

Abstract
For the past five decades the lysosome has been characterized as an unglamorous cellular recycling center. This notion has undergone a radical shift in the last ten years, with new research revealing that this organelle serves as a major hub for metabolic signaling pathways. The discovery that master growth regulators, including the protein kinase mTOR (mechanistic Target Of Rapamycin) make their home at the lysosomal surface has generated intense interest in the lysosome’s key role in nutrient sensing and cellular homeostasis. The transcriptional networks required for lysosomal maintenance and function are just being unraveled and their connection to lysosome-based signaling pathways revealed. The catabolic and anabolic pathways that converge on the lysosome connect this organelle with multiple facets of cellular function; when these pathways are deregulated they underlie multiple human diseases, and promote cellular and organismal aging. Thus, understanding how lysosome-based signaling pathways function will not only illuminate the fascinating biology of this organelle but will also be critical in unlocking its therapeutic potentials.
Keywords: lysosome, mTORC1, autophagy, metabolic signaling
Synopsis
After decades of being known as just the cellular recycling center, the lysosome has recently emerged as a central hub for metabolic signaling. Signaling pathways that call the lysosome home connect this organelle to key anabolic and catabolic processes that control many facets of cellular metabolism. Metabolic signals emanating from the lysosome are contributors to multiple human diseases, emphasizing the key role of the lysosome as a cellular site for the regulation of human health.
Introduction
As the degradative endpoint of the endosomal pathway, the lysosome has gained much notoriety as the ‘recycling center’ of the cell. Its characterization as a major catabolic center traces its origins to its discovery in the early 1950s. While investigating the mechanism of action of insulin, Christian de Duve made the serendipitous discovery of ‘sac-like structures’ that contained lytic activity 1,2. Ultrastructural characterization of these compartments by Alex Novikoff 3 led de Duve to rename them lysosomes. Following de Duve’s work, Werner Strauss traced the fate of radiolabeled extracellular proteins and discovered that these proteins localized to the lysosome and were found fragmented rather than intact 4. These pioneering studies cemented the lysosome as the cell’s degradative organelle.
The acidic environment of the lysosome maintained by the lysosomal v-ATPase 5, combined with a pantheon of luminal hydrolases, results in an organelle that is perfectly suited for the breakdown of major macromolecules, including lipids, polysaccharides and proteins 6. Once degraded, free fatty acids, monosacharrides and amino acids are transported to the cytoplasm by specific permeases, where they can be reused in anabolic processes 7. The degradative functions of the lysosome are key to maintaining cellular homeostasis, with perturbations in these processes leading to an array of human disorders collectively cataloged as lysosomal storage diseases 8. In addition to the basal level degradative processes undertaken by the lysosome, environmental stressors trigger a massive upregulation of degradation through the self-catabolic pathways known as autophagy. These pathways are critical in restoring homeostasis during times of metabolic imbalance and not surprisingly are also deregulated in multiple human maladies 9.
Our notion of the lysosome as a simple recycling center has undergone a dramatic revaluation over the last decade. With the discovery that the master regulator of cell growth, mTORC1 is localized to the lysosomal surface, we’ve now come to appreciate that the lysosome also functions as a platform for metabolic signaling. In this mini-review we focus on the lysosome as a metabolic signaling hub which integrates different environmental signals to regulate core anabolic and catabolic pathways critical in the maintenance of cellular homeostasis. We also touch on how deregulation of these pathways lead to human pathologies.
Metabolic signaling at the lysosomal surface
The mechanistic Target of Rapamycin (mTOR) is an evolutionarily conserved phosphatidylinositol-3-kinase (PI3K)-like serine/threonine protein kinase that is inhibited by rapamycin, a small molecule originally discovered in the soil of Easter Island 10. The history of rapamycin’s development as a pharmaceutical is recounted in full elsewhere; but briefly, rapamycin’s potential as pharmaceutical was immediately apparent due to its ability to slow or arrest cell proliferation. Soon after, it and several derivatives – “rapalogs”11 – were developed as immunosuppressants and later trialed as anti-cancer agents.
The 1990’s saw an intense search by several laboratories to discover the molecular mechanism by which rapamycin acts. Discoveries in yeast and in mammalian cells revealed that rapamycin acts by binding to the immunophilin FKBP12, forming a complex that then binds to and inhibits the mTOR protein kinase 12–14. Later work identified two distinct mTOR complexes 15, with shared as well as unique protein subunits and different substrates. mTOR complex 1 (mTORC1), which is acutely sensitive to rapamycin, is defined by the association of mTOR with the adaptor proteins Raptor and mLST8 16–18; while mTORC2, which is acutely rapamycin-resistant, is defined by the association of mTOR with the adaptor proteins Rictor, mLST8, and mSin1 19–22. The list of proteins that physically associate with mTORC1 or mTORC2 continues to grow, although it is likely that some of these proteins interact with mTORC1 or mTORC2 only in particular cell types or environmental conditions.
The two mTOR complexes have different cellular roles. mTORC1 functions as a key integrator of environmental and hormonal cues, sensing the availability of amino acids, glucose, cholesterol; cellular energy status; and hormones including insulin, IGF-1, leptin and adiponectin 23–28. When these states combine to form a permissive environment for cell growth and anabolism, mTORC1 localizes to the lysosome and interacts with its obligate activator Rheb-GTP to phosphorylate substrates including S6K1, 4E-BP1, and ULK1 to promote ribosomal biogenesis, translation, and lipogenesis while suppressing autophagy 19,29–32. Conversely, if one or more of these environmental or hormonal cues is not permissive for growth, mTORC1 remains inactive and these anabolic activities are inhibited, while autophagy is activated.
In contrast, mTORC2 is primarily an effector of the insulin/IGF-1/PI3K signaling pathway. While the molecular mechanism by which PI3K regulates mTORC2 has proven difficult to pin down, it was recently shown that phosphatidylinositol (3,4,5)-trisphosphate (PIP3) directly activates mTORC2 by relieving an inhibitory interaction with mSIN1 33. mTORC2 activity is also responsive to fatty acids 34, and may require interaction with ribosomal protein subunits 35. When activated, mTORC2 phosphorylates numerous AGC kinases, including AKT, SGK, and PKCα; these phosphorylations have typically been shown to promote both the activity and stability of the substrate proteins 36–38.
Work done over the last decade, and in particular over the last five years, have demonstrated that the activation of mTORC1 requires the coordinated interaction of many proteins at a previously unsuspected location, the lysosome. While previously conceived of as simply the “recycling center” of the cell, it is now clear that this hitherto unglamorous organelle serves as a critical platform for coordinating environmental and hormonal cues with mTORC1 and mTORC2 activation. The logic of this placement can be understood through an evolutionary context 39; in yeast, the TOR pathway is localized to the vacuole, the lysosomal orthologue 7, which functions as a major storage repository for nutrients. Recent technological advances 40,41 have determined that the mammalian lysosome is likewise selectively enriched in certain nutrients, including certain amino acids. Thus, the localization of mTORC1 to the lysosome allows mTORC1 to immediately gauge the nutrient status of the cell and rapidly shift its activity as required to maintain homeostasis.
mTORC1 is particularly sensitive to cellular and lysosomal levels of amino acids. mTORC1 is recruited to the lysosome in response to amino acids by the Rag family of heterodimeric small GTPases, orthologs of the yeast small GTPases Gtr1 and Gtr2 42–45. When amino acid levels drop within a cell, RagA/B bind GDP and RagC/D bind GTP (RagAGDP/RagCGTP), a state that does not interact with mTORC1. Upon amino stimulation, Rags flip their nucleotide bound state (RagAGTP/RagCGDP) and strongly interact with mTORC1 through its Raptor subunit, thus activating mTORC1 44,45. Over the last five years it has been shown that amino acids regulate mTORC1 activity by interacting with sensor proteins at the lysosomal surface that control the nucleotide loading state of the Rags (Figure 1).
Figure 1. Regulation of mTORC1 Activity at the Lysosome by Amino Acids and Growth Factors.
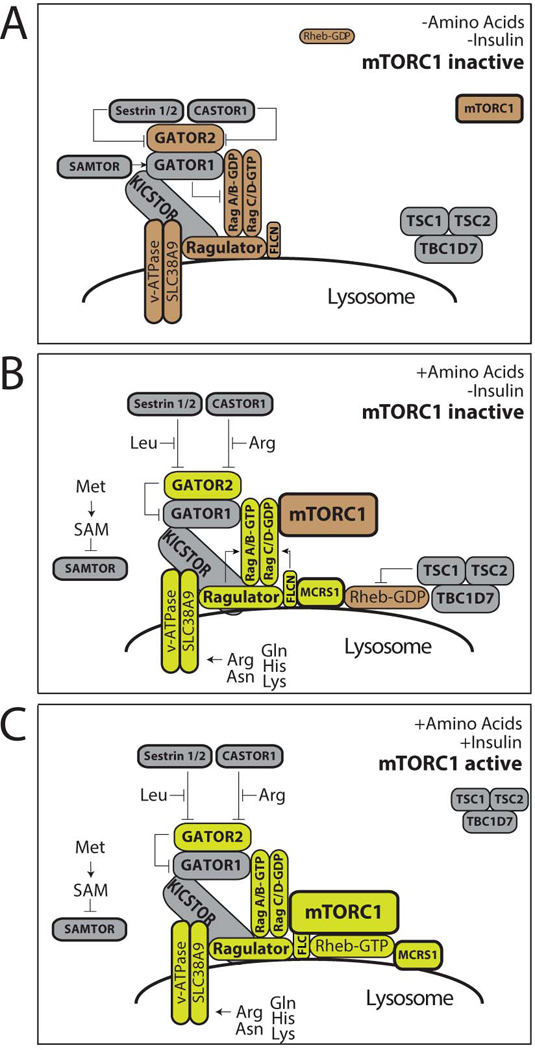
(A) In the absence of amino acids the lysosomal platform comprised of the Rag GTPases and its scaffold the Ragulator complex are inactive resulting in the cytoplasmic localization of mTORC1. GATOR1, which is localized to the lysosome by KICKSTOR and activated by SAMTOR, inactivates RagA/B. GATOR2, a negative regulator of GATOR1, is inactivated by the Sestrin and CASTOR complexes. The absence of growth factor (e.g. insulin) signaling promotes the localization of the tuberous sclerosis complex (TSC) to the lysosome. (B) Amino acid stimulation results in the inhibition of GATOR1 through multiple pathways. First, methionine catabolism raises cellular levels of SAM, which promotes the disassociation of SAMTOR from GATOR1. Secondly, binding of leucine by Sestrin and arginine by CASTOR results in the dissociation of these inhibitors from GATOR2, which is therefore free to inhibit GATOR1 activity. Amino acids also stimulate the Ragulator/SLC38A9/v-ATPase complex and FLCN to activate RagA/B and RagC/D, respectively. The active Rag heterodimer recruits mTORC1 to the lysosomal surface. MCRS1 recruits the small GTPase Rheb in an amino acid dependent manner. At the lysosome, TSC’s GAP activity inhibits Rheb, and thus mTORC1 remains inactive. (C) Insulin induces TSC to leave the lysosome, permitting Rheb to bind to GTP. GTP-bound Rheb activates the kinase activity of mTORC1. Adapted and updated from (18) with permission from Elsevier.
Regulators of the Rag GTPases
The lysosomal platform upon which mTORC1 activation occurs is known as the Ragulator, a protein complex composed of the proteins MP1, p14, HBXIP1, C7ORF59 and p18 (encoded by MAPKSP1, ROBLD3, HBXIP, c7orf59 and c11orf59, respectively), and is anchored to the lysosome by p18 46–48. The Ragulator, which is orthologous to the yeast EGO complex 49,50, tethers the Rag GTPases to the lysosomal surface and has guanine nucleotide exchange factor (GEF) activity for two of the Rag proteins, RagA and RagB 47,51, with the G-domains of the Rag proteins projecting away from the Ragulator surface 51–54. Control of Ragulator activity is one mechanism by which amino acids regulate mTORC1. Amino acids control the Ragulator in part via the lysosomal vacuolar ATPase (v-ATPase), which interacts with Ragulator in a manner sensitive to the availability of lysosomal amino acids and v-ATPase activity 55.
The importance of lysosomal metabolic signaling to human physiology has come to light by the identification of patients with partial defects in Ragulator function. Mutation in the 3’UTR of the p14 component of Ragulator leads to an introduction of an aberrant splice site 56 which reduces both p14 transcript and protein levels. Children bearing this mutation are below the 3rd percentile for height and suffer from a complex immune disorder that is marked by B-cell and cytoxic T-cell (CTL) deficiency, neutropenia and partial albinism 57. The short stature and immune disorders of these patients are characteristic of animal models where mTORC1 signaling is attenuated 58,59. Correspondingly, patient fibroblasts have a marked reduction in amino acid induced activation of mTORC1 signaling 46. The clinical phenotype associating albinism and immunodeficiency is similar to other disorders caused by defects in specialized secretory lysosomes such as melanomes and cytotoxic granules 60. Furthermore, in patients with reduced Ragulator activity late-endosome/lysosomal distribution was altered, with an increased distance from the nucleus. These results posit a lysosome-mTORC1 feedback loop, wherein inactivation of mTORC1 leads to altered distribution and function of the late-endosome/lysosomal compartment in multiple cell types including CTLs and melanocytes. Whether lysosomal function is compromised in other mTORC1 driven diseases or how mTORC1 activity is altered in lysosomal storage disorders (LSDs) represents an exciting area of research.
The nucleotide loading state of RagA and RagB are also controlled by the GATOR complexes 61,62. GATOR1, which consists of the proteins DEPDC5, NPRL2 and NPRL3, functions as a GTPase activating protein (GAP) for RagA and RagB. In contrast, GATOR2, which consists of the proteins Mios, WDR24, WDR59, Seh1L, and Sec13, acts to inhibit GATOR1 activity by unknown mechanisms. Both GATOR1 and GATOR2 are conserved in yeast (as SEACIT and SEACAT, respectively) as well as higher eukaryotes 49. The structure and regulation of the GATOR complexes is an area of active research; recent studies have taken a cryo-EM approach to shed light on how GATOR1 functions 63, and have identified a new protein complex dubbed KICSTOR (KPTN, ITFG2, C12ORF66 and SZT2) which recruits GATOR1 to the lysosomal surface and enables its interaction with the Rag proteins 64,65. KICKSTOR is required for the negative regulation of mTORC1 by amino acid or glucose deprivation, and its loss leads to hyperactivity of mTORC1; however, it is unknown if the recruitment of GATOR1 by KICSTOR is an important mTORC1 regulatory mechanism.
While much effort has focused on understanding the regulation of RagA and RagB, the nucleotide loading state of RagC and RagD are essential for this complex’s interaction with mTORC1 66. The FLCN complex, composed of FLCN, FNIP1 and FNIP2, has emerged as a positive regulator of RagC and RagD through its GAP activity for these GTPases, converting them from the GTP bound state to the GDP 66. FLCN is required for amino acids to recruit mTORC1 to the lysosome, and is itself recruited to the lysosomal surface by amino acid depletion 67. FLCN is recruited to the lysosome by interaction with RagA/BGDP and requires GATOR1 GAP activity; FLCN recruitment to the lysosome is thus likely regulated by the same set of amino acid sensors upstream of GATOR1/2 that act to regulate mTORC1 68. One possible explanation for the localization of FLCN to the lysosome following amino acid depletion was recently discovered: FLCN sequesters lysosomal leucine when amino acids are limited by blocking PAT1, a lysosomal amino acid transporter, thus helping to preserve mTORC1 activity during nutrient limitation 69.
Complicating our understanding of the role of FLCN is that it is mutated in a devastating disease, Birt-Hogg-Dube, that is characterized by large benign tumors which are the hallmark of other cancers driven by loss of negative regulators of the mTORC1 pathway. A possible explanation for this paradox may be that the function of FLCN as a tumor suppressor is context dependent. One recent study reports that FLCN is a ciliary protein that recruits LKB to activate AMPK – an inhibitor of mTORC1 signaling – in response to flow stress 70. Thus, cell type, local environmental conditions, and possibly, the subcellular compartment being investigated may impact the net effect of FLCN loss on mTORC1 activity.
Amino acid sensors, from hypothesis to reality
The greatest recent advances in understanding the response of mTORC1 to amino acids have come from identifying specific proteins which bind to amino acids and regulate the function of the GATOR complexes. The existence of these ‘amino acid sensors’ has long been hypothesized, but it has only been in the last several years that this hypothesis came to fruition with discovery of four sensing complexes. The Sestrin family of proteins (Sestrin1, Sestrin2, Sestrin3) are highly conserved and were recently found to function as negative regulators of mTORC1 71,72. Originally, the Sestrins were proposed to function as guanine dissociation inhibitors (GDI) for RagA/B, locking them in the GDP bound state 73. However, the key residues required for GDI activity are found buried in the crystal structure of Sestrin2 74–76. Purification experiments and subsequent biochemical investigation has demonstrated that Sestrins bind to and inhibit the function of GATOR2. The interaction between Sestrin1/2 and GATOR2 is regulated by leucine; however the interaction of Sestrin3 with GATOR is constitutive, suggesting another mode of regulation. In vitro binding assays demonstrate that Sestrin2 binds directly to leucine at an affinity similar to the concentrations sensed by mTORC1. Furthermore, the crystal structure of Sestrin2 prominently displays leucine bound to critical residues required for sensing, which when mutated blocks leucine sensing by mTORC1. Leucine binding relieves the inhibitory action of Sestrin2 upon GATOR2 and thus mTORC1 76,77.
The CASTOR proteins (CASTOR1 and its homolog CASTOR2) have recently been described as arginine sensors. The CASTOR proteins are found to bind to GATOR2 and inhibit its function during arginine withdrawal. In vitro binding assays confirmed that CASTOR1 can bind arginine at levels similar to media concentrations required for activation of mTORC1 and mutational studies demonstrated that CASTOR mutants that cannot bind arginine block arginine induced activation of mTORC1, designating the CASTOR proteins as bona-fide amino acid sensors 78,79.
A low-affinity lysosomal amino acid transporter, SLC38A9 interacts with both Ragulator and the v-ATPase, and acts as an amino acid sensor upstream of mTORC1 for asparagine, arginine, glutamine, histidine, and lysine 80–82. The exact mechanism by which SLC38A9 regulates mTORC1 activity is not clear; while it was initially suggested that SLC38A9 might regulate the GEF activity of Ragulator, it was shown that in response to arginine, SLC38A9 regulates the lysosomal efflux of many amino acids that then stimulate mTORC1 from the cytoplasm 40. However, this model does not explain the significance of the physical interactions between SLC38A9, the Ragulator and the v-ATPase. Curiously, SLC38A9 also enables mTORC1 activation by cholesterol via recruitment of the Niemann-Pick C1 (NPC1) protein, which acts to inhibit mTORC1 in the absence of cholesterol 83. Understanding how the amino acid sensing and transport functions of SLC38A9 regulates mTORC1 activity and the sensitivity of SLC38A9 to other environmental cues, is an important area for future research.
Finally, the SAMTOR protein was recently discovered as an evolutionarily conserved negative regulator of mTORC1 amino acid sensing that binds to GATOR1 directly to modulate its activity by an unknown mechanism. Instead of directly sensing amino acid levels, SAMTOR binds to the metabolite S-adenosylmethionine (SAM) which disrupts its interaction with GATOR1, positioning SAMTOR as an indirect sensor of methionine 84. Intriguingly, the discovery of SAMTOR provides the first molecular mechanism by which methionine restriction, a potent dietary intervention that improves metabolic health and extends rodent lifespan 85–87, may mediate mTORC1 activity 88–90. This study not only provides a key link between one carbon metabolism and the mTORC1 pathway but raises the question of whether additional intermediate metabolites in amino acid catabolism are also sensed by mTORC1. For example, mTORC1 has been shown to sense α-ketoglutarate, a product of glutaminolysis 91,92.
Rheb and Tuberous sclerosis complex
Recruitment of mTORC1 to the lysosome is a prerequisite for mTORC1 activation, because only at the lysosomal surface is mTORC1 able to interact with the Rheb-GTPase. A recent Cryo-EM structure has revealed that Rheb-GTP binds to the mTOR protein kinase and allosterically realigns and activates kinase-site residues 93. Rheb-GTP is regulated by at least two mechanisms; the first is that lysosomal recruitment of Rheb is regulated by amino acids. Amino acids stimulate the binding of Rheb to microspherule protein 1 (MCRS1), which promotes its localization to the lysosome, although the molecular mechanisms which regulate this process remain uncertain.
The second mechanism by which Rheb-GTP is regulated is the tuberous sclerosis complex (TSC), which is comprised of the proteins TSC1, TSC2, and TBC1D7 94. The TSC complex acts as a GAP for Rheb, and acts as a “mini” signaling hub upstream of mTORC1 95. Many different kinases, including AKT, AMPK, ERK, GSK3, and IKKβ phosphorylate distinct residues of TSC1 and TSC2, and thereby serve to regulate mTORC1 by altering the GAP activity of TSC towards Rheb 95–101. While it was originally thought that these posttranslational modifications directly altered the activity of TSC, it was recently shown that much like mTORC1 and Rheb, TSC is also regulated by localization. In the absence of insulin signaling, TSC is localized to the lysosome, where it can directly inhibit Rheb. However, when TSC is phosphorylated by AKT in response to insulin/PI3K signaling, TSC departs from the lysosome, removing its ability to inhibit Rheb, and permitting the activation of mTORC1 102. Another report suggests that TSC lysosomal localization is nutrient dependent, and may be recruited to the lysosome by the Rag GTPases in the absence of amino acids 103. The mechanism by which this process is mediated remain to be determined; also unknown is if relocalization of TSC is a generalized mechanism by which post-translational modifications of TSC regulate the GTP-loading of Rheb.
mTORC2
As noted above, mTORC2 is acutely resistant to rapamycin treatment, and this effect, along with the salt-sensitivity of mTORC2, lead to a delay in its discovery 15,20,21. Even after its discovery, the lack of specific chemical inhibitors of mTORC2 have slowed our understanding of the importance of this complex to a diverse set of cellular processes. The sub-cellular localization of mTORC2 in particular has been something of a mystery; while mTORC2 is an effector of PI3K signaling and sensitive to PIP3, and thus one might suspect that it is localized to the plasma membrane, work published in 2011 demonstrated that mTORC2 is physically associated with the ribosome, an interaction that is stimulated by insulin 35. While this agrees well with a model in which mTORC2 phosphorylates some motifs of AKT co-translationally, the concept that mTORC2 is associated with the ribosome also received a later boost with determination that mTORC2 localizes to the mitochondria-associated endoplasmic reticulum, interacting with a specific mitochondrial tethering complex 104. Recent studies have also placed mTORC2 at the lysosome (see below) and in immune cells chronic mTORC2 activity inhibits lysosomal acidification 105. The effect of mTORC2 on lysosomal acidification in a more acute context, such as in response to physiological levels of PI3K signaling, is unknown.
Lysosomal functions downstream of mTOR complexes
While mTORC1 was first characterized as regulator of ribosomal biogenesis and protein translation, an ever-growing set of identified substrates, both of the mTOR protein kinase itself as well as effectors such as S6K1 has linked mTORC1 to a growing list of cellular processes. While we will not attempt to enumerate all of the processes downstream of the mTOR protein kinase, we will note broadly that these processes include the regulation of apoptosis, amino acid and ion homeostasis, metabolism, and stress resistance downstream of mTORC2 19,106–109. Downstream of mTORC1, the list has grown from ribosomal biogenesis and translation to include adipogenesis, amino acid transport, ketogenesis, lipogenesis, and nucleotide synthesis 19,110–114.
In all of these processes, both mTORC1 and mTORC2 act in a common theme: to promote anabolic processes. However, both mTOR complexes also play a critical role in regulating catabolic processes. One recently discovered activity is that both mTORC2 and mTORC1 act to promote anaplerosis – the refilling of the citric acid cycle – by promoting the formation of α-ketoglutarate from glutamine 92. Here, we will focus on the lysosomal pathways that lie downstream of the mTOR complexes.
Transcriptional control of lysosomal function
Exemplifying the interdependent role of lysosome as both a catabolic and anabolic signaling hub are recent discoveries linking the transcriptional control of lysosome function to mTORC1. In work published in 2009 115, the basic helix-loop-helix transcription factor, TFEB, was found to bind to genes containing the CLEAR element, which is enriched in the promoters of lysosomal genes 115,116. TFEB is a member of the MiT/TFE transcription factor family 117, whose other members MiTF and TFE3 are now appreciated to have similar functions to TFEB. Activation of TFEB drives a major expansion of the lysosomal compartment and multiple steps in autophagy, denoting TFEB as the master transcriptional regulator of lysosomal function 118. The regulation of TFEB activity is nutrient dependent 119. During nutrient replete conditions, mTORC1 binds to TFEB at the lysosomal surface and phosphorylates two key residues 119,120 leading to 14–3-3 binding and cytoplasmic sequestration. Upon nutrient starvation, calcium is released by the lysosomal calcium channel MCOLN1, leading to the activation of a Ca2+-dependent phosphatase, calceniruin, which dephosphorylates TFEB 121 allowing its nuclear localization and activation of target genes. To suppress lysosomal gene expression, cells rely on the zinc-finger transcription factor, ZSCAN3, which sits at the promoters of multiple lysosomal genes and inhibits their expression during nutrient replete conditions 122. Upon nutrient starvation or mTORC1 inhibition, ZSCAN3 is driven from the nucleus into the cytoplasm by an unknown mechanism. Other transcription pathways including FXR also sense cellular nutrient conditions (bile acids) and coordinate lysosomal gene expression 123,124. A key challenge for future studies will be to provide an integrated portrait of how multiple nutrient sensitive transcriptional pathways coordinate lysosomal gene expression in concert with anabolic signaling pathways.
Autophagy
In response to nutrient limitation and damage to proteins or organelles, eukaryotic cells activate a highly regulated set of pathways collectively known as autophagy. Because generation of energy and metabolic intermediates under starvation and the clearance of toxic proteins and damaged organelles is a prerequisite for any cell, defects in autophagy pathways underlie human diseases ranging from neurodegenerative disorders to cancer 125.
At the heart of autophagy lies the lysosome– serving as the degradative endpoint for these pathways by providing key enzymes and a harsh environment necessary for the breakdown of toxic protein aggregates, damaged lipids or complex carbohydrates. Studies initiated in yeast have provided a solid genetic foundation for autophagy pathways with the discovery of ATG (autophagy related genes) 126 and their mammalian homologs. In mammalian cells two autophagy pathways exist: macroautophagy and chaperone mediated autophagy (CMA). Macroautophagy (herein referred to as autophagy) is the degradation of cytosolic components through a double-membrane vesicle known as the autophagosome. CMA is characterized by the degradation of specific proteins marked with a charged recognition motif. Both forms of autophagy are highly regulated processes which sense general cell stress and are sensitive to nutrient deprivation. While autophagy functions as a rapid response to starvation and is induced within minutes of nutrient withdrawal, CMA reaches its maximal activation after 1.5 days. In addition to autophagy and CMA, specialized forms of selective autophagy which target organelles (mitophagy) 127 or complexes (ribophagy) 128–130 have recently emerged.
Initiation of autophagy occurs within minutes of nutrient deprivation and central to its regulation are mTORC1 and the energy sensing AMPK pathway. During nutrient replete conditions, lysosomal mTORC1 phosphorylates sites on key components of the master autophagy regulator ULK complex, composed of ULK1/2, ATG13, FIP200 and ATG101. mTORC1 phosphorylation of the serine/threonine kinase ULK1/2 at S757 30 and ATG13 at S258 131 inactivates the ULK complex to block autophagy induction. Upon starvation, mTORC1 is inactivated, in part due to a lack of free amino acids, and AMPK phosphorylates different sites on ULK1/2, necessary for its kinase activity 30,132. Active ULK translocates to the ER 133 and phosphorylates the class III phosphatidylinositol-3 phosphate (PI3P) kinase, VPS34 134, a component of VPS34 complex I (BECN1, VPS34, ATG14L, and VPS15). Once activated, VPS34, deposits PI3P at the omegasome, a subdomain of the ER believed to give rise to the isolation membrane.
The increase in PI3P levels recruits additional ATG proteins 135, leading to the nucleation of the isolation membrane (IM) at the omegasome. IMs initially form a cup-shaped double membrane structure that is readily resolved by electron microscopy and signal autophagy induction. IM expansion is driven by vesicular traffic from different compartments (golgi 136, mitochondria 137 and plasma membrane 138), which provide a source of membranes to the growing structures. As IMs continue to expand, the ATG16L1 complex (ATG16L, ATG12 and ATG5), attaches the molecule LC3 to the IM. LC3 conjugation with phosphatidylethanolamine is a critical for downstream effector protein recognition and autophagasome closure 139 and is often used as a molecular marker for autophagy induction. Eventually the IM pinches off from the omegasome and as it closes to form the autophagasome it swallows cytosolic components.
After closure, autophagasomes merge with lysosomes to form autolysosomes, wherein the luminal contents of autophagasomes are degraded in the lysosomal lumen. While the molecular mechanisms governing autolysosome formation are not as well characterized as autophagasome initiation, it is now clear that autolysosome formation shares many components with from the lysosome-endosome fusion pathway 140. One of the best characterized components in autophagasome-lysosome fusion is the SNARE Stx17 141,142. SNARE proteins are required for membrane fusion, with a SNARE (Q-SNARE) on a vesicle forming a coiled-coil structure with a SNARE (R-SNARE) on a target membrane generating a trans-SNARE complex and driving fusion of the vesicle with the target membrane. In the context of autophagy, Stx17 is recruited to the closed autophagasome from the ER by a poorly defined mechanism and binds to another Q-SNARE, SNAP29. The Stx17/SNAP29 complex is further stabilized by ATG14 142 and interacts with the lysosomal R-SNARE, VAMP8, leading to membrane fusion and autolysosome formation 141. Additional components required for autolysosome formation are the small GTPase Rab7 143, required for late endosome/lysosome fusion events its effector the homotypic fusion and vacuolar sorting (HOPS) 144 and its GEF the Mon1-Ccz1 complex 145. In addition to the molecular recognition between Stx17 and Vamp8, PLEKHM1 serves as a tether between lysosomal HOPS and LC3, ensuring correct autophagasome-lysosome fusion 146.
Following the degradation of its contents, the lysosome needs to reform from the autolysosome to support additional rounds of autophagy through a process known as autophagic lysosome reformation (ALR) 147,148. ALR initiates with the formation of tubules in the autolysosome driven by clathrin, PI(4,5)P2 and the motor protein KIF5B 149,150. Sorting of lysosomal proteins is thought to be mediated by the lipid kinase PI4KIIIß 151 in a poorly understood manner. Curiously, ALR is regulated by mTORC1, as treatment with rapamycin inhibits this process, suggesting a model in which ALR functions as a stop-gap for autophagy once autophagy-generated nutrient levels are sufficient to reactivate mTORC1 152.
Chaperone mediated autophagy
In contrast to the degradation of cytosolic components under autophagy, CMA leads to the lysosomal degradation of only a subset of proteins that contain CMA motifs. CMA is a highly orchestrated process involving multiple steps: substrate recognition and lysosomal targeting, substrate binding and unfolding, substrate translocation and degradation in the lysosomal lumen. Key to CMA is the recognition of substrates by the cytosolic chaperone and heat shock protein HSC70 153. HSC70 binds to protein substrates with a consensus pentapeptide motif KFERQ 154 that is recognized based on its charge. Additionally, incomplete motifs may be complemented through post-translational modification of adjacent residues by phosphorylation or acetylation 155. These PTMs thus provide the necessary charge recognition by HSC70 156.
Once bound by HSC70, the protein substrate is targeted to the lysosomal surface where it interacts with the cytoplasmic tail of LAMP-2A. LAMP-2A functions as the receptor for CMA substrates 157 and multimerizers upon contact with the substrate to facilitate lysosomal entry 158. To enter the lysosomal lumen the substrate must unfold in a process mediated by HSC70 and its co-chaperones at the lysosomal membrane. Translocation of the substrate further requires a resident lysosomal luminal chaperone lys-HSC70 159. lys-HSC70 facilitates substrate entry by directly pulling on the incoming substrate or by actively blocking its departure from the lysosomal lumen. After substrate translocation, the LAMP-2A multimers are dissembled so the CMA cycle can begin again.
One regulatory point in CMA is the assembly/disassembly of LAMP-2A multimers. Stabilization of LAMP-2A multimers is enhanced by the protein GFAP in its unphosphorylated state. Recent work from the Cuervo lab has demonstrated that AKT phosphorylates GFAP, leading to the destabilization of LAMP2a multimers. This study connects CMA to a balance of AKT activation at the lysosomal surface and inhibition by mTORC2 and a protein phosphatase, PHLPP1 160. While conceivably mTORC2 at other cellular locations could also be involved in this process, Cuervo and colleagues found that mTORC2 associated with a subset of lysosomes; the mTORC2 component Rictor was found only in the subset of lysosomes engaged in chaperone mediated autophagy. In contrast, the mTORC1 subunit Raptor was enriched in lysosomes not engaged in chaperone mediated autophagy 160. In addition to being regulated by mTORC2, CMA is also regulated transcriptionally through the expression of LAMP-2A which can be dynamically modulated during hypoxia, oxidative stress, genotoxic stress or prolonged starvation. This process is mediated in part by TFEB pathway 161.
The lysosome in aging and disease
Lysosomal function is essential for the healthy functioning of both individual cells and for an entire organism. Aging represents the end result of accumulating deficits at the molecular, cellular, and organismal level, and aberrant lysosomal signaling during aging contributes to the degradation of these processes. Loss of proteostasis has been proposed as one of the nine hallmarks of aging; while many different processes contribute to the maintenance of proteostasis, an important part of this process involves the degradation of proteins by the lysosome during both CMA and macroautophagy. Proteostasis declines with aging in most organisms, with the possible exception of the exceptionally long-lived naked mole rat 162,163.
Just a decade ago, it was demonstrated that rapamycin extends the lifespan of mice, a result that has been widely reproduced 164,165. Indeed, even intermittent or transient treatment with rapamycin can extend the lifespan of mice and rejuvenate specific tissues, including the hematopoietic system, heart, immune system and kidney 166–172. In part, this may be due to the ability of rapamycin to block age-associated increases in mTOR signaling in certain tissues 173,174. However, the specific molecular and physiological mechanisms by which rapamycin extends lifespan are difficult to examine in mammals; while it is clear that inhibition of S6K1 signaling extends lifespan, and that inhibition of translation downstream of 4E-BP1 is beneficial for metabolic health, the contribution of autophagy to the effects of rapamycin on mammalian longevity have not been examined in detail 175–177. However, there is reason to believe that many of the beneficial effects of rapamycin on aging may be driven in part by increased autophagy.
First, both CMA 178 and autophagy naturally decline with aging 179. Evidence from model organisms suggests that this is detrimental. Overexpression of chaperones extends the lifespan of worms and flies 180,181. Worms and flies with defective autophagy have reduced lifespan, while conversely overexpression of autophagy genes such as Atg8a extends lifespan 182,183. Finally, experiments in both worms and flies have found that an inhibition of autophagy genes blunts the effects of reduced mTOR signaling on lifespan 184,185. In mammals, mice with genetic impairment of CMA have reduced lifespan, with an increase in age-related pathology and accelerated cellular senescence 186. While it is not yet known if upregulation of CMA can extend mammalian lifespan, an increase in hepatic CMA can rejuvenate the aging mouse liver 178. However, overexpression of Atg5, which enhances autophagy in mice, increases resistance to oxidative stress and increases lifespan 187.
Discovering safe and effective ways to regulate lysosomal function and to boost autophagy is likely to be essential to promote healthy aging and to treat a variety of age-associated diseases. Many neurodegenerative diseases, including Alzheimer’s disease, is thought to be driven in part by misfolded proteins; promoting proteostasis, either by increasing lysosomal function directly or by boosting autophagy through other means is therefore a potential mechanism to delay or treat the disease 188–190. In Alzheimer’s disease, which is thought to be partly precipitated by amyloid aggregation, the hope is that stimulating autophagy will clear these protein aggregates 191. Conversely, autophagy is believed to be beneficial for tumorigenesis and for the survival of many types of cancer cells that are under nutrient stress 192, and inhibiting this process to disrupt proteostasis has emerged as a potential therapeutic option for some malignancies, with recent pre-clinical studies targeting core autophagy proteins showing promise 193,194. Thus, autophagy has been catapulted to the forefront as a therapeutic avenue for multiple disease states, however, caution must be applied as targeting any metabolic signaling pathway must be considered in a context-dependent manner.
Summary
As a newly described center for the integration of major catabolic and anabolic pathways the lysosome is positioned as a key sensor of cellular nutrient levels. With a growing appreciation for its diverse functions within the cell, many questions still loom large. At the biochemical level, understanding whether additional nutrients such as nucleotides and lipids are sensed at the lysosome and their corresponding sensors will be critical in deciphering the inner workings of metabolic signaling at this organelle. At the cellular level, providing a systems level overview of nutrient signaling at the lysosome and exploring the crosstalk between the lysosome and other organelles will allow us to predict how different cellular stressors change nutrient flux and signaling. At the organismal level, the future lies at understanding how lysosome-based metabolic signaling translates to different tissues and organ systems. Underlying the desire to study metabolic signaling at the lysosome is the immediate realization that disruption of these pathways leads to major human pathologies. While the molecular mechanisms of lysosomal signaling in cancer, aging and immune disorders remain at their infancy, the last decade has seen the development of a numerous inhibitors targeting multiple parts of this pathway (195). While the utility of rapamycin in a handful disorders is clear, its overall success in preventing cancer growth remains poor. Whether this is due to an incomplete understanding of mTORC1’s requirement for rapid cell proliferation, the ability of cancers to acquire rapamycin-resistance 196, the reactivation of PI3K signaling due to systemic glucose-insulin feedback on most diets 197, or its mechanism of action as an allosteric inhibitor that blocks the phosphorylation of only some of the mTORC1 substrates, remains to be seen. However, there is a palpable excitement that the new generation of therapeutic agents targeting metabolic signaling at the lysosome will have substantial benefits for human health.
Acknowledgements
We thank Drs. Lynne Chantranupong and Anne Strohecker for critical reading of the manuscript. The work is supported in part by the NIH (AG050135, AG051974, AG056771 to D.W.L) and (CA215249 to L.B-P.) and by a pilot grant to D.W.L. from the Diabetes Research Center at Washington University, Grant No. 2 P30 DK020579. The Lamming laboratory is supported by the U.S. Department of Veterans Affairs (I01-BX004031). D.W.L. is an American Federation for Aging Research (AFAR) grant recipient. L.B-P. is the Lallage Feazel Wall Fellow of the Damon Runyon Cancer Research Foundation (DRG-2178–14). This work was supported using facilities and resources from the William S. Middleton Memorial Veterans Hospital. This work does not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Conflicts of Interest
D.W.L has received funding from, and is a scientific advisory board member of, Aeonian Pharmaceuticals, which seeks to develop novel, selective mTOR inhibitors for the treatment of various diseases.
References
- 1.Bainton DF. The discovery of lysosomes. J Cell Biol 1981;91(3 Pt 2):66s–76s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Duve C The lysosome turns fifty. Nat Cell Biol 2005;7(9):847–849. [DOI] [PubMed] [Google Scholar]
- 3.Essner E, Novikoff AB. Localization of acid phosphatase activity in hepatic lysosomes by means of electron microscopy. J Biophys Biochem Cytol 1961;9:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straus W Isolation and biochemical properties of droplets from the cells of rat kidney. J Biol Chem 1954;207(2):745–755. [PubMed] [Google Scholar]
- 5.Forgac M Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007;8(11):917–929. [DOI] [PubMed] [Google Scholar]
- 6.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14(5):283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol 2016;32:223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox TM, Cachon-Gonzalez MB. The cellular pathology of lysosomal diseases. J Pathol 2012;226(2):241–254. [DOI] [PubMed] [Google Scholar]
- 9.Guo JY, White E. Autophagy, Metabolism, and Cancer. Cold Spring Harb Symp Quant Biol 2016;81:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabatini DM. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A 2017;114(45):11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glass DJ, Lamming DW. Is rapamycin a rapalog? Nutrition and Healthy Aging 2018;Pre-press(Pre-press):1–2. doi: 10.3233/NHA-180045.
- 12.Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994;369(6483):756–758. [DOI] [PubMed] [Google Scholar]
- 13.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994;78(1):35–43. [DOI] [PubMed] [Google Scholar]
- 14.Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 1995;270(2):815–822. [DOI] [PubMed] [Google Scholar]
- 15.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002;10(3):457–468. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002;110(2):163–175. [DOI] [PubMed] [Google Scholar]
- 17.Kim DH, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 2003;11(4):895–904. [DOI] [PubMed] [Google Scholar]
- 18.Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002;110(2):177–189. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy BK, Lamming DW. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab 2016;23(6):990–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004;6(11):1122–1128. [DOI] [PubMed] [Google Scholar]
- 21.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004;14(14):1296–1302. [DOI] [PubMed] [Google Scholar]
- 22.Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 2006;16(18):1865–1870. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 2003;5(6):578–581. [DOI] [PubMed] [Google Scholar]
- 24.Barb D, Neuwirth A, Mantzoros CS, Balk SP. Adiponectin signals in prostate cancer cells through Akt to activate the mammalian target of rapamycin pathway. Endocr Relat Cancer 2007;14(4):995–1005. [DOI] [PubMed] [Google Scholar]
- 25.Lamming DW. Diminished mTOR signaling: a common mode of action for endocrine longevity factors. SpringerPlus 2014;3:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HK, Ahima RS. Leptin signaling. F1000prime reports 2014;6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Mao X, Wang L, et al. Adiponectin sensitizes insulin signaling by reducing p70 S6 kinase-mediated serine phosphorylation of IRS-1. J Biol Chem 2007;282(11):7991–7996. [DOI] [PubMed] [Google Scholar]
- 28.Cutler NS, Heitman J, Cardenas ME. TOR kinase homologs function in a signal transduction pathway that is conserved from yeast to mammals. Molecular and cellular endocrinology 1999;155(1–2):135–142. [DOI] [PubMed] [Google Scholar]
- 29.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331(6016):456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara K, Yonezawa K, Kozlowski MT, et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem 1997;272(42):26457–26463. [DOI] [PubMed] [Google Scholar]
- 32.Kang SA, Pacold ME, Cervantes CL, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013;341(6144):1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu P, Gan W, Chin YR, et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer discovery 2015;5(11):1194–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathy S, Jump DB. Elovl5 regulates the mTORC2-Akt-FOXO1 pathway by controlling hepatic cis-vaccenic acid synthesis in diet-induced obese mice. J Lipid Res 2013;54(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell 2011;144(5):757–768. [DOI] [PubMed] [Google Scholar]
- 36.Liu P, Wang Z, Wei W. Phosphorylation of Akt at the C-terminal tail triggers Akt activation. Cell Cycle 2014;13(14):2162–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 2008;27(14):1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005;307(5712):1098–1101. [DOI] [PubMed] [Google Scholar]
- 39.Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell 2015;161(1):67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyant GA, Abu-Remaileh M, Wolfson RL, et al. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017;171(3):642–654 e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abu-Remaileh M, Wyant GA, Kim C, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017;358(6364):807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirose E, Nakashima N, Sekiguchi T, Nishimoto T. RagA is a functional homologue of S. cerevisiae Gtr1p involved in the Ran/Gsp1-GTPase pathway. J Cell Sci 1998;111 ( Pt 1):11–21. [DOI] [PubMed] [Google Scholar]
- 43.Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem 2001;276(10):7246–7257. [DOI] [PubMed] [Google Scholar]
- 44.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008;10(8):935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008;320(5882):1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010;141(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012;150(6):1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell 2005;19(1):15–26. [DOI] [PubMed] [Google Scholar]
- 49.Panchaud N, Peli-Gulli MP, De Virgilio C. SEACing the GAP that nEGOCiates TORC1 activation: evolutionary conservation of Rag GTPase regulation. Cell Cycle 2013;12(18):2948–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kogan K, Spear ED, Kaiser CA, Fass D. Structural conservation of components in the amino acid sensing branch of the TOR pathway in yeast and mammals. J Mol Biol 2010;402(2):388–398. [DOI] [PubMed] [Google Scholar]
- 51.Su MY, Morris KL, Kim DJ, et al. Hybrid Structure of the RagA/C-Ragulator mTORC1 Activation Complex. Mol Cell 2017;68(5):835–846 e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Araujo MEG, Naschberger A, Furnrohr BG, et al. Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 2017;358(6361):377–381. [DOI] [PubMed] [Google Scholar]
- 53.Yonehara R, Nada S, Nakai T, et al. Structural basis for the assembly of the Ragulator-Rag GTPase complex. Nature communications 2017;8(1):1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang T, Wang R, Wang Z, Wang X, Wang F, Ding J. Structural basis for Ragulator functioning as a scaffold in membrane-anchoring of Rag GTPases and mTORC1. Nature communications 2017;8(1):1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011;334(6056):678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langemeier J, Schrom EM, Rabner A, et al. A complex immunodeficiency is based on U1 snRNP-mediated poly(A) site suppression. EMBO J 2012;31(20):4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bohn G, Allroth A, Brandes G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med 2007;13(1):38–45. [DOI] [PubMed] [Google Scholar]
- 58.Wu JJ, Liu J, Chen EB, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell reports 2013;4(5):913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Efeyan A, Schweitzer LD, Bilate AM, et al. RagA, but not RagB, is essential for embryonic development and adult mice. Dev Cell 2014;29(3):321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science 2004;305(5680):55–59. [DOI] [PubMed] [Google Scholar]
- 61.Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013;340(6136):1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panchaud N, Peli-Gulli MP, De Virgilio C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal 2013;6(277):ra42. [DOI] [PubMed] [Google Scholar]
- 63.Shen K, Huang RK, Brignole EJ, et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 2018;556(7699):64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolfson RL, Chantranupong L, Wyant GA, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017;543(7645):438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017;543(7645):433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsun ZY, Bar-Peled L, Chantranupong L, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 2013;52(4):495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petit CS, Roczniak-Ferguson A, Ferguson SM. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol 2013;202(7):1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meng J, Ferguson SM. GATOR1-dependent recruitment of FLCN-FNIP to lysosomes coordinates Rag GTPase heterodimer nucleotide status in response to amino acids. J Cell Biol 2018;217(8):2765–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu X, Zhao L, Chen Z, et al. FLCN Maintains the Leucine Level in Lysosome to Stimulate mTORC1. PLoS One 2016;11(6):e0157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhong M, Zhao X, Li J, et al. Tumor Suppressor Folliculin Regulates mTORC1 through Primary Cilia. J Biol Chem 2016;291(22):11689–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chantranupong L, Wolfson RL, Orozco JM, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell reports 2014;9(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parmigiani A, Nourbakhsh A, Ding B, et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell reports 2014;9(4):1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014;159(1):122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saxton RA, Knockenhauer KE, Schwartz TU, Sabatini DM. The apo-structure of the leucine sensor Sestrin2 is still elusive. Sci Signal 2016;9(446):ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim H, An S, Ro SH, et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nature communications 2015;6:10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Saxton RA, Knockenhauer KE, Wolfson RL, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016;351(6268):53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016;351(6268):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chantranupong L, Scaria SM, Saxton RA, et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016;165(1):153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016;536(7615):229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jung J, Genau HM, Behrends C. Amino Acid-Dependent mTORC1 Regulation by the Lysosomal Membrane Protein SLC38A9. Mol Cell Biol 2015;35(14):2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rebsamen M, Pochini L, Stasyk T, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015;519(7544):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang S, Tsun ZY, Wolfson RL, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015;347(6218):188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castellano BM, Thelen AM, Moldavski O, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017;355(6331):1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gu X, Orozco JM, Saxton RA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017;358(6364):813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown-Borg HM, Buffenstein R. Cutting back on the essentials: Can manipulating intake of specific amino acids modulate health and lifespan? Ageing Res Rev 2017;39:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu D, Yang SE, Miller BR, et al. Short-term methionine deprivation improves metabolic health via sexually dimorphic, mTORC1-independent mechanisms. FASEB J 2018:fj201701211R. [DOI] [PMC free article] [PubMed]
- 87.Brown-Borg HM, Rakoczy S, Wonderlich JA, Borg KE, Rojanathammanee L. Metabolic adaptation of short-living growth hormone transgenic mice to methionine restriction and supplementation. Ann N Y Acad Sci 2018;1418(1):118–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lees EK, Banks R, Cook C, et al. Direct comparison of methionine restriction with leucine restriction on the metabolic health of C57BL/6J mice. Scientific reports 2017;7(1):9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Forney LA, Stone KP, Wanders D, Gettys TW. Sensing and signaling mechanisms linking dietary methionine restriction to the behavioral and physiological components of the response. Front Neuroendocrinol 2017. [DOI] [PMC free article] [PubMed]
- 90.Green CL, Lamming DW. Regulation of metabolic health by essential dietary amino acids. Mech Ageing Dev 2018; pii: S0047–6374 (18)30079–4. doi: 10.1016/j.mad.2018.07.004. [DOI] [PMC free article] [PubMed]
- 91.Duran RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012;47(3):349–358. [DOI] [PubMed] [Google Scholar]
- 92.Arriola Apelo SI, Lamming DW. mTORC2 Puts Its Shoulder to Krebs’ Wheel. Mol Cell 2016;63(5):723–725. [DOI] [PubMed] [Google Scholar]
- 93.Yang H, Jiang X, Li B, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017;552(7685):368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dibble CC, Elis W, Menon S, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 2012;47(4):535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003;17(15):1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4(9):648–657. [DOI] [PubMed] [Google Scholar]
- 97.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115(5):577–590. [DOI] [PubMed] [Google Scholar]
- 98.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005;121(2):179–193. [DOI] [PubMed] [Google Scholar]
- 99.Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006;126(5):955–968. [DOI] [PubMed] [Google Scholar]
- 100.Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007;130(3):440–455. [DOI] [PubMed] [Google Scholar]
- 101.Tian Q, Gromov P, Clement JH, et al. RHEB1 insufficiency in aged male mice is associated with stress-induced seizures. Geroscience 2017;39(5–6):557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Menon S, Dibble CC, Talbott G, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014;156(4):771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Demetriades C, Doumpas N, Teleman AA. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014;156(4):786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A 2013;110(31):12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Monteith AJ, Vincent HA, Kang S, et al. mTORC2 Activity Disrupts Lysosome Acidification in Systemic Lupus Erythematosus by Impairing Caspase-1 Cleavage of Rab39a. Journal of immunology 2018. [DOI] [PMC free article] [PubMed]
- 106.Lamming DW, Demirkan G, Boylan JM, et al. Hepatic signaling by the mechanistic target of rapamycin complex 2 (mTORC2). FASEB J 2014;28(1):300–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sciarretta S, Zhai P, Maejima Y, et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell reports 2015;11(1):125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sengupta S, Lorente-Rodriguez A, Earnest S, et al. Regulation of OSR1 and the sodium, potassium, two chloride cotransporter by convergent signals. Proc Natl Acad Sci U S A 2013;110(47):18826–18831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gu Y, Albuquerque CP, Braas D, et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol Cell 2017;67(1):128–138 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell Metab 2013;18(4):465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013;339(6125):1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010;468(7327):1100–1104. [DOI] [PubMed] [Google Scholar]
- 113.Ji YF, Zhou L, Xie YJ, et al. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-small ka, CyrillicB cascade in astrocytic oxygen-glucose deprivation. Glia 2013;61(12):1959–1975. [DOI] [PubMed] [Google Scholar]
- 114.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4–2. Clin Sci (Lond) 2016;130(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- 116.Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011;20(19):3852–3866. [DOI] [PubMed] [Google Scholar]
- 117.Rehli M, Den Elzen N, Cassady AI, Ostrowski MC, Hume DA. Cloning and characterization of the murine genes for bHLH-ZIP transcription factors TFEC and TFEB reveal a common gene organization for all MiT subfamily members. Genomics 1999;56(1):111–120. [DOI] [PubMed] [Google Scholar]
- 118.Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci 2016;129(13):2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31(5):1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012;5(228):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17(3):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chauhan S, Goodwin JG, Chauhan S, et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell 2013;50(1):16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014;516(7529):112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Seok S, Fu T, Choi SE, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014;516(7529):108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev 2014;26:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993;333(1–2):169–174. [DOI] [PubMed] [Google Scholar]
- 127.Pickles S, Vigie P, Youle RJ. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 2018;28(4):R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 2008;10(5):602–610. [DOI] [PubMed] [Google Scholar]
- 129.An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 2018;20(2):135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wyant GA, Abu-Remaileh M, Frenkel EM, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018;360(6390):751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Puente C, Hendrickson RC, Jiang X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J Biol Chem 2016;291(11):6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011;7(6):643–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Karanasios E, Walker SA, Okkenhaug H, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nature communications 2016;7:12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013;15(7):741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009;186(6):773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yamamoto A, Masaki R, Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J Histochem Cytochem 1990;38(4):573–580. [DOI] [PubMed] [Google Scholar]
- 137.Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010;141(4):656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010;12(8):747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011;20(4):444–454. [DOI] [PubMed] [Google Scholar]
- 140.Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: Shared machinery for degradation. Bioessays 2013;35(1):34–45. [DOI] [PubMed] [Google Scholar]
- 141.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012;151(6):1256–1269. [DOI] [PubMed] [Google Scholar]
- 142.Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015;520(7548):563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004;117(Pt 20):4837–4848. [DOI] [PubMed] [Google Scholar]
- 144.Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 2014;25(8):1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nordmann M, Cabrera M, Perz A, et al. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol 2010;20(18):1654–1659. [DOI] [PubMed] [Google Scholar]
- 146.McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 2015;57(1):39–54. [DOI] [PubMed] [Google Scholar]
- 147.Chen Y, Yu L. Recent progress in autophagic lysosome reformation. Traffic 2017;18(6):358–361. [DOI] [PubMed] [Google Scholar]
- 148.Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010;465(7300):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol 2012;14(9):924–934. [DOI] [PubMed] [Google Scholar]
- 150.Du W, Su QP, Chen Y, et al. Kinesin 1 Drives Autolysosome Tubulation. Dev Cell 2016;37(4):326–336. [DOI] [PubMed] [Google Scholar]
- 151.Sridhar S, Patel B, Aphkhazava D, et al. The lipid kinase PI4KIIIbeta preserves lysosomal identity. EMBO J 2013;32(3):324–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang J, Zhou W, Lin J, et al. Autophagic lysosomal reformation depends on mTOR reactivation in H2O2-induced autophagy. Int J Biochem Cell Biol 2016;70:76–81. [DOI] [PubMed] [Google Scholar]
- 153.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989;246(4928):382–385. [DOI] [PubMed] [Google Scholar]
- 154.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990;15(8):305–309. [DOI] [PubMed] [Google Scholar]
- 155.Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 2011;42(6):719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014;24(1):92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996;273(5274):501–503. [DOI] [PubMed] [Google Scholar]
- 158.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008;28(18):5747–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 1997;137(4):825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol Cell 2015;59(2):270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev 2011;10(2):205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rodriguez KA, Valentine JM, Kramer DA, et al. Determinants of rodent longevity in the chaperone-protein degradation network. Cell Stress Chaperones 2016;21(3):453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Arriola Apelo SI, Lamming DW. Rapamycin: An InhibiTOR of Aging Emerges From the Soil of Easter Island. J Gerontol A Biol Sci Med Sci 2016;71(7):841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009;460(7253):392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE, Lamming DW. Intermittent Administration of Rapamycin Extends the Life Span of Female C57BL/6J Mice. J Gerontol A Biol Sci Med Sci 2016;71(7):876–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bitto A, Ito TK, Pineda VV, et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife 2016;5:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009;2(98):ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mannick JB, Del Giudice G, Lattanzi M, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med 2014;6(268):268ra179. [DOI] [PubMed] [Google Scholar]
- 170.Dai DF, Karunadharma PP, Chiao YA, et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 2014;13(3):529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Shavlakadze T, Zhu J, Wang S, et al. Short-term low-dose mTORC1 inhibition in aged rats counter-regulates age-related gene changes and blocks age-related kidney pathology. J Gerontol A Biol Sci Med Sci 2018. [DOI] [PMC free article] [PubMed]
- 172.Anisimov VN, Zabezhinski MA, Popovich IG, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011;10(24):4230–4236. [DOI] [PubMed] [Google Scholar]
- 173.Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell 2016;15(1):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Houtkooper RH, Argmann C, Houten SM, et al. The metabolic footprint of aging in mice. Scientific reports 2011;1:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009;326(5949):140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tsai SY, Rodriguez AA, Dastidar SG, et al. Increased 4E-BP1 Expression Protects against Diet-Induced Obesity and Insulin Resistance in Male Mice. Cell reports 2016;16(7):1903–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tsai S, Sitzmann JM, Dastidar SG, et al. Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J Clin Invest 2015;125(8):2952–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 2008;14(9):959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Martinez-Lopez N, Athonvarangkul D, Singh R. Autophagy and aging. Adv Exp Med Biol 2015;847:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell 2003;2(2):131–139. [DOI] [PubMed] [Google Scholar]
- 181.Morrow G, Samson M, Michaud S, Tanguay RM. Overexpression of the small mitochondrial Hsp22 extends Drosophila life span and increases resistance to oxidative stress. FASEB J 2004;18(3):598–599. [DOI] [PubMed] [Google Scholar]
- 182.Toth ML, Sigmond T, Borsos E, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008;4(3):330–338. [DOI] [PubMed] [Google Scholar]
- 183.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008;4(2):176–184. [DOI] [PubMed] [Google Scholar]
- 184.Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 2008;4(2):e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bjedov I, Toivonen JM, Kerr F, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 2010;11(1):35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol 2008;28(12):4018–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pyo JO, Yoo SM, Ahn HH, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nature communications 2013;4:2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fujikake N, Shin M, Shimizu S. Association Between Autophagy and Neurodegenerative Diseases. Front Neurosci 2018;12:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Peng Y, Kim MJ, Hullinger R, et al. Improved proteostasis in the secretory pathway rescues Alzheimer’s disease in the mouse. Brain 2016;139(Pt 3):937–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Peng Y, Puglielli L. N-lysine acetylation in the lumen of the endoplasmic reticulum: A way to regulate autophagy and maintain protein homeostasis in the secretory pathway. Autophagy 2016;12(6):1051–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011;134(Pt 1):258–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.White E The role for autophagy in cancer. J Clin Invest 2015;125(1):42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Egan DF, Chun MG, Vamos M, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 2015;59(2):285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer 2017;17(9):528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 2018;15(5):273–291. [DOI] [PubMed] [Google Scholar]
- 196.Wagle N, Grabiner BC, Van Allen EM, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med 2014;371(15):1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hopkins BD, Pauli C, Du X, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018; 560(7719):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]