

Abstract
Protein arginine methyltransferase 5 (PRMT5) is the main enzyme responsible for the symmetrical dimethylation of arginine residues on target proteins in both the cytoplasm and the nucleus. Though its activity has been associated with tumor progression in various cancers, the expression pattern of this oncoprotein has been scarcely studied in breast cancer. In the current work, we analyzed its expression in a large cohort of breast cancer patients, revealing higher nuclear PRMT5 levels in ERα-positive tumors and an association with prolonged disease free and overall survival. Interestingly, high PRMT5 nuclear expression was also associated with higher nuclear liver kinase B1 (LKB1), suggesting that a functional relationship may occur. Consistently, several approaches provided evidence that PRMT5 and LKB1 interact directly in the cytoplasm of mammary epithelial cells. Moreover, although PRMT5 is not able to methylate LKB1, we found that PRMT5 is a bona fade substrate for LKB1. We identified T132, 139 and 144 residues, located in the TIM-Barrel domain of PRMT5, as target sites for LKB1 phosphorylation. The point mutation of PRMT5 T139/144 to A139/144 drastically decreased its methyltransferase activity, due probably to the loss of its interaction with regulatory proteins such as MEP50, pICln and RiOK1. In addition, modulation of LKB1 expression modified PRMT5 activity, highlighting a new regulatory mechanism that could have clinical implications.
Keywords: Arginine methylation, PRMT5, LKB1, Threonine phosphorylation, Breast cancer
Introduction
Arginine methylation is a frequent post-translational modification that affects protein behavior. To date, nine protein arginine methyltransferases (PRMTs) have been identified in mammals (PRMT1–9). They are divided into three subtypes according to their enzymatic activities. Type I PRMTs catalyze ϖ−NGmonomethylarginine (MMA) and ϖ−NG, NG asymmetric dimethylarginine (ADMA), type II catalyze MMA and ϖ−NG, NG symmetric dimethylarginine (SDMA), while types III can only generate MMA (1). PRMT5 is the major type II methyltransferase responsible for depositing the SDMA mark within proteins (2). PRMT5 was first identified as the Jak-binding protein 1 (3), and shown to methylate many proteins including histones (4). R2 of Histone H3 methylation (H3R2me2s) has been correlated with transcriptional activation (5;6), while H4R3me2s and H3R8me2s repress transcription (7). In addition, PRMT5 regulates transcription through methylation of transcription factors such as NF-kB (8), p53 (9) and E2F-1 (10). PRMT5 can also methylate cytoplasmic proteins such as Sm proteins involved in the splicing of mRNA (11).
Of note, dysregulations in PRMT5 expression and protein mutations have been described in a variety of cancers (4;12). In epithelial ovarian cancer and non-small cell lung cancer, elevated PRMT5 is strongly correlated with poor survival rates (13;14). Moreover, PRMT5 overexpression causes an increase in proliferation and anchorage-independent growth (7;15) and triggers the formation of tumors in nude mice (15). PRMT5 knockdown reduces cellular proliferation in breast and lung cancer cells (7;15;16). Furthermore, in lung cancer, PRMT5 mediates a transcriptional increase in genes regulating cell adhesion, morphology and invasion, all essential for the TGFβ response and cancer metastasis (5). At the level of breast cancer, the area of expertise of our laboratory, few studies have been conducted on the prognostic value of PRMT5, through its expression has been correlated with poor patient outcome (12;17). Its expression in breast cancer stem cells (BCSCs) is crucial for maintaining stemness (18), and we speculate that a better understanding of its mechanisms of regulation either downstream (substrate identification) or upstream (regulators/interactors) may improve its therapeutic targeting.
At the molecular level, a majority of PRMT5 complexes contain MEP50, a 7-bladed WD40 repeat (tryptophan, aspartic acid) β-propeller protein. MEP50 binds PRMT5 directly and enhances its enzymatic activity, increasing its affinity for protein substrates (19;20). The interaction of PRMT5 with various partners, including pICln and the kinase RiOK1, is important to regulate its catalytic activity. These partners bind PRMT5 in a mutually exclusive manner to control substrate specificity (21). Menin, a protein found in MLL complexes, which targets histone H3K4 trimethylation, also binds to PRMT5 and targets a specific promoter of H4R3me2s (22). Post-translational modifications (PTMs) on PRMT5 or MEP50 can also modulate the methyltransferase activity. Jak2 phosphorylates PRMT5 on Tyr304 which impairs its ability to methylate histones H2A or H4 on Arg3 (23;24). Conversely, phosphorylation of MEP50 on Thr5 increases the methyltransferase activity of PRMT5-MEP50 toward histone H4 (25). In BCSCs, PRMT5 was reported to interact with FOXP1, and authors speculated that the interaction could be targeted using small inhibitor molecules (18). This was recently applied in mantle cell lymphoma, in which a selective PRMT5 inhibitor was generated offering a promising therapeutic strategy (26).
Here, based on the analysis of a large cohort of patients with breast tumors, we identified a new PRMT5 interactor, the liver kinase B1 protein (LKB1), a master kinase that acts as a key regulator of cell polarity, energy metabolism and mTOR signaling (27–29). Our results revealed that LKB1 is able to phosphorylate PRMT5 thereby modulating its enzymatic activity. We also investigated the molecular mechanisms involved, highlighting a novel way of regulating PRMT5 activity in breast cancer.
Materials and Methods
Cell culture and transfections
All of the cell lines were maintained at 37°C in the appropriate medium supplemented with 10% fetal bovine serum and 2% Penicillin/Streptomycin. For knockdown experiments, PRMT5-, LKB1- and MEP50-specific siRNAs or the scramble siRNA (Eurogentec) (50 nM) were transfected into MCF-7 cells using the lipofectamine 2000 reagent (Invitrogen). The targeted sequences are given in the supplemental data Table 1. 72 hr after transfection, proteins were analyzed. For overexpression experiments, pcDNA3.1 V5-tagged vectors and pSG5 Flag-tagged vectors were transfected using the XtremeGENE reagent (Roche). Forty eight hours after transfection, cells were analyzed.
Antibodies
All antibodies are listed in the supporting information Table 2.
Glutathione transferase pull-down assay
LKB1 expression plasmid was transcribed and translated in vitro using T7-coupled reticulocyte lysate in the presence of [35S] methionine. The different domains of PRMT5 (D1 to D4, and D1a and D1b, primers are listed in the supporting information Table 3) were cloned into the pGex4T1 vector. The experiment was then performed as previously described (30).
Immunoprecipitation and western blotting
Cells were lysed using RIPA buffer (50 mM Tris HCl, pH 8, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.25% deoxycholate) supplemented with protease inhibitor tablets (Roche Molecular Biochemicals) and phosphatase inhibitors (1 mM NaF, 1 mM Na3VO4 and 1 mM b-glycerophosphate). Protein extracts were incubated with primary antibodies overnight at 4°C on a shaker. Protein G or A-Agarose beads were added and the mixture was incubated 2 hr at 4°C. The immunoprecipitated proteins were separated on SDS-PAGE and visualized by ECL.
Proximity Ligation Assay (PLA)
This technology enables investigators to visualize protein/protein interactions in situ (31). Briefly, cells were seeded on coverslips and fixed with cold methanol. After saturation, the couples of primary antibodies were incubated for 1 hr at 37°C. The PLA probes consisting of secondary antibodies conjugated with complementary oligonucleotides, were incubated for 1 hr at 37°C. After ligation of nucleotides, the amplification lasted 100 min at 37°C. Samples were then analyzed under fluorescence microscopy. For tumors analysis, we used a bright field kit as previously described (30).
Human breast cancer sample collection
The tumors from 433 consecutive patients with invasive breast cancer, the clinical and biological data of whom were available from the regularly updated institutional database, were analyzed. Written informed consent was obtained from each patient. The study protocol was approved by the institutional ethics committee. Patients’ characteristics are presented in the supporting information Table 4.
Immunohistochemistry staining
Formalin-fixed paraffin embedded tumor tissues were used for analysis. The pathologist selected representative areas from breast invasive carcinomas. Triplicates from each tumor were inserted into TMA blocks, each containing 40 tumors. After deparaffinization and rehydration, tissue sections were boiled in 10 mM citrate buffer pH 8.0 at 95°C for 40 min. The slides were then incubated in 5% hydrogen peroxide in sterile water to block the activity of endogenous peroxidases, then at 37°C for 1 hr with the anti-LKB1 or the anti-PRMT5 antibody. The slides were subsequently incubated with a biotinylated secondary antibody bound to a streptavidin peroxidase conjugate (Envision Flex kit Ref: K800021–2, Dako). Bound antibodies were visualized by adding the substrate 3,3-diamino benzidine. Sections were counterstained with hematoxylin.
Blinded to the clinical data, PRMT5 expression was evaluated by 2 observers who assessed both the percentage and the intensity of nuclear and cytoplasmic staining separately. For scoring purposes, the intensity of staining in malignant cells was divided into 4 groups of levels (0: no staining, 1: weak staining, 2: moderate staining and 3: strong staining) and the percentage of stained cells were reported separately. Then, both intensity and percentage scores were multiplied to obtain a single H score. The most significant cutoff in terms of Disease-Free Survival (DFS) and Overall Survival (OS) was defined (H score of 70). The entire cohort of patients was divided into high nuclear PRMT5-expressing patients (> 70) and low nuclear PRMT5-expressing patients (≤ 70). During tissue preparation and staining, only 390 patients were evaluable for nuclear PRMT5 expression. Accordingly, 141 patients (36?2%) had low nuclear PRMT5 expression and 249 patients (63.8%) had high expression.
Image acquisition and analysis
The hybridized fluorescent slides were viewed under a Nikon Eclipse Ni microscope. Images were acquired under identical conditions at 60X magnification. Image acquisition was performed by imaging DAPI staining at a fixed Z Position while a Z stack of +/− 5μm at 1 μm intervals was carried out. The final image was stacked to a single level before further quantification. On each sample, at least one hundred cells were counted. Analyses and quantifications of these samples were performed using the Image J software (free access). PLA dots were quantified on 8-bit images using the ‘Analyse Particles’ command, while cell numbers were obtained using the cell counter plugin.
IHC images were acquired using a Nikon Eclipse Ni microscope at 40X magnification and PLA dots were quantified as described above.
In vitro methylation assay
Immunoprecipitated V5-PRMT5 or endogenous PRMT5 were incubated with different GST-tagged LKB1 fragments or GST-SmD3 in the presence of S-adenosyl-L [methyl-3H] methionine ([3H] SAM 85 Ci/mmol from a 10.4 μM stock solution in dilute HCl/ethanol 9/1 [pH 2.0 – 2.5]; Perkin Elmer) for 90 min at 30°C. Methylation reactions were quenched by adding 10 μL of 2X Laemmli sample buffer, heated at 95°C for 5 min, and separated by SDS-PAGE. Gels were then soaked in Amplify reagent (GE Healthcare) and visualized by autoradiography.
In vitro phosphorylation assay
The assays were performed by incubating 50 ng of LKB1/STRADα/MO25α active complex (Merck) in the presence of adenosine 5’-triphosphate, [ү−32PATP] (3000 Ci/mmol) (Perkin Elmer) and 2.5 μg of GST-PRMT5 fragments for 30 min at 30°C. Phosphorylation reactions were quenched by Laemmli sample buffer, separated by SDS-PAGE and visualized by autoradiography.
Statistical analysis
Descriptive analysis.
Correlations between PRMT5 expression and clinical and biomarker parameters (including LKB1) were conducted using Fisher’s exact test.
Survival analysis.
OS defined as time from diagnosis to death or date of last follow-up and DFS defined as time from diagnosis to death or relapse or date of last follow-up (for censored patients) were studied. Survival distributions were estimated using the Kaplan-Meier method and compared between groups according to the level of expression of the markers using the Log-Rank test. Hazard ratios for relapse or death were estimated using the cox regression model. Statistical analyses were carried out using the SPSS v20.0 software (IBN, USA). A statistically significant interaction was considered if the alpha error was less than 5%.
Results
Patient characteristics
Among the TMA of 440 patients, only 390 cases were assessable for PRMT5 expression (with countable percentage of malignant cells and intensity of expression). Median age was 58.1 years (range from 25.7 to 91 years). Three hundred and twenty patients were older than 50 years (73.9%) and 71.5% were post-menopausal. Tumors were larger than 2 cm in 181 patients (41.8%) and had positive axillary lymph nodes (LNs) in 249 patients (57.5%). Eighty two patients (18.9%) had SBR grade I tumors, 207 (47.8%) grade II and 144 (33.3%) grade III. Estrogen receptor (ERα) was positive in 377 patients (87.1%), and HER2 was over-expressed in 31 patients (7.2%).
Prognostic value of PRMT5 in breast cancer
Regarding the immunohistochemical (IHC) analysis of PRMT5, all of the tumors displayed diffuse cytoplasmic PRMT5 expression. However, only 249 tumors (63.8%) had a high nuclear localization (Fig. 1a, compare panels a and b for cytoplasmic staining and panels c and d for cytoplasmic and nuclear staining), while 141 (36,6%) had low or no nuclear expression. Higher nuclear PRMT5 expression was associated with older patients (p = 0.05), smaller tumors (p = 0.002) low SBR grade (p < 0.001), and with higher ERα (p < 0.001) and PR positivity (p < 0.001) (Table 1). Regarding the correlations with the PI3K/Akt/mTOR pathway biomarkers studied, we found that higher nuclear PRMT5 levels were correlated with high pAkt expression both in the nucleus and the cytoplasm, as well as with high nuclear LKB1 levels. Table 2 shows the correlations between nuclear PRMT5 expression and the other studied biomarkers by IHC.
Figure 1: The expression of PRMT5 in breast tumors.
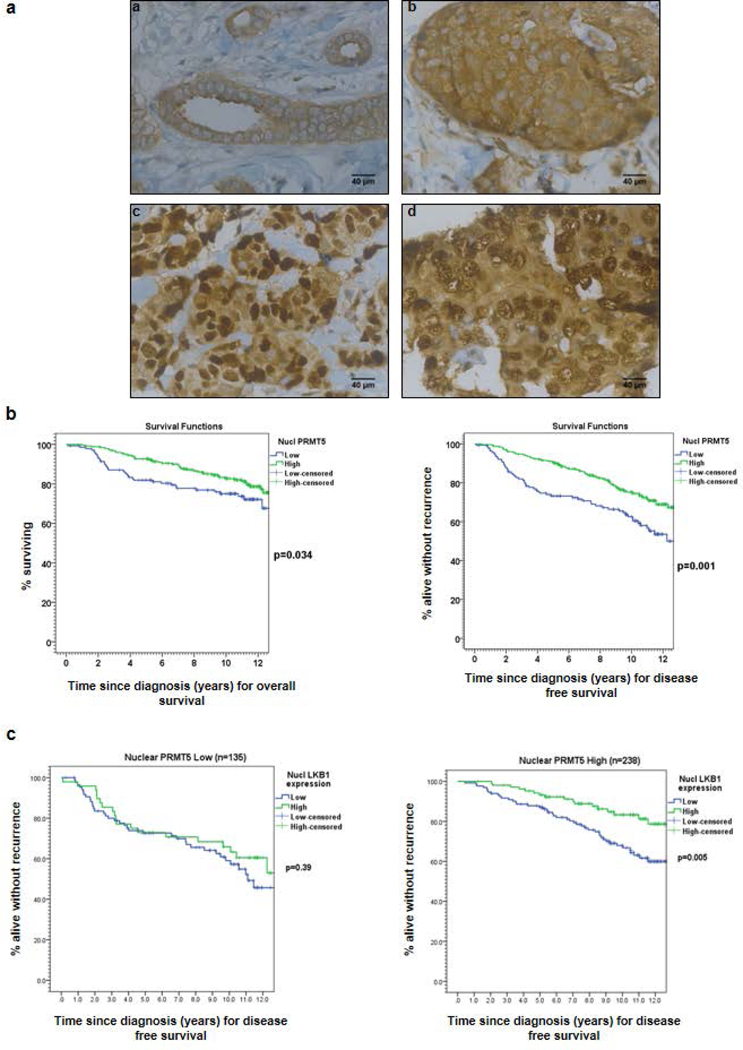
(a) PRMT5 expression was analyzed by IHC on formalin-fixed human tumors. Representative images of different IHC staining profiles are shown (panels a and b: cytoplasmic staining, panels c and d: cytoplasmic and nuclear staining) (Obj: X40). (b) Kaplan-Meier estimates of overall survival (OS) (left) and disease-free survival (DFS) (right) in patients with low (blue) versus high (green) nuclear PRMT5 expression. (c) Kaplan-Meier estimates of DFS in patients with low (blue) versus high (green) nuclear LKB1 expression in 2 groups of patients according to PRMT5 expression.
Table 1:
Correlation between Nuclear PRMT5 expression and the clinico-pathologic factors of breast cancer
| Variable | Nucl PRMT5 low | Nucl PRMT5 high | P* | |
|---|---|---|---|---|
| No. (%) | No. (%) | |||
| 141 (36.2) | 249 (63.8) | |||
| Age (Yr) | -Mean (± SD) | 57.1 (±3.6) | 59.7 (±11.8) | 0.05† |
| Age groups | - ≤50y | 45 (31.9) | 57 (22.9) | 0.05 |
| - >50y | 96 (68.1) | 192 (77.1) | ||
| BMI | - ≤25 | 81 (58.7) | 153 (64.3) | 0.281 |
| - >25 | 57 (41.3) | 85 (35.7) | ||
| T. size | - ≤2cm | 65 (46.1) | 155 (62.2) | 0.002 |
| - >2cm | 76 (53.9) | 94 (37.8) | ||
| LN invasion | -No | 64 (45.4) | 99 (39.8) | 0.28 |
| -Yes | 77 (54.6) | 150 (60.2) | ||
| SBR grade | -Gr 1 | 19 (13.5) | 53 (21.3) | <0.001 |
| -Gr 2 | 46 (35.6) | 136 (54.6) | ||
| -Gr 3 | 76 (53.9) | 60 (24.1) | ||
| ERα status | -Negative | 40 (28.4) | 10 (4.0) | <0.001 |
| -Positive | 101 (71.6) | 239 (96.0) | ||
| PR status | -Negative | 53 (37.6) | 46 (18.5) | <0.001 |
| -Positive | 88 (62.4) | 203 (81.5) | ||
| Her 2 status | -Negative | 71 (89.9) | 132 (93.0) | 0.42 |
| -Over-expressed | 8 (10.1) | 10 (7.0) | ||
Correlation by Pearson’s Chi square test unless otherwise specified
Difference between means by Student’s T test
Table 2:
Correlation between Nuclear PRMT5 expression and the different biomarkers studied by IHC
| Variable | Nucl PRMT5 low |
Nucl PRMT5 high |
P* | |
|---|---|---|---|---|
| No. (%) | No. (%) | |||
| 141 (36.2) | 249 (63.8) | |||
| Nucl. pAKT | -Low (≤50) | 87 (64.9) | 95 (39.7) | <0.001 |
| -High (>50) | 47 (35.1) | 144 (60.3) | ||
| Cytopl. pAKT | --Low (≤100) | 120 (88.9) | 189 (78.8) | 0.013 |
| -High (>100) | 15 (11.1) | 51 (21.2) | ||
| Nucl. LKB1 | -Low (=0) | 77 (57.0) | 110 (46.2) | 0.045 |
| -High (>0) | 58 (43.0) | 128 (53.8) | ||
| Cytopl. LKB1 | -Low (≤140) | 79 (58.5) | 150 (62.5) | 0.448 |
| -High (>140) | 56 (41.5) | 90 (37.5) | ||
| P4E-BP1 | -Low (≤50) | 66 (48.9) | 130 (53.7) | 0.368 |
| -High(>50) | 69 (51.1) | 112 (46.3) | ||
| P85-pS6K | -Low (≤90) | 94 (66.7) | 174 (71.9) | 0.281 |
| -High (>90) | 47 (33.3) | 68 (28.1) | ||
| PS6-RP | -Low (≤100) | 105 (77.2) | 178 (73.6) | 0.432 |
| -High (>100) | 31 (22.8) | 64 (26.4) | ||
| Menin | -Low (≤100) | 65 (50.4) | 84 (38.0) | 0.024 |
| -High (>100) | 64 (49.6) | 137 (62.0) | ||
| Cytopl IGF1-R | -Low (≤100) | 81 (60.4) | 109 (45.0) | 0.004 |
| -High (>100) | 53 (39.6) | 133 (55.0) | ||
Correlation by Pearson’s Chi square test
Regarding patient outcome based on univariate analyses, high nuclear PRMT5 levels were significantly associated with longer OS (HR = 0.63; 95% CI: 0.40–0.97, p = 0.034) and longer DFS ((HR = 0.53; 95% CI: 0.37–0.77, p = 0.001; Fig. 1b). In the multivariate analysis, and after adjustment for age (older or younger than 50), tumor size (larger or smaller than 20 mm), axillary LN invasion, SBR grade (grade 3 versus 1 and 2), ERα and PR status, PRMT5 expression remained a statistically significant predictor of breast cancer recurrence (HR = 0.64; 95% CI: 0.43–0.94, p = 0.023). It is worth mentioning that only tumor size and SBR grade were retained as independent prognostic factors in the multivariate model.
Given the correlation between high nuclear LKB1 levels and a longer survival in a previous cohort (30), we evaluated its prognostic value in this cohort. We consistently observed a significantly longer DFS (p = 0.006) in patients with higher nuclear LKB1 profiles (Supporting information Fig. S1). Interestingly, positive LKB1 prognosis was restricted to tumors with higher levels of nuclear PRMT5 (p = 0.005, Figure 1c, right panel compare with left panel), suggesting a functional link between both proteins.
LKB1 is a new partner for PRMT5
To evaluate whether these proteins interacted directly, we initially conducted a GST pull-down experiment. We found that radioactive LKB1 specifically interacts with PRMT5 and its partner MEP50, but not with the GST (Fig. 2a). By separating the PRMT5 protein into 4 fragments (Fig. 2b), we further identified the D3 fragment, containing the Middle-Rossman-fold domain, as the main site of interaction, although PRMT5 could also bind to a lesser extent to the other domains (Fig. 2c).
Figure 2: PRMT5 interacts with LKB1 in vitro.
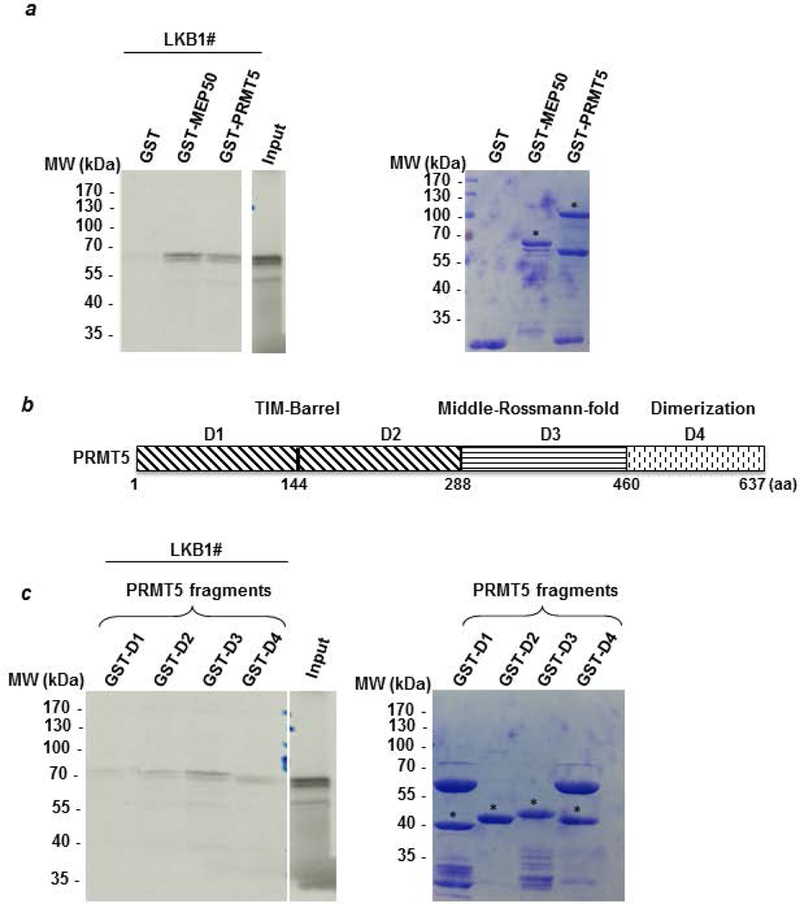
(a) A radioactive GST pull-down assay was performed by incubating labeled in vitro 35S-labeled LKB1 (LKB1#) with GST, GST-MEP50 and GST-PRMT5. The corresponding Coomassie-stained gel is shown in the right panel. * indicates the full length fusion proteins. (b) PRMT5 was divided into 4 fragments (D1 to D4). The D1 and D2 fragments cover the TIM-Barrel domain. The D3 fragment corresponds to the Middle-Rossman-fold domain and the D4 fragment contains the dimerization domain. (c) Radioactive LKB1 (LKB1#) was incubated with the different domains of PRMT5 fused to GST and the bound proteins were visualized by autoradiography. The corresponding Coomassie-stained gel is shown in the right panel. # indicates the full length fusion proteins.
In cellulo, we investigated the interaction of LKB1 with PRMT5 and its coregulatory MEP50 in several breast cancer cell lines. We first investigated their expression and Figure 3a shows that the levels of the 3 proteins were independent of the presence of ERα and their expression profiles were quite similar. For subsequent experiments, we focused on MCF-7 cells, since they naturally present high levels of the 3 proteins. Based on immunoprecipitation (IP) assays, we found that LKB1 interacts specifically with endogenous PRMT5 (Fig. 3b). Moreover, we examined to what extent and in which compartment these interactions occurred in the cells. Using different antibody pairs, we studied the interaction of LKB1 with PRMT5 and MEP50 using the proximity ligation assay (PLA). The images obtained revealed red dots in the cytoplasm of MCF-7 cells, illustrating the interaction of LKB1 with PRMT5 (Fig. 3c, panel a) and MEP50 (Fig. 3c, panel d), which were further quantified (Fig. 3d). The signals strongly decreased in MCF-7 cells following the downregulation of PRMT5 (Fig. 3c, panel b), MEP50 (Fig. 3c, panel e) and LKB1 (Fig. 3c, panels c and f) by siRNA transfection (Figs. 3d and 3e). These cytoplasmic interactions were confirmed in other breast cancer cell lines, supporting our theory of a functional link between these proteins (Supporting information Fig. S2).
Figure 3: PRMT5 interacts with LKB1 in cellulo.

(a) Expression of PRMT5, MEP50 and LKB1 was evaluated in a wide range of human breast tumor cell lines by Western blot using the corresponding antibodies. GAPDH expression was used as a loading control. (b) The PRMT5-LKB1 interaction was assessed by co-immunoprecipitation in MCF-7 cell extracts using an anti-PRMT5 antibody. A rabbit irrelevant antibody was used as a negative control. The presence of PRMT5 and LKB1 was evaluated by Western blot analysis. (c) Detection of LKB1 interaction with PRMT5 and MEP50 was performed by Proximity Ligation Assay (PLA). MCF-7 cells were transfected with Scramble siRNA or siRNA targeting LKB1, PRMT5 or MEP50 for 48 hr. After fixation, PLA experiments were performed to evaluate the interactions between LKB1/PRMT5 and LKB1/MEP50 using LKB1-, PRMT5- and MEP50-specific antibodies. The detected dimers are represented by red dots. The nuclei were counterstained with mounting medium containing DAPI (blue) (Obj: X60). (d) Quantification of the number of dots per cell was performed by computer-assisted analysis as reported in the Materials and Methods section. The mean +/− s.e.m. of one experiment representative of three experiments is shown. The P-value was determined using the Student’s t-test. *** indicates a P < 0.001. (e) The efficacy of protein inhibition was verified by Western blot analysis using the corresponding antibodies.
PRMT5 is phosphorylated by LKB1
To unravel the mechanism of interaction between PRMT5 and LKB1, we performed an in vitro methylation experiment and found that LKB1 was not methylated by PRMT5 (Supporting information Fig. S3), and thus did not serve as a substrate for the latter. We then investigated whether LKB1 could phosphorylate PRMT5. For in vitro phosphorylation experiments, purified GST-PRMT5 fragments D1 to D4 (described in Fig. 2b) were incubated with the active LKB1 complex. The D1 fragment, the only one phosphorylated by LKB1 (Fig. 4a), was then further divided into two parts (D1a and D1b, Fig. 4b), and residues on the D1b fragment were identified as phosphorylation targets. Next, since LKB1 is a Serine/Threonine kinase, we used antibodies recognizing pan phospho-Serine or -Threonine, and found that LKB1 phosphorylates PRMT5 mainly on Thr residues (Fig. 4c). Furthermore, the point-mutations of T132, T139 and T144 to A132, A139 and A144, respectively, strongly decreased PRMT5 phosphorylation, with the T144A mutation having the strongest effect (Fig. 4d). Based on the 3D structure of PRMT5, we found that the three threonine residues are close to the dimer interface (Fig. 4e), and wondered whether they are involved in its dimerization. After transfection of V5-tagged wild type or mutant (T144A, T132A/144A or T139A/144A) PRMT5 into MCF-7 cells, we immunoprecipitated V5-PRMT5 and observed the presence of endogenous PRMT5. Furthermore, neither its dimerizing capacity nor its cellular localization were affected by these point-mutations (Fig. 4f and supporting information Fig. S4). Next, to assess the effect of the mutations on its enzymatic activity, we immunoprecipitated V5-tagged wild type or mutant (T132A, T139A, T144A, T132A/144A and T139A/144A) PRMT5 from HeLa cells, and incubated the immunoprecipitates with [3H] S-adenosylmethionine and recombinant GST-SmD3, a known PRMT5 substrate. Although individual mutations had no effect on the enzymatic activity of PRMT5, the T139A/144A mutant resulted in a complete loss of methyltransferase activity (Fig. 5a). We obtained the same result using histone H4 as a substrate (Supporting information Fig. S5). To characterize this lack of activity, we conducted co-immunoprecipitation experiments and determined that the double mutant T139A/144A impaired the binding of PRMT5 to its coactivator MEP50 (Fig. 5b). In addition, the PRMT5 mutant lost its ability to bind to pICln and RiOK1, two partners involved in the recognition of substrates by PRMT5. Interestingly, the double PRMT5 mutant was further unable to bind to LKB1. In cellulo transfected V5-PRMT5 was recognized by the pan phospho-Thr antibody whereas the signal was strongly decreased in the presence of the mutant T139A/144A PRMT5 (Fig. 5c), highlighting that these residues are phosphorylated in a cellular context.
Figure 4: LKB1 phosphorylates PRMT5 on threonine residues.
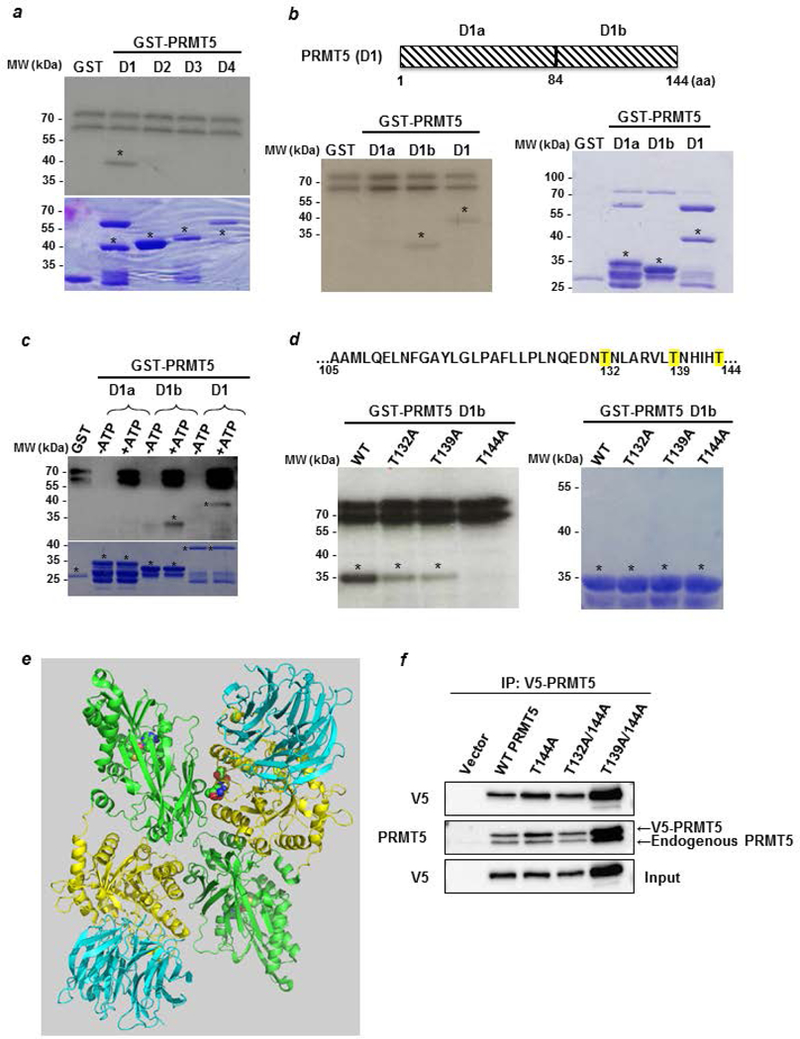
(a) An in vitro phosphorylation experiment was performed by incubating active LKB1 complex with [32Pγ] ATP and D1 to D4 PRMT5 fragments fused to GST. The phosphorylated proteins were visualized by autoradiography (upper panel). The corresponding Coomassie-stained gel is shown in the lower panel. * indicates the full length fusion proteins. (b) The PRMT5 D1 domain was divided into two parts, D1a and D1b, which were used to perform an in vitro kinase assay as described in a (left panel). The corresponding Coomassie-stained gel is shown in the right panel. * indicates the full length fusion proteins. (c) Cold in vitro phosphorylation experiments were performed by incubating active LKB1 complex with D1a, D1b and D1 PRMT5 fragments fused to GST in the absence or the presence of ATP. The phosphorylated proteins were verified by Western blot analysis using a pan phospho-Thr antibody (upper panel). The corresponding Coomassie-stained gel is shown in the lower panel. * indicates the full length fusion proteins. (d) Sequence of the PRMT5 D1b containing the potential sites for LKB1 phosphorylation. T132, 139 and 144, are highlighted in yellow. GST-D1b wild type or T/A mutants were used as substrates for radioactive LKB1 phosphorylation (left panel). The corresponding Coomassie-stained gel is shown in the right panel. * indicates the full length fusion proteins. (e) Structure of human PRMT5/MEP50 heterodimer. The N-terminal of each monomer, containing the TIM-Barrel domain, is represented in yellow and its C-terminal domain in green. The 3 mutated threonines are in the spacefill at the dimer interface. The protein represented in blue is MEP50. (Based on PDB 5fa5 from (39) and represented with the PyMOL molecular graphics system, version 2.0, Schrödinger, LLC). (f) MCF-7 cells were transfected with pcDNA3.1 V5-PRMT5 WT and mutants for 48 hr. Cell lysates analyzed for V5-PRMT5 expression with an anti-V5 antibody. Then, V5-PRMT5 was immunoprecipitated using an anti-V5 antibody and revealed by Western blot with the anti-V5 and anti-PRMT5 antibodies.
Figure 5: Phosphorylation of PRMT5 modulates its methyltransferase activity.

(a) HeLa cells were transfected with the different V5-PRMT5 mutants for 48 hr. The catalytic activity of PRMT5 was assessed by immunoprecipitation using an anti-V5 antibody, followed by a radioactive in vitro methylation assay. SmD3 was incubated with the immunoprecipitated V5-PRMT5 proteins (WT and mutants) in the presence of [methyl-3H] SAM. Reaction products were analyzed by SDS-PAGE followed by fluorography (left panel). 1/20th of the IP was analyzed for V5-PRMT5 expression and protein expression was verified by Western blot analysis (right panel). (b) Interactions of both WT and T139/144A PRMT5 with known partners were assessed by immunoprecipitation in MCF-7 cell extracts overexpressing the tagged proteins, using an anti-V5 antibody. The bound proteins were verified by Western blot using the corresponding antibodies (left panel). The IP control was analyzed for V5-PRMT5 expression and protein expression was verified by Western blot (right panel). (c) MCF-7 cells were transfected with a plasmid encoding V5-PRMT5 wild type or the mutant T139/144A PRMT5. Lysates were immunoprecipitated with pan phospho-Threonine antibody and immunoblotted with V5 antibody. PRMT5 expression was evaluated in the inputs. (d) Model for PRMT5 regulation by LKB1. In normal conditions, PRMT5 interacts with MEP50, RiOK1 and pICln in order to methylate nucleolin, histones and Sm proteins. T139 and T144 are important residues for such interactions. Following point-mutations of T139 and T144 into A139 and A144, respectively, PRMT5 no longer binds to its partners and loses its capacity to methylate substrates. M represents arginine methylation. In the presence of LKB1, PRMT5 is phosphorylated on T139 and T144 residues, thereby leading to a decrease in its catalytic activity, due to a loss of its interaction with its partners.
Having shown that LKB1 phosphorylates T139 and T144 residues, we overexpressed wild type LKB1 in MCF-7 cells. This resulted in a decreased PRMT5 activity, which remained unaffected in the presence of the catalytically inactive K78A LKB1 mutant (Supporting Information Fig. S6a). Conversely, the knock down of LKB1 in MCF-7 cells slightly increased its activity (Supporting information Fig. S6b and c).
Discussion
The expression of arginine methyltransferases is associated with various cancers, suggesting that they could constitute new therapeutic targets. However, in the case of breast cancer few studies were available on PRMTs and on their correlation with patient survival. In this study, using a large cohort of breast cancer patients, we analyzed the expression of PRMT5 alongside patient outcome and various prognostic markers, and identified a novel partner of PRMT5. Indeed, the Ser/Thr liver kinase B1 LKB1 was able to modulate its methyltransferase activity via phosphorylation.
We confirmed the dual localization of PRMT5 in the cytoplasm as well as in the nucleus of tumor cells (Fig. 1a), corroborating data obtained in testicular, breast and prostate cancers (32–34). Interestingly, it has been shown that cytoplasmic PRMT5 is essential for the growth of prostate cancer cells, whereas nuclear PRMT5 inhibits prostate cancer cell growth (35). Moreover, PRMT5 localizes in the nucleus of benign prostate epithelial cells and in the cytoplasm of cancerous prostate tissues (32). Likewise, we found that high nuclear PRMT5 was associated with better patient outcome in breast cancer. However, some recent studies showed that high PRMT5 expression is associated with shorter survival (34;36). The different subcellular localizations of PRMT5 might be responsible for these conflicting observations. Indeed, the study by Wu et al. investigated PRMT5 mRNA and not the protein (34), while that of Yang and colleagues did not take into account the nuclear localization of PRMT5 for their statistical analyses (36). Concerning the correlation with other markers analyzed by the team, we found a positive correlation between PRMT5 and nuclear LKB1 but not with cytoplasmic LKB1. Interestingly, as reported previously by our team (30), nuclear LKB1 was associated with longer patient survival. In this study this impact on the survival occurs only in tumors with high levels of nuclear PRMT5, suggesting a functional relationship between the 2 proteins. We then decided to investigate the correlation between PRMT5 and LKB1 in a cellular context. However, we were unable to find a mammary cell line in which both proteins were expressed in the nucleus (data not shown). Moreover, when we studied their interaction, we found that it occurred only in the cytoplasm of mammary tumoral cells. We cannot exclude that they could interact in the nucleus, but the interaction could be transient and consequently too difficult to detect.
LKB1 interacts with PRMT5 in the D3 fragment containing the Middle-Rossman-fold domain, which binds the SAM (23), suggesting that its binding could modify the enzymatic activity of PRMT5. In addition, the T139A and T144A point-mutations completely abolished the interaction between LKB1 and PRMT5. One can hypothesize that the phosphorylation of these residues may stabilize LKB1/PRMT5 binding.
So far, we have identified 3 threonine residues phosphorylated by LKB1. However, we cannot exclude that other residues could be modified. Indeed, when we divided the D1 domain of PRMT5 into two fragments and performed the in vitro phosphorylation experiment, we noticed that the D1a fragment is also slightly phosphorylated by LKB1 (Fig. 4b). Moreover, LKB1 also binds MEP50 and it could also be a substrate for LKB1. Phosphorylation appears to be largely involved in PRMT5/MEP50 activity, as several kinases have been described to modulate this complex activity. Mutant Jak2 phosphorylates PRMT5 at the Y304 residue, impairing the ability of PRMT5 to methylate histones. Conversely, phosphorylation of MEP50 on the T5 residue increases the methyltransferase activity toward histone H4 (25). More recently, PRMT5 was described to be phosphorylated in its C-terminus by the Akt and glucocorticoid-inducible kinases on T634. This event triggers a 14–3-3/PDZ interaction switch important for the targeting of PRMT5 to the plasma membrane (37).
In our work, we identified two threonine residues crucial for the enzymatic activity of PRMT5. We can hypothesize that these residues are important for the methylation of substrates as their mutation to alanine inhibits PRMT5 binding to MEP50, its major regulator (4), as well as to pICln, which favors its enzymatic activity towards Sm proteins (38), and to RiOK1, inducing its activity towards a broad range of substrates such as nucleolin and histones (21) (Fig. 5b). Based on our results, we can propose a model in which, in the presence of LKB1, the two threonine residues are phosphorylated, triggering the dissociation of MEP50, pICln and RioK1, thus decreasing the enzymatic activity of PRMT5 (Fig. 5d). In the future, the production of a specific antibody recognizing P-T139/P-T144 could be used as a marker of the enzymatic activity of PRMT5.
In conclusion, we identified a new PRMT5 partner, regulating its enzymatic activity. We can speculate that in breast tumors, LKB1 may maintain PRMT5 to a low level of activity, thus impeding its pro-tumoral action. However, in view of our retrospective design and heterogeneous group of patients, a prospective validation study in a larger cohort using the same cutoffs will be required to confirm the clinical role of PRMT5 expression in breast cancer patients.
Supplementary Material
Novelty and Impact.
The arginine methyltransferase PRMT5 has been largely associated with tumor progression in cancers; however its expression in breast cancer was poorly documented. Here, we found that as in prostate cancer, PRMT5 nuclear localization may have protective effects. Indeed, its expression is associated with higher patient survival. In addition, we identified two threonine residues essential to its enzymatic activity. Interestingly, the tumor suppressor LKB1 phosphorylates PRMT5 on these residues decreasing its activity. These results highlight new opportunities to measure and target PRMT5 activity in breast cancer.
Acknowledgements
We thank C. Languilaire, F. Nasri, A. Colombe, L. Odeyer, Marie-Lise Depont and Ha Thuy Pham for technical support. We thank E. Fabrizzio for the pGEX-SmD3 vector, Jocelyn Coté for the anti-PRMT5 antibody, B. Manship for proofreading the manuscript and A. Dejaegere for the PRMT5 structure.
Funding
We thank the Lebanese University for the financial support of HL and the project, Fondation Arc Cancer, INCA and DGOS, Fondation Arc Cancer and NIH R37DK055274 for CP, Fondation Arc Cancer and DGOS for JJ. We also thank the Cedre program for financial support.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to disclose.
References
- (1).Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009. January 16;33(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem 2001. August 31;276(35):32971–6. [DOI] [PubMed] [Google Scholar]
- (3).Pollack BP, Kotenko SV, He W, Izotova LS, Barnoski BL, Pestka S. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J Biol Chem 1999. October 29;274(44):31531–42. [DOI] [PubMed] [Google Scholar]
- (4).Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci 2015. June;72(11):2041–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen H, Lorton B, Gupta V, Shechter D. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017. January 19;36(3):373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC, Sahu SK, Gunaratne J, Capasso P, Bassi C, Cecatiello V, De MA, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol 2012. January 8;19(2):136–44. [DOI] [PubMed] [Google Scholar]
- (7).Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004. November;24(21):9630–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, Ghosh G, Stark GR, Lu T. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci U S A 2013. August 13;110(33):13516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol 2008. December;10(12):1431–9. [DOI] [PubMed] [Google Scholar]
- (10).Zheng S, Moehlenbrink J, Lu YC, Zalmas LP, Sagum CA, Carr S, McGouran JF, Alexander L, Fedorov O, Munro S, Kessler B, Bedford MT, et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol Cell 2013. October 10;52(1):37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Meister G, Eggert C, Buhler D, Brahms H, Kambach C, Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol 2001. December 11;11(24):1990–4. [DOI] [PubMed] [Google Scholar]
- (12).Poulard C, Corbo L, Le RM. Protein arginine methylation/demethylation and cancer. Oncotarget 2016. October 11;7(41):67532–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bao X, Zhao S, Liu T, Liu Y, Liu Y, Yang X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J Histochem Cytochem 2013. March;61(3):206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013;8(12):e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wei TY, Juan CC, Hisa JY, Su LJ, Lee YC, Chou HY, Chen JM, Wu YC, Chiu SC, Hsu CP, Liu KL, Yu CT. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci 2012. September;103(9):1640–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Scoumanne A, Zhang J, Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res 2009. August;37(15):4965–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Morettin A, Baldwin RM, Cote J. Arginine methyltransferases as novel therapeutic targets for breast cancer. Mutagenesis 2015. March;30(2):177–89. [DOI] [PubMed] [Google Scholar]
- (18).Chiang K, Zielinska AE, Shaaban AM, Sanchez-Bailon MP, Jarrold J, Clarke TL, Zhang J, Francis A, Jones LJ, Smith S, Barbash O, Guccione E, et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep 2017. December 19;21(12):3498–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Friesen WJ, Wyce A, Paushkin S, Abel L, Rappsilber J, Mann M, Dreyfuss G. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem 2002. March 8;277(10):8243–7. [DOI] [PubMed] [Google Scholar]
- (20).Ho MC, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T, Carter LG, Onikubo T, Kumar PR, Chan MK, Brenowitz M, Cheng RH, et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 2013;8(2):e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Guderian G, Peter C, Wiesner J, Sickmann A, Schulze-Osthoff K, Fischer U, Grimmler M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J Biol Chem 2011. January 21;286(3):1976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gurung B, Hua X. Menin/PRMT5/hedgehog signaling: a potential target for the treatment of multiple endocrine neoplasia type 1 tumors. Epigenomics 2013;5(5):469–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, Russell M, Sauder JM, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A 2012. October 30;109(44):17960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, Deblasio A, Hatlen M, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011. February 15;19(2):283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB, Diehl JA. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010. October 19;18(4):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, West KA, Lingaraj T, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015. June;11(6):432–7. [DOI] [PubMed] [Google Scholar]
- (27).Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem 2006;75:137–63. [DOI] [PubMed] [Google Scholar]
- (28).Forcet C, Billaud M. Dialogue between LKB1 and AMPK: a hot topic at the cellular pole. Sci STKE 2007. September 18;2007(404):e51. [DOI] [PubMed] [Google Scholar]
- (29).Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004. July;6(1):91–9. [DOI] [PubMed] [Google Scholar]
- (30).Bouchekioua-Bouzaghou K, Poulard C, Rambaud J, Lavergne E, Hussein N, Billaud M, Bachelot T, Chabaud S, Mader S, Dayan G, Treilleux I, Corbo L, et al. LKB1 when associated with methylatedERalpha is a marker of bad prognosis in breast cancer. Int J Cancer 2014. September 15;135(6):1307–18. [DOI] [PubMed] [Google Scholar]
- (31).Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006. December;3(12):995–1000. [DOI] [PubMed] [Google Scholar]
- (32).Gu Z, Zhou L, Gao S, Wang Z. Nuclear transport signals control cellular localization and function of androgen receptor cofactor p44/WDR77. PLoS One 2011;6(7):e22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liang JJ, Wang Z, Chiriboga L, Greco MA, Shapiro E, Huang H, Yang XJ, Huang J, Peng Y, Melamed J, Garabedian MJ, Lee P. The expression and function of androgen receptor coactivator p44 and protein arginine methyltransferase 5 in the developing testis and testicular tumors. J Urol 2007. May;177(5):1918–22. [DOI] [PubMed] [Google Scholar]
- (34).Wu Y, Wang Z, Zhang J, Ling R. Elevated expression of protein arginine methyltransferase 5 predicts the poor prognosis of breast cancer. Tumour Biol 2017. April;39(4):1010428317695917. [DOI] [PubMed] [Google Scholar]
- (35).Gu Z, Li Y, Lee P, Liu T, Wan C, Wang Z. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One 2012;7(8):e44033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yang F, Wang J, Ren HY, Jin J, Wang AL, Sun LL, Diao KX, Wang EH, Mi XY. Proliferative role of TRAF4 in breast cancer by upregulating PRMT5 nuclear expression. Tumour Biol 2015. August;36(8):5901–11. [DOI] [PubMed] [Google Scholar]
- (37).Espejo AB, Gao G, Black K, Gayatri S, Veland N, Kim J, Chen T, Sudol M, Walker C, Bedford MT. PRMT5 C-terminal Phosphorylation Modulates a 14–3-3/PDZ Interaction Switch. J Biol Chem 2017. February 10;292(6):2255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pesiridis GS, Diamond E, Van Duyne GD. Role of pICLn in methylation of Sm proteins by PRMT5. J Biol Chem 2009. August 7;284(32):21347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mavrakis KJ, McDonald ER III, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016. March 11;351(6278):1208–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.