essential we learn this by coupling ion transport studies with the development of ENaC blockers in vivo, rather than limit studies to only clinically relevant endpoints – otherwise we cannot fully understand or optimize treatment approaches in the case of a negative or subclinical result. Moreover, it remains to be seen whether augmenting the ion transport environment via targeting ENaC will have a major role in CF therapy should highly effective CFTR modulator therapy successfully be advanced to the overwhelming majority of CF patients and be made available for widespread use. Certainly, there is not an inconsequential risk that this may become more challenging if most CF patients obtain substantial CFTR activity through small molecule therapy. Issues related to medication cost or lack of access are unpredictable at this stage, but could obviously alter the dynamic in appreciable ways.
TRANSLATIONAL CHALLENGES
The potential for ENaC-targeted therapeutics to succeed is still controversial, given that to date, no clinical translation of ENaC inhibitors for CF has been achieved and published. Success of numerous compounds in preclinical models without clinical translation in human patients has caused some to call into question whether ENaC is an appropriate target at all. Although it remains unresolved whether ENaC activity is different in CF populations, even in the absence of overactive ENaC, inhibition of this ion channel would be expected to modulate ASL hydration by reducing ion transport and altering the electrochemical gradient to favor secretion. In addition, patients who have mutations that produce either over- or under-active ENaC who exhibit either CF-like lung disease or accelerated MCC, respectively. These genetic data suggest that pharmacologic modulation of ENaC activity could indeed have a significant impact on lung function, particularly over a long time domain. dditionally, observational data demonstrating that some CF patients who possess rare mutations in ENaC causing hypomorphic channel activity also possess a non-progressive lung function phenotype further supports that ENaC may indeed be an important adjunctive target for treating CF.
The success of CFTR modulator therapeutic development exemplifies the advances in preclinical models of CF and their utility in predicting clinical responses to CFTR modulator treatment. CF knockout animals, including mice, rats, rabbits, ferrets, and pigs, have been produced which recapitulate human CF disease with varying degrees of faithfulness [62]. Further, other models of CF lung disease such as the βENaC-transgenic mouse have also been useful for preclinical studies of CF [23,26]. Although models such as these produce CF-like lung disease as a consequence of significant airway dehydration, they do not fully recapitulate all of the potential pathologies involved in CF lung disease such as acidification of the airways and production of hyperviscous mucus. Thus, although these models are informative regarding whether targeting ENaC is effective, they may not accurately predict the effect of ENaC inhibitors in true CF lung disease.
As is common with other pharmacological development, preclinical data in ENaC-targeted therapeutics has not necessarily been predictive of clinical outcomes, nor have they been faithful in predicting safety and tolerability in patients. Moreover, the use of assays which provide insight into ENaC channel function in situ as well as functional measures related to airway surface liquid hydration and mucociliary transport rate will enable assessment of whether a putative ENaC-targeted therapeutic is truly inhibiting ENaC function and whether there is a functional effect of the therapeutic as well. In particular, benchtop assays which can also be applied in vivo in humans, such as nasal potential difference or gamma scintigraphy-based MCC assays would be particularly informative, particularly if feasibility and throughput could be improved. Monitoring ASL/PCL depth and MCT in vivo could be another approach [2,63–65]. Taken together with traditional clinical outcome measures such as lung function, exacerbation rate, sweat chloride, etc., these data would provide insight into mechanistic bases of either success or failure in clinical trials. Identification of salient biomarkers in patient populations which are sensitive to changes in ENaC channel function as well as improving the sensitivity of traditional clinical outcome measures will be particularly important as patients improve in their health, potentially from improved CFTR modulator therapies. As patients with CF become healthier, the challenge of detecting additional effects of newer treatments would be expected to become more challenging. Therefore, improving both the preclinical models for the testing of ENaC-targeted therapeutics as well the biomarkers for drug activity which can be translated to human patients will aid in successful clinical translation of future ENaC-targeted therapeutics.
ENaC INHIBITORS IN DEVELOPMENT
Summary of Current State of ENaC-Therapy Development
Despite historical challenges with ENaC-targeted therapeutics, there is still a significant effort to produce more potent, and more specifically targeted therapeutic agents to inhibit ENaC in CF, and ultimately validate this concept in CF disease. Compounds which are currently in development encompass next-generation small molecule inhibitors, rationally designed CAP-inhibitors, peptide analogs exploiting the natural autocrine regulatory mechanisms of ENaC, and molecular approaches with ASO and siRNA therapies. Table 2 summarizes these compounds which are currently in development. Many of these compounds share characteristics which distinguish them from previous drugs targeting ENaC (structures available in [33]). In particular, these compounds are intended to exhibit higher specificity to the airway epithelium, while minimizing systemic exposure and off-target effects, while retaining a similar or higher degree of efficacy. Furthermore, the duration of effect for some compounds, particularly nucleic acid therapeutics, is prolonged compared to historical ENaC inhibitors. Many of these compounds are early in development, with the only small number currently undergoing clinical trials in human patients at this time. However, the swath of promising preclinical data thus far suggests that the potency and safety profiles of many of these compounds are substantially better than previous generations of ENaC-targeted therapeutics.
Table 2.
Ongoing development of ENaC therapeutics
| Drug | Type | Mechanism | Current Stage of Development |
|---|---|---|---|
| NVP-QBE 170 (Novartis) | Small molecule | Direct inhibition | Preclinical Models [66] |
| AZD 5634 (AstraZeneca) | Small molecule | Direct inhibition | Phase I Clinical Trial clinicaltrials.gov: NCT02950805 |
| VX-371 (formerly P-1037, collaboration between Vertex and Parion) | Small molecule | Direct inhibition | Phase II Clinical Trial (combination VX-371 and Orkambi in F508del homozygotes); clinicaltrials.gov: NCT02709109 |
| QBW276 (Novartis) | Small molecule | Direct inhibition | Phase II Clinical Trials; clinicaltrials.gov: NCT02566044 |
| BI 443651 (Boehringer INgelheim) | Small molecule | Direct inhibition | Phase I Clinical Trial; clinicaltrials.gov: NCT02566044 |
| QUB-TL1 | Small molecule | Channel-Activating Protease inhibitor | Preclinical Models [74] |
| MK 104 (Mucokinetica) | Small molecule | Channel-Activating Protease inhibitor | Preclinical Models { Hall, abstract in J Cyst Fibros 2016, 15:S4-S5; R Hall, abstract in Pediatr Pulmonol 2017, 52:S239-S240} |
| ARO-ENaC | Molecular inhibition | Small interfering RNA | Preclinical Models {EW Bush abstract in Am J Respir Crit Care Med 2018, 197:A3867} |
| siRNA | Molecular inhibition | Small interfering RNA | Preclinical Models [55] |
| ENaC Inhibitory ASO (Ionis) | Molecular inhibition | Antisense oligonucleotide | Preclinical Models [81] |
| SPX-101 (Spyryx) | Peptide analogue | Indirect inhibition as a SPLUNC-1 analogue, which promotes ENaC channel internalization | Phase II Clinical Trial [53][72]; clinicaltrials.gov: NCT03229252 |
Direct Inhibitors
Next generation small molecule inhibitors have been developed by a partnership between Parion and Vertex (VX-371, formerly P-1037; clinicaltrials.gov: NCT02709109), and separately by AstraZeneca (AZD5634; clinicaltrials.gov: NCT02950805), Boehringer Ingelheim (BI 443651; clinicaltrials.gov: NCT02976519) and Novartis (QBW276; clinicaltrials.gov: NCT02566044). A potential hypothesis for the utility of ENaC inhibitors for CF is their potential to drive additional CFTR-dependent secretion through shift of membrane potential. This strategy was tested in a phase 2 clinical trial of VX-371 (formerly Parion P-1037) which compared the addition of either hypertonic saline, or hypertonic saline plus VX-371 to the treatment regimen of CF patients homozygous for the F508del mutation taking the combination CFTR modulator regimen ivacaftor/lumacaftor (Orkambi), with percent predicted forced expiratory volume in 1 s (ppFEV1) as the primary outcome measure (Vertex press release: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=1045401). Although VX-371 did not produce any clinically meaningful improvement in ppFEV1, it was well tolerated by the patients. The reasons for this are not yet known. One shortcoming of the study was that no measures of ion transport or mucus clearance, such as NPD or a radiographic measure of MCC, were recorded, thus the study will not be able to determine whether treatment with VX-371 achieved ENaC inhibition or influenced MCC in vivo. Thus, we have not yet ascertained whether the failure is mechanistically related, or due to other pharmacological problems that frequently plague drug development. For example, it is possible that the dose selected, which was shown to not cause hyperkalemia or other adverse side effects, was lower than a therapeutically meaningful dose. fundamental question which arises from these data is whether inhibition of ENaC can produce meaningful physiological improvements, which are not captured by clinical outcome measures such as ppFEV1. Regardless, at present the development of VX-371 as a CF therapy has been halted. However, a separate phase 2 study of VX-371 in patients with primary ciliary dyskinesia remains ongoing, and will assess the potential of ENaC inhibition in a disease not influenced by the more complex physiology of the CF airway.
A significant therapeutic effect in the airways must be balanced with off-target effects, particularly with potassium-sparing ENaC inhibitors, and could allow dosing to more efficacious thresholds. One such compound, NVP-QBE170, a dimeric amiloride derivative under development from Novartis, has shown to be effective in animal models in attenuating ENaC activity without inducing hyperkalemia [66]. Novartis is also exploring novel α-branched pyrazinoyl quaternary amines for their ability to act as potent ENaC inhibitors [67,68]. However, the application of both these compounds in CF therapy has not been reported yet. Therefore, at present other approaches which allow for direct or indirect airway-specific targeting of ENaC inhibition may provide a more viable approach to therapeutic ENaC inhibition.
QBW276 is a small molecule ENaC inhibitor in development by Novartis that is currently in a phase 2 clinical trial (NCT02566044) to assess multiple doses of inhaled QBW276 in three cohorts of adult CF patients [69]. Study design for cohorts 1 and 2 is a randomized, double-blind, placebo-controlled, parallel arm, multiple dose study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy in CF patients regardless of genotype. Patients in cohort 1 will receive 6 mg daily (3 mg bid) for 7 days (6:2 QBW276 and placebo, respectively). Dose and frequency will be confirmed upon completion of cohort 1 and then cohort 2 will receive QBW276 or placebo for a duration of 14 days (6:2 QBW276 and placebo, respectively). Cohort 3 will enroll 24 CF patients who are homozygous for F508del and will utilize a randomized, double-blind, placebo-controlled, cross-over design over 4 weeks. The primary and secondary outcome measures related to preliminary efficacy include ppFEV1 and lung clearance index (LCI). The inhalation administration route promises to limit systemic off-target effects and the genotype inclusive recruitment strategy for cohorts 1 and 2 should be informative about theratypes responsive to direct ENaC inhibition.
BI 443651 is another small molecule ENaC inhibitor currently in development by Boehringer Ingelheim. Boehringer Ingelheim has been developing compounds with potent ENaC activity for recently, with several patent application filings in the past 5 years [33]. Presently, their candidate molecule BI 443651 is undergoing phase 1 clinical trials in CF patients, with recruitment ongoing (NCT02976519).A separatephase1clinicaltrialofBI 443651 in patients with mild asthma recently completed, with results pending (NCT03135899).
Another novel small molecule ENaC inhibitor currently in development is AZD5634 (AstraZeneca), which is currently undergoing phase 1 clinical trials and may demonstrate improved ENaC selectivity. Preclinical data in primary CF airway epithelial cells demonstrating augmented ASL depth and enhanced mucociliary transport have been reported (E Falk Libby, abstract in Am J Respir Crit Care Med 2017, 195:A6466). Safety data from a first in human study of AZD5634 demonstrated that it was well tolerated at all doses tested, with no serious adverse events reported (P Gardiner, abstract in Am J Respir Crit Care Med 2017, 195:A7306). Importantly, no evidence of hyperkalemia was observed, possibly as a consequence of low systemic exposure and renal clearance, and current CF studies are enrolling (NCT02950805).
An innovative approach in development that circumvents the liability of small molecules and is one approach to avoid systemic absorption is the use of peptide analogs of the short palate, lung, and nasal epithelial clone 1 (SPLUNC1), which is an autocrine regulatory protein secreted by the airway epithelial cells that promotes internalization of the ENaC protein and thus reduces sodium and water resorption across the epithelial surface [70]. The N-terminal end of SPLUNC1 contains an 18-amino acid domain (S18) which interacts with ENaC and promotes channel internalization [13]. The development of a synthetic peptide analog of the S18 domain by Spyryx Biosciences, called SPX-101, represents a unique approach to inhibition of ENaC [53,71]. Preclinical data have demonstrated that SPX-101 selectively binds ENaC and promotes its internalization in airway epithelial cells. Consequently, the amiloride-sensitive current was reduced substantially in these cells and mucus transport was enhanced. Importantly, while both SPLUNC1 and S18 individually have been shown to be degraded in CF sputum, SPX-101 was shown to be stable in CF sputum and retained normal activity following exposure to CF sputum [72]. Administration of SPX-101 to neonatal transgenic mice overexpressing the β-subunit of ENaC (one of the most widely used models of CF lung disease) improved survival from roughly 45% to 92% at the most efficacious dose. Importantly, safety data in murine and canine models did not demonstrate any adverse events, even in doses up to the maximum feasible dose [73]. At therapeutically meaningful doses, SPX-101 did not produce hyperkalemia, alter body weight, urinalysis measures, or blood chemistry, as peptide delivery to the lung offers some tissue specificity. So far, doses studied have not been limited by toxicity, but rather by the capacity of the aerosol delivery system, lending further confidence to the safety profile of SPX-101. phase 2 clinical trial (HOPE-1 trial) evaluating the safety and efficacy of SPX-101 in human patients is presently underway (clinicaltrials.gov: NCT03229252). Preliminary data from the first cohort of the HOPE-1 trial reported a 5.2% improvement in ppFEV1 beginning at day 7 and persisting through day 28 in a group of 16 CF patients treated with a high dose (120 mg) of SPX-101 (Spyryx Biosciences press release: http://spyryxbio.com/2018/06/06/spyryx-spx-101-phase-2-hope-1-trial-shows-improvement-in-lung-function-in-patients-with-cystic-fibrosis-via-novel-modulation-of-enac/), although the absolute improvement from baseline (as opposed to comparison with placebo) was modest, necessitating confirmatory studies. Importantly, no major impact on blood potassium levels was reported, and only one patient experienced a slight increase in blood potassium levels at the end of the 28 day trial. While these results require validation in a phase 3 clinical trial prior to clinical translation, the potential for clinically relevant improvements in ppFEV1 suggested by these unpublished data, combined with good safety and tolerability data in a phase 1b trial in patients treated with SPX-101 represent a positive step in the development of ENaC-targeted therapeutics for CF, particularly if findings be confirmed and improvements sustained.
Next generation, rationally-designed direct ENaC inhibitors have shown promise in preclinical models, having overcome challenges that have limited previous attempts at direct ENaC inhibition. Whether or not these therapies succeed in proceeding to approved clinical use remains to be seen.
Indirect Inhibitors
Indirect inhibition of ENaC remains of interest, with several new compounds currently in development which act indirectly to reduce ENaC function. Newer-generation CAP inhibitors include the experimental compounds QUB -TL1 and MKA 104. Both of these putatively target trypsin-like serine proteases, thus reducing proteolytic cleavage and subsequent activation of ENaC.
QUB-TL1 is a potent CAP inhibitor, which has been shown to diminish ENaC-mediation sodium absorption [74]. reclinical studies of QUB-TL1 in airway epithelial cell culture demonstrated that QUB-TL1 inhibits prostasin, matriptase, and furin. Consequently, QUB-TL1 induced internalization of γ-ENaC, and reduced channel activity, measured by short-circuit current. This reduction in channel activity corresponded with an increase in ASL height, measured by confocal fluorescence microscopy, and an accelerated MCC rate, measured by microbeads applied apically to airway epithelial cells grown at an air-liquid interface. In contrast to other serine protease inhibitors such as camostat and aprotinin, QUB-TL1 inhibits furin, which may confer some protection against activation of ENaC by neutrophil elastase. Furthermore, QUB-TL1 exerted a protective effect against cell death induced by Pseudomonas aeruginosa exotoxin A, which is a virulence factor released by P. aeruginosa and is commonly found in CF airways. While this was initially thought to be due to furin inhibition, a subsequent study by the same group with a furin-specific inhibitor, QUB-F1 did not demonstrate a protective effect against P. aeruginosa exotoxin-A [75], thus the mechanism underpinning QUB-TL1’s protective effect remains unclear. Further studies utilizing primary human bronchial epithelial cells, rather than cell lines, as well as in vivo experiments characterizing the effects of QUB-TL1 are needed to understand how QUB-TL1 functions in the intact airway epithelium. Nevertheless, these data demonstrate that QUB-TL1 may be a more effective approach to therapeutic indirect ENaC inhibition than previous CAP inhibitors.
MKA 104, produced by Mucokinetica, is a repurposed anticoagulant early in development for ENaC inhibition which also acts a CAP inhibitor. To date, only rudimentary measures of “ciliary transport speed”(CTS) have been reported (R Hall, abstract in J Cyst Fibros 2016, 15:S4-S5; R Hall, abstract in Pediatr Pulmonol 2017, 52:S239-S240), with no other published supporting data such as ion transport, ASL hydration, or biochemical data to indicate that MKA-104 is indeed inhibiting ENaC activity.
A common challenge for indirect ENaC regulators is that despite their potent inhibition of specific CAPs, there are redundant proteases which can cleave the extracellular loops of ENaC, circumventing drug efficacy. For example, neutrophil elastase, a non-specific protease, is present in airway secretions and has been shown to be substantially higher in concentration in CF sputum compared to healthy normal sputum (M Webster, abstract in Pediatr Pulmonol 2017, 52:S241). While QUB-TL1 was shown to afford some protection against activation of ENaC by neutrophil elastase, it nonetheless is not capable of complete inhibition of all protease activity in the epithelial milieu. Similar limitations were reported for camostat. Thus, balancing inhibition of specific CAPs with other protease activity to achieve a level of ENaC inhibition with therapeutic benefit is a challenge that will need to be addressed.
Molecular Approaches
Advancements in the development of molecular approaches to gene replacement and gene silencing, and in particular oligonucleotide and RNA-based technologies, may offer a new avenue for ENaC-targeted therapeutics. These approaches present several potential unique advantages [54]. Firstly, these oligonucleotide-based therapies putatively allow for subunit-specific targeting, which allows for therapies directed at the ENaC subunit(s) that provide the most potent reduction in channel activity. Secondly, oligonucleotide-based therapies may allow for improved tissue-specificity, allowing for highly efficient pharmacological delivery in the lung with minimal off-target and systemic effects, assuming no systemic distribution of the molecule. Finally, oligonucleotide-based therapies may allow for a substantially longer duration of effect compared to traditional small molecule-based therapies [76].
Putatively, oligonucleotide therapies can target any or all of the subunits which compose the ENaC channel. While the β-subunit has been shown to be regulator of channel activity [6,58], the α-subunit appears to be critical for full channel function [77,78]. Thus, molecular knockdown of α-ENaC by multiple oligonucleotide therapies has repeatedly shown to reduce ENaC expression and channel activity in vitro and in vivo [55,79–84]. These approaches have the potential to overcome challenges which have limited the utility of other ENaC-targeted therapeutics such as specificity, durability, and efficacy.
Recent progress in oligonucleotide technology has dramatically enhanced stability and specificity. However, delivery to the target tissues remains a significant challenge, especially in the obstructed CF lung. Nucleic acids, by nature, are large, negatively charges particles, which makes cellular uptake weak in most cases [76,85]. Therefore, the development of effective delivery vehicles, such as lipid nanoparticles (LNPs) and targeting moieties which can be directly conjugated to the nucleotide sequences, have been critical for advancement of oligonucleotide-based therapies to the clinic [76]. Strategies to evade entrapment in the mucus layer and achieve delivery to the epithelium are also a major point of emphasis [86–88]. The potential toxicity of these delivery vehicles must also be considered. Lipid-based delivery vehicles have been shown to trigger the transient elevation of cytokines when they are internalized by cells [76]. Further, polyethylene glycol, which is commonly used to mask delivery vehicles from immune recognition and avoid binding to mucus [89], has been shown to react with antibodies, leading to anaphylaxis [76]. Accordingly, ongoing work is currently being devoted to optimize oligonucleotide therapy delivery vehicles while minimizing potential toxicity and avoiding risk of accumulation. In particular, the development of biodegradable delivery materials and new approaches which bypass lipid-based delivery vehicles are promising. These innovative approaches have made it possible to specifically and efficiently delivery oligonucleotide-based therapeutics to target tissues, producing durable knockdown of target gene expression. consideration unique to oligonucleotide-based therapies for CF is that caution must be exercised with these therapeutics in CF because many CF patients take dornase alfa (recombinant human DNase; Pulmozyme®), which is a efficacious mucolytic agent that may interfere with oligonucleotide integrity [54]. Potential oligonucleotide-based therapeutics in CF must be stable when used in combination with dornase alfa, or mitigation strategies implemented during developmental testing to avoid potential interactions between the drugs.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are short, single-stranded nucleotide sequences which can induce mRNA degradation through RNase H, as well as upregulate translation and modulate splicing [54]. ASOs can be efficiently delivered to a variety of cells and tissue types without the need for modification or conjugation, which has made ASOs a potential molecular approach to targeted, therapeutic gene silencing [54]. Recent iterations of therapeutic ASOs have exploited ligand conjugation to facilitate tissue-specific delivery by receptor-mediated uptake, which has been particularly successful in hepatic delivery [90].
An ASO targeting the α-subunit of ENaC was developed in 1999 by Jain et al. [79], who demonstrated that targeting the α-subunit resulted in substantially reduced nonselective cation channel activity in rat alveolar type II cells measured by patch-clamp. These findings have been replicated by others [80], and more recently Ionis Pharmaceuticals reported similar findings in mice following either aerosolized administration or orotracheal instillation of ASOs targeting α-ENaC [81]. The latter study represents the first in vivo application of ASO therapy targeting ENaC in a CF model. Using two distinct mouse models of CF-like lung disease (the Nedd4L knockout mouse, and the β-ENaC transgenic mouse), aerosolized ENaC ASO treatment substantially reduced α-ENaC expression, reduced mucin expression, goblet cell metaplasia, lung inflammation, neutrophil infiltration, and normalized lung function and sodium channel activity. Survival of Nedd4L knockout mice is roughly 3 weeks, therefore the authors developed an ASO targeting Nedd4L, which, when administered to adult mice, recapitulated CF-like lung disease. Administration of ENaC ASOs in these mice, which were also treated with the Nedd4L ASO, prevented the development of CF-like lung disease when administered before Nedd4L ASO administration. Similarly, treatment with ENaC ASOs also ameliorated CF-like symptoms when administered after Nedd4L ASO-induced lung disease was already established. Evaluation in model systems with CFTR-induced disease represents the next logical test.
siRNA
Small-interfering RNAs (siRNAs) are small nucleotide sequences (21–23 nucleotides) containing an mRNA sequence along with its complement, which trigger the natural pathway of RNA interference (RNAi) leading to degradation of mRNA from a specific target gene [76]. This therapeutic gene silencing is mediated by the RNA-induced silencing complex (RISC), which associates with synthetic siRNAs, leading to recognition and base-pairing with target mRNAs and degradation by Argonaute 2 in the RISC complex. Once loaded into RISC, the antisense strand of siRNA can be stable for weeks, thus reducing the frequency of dosing necessary for therapeutic gene knockdown. Of note, there are pulmonary-intrinsic barriers to efficient local siRNA delivery into the lung such as anatomy, cough, mucociliary clearance, and clearance by alveolar macrophages, which is reviewed elsewhere [91–95].
Identifying siRNAs to inhibit ENaC in the airways selectively has been of great interest in the past decade, with several independent groups developing various siRNAs against ENaC [55,82,83,96], or regulators of ENaC [97]. The latter study utilized a large-scale screen to identify candidate genes which may be involved in the regulation of ENaC, particularly ENaC activators. Subsequent investigation demonstrated that several genes, including the ciliary neurotrophic factor receptor and diacylglycerol kinase, iota (DGKι) both modulated ENaC activity. In particular, DGKι appeared to be a potential novel drug target for CF because inhibition of DGKι resulted in reduced ENaC activity. However, this approach to ENaC inhibition requires further investigation and to date, no further studies have pursued DGKι inhibition as an indirect means of inhibiting ENaC.
Other studies have identified synthetic siRNAs which directly target ENaC, which have been transfected into airway epithelial cell cultures [83,96], and also tested in vivo in murine models [55,82]. Following delivery of siRNAs targeting either α-, β-, γ-ENaC, or the CAP prostasin by transfection to bronchial epithelial cells, Gianotti et al. [83] found that a combination of siRNAs for either α- or β-ENaC substantially reduced ENaC activity and expanded ASL depth. Of note, the increase in ASL depth was similar in magnitude to what is observed in CF cells with the F508del mutation following treatment with a CFTR corrector and potentiator. Similarly, Caci et al [96] demonstrated that siRNAs individually targeting either α-, β-, γ-ENaC all substantially reduced ENaC mRNA expression and inhibited ENaC function measured by short-circuit current. A concomitant reduction in transepithelial fluid transport was also observed with siRNA knockdown of any of the three ENaC subunits.
Separately, siRNAs against α-ENaC delivered via LNPs have been shown in vivo to reduce ENaC mRNA expression [55,82], although no functional measures of ion transport, ASL hydration, or MCC either in vivo or ex vivo have been reported following treatment with siRNAs against α-ENaC. ata from primary human bronchial and nasal epithelial cell culture experiments largely confirm previous findings of substantial ENaC knockdown and reduced channel activity. While these results indicate that siRNAs against ENaC can efficiently reduce channel activity, translation of ENaC inhibition to physiologically relevant outcomes such as increased ASL hydration or enhanced MCC will be crucial in advancing the development of ENaC siRNAs for CF. Thus, future work should focus on recapitulating these findings in CF models with the additional of relevant biomarkers to predict potential treatment effects in CF.
A recently reported study utilizing a novel delivery platform consisting of an siRNA trigger against αENaC directly conjugated to a lung epithelial-specific targeting ligand developed by Arrowhead Pharmaceuticals (ARO-ENaC) produced significant molecular knockdown of αENaC mRNA expression and protein levels in the lung in both single and multiple dose regimens in wild-type rats (EW Bush abstract in Am J Respir Crit Care Med 2018, 197:A3867). Even after 21 days post-dose administration, the relative expression of αENaC remained below 50%, suggesting a durable duration of effect. Importantly, no changes in lung CFTR expression, nor kidney ENaC expression were detected, indicating that off-target effects of ARO-ENaC were minimized. Localization of delivery to the lung tissue was achieved using an epithelial targeting ligand specific to the integrin αvβ6 ligand, which facilitated pulmonary epithelial uptake of the siRNA triggers, but not in other tissues. As with previous studies utilizing siRNA against ENaC, further studies are necessary to quantify the effects of RO-ENaC on airway epithelial ion transport, hydration, and MCC. These preliminary data from this study and others are encouraging however, and collectively these findings are supportive of utilizing siRNA to inhibit ENaC activity. Future studies translating these into in vitro and in vivo models of CF in conjunction with informative biomarkers examining physiologic and functional effects of these compounds should provide additional insight into the utility of siRNA and other oligonucleotide targeting mechanisms therapeutics in CF.
CONCLUSION AND FUTURE DIRECTIONS
ENaC-targeted therapeutics remain a substantial area of interest, and ongoing development of compounds utilizing a diverse array of strategic approaches provides a promising outlook for the future of ENaC-targeted therapeutics for CF. We are hopeful that these approaches will help us discover where the rubber meets the road for ENaC therapies, and how they fit in to the armamentarium of ion transport modalities useful to CF, most notably CFTR modulators, along with other methods to hydrate the airway, such as hypertonic saline or inhaled mannitol. These novel compounds must successfully address concerns regarding off-target effects, sufficient duration of ENaC inhibition in the airways, and the potential for influencing the function of other ion channels that contribute to ASL homeostasis. Nevertheless, advances in oligonucleotide-based therapies, peptide analogs mimicking the natural autocrine regulatory mechanisms of ENaC, and next generation small molecules that directly or indirectly inhibit ENaC have brought ENaC-targeted therapeutics closer than ever to successful translation into the clinic.
HIGHLIGHTS.
ENaC contributes to airway surface liquid dehydration in CF lung disease; thus, ENaC inhibitors have been developed as CF therapeutics.
The use of ENaC as a therapeutic target in CF has yet to be successfully translated to CF patients, with conflicting reports regarding whether ENaC activity is abnormal in CF.
Early attempts by small molecule direct and indirect inhibition were unsuccessful, due to off-target effects and potential adverse events; however improved molecules are on the horizon.
New approaches with peptide analogs and molecular strategies are promising and could promise enhanced efficacy with a reduced risk of adverse effects.
Acknowledgments
Funding Sources:
This work was supported by the National Institutes of Health [R-JS: T32HL105346 (PI Victor J. Thannickal]; [JEP: T32HL134640 (PI William E. Swords)]; [SMR: R35HL135816 and P30DK072482].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
of special interest
of outstanding interest
REFERENCES
- 1.Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A: Cystic fibrosis. Nature Reviews Disease Primers 2015, 1:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, Dierksen G, Mazur M, Shastry S, Li Y, et al. : A Functional Anatomic Defect of the Cystic Fibrosis Airway. American Journal of Respiratory and Critical Care Medicine 2014, 190:421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, et al. : Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345:818–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, et al. : Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shamsuddin AKM, Quinton PM: Surface fluid absorption and secretion in small airways. The Journal of Physiology 2012, 590:3561–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanukoglu I, Hanukoglu A: Epithelial sodium channel (ENaC) family: Phylogeny, structure–function, tissue distribution, and associated inherited diseases. Gene 2016, 579:95–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *7.Livraghi-Butrico A, Wilkinson KJ, Volmer AS, Gilmore RC, Rogers TD, Caldwell RA, Burns KA Jr. CRE, Mall MA, Boucher, et al. : Lung disease phenotypes caused by overexpression of combinations of α-, β-, and γ-subunits of the epithelial sodium channel in mouse airways. American Journal of Physiology-Lung Cellular and Molecular Physiology 2018, 314:L318–L331. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study utilized transgenic mouse models of double and triple transgenic mice overexpressing various ENaC subunits and characterized their phenotypes. Findings indicated that over-expression of βENaC is rate limiting for pathologic airway dehydration, and co-overexpressin of βγENaC had additive effects on sodium transport and disease severity.
- 8.Collawn JF, Matalon S: CFTR and lung homeostasis. American Journal of Physiology-Lung Cellular and Molecular Physiology 2014, 307:L917–L923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collawn JF, Lazrak A, Bebok Z, Matalon S: The CFTR and ENaC debate: how important is ENaC in CF lung disease? American Journal of Physiology-Lung Cellular and Molecular Physiology 2012, 302:L1141–L1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stutts M, Canessa C, Olsen J, Hamrick M, Cohn J, Rossier B, Boucher R: CFTR as a cAMP-dependent regulator of sodium channels. Science 1995, 269:847–850. [DOI] [PubMed] [Google Scholar]
- 11.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, Boucher RC, Gabriel SE: Regulation of the Epithelial Sodium Channel by Serine Proteases in Human Airways. Journal of Biological Chemistry 2002, 277:8338–8345. [DOI] [PubMed] [Google Scholar]
- 12.Guggino WB, Stanton BA: New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 2006, 7:426–436. [DOI] [PubMed] [Google Scholar]
- 13.Hobbs CA, Blanchard MG, Alijevic O, Tan CD, Kellenberger S, Bencharit S, Cao R, Kesimer M, Walton WG, Henderson AG, et al. : Identification of the SPLUNC1 ENaC-inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airway epithelial cultures. American Journal of Physiology-Lung Cellular and Molecular Physiology 2013, 305:L990–L1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donaldson SH, Poligone EG, Stutts MJ: CFTR Regulation of ENaC. In Cystic Fibrosis Methods and Protocols Edited by Skach WR: Humana Press; 2002:343–364. [DOI] [PubMed] [Google Scholar]
- *15.Cholon DM, Gentzsch M: Recent progress in translational cystic fibrosis research using precision medicine strategies. Journal of Cystic Fibrosis 2018, 17:S52–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent review detailing the state-of-the-art of precision medicine strategies in CF therpeutic development.
- 16.Donaldson SH, Galietta L: New Pulmonary Therapies Directed at Targets Other than CFTR. Cold Spring Harbor Perspectives in Medicine 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *17.Agrawal PB, Wang R, Li HL, Schmitz-Abe K, Simone-Roach C, Chen J, Shi J, Louie T, Sheng S, Towne MC, et al. : The Epithelial Sodium Channel Is a Modifier of the Long-Term Nonprogressive Phenotype Associated with F508del CFTR Mutations. American Journal of Respiratory Cell and Molecular Biology 2017, 57:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study, investigators identified an association between mutations in the SCNN1D gene encoding the δ-subunit of ENaC, which cause hypomorphic channel activity, and a long-term, nonprogressive lung phenotype in CF patients. This finding suggests a potential genetic modifier in CF patients who possess these rare SCNN1D mutations.
- 18.Chen J-H, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, et al. : Loss of Anion Transport without Increased Sodium Absorption Characterizes Newborn Porcine Cystic Fibrosis Airway Epithelia. Cell 2010, 143:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itani OA, Chen J-H, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ: Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proceedings of the National Academy of Sciences 2011, 108:10260–10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TIA, Zhang Y, Zhou W, Tyler SR, Fisher JT, et al. : Lung Phenotype of Juvenile and Adult Cystic Fibrosis Transmembrane Conductance Regulator–Knockout Ferrets. American Journal of Respiratory Cell and Molecular Biology 2014, 50:502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher JT, Tyler SR, Zhang Y, Lee BJ, Liu X, Sun X, Sui H, Liang B, Luo M, Xie W, et al. : Bioelectric Characterization of Epithelia from Neonatal CFTR Knockout Ferrets. American Journal of Respiratory Cell and Molecular Biology 2013, 49:837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoltz DA, Meyerholz DK, Welsh MJ: Origins of Cystic Fibrosis Lung Disease. New England Journal of Medicine 2015, 372:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.McCarron A, Donnelley M, Parsons : Airway disease phenotypes in animal models of cystic fibrosis. Respiratory Research 2018, 19:54. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent review discussing the current state of animal models of CF. This is particularly important in the context of translational challenges for therapeutic development.
- 24.Nur N, Lang C, Hodax JK, Quintos JB: Systemic Pseudohypoaldosteronism Type I: A Case Report and Review of the Literature. Case Reports in Pediatrics 2017, 2017:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mall MA, Graeber SY, Stahl M, Zhou-Suckow Z: Early cystic fibrosis lung disease: Role of airway surface dehydration and lessons from preventive rehydration therapies in mice. The International Journal of Biochemistry & Cell Biology 2014, 52:174–179. [DOI] [PubMed] [Google Scholar]
- 26.Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC: Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 2004, 10:487–493. [DOI] [PubMed] [Google Scholar]
- 27.Rauh R, Soell D, Haerteis S, Diakov A, Nesterov V, Krueger B, Sticht H, Korbmacher C: A mutation in the β-subunit of ENaC identified in a patient with cystic fibrosis-like symptoms has a gain-of-function effect. American Journal of Physiology-Lung Cellular and Molecular Physiology 2013, 304:L43–L55. [DOI] [PubMed] [Google Scholar]
- 28.Mutesa L, Azad AK, Verhaeghe C, Segers K, Vanbellinghen J-F, Ngendahayo L, Rusingiza EK, Mutwa PR, Rulisa S, Koulischer L, et al. : Genetic Analysis of Rwandan Patients With Cystic Fibrosis-Like Symptoms. Chest 2009, 135:1233–1242. [DOI] [PubMed] [Google Scholar]
- 29.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, et al. : Pulmonary Epithelial Sodium-Channel Dysfunction and Excess Airway Liquid in Pseudohypoaldosteronism. New England Journal of Medicine 1999, 341:156–162. [DOI] [PubMed] [Google Scholar]
- 30.Althaus M: ENaC Inhibitors and Airway Re-hydration in Cystic Fibrosis: State of the Art. Current Molecular Pharmacology 2013, 6:3–12. [DOI] [PubMed] [Google Scholar]
- 31.Bangel-Ruland N, Tomczak K, Weber W-M: Targeting ENaC as a Molecular Suspect in Cystic Fibrosis. Current Drug Targets 2015, 16:951–957. [DOI] [PubMed] [Google Scholar]
- 32.Butler R, Hunt T, Smith NJ: ENaC inhibitors for the treatment of cystic fibrosis. Pharmaceutical Patent Analyst 2015, 4:17–27. [DOI] [PubMed] [Google Scholar]
- 33.Smith NJ, Solovay CF: Epithelial Na+ channel inhibitors for the treatment of cystic fibrosis. Pharmaceutical Patent Analyst 2017, 6:181–190. [DOI] [PubMed] [Google Scholar]
- 34.Farinha CM, Matos P: Repairing the basic defect in cystic fibrosis – one approach is not enough. FEBS Journal 2016, 283:246–264. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Salomon JJ, Sheppard DN, Mall MA, Galietta LJV: Bypassing FTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Current Opinion in Pharmacology 2017, 34:91–97. [DOI] [PubMed] [Google Scholar]
- *36.Harutyunyan M, Huang Y, Mun K-S, Yang F, Arora K, Naren AP: Personalized medicine in CF: from modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. American Journal of Physiology-Lung Cellular and Molecular Physiology 2018, 314:L529–L543. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review details the state of modulator therapy for CF patients with rare mutations. Should ENaC-targeted therapeutics successfully translate to the clinic, these patients would likely benefit from these types of therapies.
- 37.Qadri YJ, Rooj AK, Fuller CM: ENaCs and ASICs as therapeutic targets. American Journal of Physiology-Cell Physiology 2012, 302:C943–C965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.App EM, King M, Helfesrieder R, Köhler D, Matthys H: Acute and Long-term Amiloride Inhalation in Cystic Fibrosis Lung Disease: A Rational Approach to Cystic Fibrosis Therapy. American Review of Respiratory Disease 1990, 141:605–612. [DOI] [PubMed] [Google Scholar]
- 39.Knowles MR, Church NL, Waltner WE, Yankaskas JR, Gilligan P, King M, Edwards LJ, Helms RW, Boucher RC: A Pilot Study of Aerosolized Amiloride for the Treatment of Lung Disease in Cystic Fibrosis. New England Journal of Medicine 1990, 322:1189–1194. [DOI] [PubMed] [Google Scholar]
- 40.Riedler J, Huttegger I: Pilot study of amiloride inhalation in children with cystic fibrosis. Klinische Padiatrie 1992, 204:158–162. [DOI] [PubMed] [Google Scholar]
- 41.Canessa CM, Horisberger J-D, Rossier BC: Epithelial sodium channel related to proteins involved in neurodegeneration. Nature 1993, 361:467. [DOI] [PubMed] [Google Scholar]
- 42.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger D, Rossier BC: Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367:463. [DOI] [PubMed] [Google Scholar]
- 43.Graham A, Hasani A, Alton E, Martin G, Marriott C, Hodson M, Clarke S, Geddes D: No added benefit from nebulized amiloride in patients with cystic fibrosis. European Respiratory Journal 1993, 6:1243–1248. [PubMed] [Google Scholar]
- 44.Cuthbert AW: New horizons in the treatment of cystic fibrosis. British Journal of Pharmacology 2011, 163:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirsh AJ, Sabater JR, Zamurs A, Smith RT, Paradiso AM, Hopkins S, Abraham WM, Boucher RC: Evaluation of Second Generation Amiloride Analogs as Therapy for Cystic Fibrosis Lung Disease. Journal of Pharmacology and Experimental Therapeutics 2004, 311:929–938. [DOI] [PubMed] [Google Scholar]
- 46.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC: Mucus Clearance and Lung Function in Cystic Fibrosis with Hypertonic Saline. New England Journal of Medicine 2006, 354:241–250. [DOI] [PubMed] [Google Scholar]
- 47.Levin MH, Sullivan S, Nielson D, Yang B, Finkbeiner WE, Verkman AS: Hypertonic Saline Therapy in Cystic Fibrosis: Evidence Against the Proposed Mechanism Involving Aquaporins. Journal of Biological Chemistry 2006, 281:25803–25812. [DOI] [PubMed] [Google Scholar]
- 48.O’Riordan TG, Donn KH, Hodsman P, Ansede JH, Newcomb T, Lewis SA, Flitter WD, White VS, Johnson MR, Montgomery AB, et al. : Acute Hyperkalemia Associated with Inhalation of a Potent ENaC Antagonist: Phase 1 Trial of GS-9411. Journal of Aerosol Medicine and Pulmonary Drug Delivery 2014, 27:200–208. [DOI] [PubMed] [Google Scholar]
- 49.Vallet V, Chraibi A, Gaeggeler H-P, Horisberger J-D, Rossier BC: An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 1997, 389:607. [DOI] [PubMed] [Google Scholar]
- 50.Rowe SM, Reeves G, Hathorne H, Solomon GM, Abbi S, Renard D, Lock R, Zhou P, Danahay H, Clancy JP, et al. : Reduced Sodium Transport With Nasal Administration of the Prostasin Inhibitor Camostat in Subjects With Cystic Fibrosis. Chest 2013, 144:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hobbs CA, Da Tan C, Tarran R: Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? The Journal of Physiology 2013, 591:4377–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huggins DJ, Sherman W, Tidor B: Rational pproaches to Improving Selectivity in Drug Design. Journal of Medicinal Chemistry 2012, 55:1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **53.Scott DW, Walker MP, Sesma J, Wu B, Stuhlmiller TJ, Sabater JR, Abraham WM, Crowder TM, Christensen DJ, Tarran R: SPX-101 Is a Novel Epithelial Sodium Channel–targeted Therapeutic for Cystic Fibrosis That Restores Mucus Transport. American Journal of Respiratory and Critical Care Medicine 2017, 196:734–744. [DOI] [PubMed] [Google Scholar]; This study first reported a therapeutic effect of SPX-101, which is a peptide analog of SPLUNC1, and was shown to cause NaC channel internalization. This significantly reduced sodium transport, increased mucus transport, and improved survival in the βENaC-transgenic mouse model of CF lung disease.
- *54.Sasaki S, Guo S: Nucleic Acid Therapies for Cystic Fibrosis. Nucleic Acid Therapeutics 2018, 28:1–9. This review details the utility of nucleic acid based therapies, including ASOs and siRNA for CF. [DOI] [PubMed] [Google Scholar]
- **55.Manunta MDI, Tagalakis AD, Attwood M, Aldossary AM, Barnes JL, Munye MM, Weng A, McAnulty RJ, Hart SL: Delivery of ENaC siRNA to epithelial cells mediated by a targeted nanocomplex: a therapeutic strategy for cystic fibrosis. Scientific Reports 2017, 7:700. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrated that a self-assembling nanocomplex formulation for a delivery vehicle containing siRNA was effective at silencing expression of αENaC both in vitro and in vivo. Treatment with this siRNA against αENaC significantly reduced measured protein levels in cell models and in mice.
- 56.Jiang Q, Li J, Dubroff R, Ahn YJ, Foskett JK, Engelhardt J, Kleyman TR: Epithelial Sodium Channels Regulate Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channels in XenopusOocytes. Journal of Biological Chemistry 2000, 275:13266–13274. [DOI] [PubMed] [Google Scholar]
- 57.Sharma N, LaRusch J, Sosnay PR, Gottschalk LB, Lopez AP, Pellicore MJ, Evans T, Davis E, Atalar M, Na C-H, et al. : A sequence upstream of canonical PDZ-binding motif within CFTR COOH-terminus enhances NHERF1 interaction. American Journal of Physiology-Lung Cellular and Molecular Physiology 2016, 311:L1170–L1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kleyman TR, Kashlan OB, Hughey RP: Epithelial Na+ Channel Regulation by Extracellular and Intracellular Factors. Annual Review of Physiology 2018, 80:263–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ji H-L, Chalfant ML, Jovov B, Lockhart JP, Parker SB, Fuller CM, Stanton BA, Benos DJ: The Cytosolic Termini of the β- and γ-ENaC Subunits Are Involved in the Functional Interactions between Cystic Fibrosis Transmembrane Conductance Regulator and Epithelial Sodium Channel. Journal of Biological Chemistry 2000, 275:27947–27956. [DOI] [PubMed] [Google Scholar]
- 60.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. : Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences 2009, 106:18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rowe SM, Liu B, Hill A, Hathorne H, Cohen M, Beamer JR, Accurso FJ, Dong Q, Ordoñez CL, Stone AJ, et al. : Optimizing Nasal Potential Difference Analysis for CFTR Modulator Development: Assessment of Ivacaftor in CF Subjects with the G551D-CFTR Mutation. PLOS ONE 2013, 8:e66955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, Engelhardt JF: Animal and model systems for studying cystic fibrosis. Journal of Cystic Fibrosis 2018, 17:S28–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Birket SE, Chu KK, Houser GH, Liu L, Fernandez CM, Solomon GM, Lin V, Shastry S, Mazur M, Sloane PA, et al. : Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. American Journal of Physiology-Lung Cellular and Molecular Physiology 2016, 310:L928–L939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Birket SE, Davis JM, Fernandez CM, Tuggle KL, Oden AM, Chu KK, Tearney GJ, Fanucchi MV, Sorscher EJ, Rowe SM: Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu L, Shastry S, Byan-Parker S, Houser G, Chu KK, Birket SE, Fernandez CM, Gardecki JA, Grizzle W , Wilsterman EJ, et al. : An Autoregulatory Mechanism Governing Mucociliary Transport Is Sensitive to Mucus Load. American Journal of Respiratory Cell and Molecular Biology 2014, 51:485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coote KJ, Paisley D, Czarnecki S, Tweed M, Watson H, Young A, Sugar R, Vyas M, Smith NJ, Baettig U, et al. : NVP-QBE170: an inhaled blocker of the epithelial sodium channel with a reduced potential to induce hyperkalaemia. British Journal of Pharmacology 2015, 172:2814–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hunt T, Atherton-Watson HC, Axford J, Collingwood SP, Coote KJ, Cox B, Czarnecki S, Danahay H, Devereux N, Howsham C, et al. : Discovery of a novel chemotype of potent human ENaC blockers using a bioisostere approach. Part 1: Quaternary amines. Bioorganic & Medicinal Chemistry Letters 2012, 22:929–932. [DOI] [PubMed] [Google Scholar]
- 68.Hunt T, Atherton-Watson HC, Collingwood SP, Coote KJ, Czarnecki S, Danahay H, Howsham C, Hunt P, Paisley D, Young A: Discovery of a novel chemotype of potent human ENaC blockers using a bioisostere approach. Part 2: α-Branched quaternary amines. Bioorganic & Medicinal Chemistry Letters 2012, 22:2877–2879. [DOI] [PubMed] [Google Scholar]
- *69.Gentzsch M, Mall MA: Ion Channel Modulators in Cystic Fibrosis. Chest IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent review detailing the status of various ion channel modulators in CF, including CFTR modulators, ENaC inhibitors, as well as Calcium-activated Chloride Channel modulators.
- 70.Garcia-Caballero A, Rasmussen JE, Gaillard E, Watson MJ, Olsen JC, Donaldson SH, Stutts MJ, Tarran R: SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proceedings of the National Academy of Sciences 2009, 106:11412–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *71.Terryah ST, Fellner RC, Ahmad S, Moore PJ, Reidel B, Sesma JI, Kim CS, Garland AL, Scott DW, Sabater JR, et al. : Evaluation of a SPLUNC1-derived peptide for the treatment of cystic fibrosis lung disease. American Journal of Physiology-Lung Cellular and Molecular Physiology 2018, 314:L192–L205. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the development of a synthetic peptidomic mimicking the S18 regulatory domain of SPLUNC1 for therapeutic inhibition of ENaC.
- 72.Sesma JI, Wu B, Stuhlmiller TJ, Scott DW: SPX-101 is stable in and retains function after exposure to cystic fibrosis sputum. Journal of Cystic Fibrosis IN PRESS. [DOI] [PubMed] [Google Scholar]
- 73.Walker MP, Cowlen M, Christensen D, Miyamoto M, Barley P, Crowder T: Nonclinical safety assessment of SPX-101, a novel peptide promoter of epithelial sodium channel internalization for the treatment of cystic fibrosis. Inhalation Toxicology 2017, 29:356–365. [DOI] [PubMed] [Google Scholar]
- **74.Reihill JA, Walker B, Hamilton RA, Ferguson TEG, Elborn JS, Stutts MJ, Harvey BJ, Saint-Criq V, Hendrick SM, Martin SL: Inhibition of Protease–Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. American Journal of Respiratory and Critical Care Medicine 2016, 194:701–710. [DOI] [PubMed] [Google Scholar]; This study reports the findings of a novel CAP inhibitor, QUB-TL1, which acts as an indirect ENaC inhibitor. In vitro studies demonstrated significantly reduced sodium transport and internalization of γENaC. Consequently, airway hydration and mucociliary clearance were improved. Importantly, QUB- L1 was shown to inhibit neutrophil elastase, which has not been achieved by other serine protease inhibitors.
- 75.Ferguson TEG, Reihill JA, Walker B, Hamilton RA, Martin SL: A Selective Irreversible Inhibitor of Furin Does Not Prevent Pseudomonas Aeruginosa Exotoxin A-Induced Airway pithelial Cytotoxicity. PLOS ONE 2016, 11:e0159868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wittrup A, Lieberman J: Knocking down disease: a progress report on siRNA therapeutics. Nature Reviews Genetics 2015, 16:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E, Rossier B, Boucher R , Koller B: Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. The Journal of Clinical Investigation 1998, 102:1634–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hummler Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC: Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nature Genetics 1996, 12:325. [DOI] [PubMed] [Google Scholar]
- 79.Jain L, Chen X-J, Malik B, Al-Khalili O, Eaton DC: Antisense oligonucleotides against the α-subunit of ENaC decrease lung epithelial cation-channel activity. American Journal of Physiology-Lung Cellular and Molecular Physiology 1999, 276:L1046–L1051. [DOI] [PubMed] [Google Scholar]
- 80.Sobczak K, Segal A, Bangel-Ruland N, Semmler J, Van Driessche W, Lindemann H, Heermann R, Weber W-M: Specific inhibition of epithelial Na+ channels by antisense oligonucleotides for the treatment of Na+ hyperabsorption in cystic fibrosis. The Journal of Gene Medicine 2009, 11:813–823. [DOI] [PubMed] [Google Scholar]
- **81.Crosby JR, Zhao C, Jiang C, Bai D, Katz M, Greenlee S, Kawabe H, McCaleb M, Rotin D, Guo S, et al. : Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. Journal of Cystic Fibrosis 2017, 16:671–680. [DOI] [PubMed] [Google Scholar]; This study demonstrates the efficacy of an inhaled ASO against αENaC in vitro and in vivo. Significant reduction of ENaC expression was observed, and pre-treatment with the ASO against αENaC prevented the development of lung disease in an induced model of CF-like lung disease (ASO against Nedd4L in mice, which produces a CF-like phenotype).
- 82.Clark KL, Hughes SA, Bulsara P, Coates J, Moores K, Parry J, Carr M, Mayer RJ, Wilson P, Gruenloh C, et al. : Pharmacological Characterization of a Novel ENaCα siRNA (GSK2225745) With Potential for the Treatment of Cystic Fibrosis. Molecular Therapy. Nucleic Acids 2013, 2:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gianotti A, Melani R, Caci E, Sondo E, Ravazzolo R, Galietta LJV, Zegarra-Moran O: Epithelial Sodium Channel Silencing as a Strategy to Correct the Airway Surface Fluid Deficit in Cystic Fibrosis. American Journal of Respiratory Cell and Molecular Biology 2013, 49:445–452. [DOI] [PubMed] [Google Scholar]
- 84.Li T, Folkesson HG: RNA interference for α-ENaC inhibits rat lung fluid absorption in vivo. American Journal of Physiology-Lung Cellular and Molecular Physiology 2006, 290:L649–L660. [DOI] [PubMed] [Google Scholar]
- 85.Kole R, Krainer AR, Altman S: RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nature Reviews Drug Discovery 2012, 11:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duncan GA, Jung J, Hanes J, Suk JS: The Mucus Barrier to Inhaled Gene Therapy. Molecular Therapy 2016, 24:2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sanders N, Rudolph C, Braeckmans K, De Smedt SC, Demeester J: Extracellular barriers in respiratory gene therapy. Advanced Drug Delivery Reviews 2009, 61:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kim N, Duncan GA, Hanes J, Suk JS: Barriers to inhaled gene therapy of obstructive lung diseases: A review. Journal of Controlled Release 2016, 240:465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kanasty R, Dorkin JR, Vegas A, Anderson D: Delivery materials for siRNA therapeutics. Nature Materials 2013, 12:967. [DOI] [PubMed] [Google Scholar]
- 90.Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, et al. : Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Research 2014, 42:8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Merkel OM, Rubinstein I, Kissel T: siRNA Delivery to the lung: What’s new? Advanced Drug Delivery Reviews 2014, 75:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Merkel OM, Kissel T: Nonviral Pulmonary Delivery of siRNA. Accounts of Chemical Research 2012, 45:961–970. [DOI] [PubMed] [Google Scholar]
- 93.Qiu Y, Lam J, Leung S, Liang W: Delivery of RNAi Therapeutics to the Airways—From Bench to Bedside. Molecules 2016, 21:1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lam JK-W, Liang W, Chan H-K: Pulmonary delivery of therapeutic siRNA. Advanced Drug Delivery Reviews 2012, 64:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujita Y, Takeshita F, Kuwano K, Ochiya T: RNAi Therapeutic Platforms for Lung Diseases. Pharmaceuticals 2013, 6:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Caci E, Melani R, Pedemonte N, Yueksekdag G, Ravazzolo R, Rosenecker J, Galietta LJV, Zegarra-Moran O: Epithelial Sodium Channel Inhibition in Primary Human Bronchial Epithelia by Transfected siRNA. American Journal of Respiratory Cell and Molecular Biology 2009, 40:211–216. [DOI] [PubMed] [Google Scholar]
- 97.Almaça J, Faria D, Sousa M, Uliyakina I, Conrad C, Sirianant L, Clarke Luka A, Martins José P, Santos M, Heriché J-K, et al. : High-Content siRNA Screen Reveals Global ENaC Regulators and Potential Cystic Fibrosis Therapy Targets. Cell 2013, 154:1390–1400. [DOI] [PubMed] [Google Scholar]