

Abstract
The APOBEC family of single-stranded (ss)DNA cytosine deaminases provides innate immunity against virus and transposon replication1–4. A well-studied mechanism is APOBEC3G restriction of HIV-1, which is counteracted by a virus-encoded degradation mechanism1–4. Accordingly, most work has focused on retroviruses with obligate ssDNA replication intermediates and it is unclear whether large double-stranded (ds)DNA viruses may be similarly susceptible to restriction. Here, we show that the large dsDNA herpesvirus Epstein-Barr virus (EBV), which is the causative agent of infectious mononucleosis and multiple cancers5, utilizes a two-pronged approach to counteract restriction by APOBEC3B. The large subunit of the EBV ribonucleotide reductase, BORF26,7, bound to APOBEC3B in proteomics studies and immunoprecipitation experiments. Mutagenesis mapped the interaction to the APOBEC3B catalytic domain, and biochemical studies demonstrated that BORF2 stoichiometrically inhibits APOBEC3B DNA cytosine deaminase activity. BORF2 also caused a dramatic relocalization of nuclear APOBEC3B to perinuclear bodies. Upon lytic reactivation, BORF2-null viruses were susceptible to APOBEC3B-mediated deamination as evidenced by lower viral titers, lower infectivity, and hypermutation. The Kaposi’s sarcoma herpesvirus (KSHV) homolog, ORF61, also bound APOBEC3B and mediated relocalization. These data support a model in which the genomic integrity of human γ-herpesviruses is maintained by active neutralization of the antiviral enzyme APOBEC3B.
During the course of an EBV proteomics analysis, A3B was identified as the dominant cellular protein co-purifying with Flag-tagged EBV BORF2 in three independent affinity purification-mass spectrometry experiments in 293T cells (Fig. 1a), whereas no other APOBECs were recovered. A3B was identified based on recovery of 9 unique peptides spanning 36% of this 382 amino acid protein (Supplementary Fig. 1a). This result was surprising because 293T cells express endogenous A3B at levels that are undetectable by immunoblot and barely detectable by RT-qPCR (Fig. 1b, Supplementary Fig. 1b). Co-immunoprecipitation experiments confirmed that BORF2-Flag binds endogenous A3B in 293T cells as well as in AGS gastric carcinoma cells, an established model for EBV studiese.g.,8–10 (Fig. 1b, Supplementary Fig. 1c-d). Mechanistic conservation was indicated by similarly strong interaction with the large ribonucleotide reductase (RNR) subunit from KSHV, ORF61, and undetectable interactions with homologous proteins from more distantly related herpesviruses (HSV1 UL39 and HCMV UL45) or human RRM1 (Supplementary Fig. 2a). In addition, a BORF2 deletion analysis mapped the A3B interaction to the conserved core RNR domain, and also showed that the required region is more extensive than that needed for interaction with the EBV RNR small subunit BaRF1 (Supplementary Fig. 3a-d). Furthermore, BORF2 catalytic residues were dispensable for interacting with A3B suggesting an RNR-independent function (Supplementary Fig. 3e).
Figure 1 |. EBV BORF2 interacts with cellular A3B.
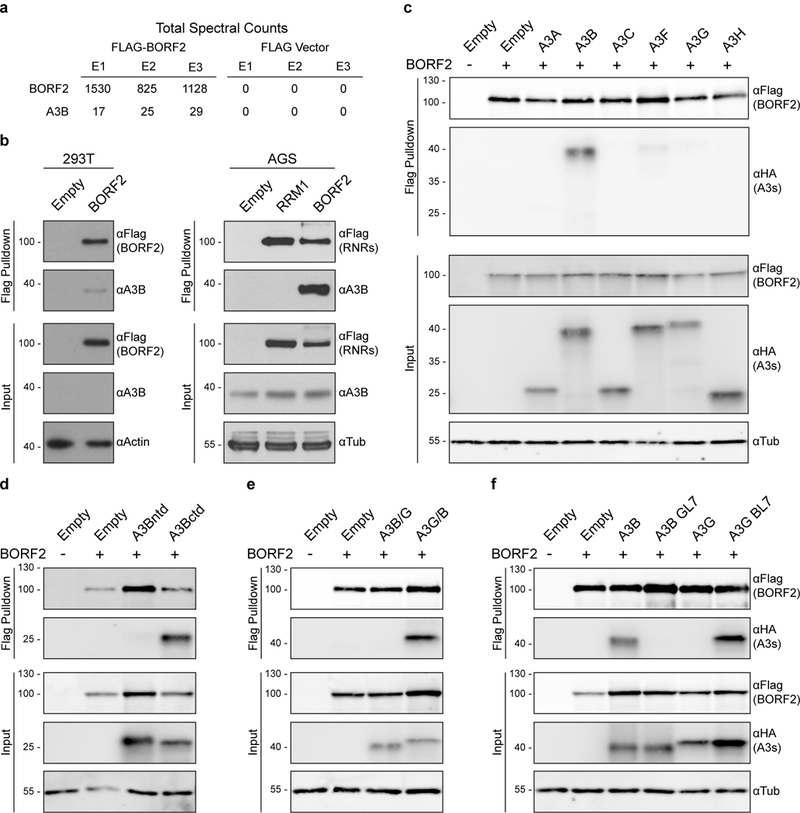
a, Total spectral counts from three independent affinity purification-mass spectrometry experiments using transfected BORF2-Flag as bait and empty Flag vector as negative control in 293T cells.
b, Co-immunoprecipitation of endogenous A3B in 293T and AGS cells with BORF2-Flag, RRM1-Flag, or an empty vector control.
c-f, Co-immunoprecipitation of indicated HA-tagged A3 constructs in 293T cells with BORF2-Flag. These data (a-f) are each representative of at least n = 3 biologically independent experiments.
Interaction specificity was further evidenced by comparisons with related APOBEC family members, which revealed a prominent interaction with A3B-HA, barely detectable interactions with A3A-HA and A3F-HA, and undetectable interactions with A3C-HA, A3D-HA, A3G-HA, and A3H-HA (Fig. 1c, Supplementary Fig. 4). The interaction mapped to the A3B C-terminal domain by co-immunoprecipitation experiments using BORF2-Flag and individually expressed HA-tagged N- and C-terminal domains (ntd, ctd) (Fig. 1d). Additional co-immunoprecipitation experiments using chimeras of A3B and A3G (A3Bntd-A3Gctd or A3Gntd-A3Bctd) confirmed the C-terminal specificity of this interaction (Fig. 1e). A3Bctd and A3Gctd belong to the same deaminase subgroup11 and the majority of amino acid differences are confined to surface exposed regions, which enabled the construction of a series of chimeras with exchanged loop regions. The BORF2 interaction was abolished using an A3B construct with loop 7 (L7) from A3G and, conversely, it was enabled using an A3G construct with L7 from A3B (Fig. 1f). These data indicated that BORF2 interacts preferentially with A3Bctd and specifically with a region involving L7 residues.
Recent structural studies have shown that A3Bctd L7 is essential for binding to 5’-TC containing single-stranded DNA substrates12. The specificity of the BORF2 interaction to the L7 region of A3B suggested that BORF2 may function by directly inhibiting ssDNA deaminase activity. To test this mechanism, recombinant BORF2 and A3Bctd were purified from E. coli and used in a series of in vitro ssDNA deaminase assays (Fig. 2a, Supplementary Fig. 5). The related enzyme, A3H, which also prefers 5’-TC ssDNA substrates13 and does not interact with BORF2 (above), was purified and tested in parallel as a negative control. A clear dose-dependent inhibition of A3Bctd catalytic activity, approaching near complete inhibition at equimolar concentrations, was observed in multiple independent BORF2 titration experiments (p<2.7×10−5, one sample t-test; Fig. 2b-c). In comparison, BORF2 had no significant effect on A3H enzymatic activity even at 8-fold molar excess concentrations (p<0.1575, one sample t-test). These results demonstrated that an alternative function of BORF2 is specific inhibition of A3B catalytic activity and that other viral (e.g., BaRF1) or cellular factors are not required for this activity.
Figure 2 |. EBV BORF2 inhibits A3B catalytic activity specifically.
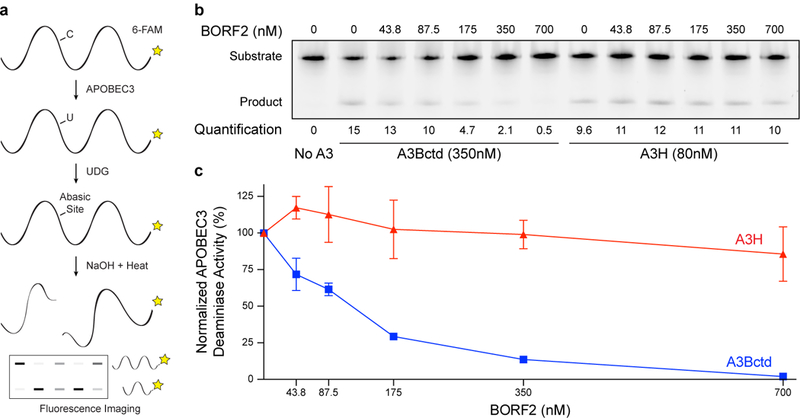
a, Schematic of deaminase activity assay in which A3-mediated deamination of C-to-U in single-stranded DNA substrate, uracil excision by uracil DNA glycosylase (UDG), and abasic site cleavage by NaOH treatment yields a shorter product (6-FAM labeled for quantification by fluorescence scanning).
b, Representative TBE-urea PAGE analysis of A3Bctd and A3H deaminase activity in the presence of increasing concentrations of EBV BORF2 (product percentage indicated below each lane).
c, Quantification of the DNA deaminase activity data in panel (b) and two additional biologically independent experiments (normalized mean +/− SD with some error bars smaller than the symbols). These data (b-c) are representative of n = 3 biologically independent experiments.
The archetypal mechanism for virus-mediated neutralization of APOBEC3 enzymes is target engagement followed by polyubiquitination and proteasomal degradation1–4. In contrast, BORF2 expression stabilizes steady-state levels of cellular A3B, suggesting a degradation-independent mechanism (Fig. 3a). In support, proteasome inhibition by MG132 had little effect on cellular A3B levels with or without BORF2 (Supplementary Fig. 6). To gain further insights into this unique mechanism and to assess the interaction of these proteins in the context of EBV lytic replication, immunofluorescence microscopy was used to examine the subcellular localization of endogenous A3B and BORF2 produced after reactivation of AGS cells latently infected with EBV (AGS-EBV)14,15. A3B was predominantly pan-nuclear in latent AGS-EBV cells prior to reactivation (Fig. 3b, upper panel), consistent with prior reports for A3B localization in a variety of non-infected cell typese.g.,16,17. After reactivation to the lytic cycle, nuclear and perinuclear bodies containing both A3B and BORF2 accumulated rapidly and, as the lytic phase progressed, additional cytoplasmic bodies containing BORF2 and A3B increased in abundance (Fig 3b, bottom panels and Fig. 3c). Additional imaging studies with markers for cytoplasmic organelles indicated that the BORF2/A3B bodies accumulated within the endoplasmic reticulum (Fig. 3d). In particular, three-dimensional reconstructions of image z-stacks showed that these aggregates are surrounded on all sides by the integral endoplasmic reticulum component BiP/GRP-78 (Supplementary Video 1). A similar result was obtained in reactivated AGS-EBV cells using a second endoplasmic reticulum marker, TRAPα (Supplementary Video 2). BORF2 alone also caused A3B relocalization in other cell types, including U2OS, HeLa, 293T, and AGS (Fig. 3d-e, Supplementary Fig. 7 & Videos 3-4). In addition, relocalization of endogenous A3B and co-localization with BORF2 was observed in M81 EBV-transformed B cells that spontaneously enter the lytic cycle18 (Fig. 3f, Supplementary Fig. 7g-h). Importantly, ORF61 of KSHV also sequestered A3B in perinuclear and cytoplasmic bodies, further indicating mechanistic conservation (Supplementary Fig. 2b-c).
Figure 3 |. BORF2 relocalizes A3B from the nuclear compartment to the endoplasmic reticulum.
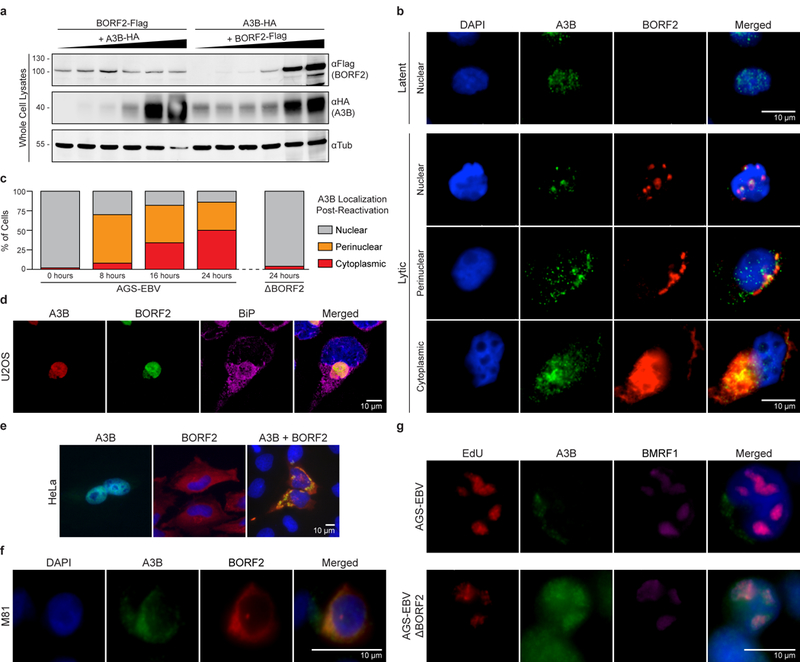
a, Immunoblots of lysates from 293T cells transfected with equal amounts of BORF2-Flag and increasing amounts of A3B-HA (left) or the reciprocal set-up (right).
b, Representative immunofluorescence microscopy images of latent (top panel) or lytic (bottom 3 panels) AGS-EBV stained with DAPI (blue) and antibodies against endogenous A3B (green) or BORF2 (red). A 10 μm scale is shown in the merged panel images.
c, Quantification of A3B localization in AGS-EBV cells grown under latent (0 hrs) or lytic (8, 16, 24 hrs) conditions (n = 50 cells per condition). Parallel quantification of A3B localization in lytic BORF2-null AGS-EBV cells (24 hrs; n = 50 cells).
d, Representative immunofluorescence microscopy images of U2OS expressing A3B-mCherry and BORF2-eGFP, and stained with an antibody against the endoplasmic reticulum protein, BiP/GRP-78 (purple; also see Supplementary Video 1).
e, Representative immunofluorescence microscopy images of HeLa transiently expressing A3B-eGFP alone, BORF2-Flag alone, or both proteins together.
f, Representative immunofluorescence microscopy images of endogenous A3B (green) and BORF2 (red) in a M81-transformed B cell that has spontaneously entered the lytic cycle.
g, Representative immunofluorescence microscopy images of endogenous A3B (green) in AGS-EBV and ΔBORF2 derivative pools 24 hrs after lytic reactivation. Anti-BMRF1 (purple) marks sites of viral DNA replication in lytic cells and EdU (red) shows newly synthesized DNA. These data are representative of n = 2 (a,d,f,g) or n = 3 (b,c,e,f) biologically independent experiments.
To directly test whether A3B relocalization is dependent on BORF2, CRISPR/Cas9 was used to disrupt the BORF2 gene in AGS-EBV cells and in AGS cells containing GFP-tagged viral genomes [AGS-EBV(Bx1g)15,19; Supplementary Fig. 8a]. Despite numerous EBV genome copies in these cells, a near complete ablation of BORF2 expression was achieved as evidenced by immunoblotting and by sequencing the DNA region targeted by Cas9/gRNA complexes (Supplementary Fig. 8b-c). BORF2 knockout viruses were then reactivated to the lytic cycle, and cells containing lytic replicating EBV were identified by staining for the EBV DNA polymerase processivity factor BMRF1 and for new viral DNA synthesis by EdU incorporation. In these cells, perinuclear and cytoplasmic A3B bodies were not observed, indicating that A3B relocalization from the nucleus is completely dependent on BORF2 (Fig. 3g). In addition, a proportion of A3B colocalized with BMRF1 and EdU indicating that A3B associates with viral DNA replication intermediates in the absence of BORF2.
Our findings that BORF2 directly inhibits and relocalizes A3B away from viral replication centers strongly suggested that this DNA cytosine deaminase could be a threat to EBV genomic integrity. Based on the hallmark DNA deamination activity of A3B, we hypothesized that single-stranded viral DNA replication intermediates would become susceptible to A3B-catalyzed DNA cytosine deamination in the absence of BORF2. To test this possibility, we analyzed viral genomic DNA C/G-to-T/A hypermutation by sequencing individual differential DNA denaturation (3D)-PCR products representing multiple regions of the viral genome. 3D-PCR estimates the lowest denaturation temperature required for amplification of any DNA substrate, and it thereby enables recovery of C/G-to-T/A hypermutated sequences by virtue of preferential amplification at lower than wild-type DNA denaturation temperatures20,21 (Fig. 4a). Indeed, in a series of pilot experiments, a segment of the BRRF2 gene amplified at lower denaturation temperatures from reactivated AGS-EBV cells containing BORF2-null EBV genomes as compared to cells containing wild-type EBV genomes (Supplementary Fig. 9a). Low temperature amplicons were not recovered from latently infected cell pools (prior to reactivation). Cloning and sequencing of individual PCR products revealed the highest level of mutation in amplicons from lytic replication-induced BORF2-null conditions (Supplementary Fig. 9b).
Figure 4 |. BORF2 functions to preserve EBV genome integrity from A3B.
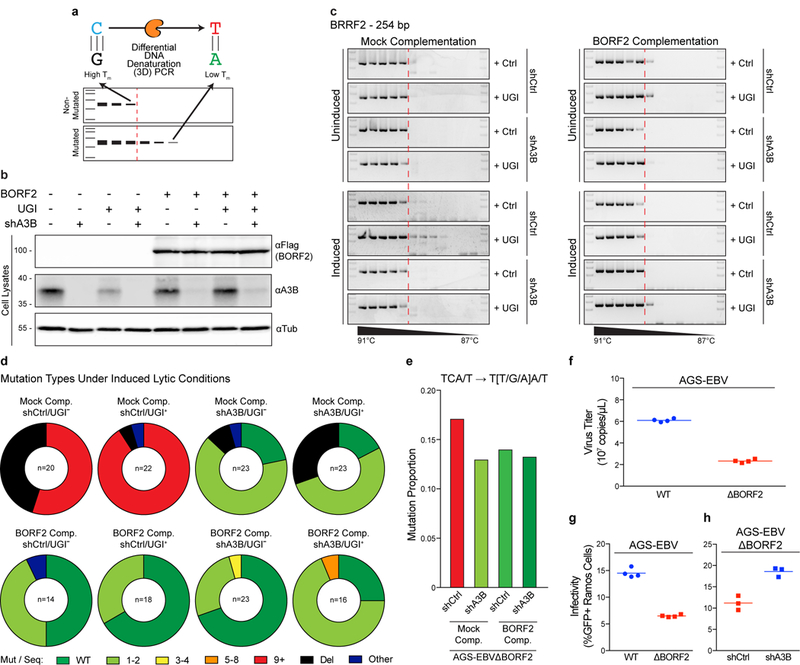
a, 3D-PCR differentiates between non-mutated and mutated DNA substrates by virtue of product accumulation at higher vs lower denaturation temperature (Tm) thresholds (e.g., A3B, orange enzyme, causes C/G-to-T/A mutations through uracil intermediates).
b, Immunoblots of an AGS-EBV(Bx1g)ΔBORF2 clone engineered to express BORF2-Flag or vector control, shA3B or shControl, and UGI or vector control (see text for details).
c, Representative agarose gel images showing the results of 3D-PCR experiments involving a 254 bp BRRF2 gene segment of AGS-EBV(Bx1g)ΔBORF2. The dashed red line shows the point at which non-mutated BRRF2 DNA fails to amplify under high denaturation conditions; visible PCR products below this Tm threshold represent lower temperature amplicons (i.e., mutated sequences). The right panels show the effect of complementing each condition with BORF2 expression.
d, Pie charts showing the types of mutational events observed in Sanger sequences of cloned lower temperature amplicons from the 8 induced conditions shown in the bottom half of panel (c). Wild-type non-mutated sequences are depicted in green; base substitutions with number of mutations per sequence depicted in light green, yellow, orange, and red; deletions are depicted in black; other types of mutations (e.g., combination of base substitution and deletion) are depicted in navy.
e, A summary of cytosine mutations detected in multiple EBV DNA regions under the indicated conditions following recovery by high-fidelity PCR (high temperature) and deep-sequencing.
f, Titers of wild-type and ΔBORF2 viruses after lytic induction of AGS-EBV(Bx1g). Each symbol represents data from 4 independent cultures and the horizontal line shows the mean.
g, Infectivity of wild-type and ΔBORF2 viruses produced by lytic replication in AGS-EBV(Bx1g). Each symbol represents the percent of GFP-positive Ramos reporter cells from n = 4 independent infections, and the horizontal line shows the mean.
h, Infectivity of ΔBORF2 EBV produced by lytic replication in AGS-EBV(Bx1g) with endogenous A3B intact (shCtrl) or depleted (shA3B). Each symbol represents the percent of GFP-positive Ramos reporter cells from n = 3 independent infections, and the horizontal line shows the mean. These data are representative of n = 2 (f,g) or n = 3 (b,c,h) biologically independent experiments.
We next generated a clonal system to extend these studies. Virus-containing supernatants from pooled AGS-EBV(Bx1g) cells with BORF2-null EBV were harvested and used to infect new parental AGS cells. A clonal AGS cell line with a single isogenic BORF2-null EBV was generated [AGS-EBV(Bx1g)ΔBORF2]. These cells were first stably complemented with control or BORF2-Flag expressing lentivirus and, second, transduced with shRNA to knockdown endogenous A3B or with a non-targeting control (Fig. 4b). Third, a control vector or uracil glycosylase inhibitor (UGI) was added by transduction of cells from each condition in order to inhibit the repair of A3B-mediated uracil lesions and potentially exacerbate the effects of A3B-mediated viral hypermutation. UGI uses a structurally conserved mechanism to inhibit uracil DNA glycosylases across kingdoms, including bacterial, human, and EBV enzymes22,23 (Supplementary Fig. 10). Cells from each of these 8 conditions were cultured to maintain EBV latency (uninduced) or treated with TPA and sodium butyrate for 48 hrs to reactivate EBV lytic replication (induced), and then DNA was harvested and mutations analyzed by sequencing individual 3D-PCR products.
Under normal cell culture conditions that maintain EBV latency, low temperature BRRF2 amplicons were rarely observed for any of the 8 conditions (Fig. 4c). Similarly, BORF2 complemented cells, regardless of cell culture treatments or other genetic manipulations, did not yield lower temperature BRRF2 PCR amplicons. In contrast, after induction of EBV lytic replication, lower temperature PCR amplicons could be generated from AGS-EBV(Bx1g)ΔBORF2 cells expressing endogenous levels of A3B, and this phenotype was exacerbated by UGI treatment. The accumulation of lower temperature amplicons was completely dependent upon A3B activity as no lower temperature BRRF2 amplicons were recovered from A3B knockdown cells that were otherwise treated identically. Similar results were obtained by 3D-PCR analysis of a region of EBNA2 (Supplementary Fig. 9c).
A sequence analysis of individual 3D-PCR products confirmed these results showing extensive mutagenesis of BRRF2 sequences from AGS-EBV(Bx1g)ΔBORF2 cells expressing endogenous A3B, both with and without UGI treatment (Fig. 4d, Supplementary Figs. 11a & 12). Interestingly, diverse mutational events were observed including strong enrichments for C/G-to-T/A mutations and deletions. A high deletion frequency was unexpected and, based on precedents with AID in antibody diversification24,25, likely due to processing of A3B-induced lesions by cellular DNA damage response mechanisms. This is supported by a trend toward more deletions without UGI, as uracil excision can readily lead to single-stranded nicks and broken DNA. However, A3B-attributable hypermutation was not observed in Sanger sequences of >20 high-temperature BRRF2 amplicons from BORF-null conditions (Supplementary Fig. 11b), but enrichments for mutations in A3B-preferred 5’-TCA/T motifs17 were apparent by deep-sequencing high-temperature amplicons representing 4 different viral genomic DNA regions (Fig. 4e). We therefore inferred that the hypermutation frequency is relatively low for a given region of EBV, and that this may be explained by low reactivation rates and by a viral DNA replication mechanism that may only periodically expose single-stranded DNA to deamination by A3B.
The potential for this viral DNA hypermutation mechanism to exert a cumulative effect is supported by BORF2-null viruses showing 60-70% lower titers and, even upon titer normalization, a further 50-60% less infectivity after one round of lytic replication in AGS-EBV(Bx1g) cells (Fig. 4f-g). Specific knockdown of endogenous A3B resulted in a near-full restoration of viral infectivity (Fig. 4h). A dependence on A3B is further evidenced by Akata cells, which we found are homozygous for a 29.5 kbp deletion spanning the entire A3B gene26, producing BORF2-null and wild-type viruses with similar titers and infectivity (Supplementary Fig. 13). Combined, these hypermutation and infectivity results indicated that BORF2 is dispensable during EBV latency, but required to protect viral DNA from A3B mutagenesis during conditions of induced lytic replication.
The A3B neutralization mechanism described here for γ-herpesviruses proteins, EBV BORF2 and KSHV ORF61, is fundamentally different than the A3 degradation mechanism used by lentiviral Vif proteins1–4. In addition to the stark differences of direct inhibition and relocalization versus proteasomal degradation, the replication cycles of EBV and KSHV may be fundamentally less tolerant of APOBEC mutagenesis. This inference is supported by bioinformatics analyses of all sequenced EBV isolates indicating that BORF2 is under negative selection and that the domain of BORF2 that binds A3B contains few non-synonymous mutations (Supplementary Fig. 14a-c). APOBEC hypermutated sequences are also absent from databases (only one prior publication detected hypermutation in EBV by 3D-PCR and implicated a different APOBEC family member27). Nevertheless, A3B-preferred 5’-TCA deamination substrate motifs are depleted from EBV genomes and, correspondingly, 5’-TTA product motifs are enriched, consistent with periodic A3B-catalyzed mutation events (despite BORF2) and long-term evolutionary pressure (Supplementary Fig. 14e).
In comparison to EBV, KSHV, and related large DNA viruses with low mutation rates28, retroviruses have much higher mutation rates and frequent scars from APOBEC mutagenesis29,30 (e.g., ~10% of patient samples contain HIV-1 sequences with APOBEC signature hypermutations). This mechanistic difference may be due, at least partly, to γ-herpesviruses using a coordinated leading/lagging strand replication mechanism to copy viral double-stranded DNA, whereas HIV-1 uses reverse transcription to copy genomic RNA into an obligate single-stranded cDNA intermediate prior to conversion into double-stranded DNA for integration into the host genome. EBV and KSHV may therefore have fewer exposed single-stranded DNA replication intermediates during the lytic phase that can be attacked by A3B. Thus, although the frequency of BORF2-null EBV hypermutation is lower than that documented for Vif-null HIV-1, the overall effect has the potential to be similarly catastrophic because even localized hypermutation could inactivate an essential viral gene or trigger degradation of the full episome. Furthermore, cumulative viral DNA deamination over the lifespan of an infected cell is likely to lead to depletion and potentially even clearance of viral episomes. Disrupting the BORF2/A3B and ORF61/A3B interactions in vivo may have merit for compromising the genetic integrity of EBV and KSHV, thereby limiting lytic infection that induces infectious mononucleosis and contributes to EBV- and KSHV-driven tumors.
METHODS
DNA constructs for expression in human cell lines.
The full set of pcDNA3.1(+) human APOBEC-HA expression constructs has been described31 [A3A (GenBank accession NM_145699), A3B (NM_004900), A3C (NM_014508), A3D (NM_152426), A3F (NM_145298), A3G (NM021822), A3H (haplotype II; FJ376615)]. A3Bntd1-190, A3Bctd191-382, A3Bntd-A3Gctd, and A3Gntd-A3Bctd constructs were cloned by overlap extension PCR as described32. The HIV-1IIIB Vif-Myc construct was also described previously33. A3B and A3G loop 7 swaps were generated by overlap extension PCR on A3B-HA and A3G-HA constructs with primers introducing mutations in loop 7 to generate the reciprocal amino acid sequence (A3B GL7YDPLYK(132-137)DQGRCQ and A3G BL7DQGRCQ(132-137)YDPLYK). The primers used for A3B GL7 were RSH5336 5’-TAC GAC CCC CTA TAT AAG GAG GGG CTG CGC ACC CT-3’ and RSH5337 5’-CTT ATA TAG GGG GTC GTA ATC ATA GAT GCG GGC-3’ and A3G BL7 were RSH5338 5’-GAT CAA GGA AGA TGT CAG GAG GCG CTG CAA ATG C-3’ and RSH5339 5’-CTG ACA TCT TCC TTG ATC ATC ATA GAT GCG GGC-3’. A3B with a C-terminal 2x-Strep (WSHPQFEK) tag was subcloned using high fidelity PCR from pcDNA3.1(+)-A3B-3xHA using primers (5’-NNA AGC TTA TGA ATC CAC AGA TCA GA-3’ and 5’-NNG CGG CCG CCC GTT TCC CTG ATT CTG GA-3’). PCR products were digested with HindIII-HF (NEB R3104) and NotI-HF (NEB R3189) and ligated into pcDNA4/TO (Invitrogen V102020) with a C-terminal 2x-Strep tag. Tetracycline-inducible A3B with a C-terminal mCherry tag was cloned by high-fidelity PCR of previously described A3B-mCherry34 using primers (RSH7018 5’-NNN NNA AGC TTA CCA CCA TGA ATC CA-3’ and RSH7016 5’-AGA GTC GCG GCC GCT TAC TTG TAC A-3’). PCR products were digested with HindIII-HF and NotI-HF and ligated into similarly digested pcDNA5/TO (Invitrogen V103320). The A3B-eGFP construct has been described34. The pLKO construct expressing an A3B-specific shRNA has been described and validated17,35–37, and the construct used here has a blasticidin resistance gene in place of the puromycin resistance gene. The pLKO construct expressing a non-targeting shRNA control38 (5’-CCT AAG GTT AAG TCG CCC TCG-3’) was a gift from Keith Mostov (Addgene plasmid #26701).
BORF2 (GenBank accession V01555.2) with a C-terminal 3x-Flag (DYKDDDDK) tag was subcloned by high-fidelity PCR from the previously described PMZS3F-BORF239 using primers (RSH12971 5’-NNN NGA TAT CGC CGA CAC CAT GGC AAC GAC C-3’ and RSH12848 5’-NNN NGC GGC CGC CCT TGG CAA GAT TCA CAG GC-3’). PCR products were digested with EcoRV-HF (NEB R1395) and NotI-HF and ligated into pcDNA4 (Invitrogen V102020) with a C-terminal 3x-Flag40. BORF2 with a C-terminal eGFP tag was generated by high-fidelity PCR of pcDNA4-BORF2-3xFlag using primers RSH13422 5’-NNN NAT GCA TCA TGG CAA CGA CCA GTC ATG TC-3’ and RSH13424 5’-NNN NAC GCG TCC TTG GCA AGA TTC ACA GGC TCG-3’. PCR products were digested with NsiI-HF (NEB R3127) and MluI-HF (NEB R3198) and cloned into the previously described pQCXIP (Clontech) with a C-terminal eGFP tag41. BORF2 with C-terminal 3x-Flag tag was cloned into an MLV-based pQCXIP lentivirus vector (Clontech) for complementation experiments by PCR of pcDNA4-BORF2-3xFlag using primers RSH13422 5’-NNN NAT GCA TCA TGG CAA CGA CCA GTC ATG TC-3’ and RSH13423 5’-NNN NTT AAT TAA TTA AAC GGG CCC CTT GTC GTC-3’. PCR product was digested into NsiI-HF and PacI (NEB R0547) and ligated into pQCXIP digested with SbfI-HF (NEB R3642) and PacI. BORF2 truncation mutants were generated by subcloning portions of pcDNA4-BORF2-3xFlag by high fidelity PCR, digesting the PCR products with EcoRV-HF and NotI-HF, and ligating into pcDNA4-3xFlag as described above. The PCR primers are as follows: N-terminal BORF2 25-826 mutant (RSH14011 5’-NNN NGA TAT CGC CAC CAT GTC AGA CCC CGA GGC TGA TGT C-3’ and RSH12848 above), BORF2 50-826 (RSH14012 5’-NNN NGA TAT CGC CAC CAT GGC CGA ATA TCT GGA GGT CTT C-3’ and RSH12848), C-terminal BORF2 1-739 mutant (RSH12971 above and RSH13396 5’-NNN NGC GGC CGC TCC ATC ACC CCC AGA TCG GCG GC-3’), and BORF2 1-687 (RSH12971 and RSH13612 5’-NNN NGC GGC CGC ACA AAG GGG GCC CTG TCC C-3’). BORF2 catalytic mutants were generated in pcDNA4-BORF2-3xFlag by site-directed mutagenesis using PfuUltra II Fusion HS DNA polymerase (Agilent 600670) with the following primers for YY725/6AA (RSH13776 5’-GGC CTG AAG ACT ATC ATG GCG CTT TGT CGC ATT GAG AAG GC-3’ and RSH13777 5’-GCC TTC TCA ATG CGA CAA AGC GCC ATG ATA GTC TTC AGG CC-3’) and YY725/6FF (RSH13774 5’-GGC CTG AAG ACT ATC ATG TTC TTT TGT CGC ATT GAG AAG GC-3’ and RSH13775 5’-GCC TTC TCA ATG CGA CAA AAG AAC ATG ATA GTC TTC AGG CC-3’).
BaRF1 (Genbank accession V01555.2) with C-terminal 3x-HA (YPYDVPDYA) tag was cloned by high fidelity PCR from a gBlock purchased from IDT using primers (RSH13621 5-NNN NGA ATT CGC CGC CAC CAT GTC CAA GTT G-3’ and RSH13622 5’- NNN NCT CGA GAA GGT CAT CTA CCA CCA GCA T-3’). PCR products were digested with EcoRI-HF (NEB R3101S) and XhoI (NEB R0146) and cloned into pcDNA3.1(+) (Invitrogen V79020) with a C-terminal 3x-HA tag.
The LentiCRISPRv242 vector, which was a gift from Feng Zhang (Addgene plasmid #52961), was modified to introduce a loxP site into the viral long-terminal repeat (LentiCRISPRv2-loxP; Carpenter et al., manuscript in preparation). A gRNA targeting BORF2 was designed using crispr.mit.edu with PAM site of AGG at +119 and predicted cut site at +122 on the antisense strand (relative to BORF2 start codon). gRNA primers (RSH13690 5’-CAC CGG TGT AAC TGA CTC GGC CTT A-3’ and RSH13691 5’-GAA CTA AGG CCG AGT CAG TTA CAC C-3’) were annealed, cut with BsmBI, and ligated into similarly digested LentiCRISPRv2-loxP.
UGI (GenBank accession J04434.1) was amplified with high fidelity polymerase from pcDNA3.1(+)-UGI43 using primers (RSH7505 5’-NNN NCT CGA GAC CAT GAC AAA TTT ATC TGA CAT CAT TG-3’ and RSH7506 5-NNN ACG CGT TTA TAA CAT TTT AAT TTT ATT TTC TCC-3’), digested with XhoI and MluI-HF, and ligated into similarly digested pLenti4/TO (Invitrogen). EBV uracil DNA glycosylase (BKRF3) (GenBank accession MG021307.1) was amplified with a high fidelity polymerase from AGS-EBV(Bx1g) genomic DNA using primers (RSH15814 5’-GAA GCT TGG TAC CAT GGC ATC GCG GG-3’ and RSH15815 5’-TCT AGA CTC GAG CTA CAG CCT CCA ATC-3’), followed by restriction digest with KpnI-HF (NEB3142S) and XhoI and ligation into similarly digested pcDNA3.1-V544. A gRNA targeting human UNG2 was designed using crispr.mit.edu with a PAM site of TGG at +274 and predicted cut site at +271 on the sense strand (relative to UNG2 start codon). gRNA primers (RSH11189 5’-CAC CGC GCG GCC CGC AAC GTG CCC G-3’ and RSH11190 5’-GAA CCG GGC ACG TTG CGG GCC GCG C-3’) were annealed, cut with BsmBI, and ligated to similarly digested LentiCRISPRv2-loxP.
All RNR large subunit homologs were cloned into the previously described pCMV-3F plasmid45. Human RRM1 (GenBank accession BC006498) was amplified by high-fidelity PCR from a plasmid provided by the SPARC BioCentre using primers 5’-GTA CGA GCT CGC CAC CAT GCA TGT GAT CAA GCG AGA TGG-3’ and 5’-GAC TTC TAG AGG ATC CAC ACA TCA GAC ATT CAT C-3’, digested with SacI (NEB R0156S) and XbaI (NEB R0145S), and ligated into similarly digested pCMV-3F. KSHV ORF6146 (GenBank accession U75698.1) was kindly provided by Britt Glausinger (UCSF), amplified using 5’-GTA CAG ATC TGC CAC CAT GTC TGT CCG GAC ATT TTG-3’ and 5’-GAC TTC TAG ACT GAC AGA CCA GGC ACT CG-3’, digested with BglII and XbaI, and ligated into similarly digested pCMV-3F. HSV-1 UL39 (GenBank accession JN555585.1) was amplified from a library described previously39 using primers 5’-GAC TGA ATT CGC CAC CAT GGC CAG CCG CCC AGC-3’ and 5’-GCA TTC TAG ACA GCG CGC AGC TCA TGC-3’, digested with EcoRI (NEB R0101S) and XbaI, and ligated into similarly digested pCMV-3F. HCMV UL45 (GenBank accession FJ527563.1) was amplified from a library described previously39 using primers 5’-GTA CAG ATC TGC CAC CAT GAA TCC GGC TGA CGC GGA-3’ and 5’-GAC TTC TAG AAG AGG CAC AGT ACT TAT ATA CTC-3’, digested with BglII (NEB R0144S) and XbaI, and ligated into similarly cut pCMV-3F. All new plasmid constructions were confirmed by restriction fragment analysis and DNA sequencing.
Human cell culture.
Unless indicated, cell lines were derived from established lab collections. All cell cultures were supplemented with 10% heat-inactivated fetal bovine serum (Gibco 16140-063), 1x Pen-Strep (Thermo Fisher 15140122), and periodically tested for mycoplasma (Lonza MycoAlert PLUS LT07-710). 293T cells were cultured in high glucose DMEM (Hyclone), U2OS cells in McCoy’s 5A media (Hyclone), HeLa, AGS (gastric carcinoma), AGS-EBV, and Ramos (EBV-negative Burkitt’s lymphoma) cells were cultured in RPMI 1640 (Corning). The Akata B cell line (EBV-positive Burkitt’s lymphoma) used here was a kind gift from Kristin Hogquist and Samantha Dunmire (University of Minnesota). The EBV strain in Akata was engineered previously to express neomycin resistance47 and GFP19. The neomycin resistant Akata EBV strain was introduced previously into AGS cells to yield the AGS-EBV and the neomycin resistant, GFP-expressing virus was used to derive the AGS-EBV(Bx1g) line used here14,15,19. Akata EBV containing cell cultures were additionally supplemented with 0.4 mg/mL G418. B cells transformed with the M81 EBV strain18 were a kind gift from Henri-Jacques Delecluse (DKFZ) and were cultured in RPMI. This EBV strain spontaneously reactivates to the EBV lytic cycle without external treatment. Cells transduced with shControl or shA3B lentivirus were selected with 5 μg/mL blasticidin (GoldBio B-800-500). Cells transduced with pLentiCRISPR targeting BORF2 or pQCXIP-BORF2 complementation lentiviruses were selected with 1 μg/mL puromycin. Cells transduced with pLenti/TR to express tetracycline repressor were selected with 5 μg/mL blasticidin.
Affinity purification and mass spectrometry.
Five 10 cm dishes of 293T cells were transfected with 6 μg of PMZS3F-BORF239 using PolyJet at a 1:2 ratio. 48 hrs after transfection cells were harvested and pooled. Cells were washed two times in PBS followed by lysis [50 mM Tris pH 7.9, 420 mM NaCl, 10% glycerol, 0.1% NP-40, protease inhibitors (Sigma P8340), 1 mM NaF]. Lysates were subjected to three cycles of freeze-thaw followed by sonication. Benzonase (Sigma E1014, 25 U/mL) was added to the lysate and incubated on ice for 30 min. Lysates were then cleared by centrifugation (15K rpm) in a table top microcentrifuge for 30 min. Cleared lysate was then added to 50 μL of anti-Flag M2 resin (Sigma, A2220) followed by end-over-end rotation for 3 hrs at 4ºC. Following immunoprecipitation, the anti-Flag resin was washed two times in lysis buffer, followed by three additional washes in lower salt, detergent free wash buffer (10 mM Tris pH 7.9, 150 mM NaCl, 10% glycerol). Protein was eluted from the resin with three washes of 0.5 M ammonium hydroxide (15 minutes each at 25ºC with rotation). Samples were lyophilized in a speed-vac (Savant DNA120, Thermo Electron Corp.) and washed with 400 μL HPLC-grade water (Gibco). Samples were lyophilized once more to remove traces of ammonium hydroxide, and then subjected to TCA precipitation, tryptic digestion, and tandem mass spectrometry (MS/MS) as described48. RAW files were submitted for database searching using X! TANDEM (version: 2007.07.01.3) and TPP (version: 4.3) under standard workflow and a modified UniProt/Swiss-Prot protein database FASTA file. The modification consisted of adding BSA (Swiss-Prot accession number P02769). Search parameters were set to allow for two missed cleavage sites, variable modification by methionine oxidation, and one fixed modification by cysteine carbamidomethylation. A 10 ppm filter was used for peptide identification. The search results were uploaded to ProHits49 and compared using at least 99% TPP probability.
RNA isolation, cDNA synthesis and RT-qPCR.
RNA isolation, cDNA synthesis, and qPCR primers were described50. In brief, RNA was harvested from 2×106 cells using High Pure RNA Isolation Kit. cDNA libraries were generated with reverse transcriptase (Roche). qPCR was performed on a Roche LightCycler 480 instrument. The housekeeping gene TBP was used for normalization.
Co-immunoprecipitation experiments and immunoblots.
Semi-confluent 293T cells were grown in 6-well plates and transfected with plasmids and 0.6 μL TransIT-LT1 (Mirus 2304) per 100 ng DNA in 100 μL serum-free Opti-MEM (Thermo Fisher 31985062). In most experiments, 100 ng DNA was used for each unique plasmid transfection (e.g., Fig. 1b, 1c-f, Supplementary Figs. 3 & 4). For the A3 panel and RNR homolog co-IP experiments (Fig. 1c and Supplementary Fig. 2a), a titration series was performed to achieve roughly equivalent protein expression by immunoblot. Growth medium was removed after 48 hrs and whole cells were harvested in 1 mL PBS-EDTA by pipetting. Cells were spun down, PBS-EDTA was removed, and cells were resuspended in 300 μL of ice-cold lysis buffer [150 mM NaCl, 50mM Tris-HCl, 0.5% Tergitol, Roche cOmplete EDTA-free protease inhibitor cocktail tablet (Roche 5056489001), pH 7.4]. Cells were vortexed vigorously and left on ice for 30 minutes, then sonicated for 5 seconds in an ice water bath. 30 μL of whole cell lysate was aliquoted for immunoblot. Lysed cells were spun down at 13,000 rpm for 15 minutes to pellet debris and supernatant was added to clean tube with 25 μL resuspended anti-Flag M2 Magnetic Beads (Sigma M8823) for overnight incubation at 4 °C with gentle rotation. Beads were then washed three times in 700 μL of ice-cold lysis buffer. Bound protein was eluted in 30 μL of elution butter [0.15 mg/mL Flag peptide (Sigma) in 150 mM NaCl, 50 mM Tris-HCl, 0.05% Tergitol, pH 7.4]. Proteins were analyzed by immunoblot and antibodies used include mouse anti-Flag 1:5000 (Sigma F1804), mouse anti-BORF2 1:1000 (Santa Cruz sc56979), mouse anti-BZLF1 1:3000 (Santa Cruz sc53904), mouse anti-tubulin 1:10,000 (Sigma T5168), mouse anti-β-actin 1:10,000 (Santa Cruz sc47778), rabbit anti-HA 1:3000 (Cell Signaling C29F4), rabbit anti-Strep 1:5000 (Abcam ab76949), and rabbit anti-A3B 1:1000 (5210-87-1336).
Protein purification from E. coli and DNA deaminase activity assays.
pE-6xHis-SUMO-A3Bctd187-378-DM (L230K/F308K)51 and pE-6xHis-SUMO A3H Hap II1-18352 have been reported previously for E. coli expression and protein purification. Codon-optimized BORF2 was synthesized by Bio Basic, digested with BsmBI (NEB R0580), and ligated into pE-6xHis-SUMO (LifeSensors) for E. coli expression and protein purification. CaCl2-competent BL21(DE3) E. coli were transformed, grown overnight on LB-kanamycin plates, and single cell colonies were inoculated in 1 L 2xYT media with 50 μg/mL kanamycin and grown at 37 °C until ~OD600 0.8. Cells were then induced with 0.5 mM IPTG and grown at 16 °C overnight for protein expression. Cells were centrifuged and resuspended in 30mL of ice cold lysis buffer (300 mM NaCl, 50 mM Tris-HCl, 20 mM imidazole, 200 μg/mL RNase A, 100 μg/mL lysozyme, Roche cOmplete EDTA-free protease inhibitor, pH 7.4). Cells were incubated on ice for 30 minutes, then lysed by pulse sonication two times for 2 minutes in an ice water bath (Branson Sonifer). Lysed cells were spun down at 13,000g for 30 minutes to remove debris. The supernatant was added to 2 mL of Ni-NTA agarose beads (Qiagen 30230) and incubated at 4 °C for 30 minutes with gentle rocking. Beads were then washed twice with 10 mL of wash buffer (300 mM NaCl, 50mM Tris-HCl, 40 mM imidazole, pH 7.4). Bound protein was eluted twice with 500 μL of elution buffer (300 mM NaCl and 250 mM imidazole).
Recombinant purified proteins were mixed with 2x reducing sample buffer (100 mM Trish-HCl pH 6.8, 20% glycerol, 4% SDS, 5% β-mercaptoethanol, 0.05% bromophenol blue), run on 4-20% SDS-PAGE gels, stained with Coomassie stain (40% methanol, 10% acetic acid, 0.1% Coomassie R250), then quantified by densitometric analyses on ImageJ using bovine serum albumin as a standard. A3 proteins were titrated to achieve equivalent enzymatic cleavage of a fluorescent oligo substrate (RSH5194 5’-ATT ATT ATT ATT CAA ATG GAT TTA TTT ATT TAT TTA TTT ATT T-fluorescein-3’) by mixing together 1 μL recombinant A3, 1 μL 10.7 μM oligo, 0.5 μL 1 mg/mL RNase, 0.025 μL UDG (NEB M0280), and 7.47 μL modified HED buffer (20 mM HEPES, 50 mM NaCl, 0.1mM EDTA, 0.1 mg/mL BSA, pH 7.4) and incubating at 37 °C for 30 minutes. 1 μL of 1.1 M NaOH (100 mM final) was then added and heated to 98 °C for 5 minutes to cleave the DNA at abasic sites. The reaction was then mixed with 11 μL 2x formamide buffer (80% formamide, 1x TBE, bromophenol blue, and xylene cyanol) and run on a 15% TBE-urea PAGE gel. Separated DNA fragments were visualized on a Typhoon FLA-7000 scanner on fluorescence mode (GE Healthcare). A3 deaminase activity was quantified by densitometric analyses on ImageJ by dividing product band intensity by sum of product and substrate band intensities. Approximately equivalent deaminase activity was achieved at 350nM A3Bctd and 80nM A3H. Recombinant proteins were then diluted to specified concentrations in HED buffer. 5 μL of recombinant BORF2 was equilibrated with 2 μL of A3 proteins for 15 minutes and added to 3 μL of an oligo master mix containing 1 μL of 10.7 μM fluorescent oligo, 0.5 μL 1 mg/mL RNase, 0.025 μL UDG (NEB M0280), and 1.475 μL HED buffer for a total reaction volume of 10 μL, which was incubated at 37 °C for 30 minutes. Deaminase activity assay then proceeded as above.
Normalized percent A3 deaminase activity was calculated for n = 3 independent biological replicates by defining 100% deaminase activity as the no BORF2 condition for both A3Bctd and A3H. Normalized percent activities were averaged and standard deviations were calculated. Assuming normal data distributions, one-sample t-tests were performed at each concentration of BORF2 to determine if mean A3Bctd or A3H deaminase activity differed from null hypothesis μ=1 (100% activity) with alternative hypothesis μ<1, df=2. p-values for A3Bctd are as follows for the following concentrations of BORF2 (p=0.0237 at 43.75 nM; p=0.00206 at 87.5 nM; p=9.56×10−5 at 175 nM; p=2.51×10−6 at 350 nM; p=2.685×10−5 at 700 nM) and p-values for A3H are (p=0.970 at 43.75 nM; p=0.816 at 87.5 nM; p=0.424 at 175 nM; p=0.440 at 350nM; p=0.1575 at 700 nM).
MG132 experiments.
Semi-confluent 293T cells were transfected with 100 ng of each indicated plasmid and 1.8 μL TransIT-LT1 in 100 μL serum-free Opti-MEM in 6-well. After 42 hrs post-transfection, media was removed and replaced with fresh media containing 10 μM MG132 (Sigma M7449) or DMSO control. Cells were harvested after 4 hrs of MG132 treatment, washed twice with PBS, and analyzed by immunoblot. Antibodies used include rabbit anti-Myc 1:3000 (Sigma C3956), rabbit anti-HA, mouse anti-Flag 1:5000, and mouse anti-tubulin 1:10,0000.
Immunofluorescence microscopy.
For immunofluorescence imaginga of endogenous proteins, AGS-EBV or BORF2-null AGS-EBV cells were grown on acid-washed and poly-lysine-treated 22×22×1.5mm coverslips (Fisherbrand 12-541-B) until they reached 80% confluency. Cells were either left untreated (latent infection samples) or treated with 20 ng/mL TPA and 3 mM sodium butyrate (NaB) for 8, 16, or 24 hrs to reactivate the virus into the lytic cycle. Coverslips were then washed with PBS, fixed in 4% methanol-free formaldehyde (Thermo Scientific 28906) for 15 minutes, and rinsed 3 times for 5 minutes in PBS with gentle rocking. Cells were permeabilized with 0.2% Triton X-100 in PBS for 10 minutes, washed three time for 5 minutes in PBS, then blocked overnight in 5% bovine serum albumin (BSA). Cells were incubated with rabbit anti-A3B 1:1000 (5210-87-13)36 and mouse anti-BORF2 1:400 (Santa Cruz sc56979) in AGS-EBV cells, or mouse anti-BMRF1 1:400 (Millipore MAB8186) in BORF2-null AGS-EBV cells to indicate reactivated cells for 48 hrs in a humid chamber at 4 °C. Coverslips were washed 3 times with PBS, followed by incubation with secondary antibodies, goat anti-rabbit AlexaFluor 488 1:1000 (Invitrogen A11034) or donkey anti-mouse AlexaFluor 647 1:1000 (Invitrogen A31571) in 5% BSA for 1 hr. After washing in PBS, coverslips were mounted onto slides using ProLong Gold antifade medium containing DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen). Images were captured using the 40x oil objective on a Leica inverted fluorescence microscope and analyzed using the Leica Application Suite X (LAS X, version 3.3.0) software. For each TPA/NaB-treated sample, 50 reactivated cells were analyzed for co-localization of BORF2 and A3B. For latent AGS-EBV cells and reactivated cells containing the BORF2 knockout virus, the overall localization of A3B was scored for 50 cells. For immunofluorescence of M81 cells, approximately 5×106 cells were harvested during log-phase growth. Immunofluorescence protocol then proceeded as above except for permeabilization in 0.5% instead of 0.2% Triton X-100 and primary antibody incubation for 24 hrs in constant rotation at 4 °C.
For EdU staining of AGS-EBV, cells were grown on coverslips until they reached 80% confluency. Cells were then treated with 10 μM EdU (Thermo Fisher C10338) for 1 hr prior to harvesting and fixed in 4% formaldehyde followed by washing in PBS, permeabilization in 0.5% Triton X-100, and blocking in 5% BSA for 1 hr. Staining of EdU was done according to the manufacturer’s protocol. Cells were incubated with rabbit anti-A3B 1:1000 (5210-87-1327) and anti-BMRF1 (Millipore MAB8186) 1:1000 for 48 hrs in a humid chamber. Immunofluorescence protocol then proceeded as above.
For immunofluorescence imaging of transfected cells, 293T, HeLa, or AGS cells were plated on coverslips and, after 24 hrs, transfected with 200 ng pcDNA4-BORF2-3xFlag, 200 ng pcDNA5/TO-A3B-eGFP, or both. After 48 hrs, cells were harvested, fixed in 4% formaldehyde, and permeabilized in 0.2% Triton X-100 as above. Cells were then incubated in blocking buffer (0.0028 M KH2PO4, 0.0072 M K2HPO4, 5% goat serum (Gibco), 5% glycerol, 1% cold water fish gelatin (Sigma), 0.04% sodium azide, pH 7.2) for 1 hr. Cells were then incubated in blocking buffer with primary mouse anti-Flag 1:1000 for 2 hrs to detect BORF2-Flag. Cells were washed 3 times for 5 minutes with PBS then incubated in secondary antibodies diluted in blocking buffer for 1 hr at room temperature with goat anti-mouse AlexaFluor 594 1:1000 in the dark. Cells were then counterstained with 1 μg/mL Hoescht 33342 for 10 minutes, rinsed twice for 5 minutes in PBS, and once in sterile water. Coverslips were mounted on pre-cleaned slides (Gold Seal Rite-On) using one drop (10-15 μL) of mounting media (dissolve 1g n-propyl gallate (Sigma) in 30 mL glycerol overnight, add 0.35 mL 0.1M KH2PO4, then pH to 8-8.5 with K2HPO4, Q.S. to 50mL with water). Slides were imaged on a Nikon Inverted Ti-E Deconvolution Microscope instrument and analyzed using NiS Elements.
For KSHV ORF61 experiments, 293T and AGS cells were transfected with 500 ng pcDNA5/TO-A3B-eGFP, 500 ng pCMV3F-ORF61, or both. After 48 hrs, cells were harvested, fixed in 3.7% formaldehyde, and permeabilized in 0.2% Triton X-100 as above. Cells were then incubated blocked in 5% BSA for 1 hr. Cells were then incubated with mouse anti-Flag 1:1000 for 2 hrs and then immunofluorescence protocol proceeded as above. Cell images were taken using Leica inverted fluorescence microscope and analyzed using the Leica Application Suite X (LAS X, version 3.3.0) software.
For live cell imaging experiments, 293T cells were transfected with pLenti6/TR (Thermo Fisher V48020), gag/pol, and VSVG to produce lentivirus expressing tetracycline repressor. Other 293T cells were transfected with MLV-based pQCXIP-BORF2-eGFP, MLV gag/pol, and VSVG to produce lentivirus expression for BORF2-eGFP transduction. U2OS cells were first transduced with pLenti6/TR and after 48 hrs, virus was washed off and cells were selected with 5 μg/mL blasticidin for 2 weeks. Limiting dilution was used to generate single cell clones and screened for TR expression and gene repression. U2OS-TR cells were then transfected with pcDNA5/TO-A3B-mCherry and put under 200 μg/mL hygromycin selection. After 2 weeks, limiting dilution was used to generate a second generation of single cell clones and screened. U2OS-TR-A3B-mCherry cells were plated on coverslips in 6-well plates for live cell imaging. A3B-mCherry expression was induced after 24 hrs with 10 ng/mL doxycycline, and then transduced with pQCXIP-BORF2-eGFP lentivirus 24 hrs after induction. One hr after transduction, cells were imaged on a Nikon BioStation IM machine every 15 minutes for 48 hr. Images were analyzed and stitched into a movie using NIS Elements software. In the reciprocal experiment, U2OS-TR-A3B-mCherry cells were plated, transduced with pQCXIP BORF2-eGFP, and after 24 hrs, induced with 10 ng/mL doxycycline. Image acquisition began one hr after A3B-mCherry induction as above.
For immunofluorescence imaging of BiP, U2OS-TR-A3B-mCherry cells (above) were plated on coverslips and induced with 10 ng/mL doxycycline for A3B-mCherry expression and transduced with pQCXIP-BORF2-eGFP lentivirus. After 48 hrs, cells were fixed and blocked as above and stained with rabbit anti-BiP/GRP-78 1:1000 (Abcam ab21685) overnight to detect endoplasmic reticulum. Cells were washed 3 times for 5 minutes each with PBS then incubated in goat anti-rabbit AlexaFluor 647 1:1000 (Invitrogen A32733) diluted in blocking buffer for 1hr at room temperature in the dark. Immunofluorescence protocol then proceeded as above for transfected proteins. Slides were imaged on a Nikon Inverted Ti-E Deconvolution Microscope instrument and analyzed using NiS Elements including deconvolution of images. For 3D-reconstruction of endoplasmic reticulum experiments, z-stacks were captured every 0.6 microns throughout the entire cell depth. Deconvolution was performed for each z-stack and then a movie of the 3D-reconstruction was generated using NiS Elements (Supplementary Video 1). For endoplasmic reticulum colocalization with TRAPα, reactivated AGS-EBV cells were stained with primary mouse anti-BORF2 1:400, rabbit anti-TRAPα 1:1000, or both for 1 hr (kind gift from Alexander Palazzo53) and then immunofluorescence protocol proceeded as above. z-stacks were captured every 0.3 microns throughout the entire cell depth and stitched together into a z-series movie (Supplementary Video 2).
Generation of BORF2-null EBV with CRISPR/Cas9.
Guide RNAs targeting BORF2 were generated using CRISPR Design Tool on crispr.mit.edu. Oligonucleotides were purchased from IDT and cloned into a pLentiCRISPRv2-loxP vector (see DNA constructs). 293T cells were transfected with pLentiCRISPR, gag/pol, and VSVG. Supernatant containing knockout lentivirus was collected after 48 hrs and used to transduce AGS-EBV, AGS-EBV(Bx1g), or Akata B cells. After 48 hrs, lentivirus was washed off and cells were subjected to antibiotic selection with media supplemented with 1 μg/mL puromycin for 4 weeks. Pooled cells were screened for successful BORF2 knockout from EBV genomes by immunoblot with mouse anti-BORF2 1:1000. Sanger sequencing was completed on viral genomic DNA collected from BORF2-null AGS-EBV(Bx1g) cells (Supplementary Fig. 4).
To generate a clonal AGS cell line with isogenic BORF2-null EBV(Bx1g), described here as AGS-EBV(Bx1g)ΔBORF2, pooled cells were supplemented with exogenous dNMPs (dAMP/dCMP/dGMP/dTMP each 400 μM) for 72 hrs and then induced with 3 mM sodium butyrate and 20 ng/ml TPA for production of EBV particles. After 48 hrs, virus-containing supernatant was harvested, filtered, and ultra-centrifuged at 22,000 rpm for 2 hrs to obtain concentrated cell-free BORF2-null EBV stock. This virus stock was used infect non-EBV-infected AGS cells at various empirically determined dilutions. Virus was washed off after 48 hrs and AGS cells were subjected to antibiotic supplementation with 0.4 mg/mL G418 for 2 weeks to select for successful EBV infection. These EBV-infected AGS cells were split into 96-well plates to obtain single cell colonies by limiting dilution. Colonies were then screened for BORF2-null virus infection by immunoblot. Knockout clones were screened by Sanger sequencing for successful isogenic EBV infection (n>10 sequences per clone). The isogenic AGS-EBV(Bx1g)ΔBORF2 clone used here has a 1426 bp deletion spanning nucleotides +88 to +1514 relative to the start codon.
Complementation, shRNA knockdown, and UGI experiments.
293T cells were transfected with MLV-based pQCXIP-BORF2-Flag or empty pQCXIP vector, MLV gag/pol, and VSVG to produce complementation lentivirus. Other 293T cells were transfected with pLKO constructs expressing A3B-targeting shRNA or control non-targeting shRNA, gag/pol, and VSVG to produce knockdown lentivirus. Other 293T cells were transfected with pLenti4-UGI or empty pLenti4 vector, gag/pol, and VSVG to produce UGI lentivirus.
The AGS-EBV(Bx1g)ΔBORF2 clone was first transduced with BORF2 or control complementation lentivirus. After 48 hrs, virus was washed off and cells were selected with 1 μg/mL puromycin for 2 weeks. These cells were then transduced with either A3B or control knockdown lentivirus. After 48 hrs, virus was washed off and cells were selected with 5 μg/mL blasticidin for 2 weeks. These cells were then transduced with either UGI or control treatment lentivirus. 48 hrs after transduction, virus was washed off and split into two arms for either virus induction with TPA/NaB or mock induction. After 48 hrs of lytic reactivation, DNA and protein was harvested and analyzed by 3D-PCR (see below) or immunoblot, respectively.
Generation of AGSΔUNG2 cell line with CRISPR/Cas9.
Guide RNAs targeting human UNG2 were generated using CRISPR Design Tool on crispr.mit.edu. Oligonucleotides were purchased from IDT and cloned into a pLentiCRISPRv2-loxP vector. 293T cells were transfected with pLentiCRISPRv2-loxP, gag/pol, and VSVG. Supernatant containing knockout lentivirus was collected after 48 hrs and used to transduce AGS cells. After 48 hrs, lentivirus was washed off and cells were subjected to antibiotic selection with media supplemented with 1 μg/mL puromycin for 2 weeks. Limiting dilution was then used to generate single cell clones, which were screened for successful UNG2 knockout by immunoblot and uracil DNA glycosylase activity (see below). Antibodies used include rabbit anti-UNG2 1:1000 (kind gift from Sal J. Caradonna, Rowan University) and anti-tubulin 1:10,000.
UDG activity assays.
AGS or AGSΔUNG cells were transfected with 1 μg BKRF3 or vector control using 3 μL TransIT-LT1 in serum free RPMI. Cells were harvested after 30 hrs and were lysed in 300 μL modified HED buffer (20 mM HEPES, 15 mM EDTA, Roche cOmplete EDTA-free protease inhibitor cocktail tablet, pH 7.4) per 106 cells. Activity assays were carried out as described12 using a 10 minute incubation with a dU-containing oligo (RSH12955 5’-AAA AAA AAA UCG GGA AAA AAA-fluorescein-3’). 2 μL of UGI (4 units, NEB M0281S) were used to inhibit UDG. Products were separated by a 20% TBE-Urea PAGE. Separated DNA fragments were visualized on a Typhoon FLA-7000 scanner on fluorescence mode.
3D-PCR and sequencing.
Total genomic DNA was collected from cells using Gentra Puregene Cell Kit (Qiagen 158689). Genomic DNA was used as template for first round PCR with ChoiceTaq DNA polymerase using manufacturer’s protocol (Denville Scientific CB4050) with primers for BRRF2 (5’-GTA GCA TCT CTG TCT GGT GAC CTT GAA-3’ and 5’-TTT TGG GGT CTC CGG ACA CCA TCT CTA-3’) and EBNA2 (5’-TAA CGT GCA AGA CGC TAA ACT TAA CCA A-3’ and 5’-AGC CTC GGT TGT GAC AGA GGT GAC AA-3’). First round PCR products were run on a 3% agarose gel by gel electrophoresis, normalized by densitometry, and then used for second round PCR. Second round PCR used Phusion High Fidelity DNA Polymerase (NEB M0530) using primers for BRRF2 (5’-AGG CCT GGC TTG AGG CTC AGG ACG CAA-3’ and 5’-GAC ATG ATT CAC ACT AAA AGA GAT CAA-3’) and EBNA2 (5’-ACA ATC TTT GTT GGG GAA AAC ACG GGG G-3’ and 5’-CGT CAT ATC CTA GCG GAT CCC TAT CAA-3’). The following PCR conditions were used for BRRF2 (87-91°C melting, 60 °C annealing, 72 °C extension) and EBNA2 (84-88 °C melting, 60 °C annealing, 72 °C extension). Second round PCR products were run on a 3% agarose gel and lowest temperature PCR amplicons were gel extracted using GeneJET Gel Extraction Kit (Thermo Fisher K0691), blunt-end cloned using CloneJET PCR Cloning Kit (Thermo Fisher K1231), and transformed into CaCl2-competent DH10B E. coli. Single cell colonies were picked for colony PCR using Phusion DNA polymerase with provided primers from CloneJET (5’-CGA CTC ACT ATA GGG AGA GCG GC-3’ and 5’-AAG AAC ATC GAT TTT CCA TGG CAG-3’). PCR products were enzymatically purified using Exonuclease I (NEB M0293) and rSAP (NEB M0371) treatment. Sanger sequencing of purified PCR products used 5’-AAG AAC ATC GAT TTT CCA TGG CAG-3’ primer, sequenced by GeneWiz, and results were analyzed with SnapGene software.
A3B genotyping.
The PCR breakpoint assay to identify wild-type vs the 29.5 kbp A3B deletion genotype was previously described26. Briefly, PCR primers within the A3B gene (blue arrows in Supplementary Fig. 13b) generate a 490 bp PCR product while the primers flanking the A3A/B fusion (red arrows) generate a 700 bp PCR product.
Deep sequencing analysis.
The AGS-EBVΔBORF2 clone was complemented with mock vector or BORF2-Flag, and transduced with shControl or shA3B lentivirus. Genomic DNA was harvested from cells 3 days after induction with PMA/NaB and portions of the EBV genome were PCR amplified using PfuTurbo Cx Hotstart DNA Polymerase (Agilent 600410). PCR products correspond to regions near the genes BRRF2 (95085-95584), LMP1 (167979-168514), BHRF1 (42090-42595), and BcLF1 (124082-124598). Base numbers correspond to the Akata reference genome (GenBank accession KC207813), which was also used for both the assembly and variant calling described below. Primers were designed with Nextera-compatible adapter sequences: Forward 5’-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-[locus-specific]-3’ and Reverse 5’-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-[locus-specific]-3’. Locus-specific primers were designed as follows: BRRF2 (RSH15191 5’-ACC GTC CAG CAA AAA GGG-3’ and RSH15192 5’-CCC CTT TGC AGC CAA TGC-3’), LMP1 (RSH15195 5’-CTG CCA CAC TAC CCT GAC-3’ and RSH15196 5’-AGC CGC CAG AGA ATC TCC-3’), BHRF1 (RSH15205 5’-GTG TTG GAG CTA GCA GCA AGA G-3’ and RSH15206 5’-CCG CAG GCC CAA TGA CCC-3’), and BcLF1 (RSH15207 5’-CCT GCT GGT GGG CAA GGA-3’ and RSH15208 5’-AGA TGC CTC TTG AAC ATG GC-3’). PCR products were enzymatically purified using Exonuclease I and rSAP treatment.
Illumina amplicon sequencing was performed at the University of Minnesota Genomics Center (UMGC) as a ≥16 million read, 2×300 bp paired-end MiSeq Version 3 run. Sequences were aligned and assembled using the Burrows-Wheeler Aligner (BWA)54, GATK toolkit55, Picard tools (Picard Toolkit. 2018. Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/), and SAMtools56,57,VarScan258 was used to call variation compared to the Akata reference genome. SAMtools and R were used for subsequent read and sequence filtering, processing, and visualization (R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org).
Viral infectivity assays.
AGS-EBV(Bx1g) or BORF2-null cells were plated on ten 15 cm plates each at equal density and grown until 70-80% confluency. Media was then replaced with fresh RPMI containing 10% FBS, 1x Pen/Strep, 20ng/mL TPA, and 3 mM sodium butyrate to induce lytic reactivation (without G418). Cells were grown for 4 days and media was collected, centrifuged briefly to pellet cells, and resulting supernatant was passed through a 0.45 μm filter (VWR 10040-470). Filtered supernatant was centrifuged at 25,000 rpm for 2.5 hrs using a Beckman Coulter JLA-16.250 rotor at 4 °C in a floor model centrifuge. Supernatant was then removed and bottle walls were washed extensively with PBS to remove residual TPA/NaB using care to not disturb the visible viral pellet. The virus pellet was then resuspended in 1 mL RPMI with 10% FBS and 1x Pen/Strep. 19 μL concentrated virus was then mixed with 1 μL of DNase (Qiagen 79254) and incubated at 37 °C for 10 minutes followed by 10 minutes of heat inactivation at 98 °C. 1 μL of DNase-treated virus was used to quantify viral titers in quadruplicate by RT-qPCR using SsoFast EvaGreen Supermix (Biorad 172-5202) and the primer set for EBV gp350 (5’-GTC AGT ACA CCA TCC AGA GCC-3’ and 5’-TTG GTA GAC AGC CTT CGT ATG-3’). Known quantities of EBV B98.5 BAC DNA were used as a standard curve (kind gift from Ya-Fang Chiu, Chang-Gung University). Equal titers of viral DNA were then used to infect 50,000 Ramos cells in 200 μL in a 96-well plate. Fresh RPMI was used as mock infection. After 22 hrs, Ramos cells were harvested, washed twice with PBS, resuspended in 200 μL PBS-EDTA, and analyzed by flow cytometry.
Akata or Akata BORF2-null cells were grown in 400 mL media until a confluency of ~1-2×106 cells/mL. Cells were pelleted and resuspended in at a density of 4×106 cells/mL in 100 mL of RPMI with 10% FBS, 1x Pen/Strep, and 6 μg/mL anti-Human IgG (MP Biomedicals 682651) to induce lytic reactivation (without G418). After 2 days, media volume was doubled with RPMI supplemented with 10% FBS and 1x Pen/Strep. After 4 days post-induction, cells were briefly centrifuged to pellet cells and resulting supernatant was passed through a 0.42 μm filter. Filtered supernatant was treated similar to above for virus produced from AGS-EBV cells. All infectivity studies were performed with at least 2 independent biological replicates for virus production, each quantified by n = 3-4 independent reporter cell infections.
Flow cytometry.
Analysis was done a BDFACS Canto II instrument immediately after cell collection and completed within 1 hr. Acquisition occurred for 75 sec or until 10,000 cells were collected. Cells were gated on live cells using SSC-A vs FSC-A, then mock-infected cells were gated on GFP vs FSC-A to determine threshold for GFP-negative gates.
Bioinformatics analysis.
EBV whole genome sequences previously isolated from patient gastric carcinoma and normal samples were analyzed for the evidence of APOBEC-related mutation59. Phylogenetic analyses were used to determine novel mutations that only occur once on an EBV sequence compared to all other whole genome sequences in the dataset (n = 142). This phylogeny was constructed from an alignment to a consensus sequence using the MAFFT (v7) software and classified into a neighbor-joining tree using an HKY model60. All single base substitutions compared to an ancestral EBV sequence (GenBank accession NC_007605.1) were used to calculate dN/dS ratios.
Whole genomes for EBV strain Akata (GenBank accession KC207813), rhesus lymphocryptovirus (Genbank accession NC_006146.1), and murine herpesvirus 68 WUMS strain (Genbank accession NC_001826.2) were used for trinucleotide motif enrichment analyses (substrate 5’-TCA and product 5’-TTA) based on Markov modeling61,62. Sliding window analyses across herpesvirus genomes was calculated using 15 kbp windows and an overlapping interval rate of 1 kbp. Smoothed fitted lines and 95% confidence intervals of these densities were calculated and plotted using the ggplot2 package in the R statistical environment.
Supplementary Material
Acknowledgements
We thank B. Anderson for technical advice and RT-qPCR data, Y.F. Chiu for sharing BAC EBV B95.8, H.-J. Delecluse for M81 transformed B cells, K. Hogquist and S. Dunmire for providing Akata cells, T. Ikeda and C. Richards for cell culture assistance, M. Sanders and staff at the University of Minnesota Imaging Center for assistance with fluorescence microscopy and live cell imaging, A. Serebrenik for the gRNA construct targeting UNG2, G. Starrett for technical programming advice, and R. Khanna, S. Rice, S. Simon, and P. Southern for thoughtful comments. This work was supported by NCI R21-CA206309 (RSH), the University of Minnesota (College of Biological Sciences, Academic Health Center, and Masonic Cancer Center to RSH), and Canadian Institutes for Health Research grant 153014 (to LF). NIH training grants provided salary support for AZC (F30 CA200432 and T32 GM008244) and MCJ (T32 CA009138). Salary support for JLM was provided by a National Science Foundation Graduate Research Fellowship. JY-M was funded by Secretaría Nacional de Educación Superior, Ciencia, Tecnología e Innovación (SENESCYT). LF is a tier 1 Canada Research Chair in Molecular Virology. RSH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished University McKnight Professor, and an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing Interests RSH is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc. The other authors declare no competing interests.
Data availability. The data that support the findings of this study are available upon request from the corresponding authors.
References
- 1.Simon V, Bloch N & Landau NR Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16, 546–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS & Dudley JP APOBECs and virus restriction. Virology 479-480C, 131–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malim MH & Emerman M HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe 3, 388–398 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Yang B, Li X, Lei L & Chen J APOBEC: from mutator to editor. J Genet Genomics 44, 423–437 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Rickinson A & Kieff E in Virology Fields (eds Knipe DM & Howley PM) 2655–2700 (Lippincott Williams & Wilkins, 2007). [Google Scholar]
- 6.Whitehurst CB, Ning S, Bentz GL, Dufour F, Gershburg E, Shackelford J, Langelier Y & Pagano JS The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J Virol 83, 4345–4353 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lembo D & Brune W Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci 34, 25–32 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Kraus RJ, Yu X, Cordes BA, Sathiamoorthi S, Iempridee T, Nawandar DM, Ma S, Romero-Masters JC, McChesney KG, Lin Z, Makielski KR, Lee DL, Lambert PF, Johannsen EC, Kenney SC & Mertz JE Hypoxia-inducible factor-1alpha plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog 13, e1006404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagemeier SR, Barlow EA, Kleman AA & Kenney SC The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. J Virol 85, 4318–4329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma D, Thompson J & Swaminathan S Spironolactone blocks Epstein-Barr virus production by inhibiting EBV SM protein function. Proc Natl Acad Sci U S A 113, 3609–3614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LaRue RS, Andrésdóttir V, Blanchard Y, Conticello SG, Derse D, Emerman M, Greene WC, Jonsson SR, Landau NR, Lochelt M, Malik HS, Malim MH, Munk C, O’Brien SJ, Pathak VK, Strebel K, Wain-Hobson S, Yu XF, Yuhki N & Harris RS Guidelines for naming nonprimate APOBEC3 genes and proteins. J Virol 83, 494–497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi K, Carpenter MA, Banerjee S, Shaban NM, Kurahashi K, Salamango DJ, McCann JL, Starrett GJ, Duffy JV, Demir O, Amaro RE, Harki DA, Harris RS & Aihara H Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol 24, 131–139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law EK, Carpenter MA & Harris RS The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun 7, 12918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takada K Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int J Cancer 33, 27–32 (1984). [DOI] [PubMed] [Google Scholar]
- 15.Borza CM & Hutt-Fletcher LM Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8, 594–599 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV & Cullen BR Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A 103, 8780–8785 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK & Harris RS APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R & Delecluse HJ Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5, 458–470 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Molesworth SJ, Lake CM, Borza CM, Turk SM & Hutt-Fletcher LM Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol 74, 6324–6332 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suspène R, Henry M, Guillot S, Wain-Hobson S & Vartanian JP Recovery of APOBEC3-edited human immunodeficiency virus G->A hypermutants by differential DNA denaturation PCR. J Gen Virol 86, 125–129 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Stenglein MD, Burns MB, Li M, Lengyel J & Harris RS APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol 17, 222–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geoui T, Buisson M, Tarbouriech N & Burmeister WP New insights on the role of the gamma-herpesvirus uracil-DNA glycosylase leucine loop revealed by the structure of the Epstein-Barr virus enzyme in complex with an inhibitor protein. J Mol Biol 366, 117–131 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW & Tainer JA Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell 82, 701–708 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Robbiani DF & Nussenzweig MC Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol 8, 79–103 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Di Noia JM & Neuberger MS Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76, 1–22 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Kidd JM, Newman TL, Tuzun E, Kaul R & Eichler EE Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet 3, e63 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suspene R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, Gaertner B, Vartanian JP, Meyerhans A & Wain-Hobson S Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol 85, 7594–7602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanjuan R & Domingo-Calap P Mechanisms of viral mutation. Cell Mol Life Sci 73, 4433–4448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janini M, Rogers M, Birx DR & McCutchan FE Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. J Virol 75, 7973–7986 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim EY, Lorenzo-Redondo R, Little SJ, Chung YS, Phalora PK, Maljkovic Berry I, Archer J, Penugonda S, Fischer W, Richman DD, Bhattacharya T, Malim MH & Wolinsky SM Human APOBEC3 induced mutation of human immunodeficiency virus type-1 contributes to adaptation and evolution in natural infection. PLoS Pathog 10, e1004281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL & Harris RS Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85, 11220–11234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stenglein MD & Harris RS APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem 281, 16837–16841 (2006). [DOI] [PubMed] [Google Scholar]
- 33.LaRue RS, Lengyel J, Jónsson SR, Andrésdóttir V & Harris RS Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J Virol 84, 8193–8201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lackey L, Law EK, Brown WL & Harris RS Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 12, 762–772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, Vogel RI, Nikas JB, Law EK, Brown WL, Li Y, Zhang Y, Maurer MJ, Oberg AL, Cunningham JM, Shridhar V, Bell DA, April C, Bentley D, Bibikova M, Cheetham RK, Fan JB, Grocock R, Humphray S, Kingsbury Z et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res 73, 7222–7231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leonard B, McCann JL, Starrett GJ, Kosyakovsky L, Luengas EM, Molan AM, Burns MB, McDougle RM, Parker PJ, Brown WL & Harris RS The PKC/NF-kappaB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res 75, 4538–4547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, Temiz NA, Vogel RI, Meijer-van Gelder ME, Sweep FC, Span PN, Foekens JA, Martens JW, Yee D & Harris RS The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv 2, e1601737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F & Mostov KE A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 12, 1035–1045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salsman J, Zimmerman N, Chen T, Domagala M & Frappier L Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4, e1000100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD & Krogan NJ Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salamango DJ & Johnson MC Characterizing the murine leukemia virus envelope glycoprotein membrane-spanning domain for its roles in interface alignment and fusogenicity. J Virol 89, 12492–12500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Noia J & Neuberger MS Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419, 43–48 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Albin JS, Hache G, Hultquist JF, Brown WL & Harris RS Long-term restriction by APOBEC3F selects human immunodeficiency virus type 1 variants with restored Vif function. J Virol 84, 10209–10219 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagannathan M, Nguyen T, Gallo D, Luthra N, Brown GW, Saridakis V & Frappier L A role for USP7 in DNA replication. Mol Cell Biol 34, 132–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ & Glaunsinger BA Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57, 349–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu N, Yoshiyama H & Takada K Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J Virol 70, 7260–7263 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni Z, Olsen JB, Emili A & Greenblatt JF Identification of mammalian protein complexes by lentiviral-based affinity purification and mass spectrometry. Methods Mol Biol 781, 31–45 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Liu G, Zhang J, Larsen B, Stark C, Breitkreutz A, Lin ZY, Breitkreutz BJ, Ding Y, Colwill K, Pasculescu A, Pawson T, Wrana JL, Nesvizhskii AI, Raught B, Tyers M & Gingras AC ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat Biotechnol 28, 1015–1017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL & Harris RS Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res 38, 4274–4284 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi K, Carpenter MA, Kurahashi K, Harris RS & Aihara H Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J Biol Chem 290, 28120–28130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaban NM, Shi K, Lauer KV, Carpenter MA, Richards CM, Salamango D, Wang J, Lopresti MW, Banerjee S, Levin-Klein R, Brown WL, Aihara H & Harris RS The antiviral and cancer genomic DNA deaminase APOBEC3H is regulated by an RNA-mediated dimerization mechanism. Mol Cell 69, 75–86 e79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cui XA, Zhang H & Palazzo AF p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol 10, e1001336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M & DePristo MA The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G & Durbin R The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L & Wilson RK VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Borozan I, Zapatka M, Frappier L & Ferretti V Analysis of Epstein-Barr virus genomes and expression profiles in gastric adenocarcinoma. J Virol 92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katoh K & Standley DM MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verhalen B, Starrett GJ, Harris RS & Jiang M Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J Virol 90, 6379–6386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ebrahimi D, Anwar F & Davenport MP APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J Virol 85, 9139–9146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.