

Abstract
In the Mexican axolotl (Ambystoma mexicanum), sex is determined by a single Mendelian factor, yet its sex chromosomes do not exhibit morphological differentiation typical of many vertebrate taxa that possess a single sex-determining locus. As sex chromosomes are theorized to differentiate rapidly, species with undifferentiated sex chromosomes provide the opportunity to reconstruct early events in sex chromosome evolution. Whole genome sequencing of 48 salamanders, targeted chromosome sequencing and in situ hybridization were used to identify the homomorphic sex chromosome that carries an A. mexicanum sex-determining factor and sequences that are present only on the W chromosome. Altogether, these sequences cover ~300 kb of validated female-specific (W chromosome) sequence, representing ~1/100,000th of the 32 Gb genome. Notably, a recent duplication of ATRX, a gene associated with mammalian sex-determining pathways, is one of few functional (non-repetitive) genes identified among these W-specific sequences. This duplicated gene (ATRW) was used to develop highly predictive markers for diagnosing sex and represents a strong candidate for a recently-acquired sex determining locus (or sexually antagonistic gene) in A. mexicanum.
Introduction
In many species, sex is determined by the inheritance of highly differentiated (heteromorphic) sex chromosomes, which have evolved independently many times throughout the tree of life1–3. Often these chromosomes differ dramatically in morphology and gene content4–6. In mammals, males have a large, gene rich X-chromosome and a degraded, gene poor Y-chromosome, while females have two X chromosomes. In birds and many other eukaryotes, females are the heterogametic sex with a large Z and smaller W chromosome, while males are homozygous, carrying two Z chromosomes. Differentiated sex chromosomes are thought to arise through a relatively stereotypical process that begins when a sex-determining gene arises on a pair of homologous autosomes5,6. The acquisition of sexually antagonistic alleles, alleles that benefit one sex and are detrimental to the other, favors the fixation of mutational events that suppress recombination in the vicinity of the sex-determining locus7,8. Recombination suppression can lead to the accumulation of additional sexually antagonistic mutations and repetitive elements, and over time this results in the loss of nonessential parts of the Y or W chromosome, resulting in the formation of heteromorphic sex chromosomes9.
Unlike the majority of mammals and birds with stable sex-determining systems and heteromorphic sex chromosomes, amphibians have undergone numerous evolutionary transitions between XY and ZW-type mechanisms and may possess morphologically indistinguishable (homomorphic) sex chromosomes, like those of the axolotl10–13. Homomorphic sex chromosomes are not altogether rare among animals, with examples in fish14, birds15, reptiles16 and amphibians17. Among most amphibians that have been investigated, homomorphy is prevalent17–19. It has been suggested that a majority of salamanders have homomorphic sex chromosomes18,20, however, evidence for genetic sex determination in most species is largely based on the observation of 1:1 sex ratios from clutches without thorough demonstration of Mendelian inheritance.
Early developmental/genetic experiments revealed a ZW type sex-determining mechanism for A. mexicanum21–23. The first experiment to test for female heterogamety involved sex reversal through implantation of a testis preprimordium from a donor embryo to a host female embryo. The prospective ovary developed instead into a functional testis. This sex-reversed male was then crossed with a normal female24. It was expected that if the female were homozygous for sex (XX), the offspring would all be female. If the female were heterozygous for sex (ZW), however, the offspring would have an approximate female to male ratio of 3:1. Two matings with the sex-reversed animals produced a combined 26.1% males, consistent with the hypothesis that the male was indeed a sex-reversed female with ZW chromosomes21,24. Subsequent studies showed normal sex ratios from matings with the F1 males and most of the F1 females, but several of the F1 females produced spawns of all females, suggesting they carried the unusual WW genotype24.
Following these foundational studies, early genetic mapping studies used cold shock to inhibit meiosis II and assessed triploid phenotypes to estimate the frequencies of equatorial separation and map distances between recessive mutations and their linked centromeres25. Based on these analyses, the sex determining locus was predicted to occur near the end of an undefined chromosome25 and later estimated to be 59.1 cM distal to the centromere (essentially, freely recombining)23.
Karyotypic analyses later indicated that the smallest chromosomes were heteromorphic in Ambystoma species, suggesting that the smallest pair of chromosomes carried the Mendelian sex determining factor in A. mexicanum26 and in the A. jeffersonianum species complex27. However, more recent linkage mapping studies indicated that sex was determined by a locus on one of the larger linkage groups26,28, and chromosome sequencing studies have demonstrated that the smallest chromosomes do not carry the sex determining region29,30. Notably, extensive cytogenetic studies performed by Callan31, including the use of cold treatments to add constrictions to chromosomes and examination of lampbrush chromosomes from developing oocytes, revealed no features that could be associated with differentiated sex chromosomes. These analyses not only indicated that the sex chromosomes were apparently identical to one another, but also revealed that mitotic chromosomes 9, 10 and 11 were essentially indistinguishable from one another31.
More recently, meiotic mapping of polymorphisms within controlled crosses localized the sex-determining region to the tip of Ambystoma LG9 (previously designated LG5) distal to the marker E24C329. These crosses also revealed no difference in recombination frequencies between the sexes. However, these studies were somewhat limited by the fact that they did not sample large numbers of markers in close proximity to the sex locus or W-specific sequences29. Taken together, analyses of the Ambystoma sex determination suggest that the sex chromosomes are largely undifferentiated and that, presumably, the sex chromosomes arose recently within the tiger salamander species complex.
To identify sex-linked (W-specific) regions in these relatively undifferentiated sex chromosomes, we generated sequence reads for 48 individuals of known sex that were derived from a backcross (A. mexicanum/A. tigrinum X A. mexicanum). These reads were then aligned to an existing reference genome from a female axolotl30,32 (www.ambystoma.org). Analyses of read depth of coverage identified 152 putative W-linked sequences, including two genes, an ATRX paralog and an ortholog of MAP2K3. The W-linked ATRX paralog, ATRW, is estimated to have duplicated within the last 20 million years, providing an estimate of the possible origin of the sex-determining locus in the tiger salamander species complex. In addition, we anticipate that these sex-linked markers will be useful for identifying sex in juvenile axolotls within lab-reared populations, where sex is an important covariate for experimental studies, including studies of metamorphosis and regeneration28,33.
Results
Identification of the sex-bearing chromosomes by FISH
Previous studies have demonstrated that sex is linked to the marker E24C3, at a distance of ~5.9 cM distal to the terminal marker on LG929. Consistent with linkage analyses, E24C3 was detected near the tip of an average-sized chromosome (Fig. 1). A second BAC corresponding to a marker from the opposite end of LG9 (E12A6) localized to the opposite tip of the same chromosome, indicating that this chromosome corresponds precisely to LG9 (Fig. 1). Notably, the BAC carrying E12A6 also cross-hybridized with the centromere of all chromosomes, a feature that could potentially be useful in estimating distances of genes to their respective centromeres.
Figure 1.
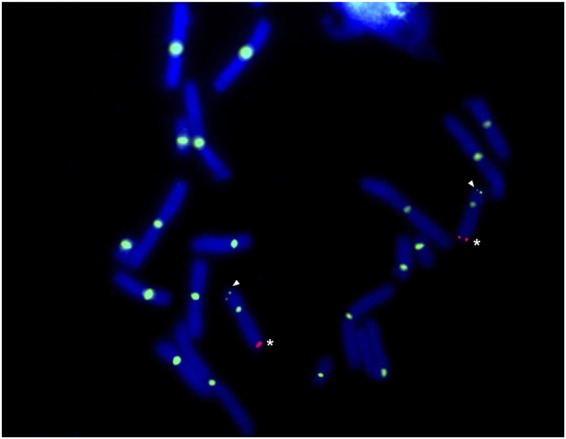
FISH of sex-linked BACs. FISH localizes two markers (E24C3 and E12A6) associated with the sex locus, ambysex, on a DAPI stained metaphase spread of chromosomes from an axolotl embryo of unknown sex. E24C3 is labeled with cy3 (red) and E12A6 is labeled with fluorescein (green). White asterisks show labeling of E24C3, and white arrows point to the labeling of E12A6 on the opposite end of the same chromosomes.
Laser capture, sequencing and assembly of the Z chromosome
In an attempt to increase the number of markers that could be associated with the sex chromosome, we performed laser-capture sequencing on a chromosome corresponding to LG9. This library was generated from a single dyad that was collected in a larger series of studies on laser capture microscopy of axolotl chromosomes34. The sex chromosome library contained a total of ~143 M reads between 40 and 100 bp after trimming and contained 995 reads that mapped to 23 distinct markers (transcripts) that had been previously placed on LG9 (Fig. 2). In total, this initial sequencing run accounted for 40% of the markers that are known to exist on the linkage group, which was considered strong evidence that this library sampled the sex chromosome. Given this support, an additional lane of sequencing was performed, yielding ~936 M additional reads (for a total of 1,078,893,614 reads). After trimming, ~542 M reads remained. Alignment to human and bacterial genomes revealed that 1.7% and 0.1% of trimmed reads aligned concordantly to the human genome and bacterial genomes, respectively. These were considered contaminants and were removed from subsequent analyses. Of the remaining reads, 68,844 aligned to 40 LG9 contigs representing 70% of the known markers on LG9 (Fig. 2). An error-corrected assembly of these data yielded a total of 1,232,131 scaffolds totaling 242.4 Mb with a scaffold N50 length of 295 bp, and contig N50 length of 126 bp. (Table 1: results from other chromosomes are shown for comparison purposes). We also used this library to identify a set of scaffolds from a recently published assembly of a male axolotl genome that could be assigned to the Z chromosome on the basis of sequence read depth of coverage. This analysis yielded 2531 scaffolds spanning a total of 1.02 Gb (Supplementary Table 1).
Figure 2.

Individual sex chromosome dyad alignment results on LG9. Read mapping was used to assess the specificity of the laser capture, amplified library of the sex chromosome dyad. (A) A partial metaphase spread of axolotl chromosomes stained with Giemsa on a membrane slide. The sex chromosome is circled in green. (B) The distribution of markers sampled from the sex chromosome (LG9) via targeted sequencing of individual chromosomes. LG9 is based on a previously published linkage map for the axolotl35. Individual gene markers are designated by labels to the right of their corresponding map position and their predicted position (in centiMorgans) is provided by numerical labels to the left. Dots represent markers with mapped reads from a single library. Red denotes the first sequencing attempt using the DNA-seq kit with 48 total barcoded samples on a single lane of an Illumina HiSeq flowcell. Blue denotes re-sequencing of the same chromosome library on a single lane.
Table 1.
Summary statistics for LG9, AM13 and AM14 chromosome assemblies.
| Assembly | Contig | Scaffold | |||||
|---|---|---|---|---|---|---|---|
| Length (Mb) | Number of Scaffolds | Number of Singletons | N50 Length Improvement | Proportion Scaffolded | N50 Length Improvement | Number | |
| >N50 | |||||||
| LG9 (R) | 189.7 | 1,054,224 | 760,174 | 118 | 0.352 | 256 | 285,628 |
| LG9 (EC) | 242.4 | 1,232,131 | 866,817 | 126 (6.8%) | 0.429 | 295(15.2%) | 335,062 |
| LG15/17 (R) | 302.5 | 604,617 | 243,354 | 231 | 0.598 | 705 | 136,682 |
| LG15/17 (EC) | 210.9 | 353,381 | 126,169 | 295 (28%) | 0.643 | 830 (18%) | 82,835 |
| LG14 (R) | 180.4 | 367,575 | 145,951 | 232 | 0.603 | 686 | 83,979 |
| LG14 (EC) | 143.0 | 258,214 | 93,931 | 290 (25%) | 0.636 | 765 (12%) | 62,022 |
Summary statistics for de novo assembly of sequence data from the sex chromosome, which corresponds to linkage group 9 (LG9) as well as AM13 and AM14 for comparison as previously published30. Chromosomes 13 and 14 correspond to A. mexicanum linkage groups 15/17 (LG15/17) and linkage group 14 (LG14), respectively. Statistics are presented for assemblies of raw sequence data (R) and assemblies post error correction (EC).
Alignments between the sex chromosome assembly and Ambystoma reference transcripts (www.ambystoma.org) were used to identify genes that are encoded on the sex chromosome. These genes were aligned to the chicken genome assembly to confirm that homologs from the axolotl sex chromosome were heavily enriched on chicken chromosomes 7, 19 and 24, and similar enrichment was observed among scaffolds assigned to the Z from the male assembly, consistent with previous findings (Fig. 3A, Supplementary Table 1)35. Alignments to the newt (Notophthalmus viridescens) linkage map support previous analyses demonstrating that axolotl LG9 is homologous to newt LG736, revealing strong conservation of the chromosome’s gene content over the last 150 million years (Fig. 3B). While a ZW-type mechanism for sex determination has been inferred for the newt37, the exact chromosome that determines sex is unknown and no candidate genes currently exist.
Figure 3.
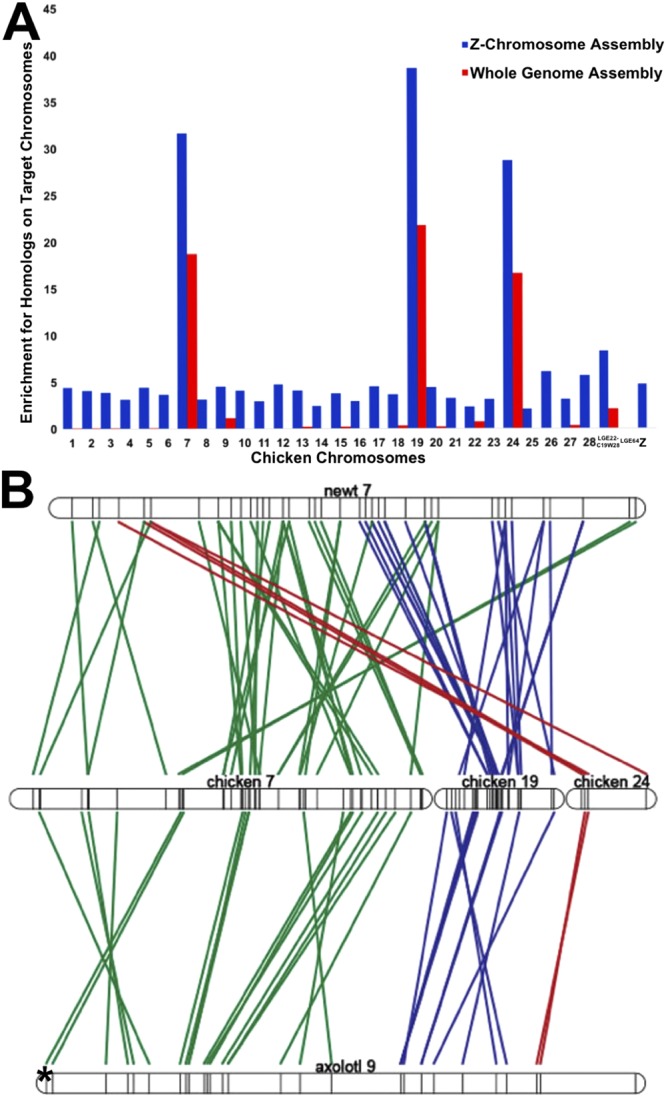
Conserved synteny for A. mexicanum sex chromosome. (A) Conserved synteny between assembled A. mexicanum Z chromosome and the chicken genome. Tests for enrichment of Z chromosome homologs with 99% identity from read mapping-based (blue) and assembly-based (red) methods across all assembled chicken chromosomes. Enrichment scores are calculated by dividing the observed number of homologs by the total number of genes annotated to the individual chicken chromosomes86. (B) Conserved synteny studies show syntenic regions shared between newt (Notophthalmus viridescens) linkage group 7 (top), chicken chromosomes 7,19, and 24 (middle), and axolotl LG9 (bottom). Each line corresponds to an alignment between a pair of presumptive chicken and salamander (newt or axolotl) orthologs, and the asterisk denotes the sex-specific region. Alignments involving orthologs on chicken chromosome 7 are colored green, chromosome 19 are colored blue, and chromosome 24 are red. More alignments were found between newt and chicken, as the linkage map of the newt is denser than that of the axolotl36.
In silico identification of female-specific regions
To identify sex-specific regions of the genome, we aligned low coverage sequence data from 26 males and 22 females to both the LG9 assembly and the first public draft assembly of the axolotl genome30,32 (www.ambystoma.org). Males and females were drawn from a backcross that was generated by crossing a male A. mexicanum to a female A. tigrinum X A. mexicanum hybrid that had been previously generated by crossing a male A. mexicanum to a female A. tigrinum29. Thus, all backcross progeny possessed a W chromosome inherited from A. tigrinum. The draft assembly was generated using a modified version of SparseAssembler38 from 600 Gb of HiSeq paired end reads and 640 Gb of HiSeq mate pair reads. Sequencing data were produced using DNA from a female axolotl, which should contain genomic regions from both Z and W chromosomes. Notably, a recently published draft genome was generated from a male and is not expected to represent W-specific regions39. Males and females used for re-sequencing efforts were drawn from a previously published meiotic mapping panel, which was used in the initial mapping of the sex locus29. Each individual was sequenced to ~1X coverage with Illumina HiSeq short paired-end reads (125 bp) resulting in ~7.4 billion total male reads and 6.4 billion total female reads. The ratio of female to male read depth of coverage was calculated across ~10.5M intervals covering ~19 Gb of the draft assembly. Genome-wide read coverage ratios generally fell within a tight distribution centered on equal coverage, after accounting for initial differences in average read depth of coverage (Fig. 4). Intervals were considered to be candidate sex-specific regions if enrichment scores [log2 (female coverage/adjusted male coverage)] exceeded two. In total, these analyses identified only 201 candidate female-specific intervals that were contained within 109 genomic scaffolds, with 20 genomic scaffolds having 2 or more intervals (Supplementary Table 2). The combined size of these intervals is approximately 300Kb or ~0.0094% of the genome. 47 intervals were represented by zero male reads, and the average male coverage of male reads for other intervals ranged from 0.002 to 8.63.
Figure 4.
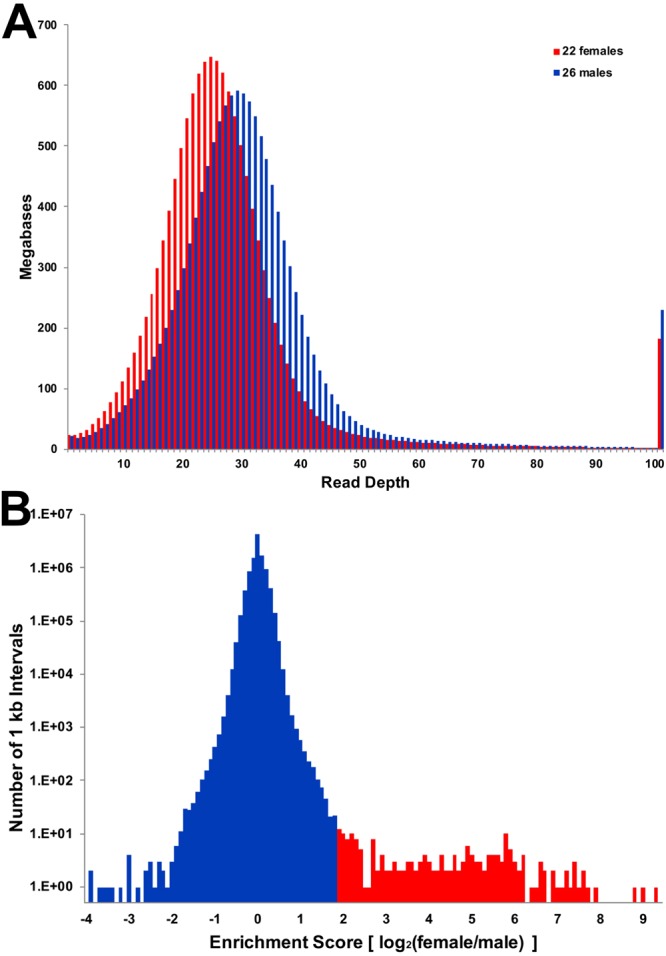
Distribution of read depth from combined female and males sequencing data. (A) Sequence reads from 48 individuals were mapped separately to the female whole genome assembly, then alignment files were merged across all individuals of a given sex (22 females and 26 males). Values represent the number of base pairs of the reference assembly that were sampled at a given depth of coverage. These distributions reveal that the modal coverage of reads from females was lower than the coverage of males, ~25X and ~29X, respectively, consistent with random sampling of sequences across individuals. There is no overtly visible evidence that female sequences map to a larger proportion of the approximate single copy sequence within the female genome. (B) The distribution of coverage ratios is tightly centered on equal coverage and only a small tail corresponds to intervals with higher sequence coverage in female relative to male.
PCR validation of candidate regions in A. mexicanum
PCR primers were designed for all candidate scaffolds and subjected to initial PCR validation using a panel of six females and six males from different crosses, corresponding to an expected false-positive rate of 2E-4 (Supplementary Table 3). In total, primers from 42 of the 109 scaffolds yielded specific amplicons in all females and no amplicons from males and were considered sex-specific. The combined size of these scaffolds is approximately 174Kb or ~0.0054% of the genome. Aside from the PCR validated female-specific scaffolds, primers from one scaffold were present in all females and one male, two were present in four females and no males, and four were present in a subset of the animals with no specific trend toward one sex or the other. Presumably these represent structural (insertion/deletion) variants that are segregating within the lab population of A. mexicanum, perhaps representing tiger salamander (A. tigrinum) DNA remnants that were introgressed in 196240. Primers for another 46 scaffolds yielded amplification in both sexes with 14 showing brighter bands in females and two showing varying brightness across all individuals. Primers for seven other scaffolds yielded no amplification in either sex. To further investigate our PCR validation results, we retrospectively aligned predicted W-specific regions to the recently published A. mexicanum (male) genome. These revealed that several predicted W-specific contigs correspond to copies of repetitive elements with highly similar sequences elsewhere in the genome, which appears to explain a majority of cases wherein primers yield amplicons in both sexes or are polymorphic among males and females.
Identifying W-specific genes
To search for evidence of sex-specific genes, all 42 validated sex-specific scaffolds were aligned (blastx) to the NCBI nonredundant protein database41. In total, these searches yielded alignments to 17 protein-coding genes (Table 2), several of which involved weak alignments to uncharacterized proteins (N = 4) or transposable elements (N = 5). However, two scaffolds yielded strong alignments to human protein coding genes. Specifically, Scaffold SuperContig_990642 aligned to transcriptional regulator ATRX (ATRX: 65% amino acid identity) and scaffold SuperContig_1084421 aligned to mitogen-activated protein kinase kinase kinase 2-like (MAP3K2: 97% amino acid identity). Notably, a conserved syntenic ortholog of MAP3K2 would be expected to occur on LG9 and thus it seems likely that MAP3K2 resided on the ancestral LG9 sex chromosome prior to the origin of the A. mexicanum sex-determining locus. However, a syntenic ortholog of ATRX would be expected to occur within a conserved synteny on a different chromosome (LG2, LG8/12), corresponding to a large region of conservation with mammalian X chromosomes and chicken chromosome 442–44.
Table 2.
Blast results to nonredundant protein NCBI database.
| Sex-specific Scaffold | Scaffold length (bp) | NCBI Best Hit | Query Cover | E value | % identity | Accession# |
|---|---|---|---|---|---|---|
| SuperContig1084421 | 991 | PREDICTED: mitogen-activated protein kinase kinase kinase 2-like [Phaethon lepturus] | 18% | 6.00E-33 | 98% | XP_010292439.1 |
| SuperContig_990642 | 1488 | PREDICTED: transcriptional regulator ATRX isoform X2 [Alligator sinensis] | 17% | 6.00E-13 | 64% | XP_006032758.2 |
| SuperContig_1201750 | 725 | PREDICTED: uncharacterized protein LOC101734340 [Xenopus tropicalis] | 14% | 0.13 | 50% | XP_017945915.1 |
| SuperContig_1270996 | 631 | hypothetical protein [Rhodopirellula baltica] | 12% | 9.7 | 50% | WP_011119337.1 |
| SuperContig_1240926 | 668 | PREDICTED: uncharacterized protein LOC106589496 [Salmo salar] | 39% | 2.00E-16 | 47% | XP_014035031.1 |
| SuperContig_481414 | 11464 | PREDICTED: dynein heavy chain 11, axonemal [Xenopus tropicalis] (reverse transcriptase) | 5% | 1.00E-32 | 43% | XP_017952780.1 |
| SuperContig_1139773 | 843 | aminotransferase class I and II [Streptomyces sp. CB00455] | 17% | 6.2 | 42% | WP_073917349.1 |
| SuperContig_1398497 | 510 | hypothetical protein [Massilia sp. BSC265] | 36% | 4.1 | 40% | WP_051933638.1 |
| SuperContig_1136461 | 850 | flagellar autotomy protein [Micromonas pusilla CCMP1545] (reverse transcriptase) | 12% | 0.55 | 39% | XP_003062983.1 |
| SuperContig_1105317 | 928 | hypothetical protein A2Z37_15870 [Chloroflexi bacterium RBG_19FT_COMBO_62_14] | 12% | 6.3 | 37% | OGO67717.1 |
| SuperContig_960617 | 1857 | PREDICTED: uncharacterized protein LOC106605384 [Salmo salar] | 20% | 1.8 | 36% | XP_014056412.1 |
| SuperContig_446459 | 12684 | ORF2 protein [Salmo salar] (reverse transcriptase) | 8% | 1.00E-36 | 35% | AKP40998.1 |
| SuperContig556195 | 9021 | PREDICTED: uncharacterized protein LOC108708171 [Xenopus laevis] | 19% | 2.00E-70 | 34% | XP_018102087.1 |
| SuperContig_981147 | 1581 | PREDICTED: LOW QUALITY PROTEIN: dynein heavy chain domain-containing protein 1 [Orcinus orca] | 13% | 8.4 | 32% | XP_004279330.1 |
| SuperContig_1025868 | 1238 | DNA primase [Pseudaminobacter manganicus] | 23% | 9.2 | 32% | WP_080921700.1 |
| SuperContig_1035909 | 1185 | hypothetical protein T12_433 [Trichinella patagoniensis] | 16% | 5.5 | 31% | KRY11477.1 |
| SuperContig_1196200 | 734 | DUF948 domain-containing protein [Lactobacillus buchneri] | 41% | 4.7 | 27% | WP_014939867.1 |
The table shows best match amino acid alignments for blast (blastx)78 hit results for all 42 sex-specific scaffolds. 17 scaffolds aligned to a protein-coding gene, and most shared <40% identity. The two highest identity hits to genes were to transcriptional regulator ATRX by SuperContig_990642 and mitogen-activated kinase kinase kinase 2 by SuperContig_108441.
The identification of a sex-linked ATRX homolog is notable as ATRX is known to play contribute to sex differentiation in mammals and other vertebrates45–48. Alignments between scaffold SuperContig_990642 and the autosomal ATRX homolog revealed that two distinct ATRX homologs exist in axolotl (Fig. 5). Alignments between ATRX and its sex-specific duplicate show polymorphisms in the ATRX gene that are not present in sex-linked ATRX, characteristic of a hemizygously-inherited duplication (Supplementary Fig. 1). Henceforth, we refer to the conserved syntenic homolog on LG2 as ATRX and the W-specific homolog as ATRW. Notably, presence vs. absence of ATRW is highly predictive of gonadal sex. Follow-up PCRs using sex-specific primers for ATRW have been used to sex more than 50 individuals, with no errors, as verified by dissection and examination of differentiated gonads. A nucleotide alignment between the axolotl ATRX and ATRW genes shows that the genes share 90% identity across 1089 aligned nucleotides, and as such it appears that the two genes diverged relatively recently by transposition of a duplicate gene copy to the W chromosome. To further test this idea and better define the timing of this duplication, several trees were generated using ATRX homologs from multiple vertebrate taxa (Fig. 6, Supplementary Fig. 2). Based on these trees, we infer that a duplication event gave rise to ATRW within Ambystoma, after divergence from its common ancestor with newt (the two lineages shared a common ancestor ~151 MYA)49. Considering the degree of sequence divergence and the relative length of shared vs. independent branches we estimate that the ATRW homolog may have arisen sometime in the last 20 MY (Fig. 6B), a timing that roughly coincides with a major adaptive radiation in the tiger salamander lineage50,51. Species within this complex may therefore represent biological replicates for understanding early sex chromosome evolution after the acquisition of ATRW.
Figure 5.

Alignment of translated nucleotides from ATRX in multiple vertebrate taxa. The alignment from MEGA784 of 84 amino acids of ATRX with conservation in 12 vertebrates, including ATRX and ATRW from axolotl show the relative number of changes in codons specific to all amphibians, salamanders (the newt, Notophthalmus viridescens), axolotl and axolotl ATRW. A total of two out of nine nucleotide substitution events specific to the ATRW have altered the predicted codon.
Figure 6.

Neighbor-Joining trees for vertebrate ATRX with bootstrap and divergence time estimations. (A) Evolutionary relationships among ATRX homologs were inferred using the Neighbor-Joining method87. The bootstrap consensus tree inferred from 10000 replicates is taken to represent the evolutionary history of the taxa analyzed88. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (10000 replicates) are shown next to the branches88. Evolutionary distances were computed using the Maximum Composite Likelihood method89 and are in the units of the number of base substitutions per site. The analysis involved 13 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions containing gaps and missing data were eliminated resulting in the inclusion of 251 positions in the final dataset. Evolutionary analyses were conducted in MEGA784. The newt represents sequence from Notophthalmus viridescens. (B) A time-scaled phylogenetic tree inferred using the Reltime method90 and estimates of branch lengths inferred using the Neighbor-Joining method as in A87. The tree was computed using 10 calibration constraints. Divergence times estimated by Timetree were added manually and are marked with gray arrows49. This tree indicates that the duplication event giving rise to ATRW in axolotl may have occurred ~20MYA.
To shed further light on the evolution of ATRX and ATRW within the Ambystoma lineage, we examined patterns of derived substitutions in ATRX and ATRW. Across the 251 bp alignment, 9 nucleotide substitutions can be attributed to ATRW since the divergence of axolotl, and these are associated with changes in 2 amino acids. By comparison, ATRX on LG2 shows only 1 nucleotide substitution since the duplication event (Fig. 6). This suggests that ATRW may be evolving at a faster rate than ATRX, in which case 20 MY may represent a substantial overestimate for the origin of the duplication that gave rise to ATRW.
Discussion
The results from this study show that the homomorphic sex chromosomes of the axolotl contain a small non-recombining region that is specific to the female W chromosome. The female-specific sequence is estimated to be approximately 300Kb, or roughly 1/100,000th of the enormous axolotl genome. It is not surprising that the differences in recombination were not initially evident due to the physical size of the genome and marker density in the Ambystoma meiotic map29. With respect to the current fragmented female genome assembly, it is still not possible to predict gene orders within this region or locate possible inversions; however, the data are sufficient to identify robust markers for sex and genes that exist in the non-recombining region. Of the few protein-coding genes found within the validated sex-specific scaffolds, two appear to represent non-repetitive coding sequences, including one that represents a relatively recent duplication of the transcriptional regulator ATRX.
The ATRX gene is located in the non-recombining region of the X chromosome in mammals. The gene encodes a chromatin remodeling protein that belongs to the SWI/SNF family. It is linked to the rare recessive disorder, alpha-thalassemia X-linked intellectual disability, which is characterized by severe intellectual disability, developmental delays, craniofacial abnormalities, and genital anomalies in humans. In some cases, a mutation in the ATRX gene can lead to female sex reversal due to early testicular failure52,53. Gene expression studies performed in a marsupial and eutherian showed that ATRX expression was highly conserved between the two mammals and was necessary for the development of both male and female gonads48. Because ATRX is one of the few protein-coding genes present in the region of W-specific sequence and has been characterized in the sex differentiation of mammals, we propose ATRW as a candidate sex gene for axolotl, or alternately a strong candidate for an acquired, sexually antagonistic gene.
Reanalysis of expression data from recent published tissue-specific transcriptomes showed expression of the ATRX gene (from LG2) in all major tissues and developing embryos, however, they showed no evidence of expression of the ATRW gene54. The tissues represented in the study included whole limb segments, blastemas from regenerating limbs, bone and cartilage, muscle, heart, blood vessel, gill, embryos, testis, and notably, ovaries. It is not clear at what stage the ovarian tissue was taken; however, the author suggests multiple ovaries were sequenced from an adult, and multiple libraries exist for the tissue. It is possible that this sex-specific gene is simply not highly expressed at this specific stage (or in the adult stage, in general) and may only be expressed during early gonadogenesis. Examining expression profiles and isoforms of ATRW before and throughout gonadogenesis may reveal interesting sexually dimorphic gene expression profiles in male and female genes, including those that are sex-linked. Similarly, W-linked genes in chicken were unknown until RNAseq studies were performed prior to and during gonadogenesis55.
If ATRW is the primary sex-determining gene in axolotl, then the origin of this gene marks the origin of sex chromosomes in the tiger salamander lineage. A time-scaled gene tree based on sequence substitution rates of ATRX genes in multiple vertebrate placed the ATRX duplication event at ~20 MYA (Supplementary Fig. 1). This estimate places the ATRX duplication event within the Ambystoma clade but suggests that not all ambystomatids necessarily share the sex chromosome. Based on the Ambystoma species tree49, we expect the same sex chromosomes and sex locus to be present in the tiger salamander species complex but not necessarily in the more distantly related A. jeffersonianum complex or deeper ambystomatid lineages (Fig. 7).
Figure 7.

A species tree for the genus Ambystoma. The gray shaded region shows the approximate timing of the ATRW duplication event. The tiger salamander complex consists of 7 named species that occur in the same monophyletic clade as A. californiense, A. mexicanum, and A. tigrinum56,91. This tree was generated using Timetree49 with modification to the position of A. californiense based on previously published tiger salamander complex tree56,91.
Given the relatively recent origin of ATRW, species within the tiger salamander complex are predicted to contain the same sex chromosomes. The tiger salamander species complex consists of more than 30 named species that encompass a range of diversification dates50,56. Further analyses of sex determination within this complex should therefore facilitate future studies aimed at more precisely characterizing the timing of the ATRX/W duplication and the evolution of other W-specific sequences. Ongoing improvements to the Ambystoma genome assembly and development of genome assemblies for other salamander taxa should improve our ability to assess hypotheses related to the presence of homomorphic sex chromosomes (e.g. recent evolution, high-turnover, and fountain of youth)1,17,57–62. Additionally, recent efforts to develop genetic tools for the axolotl model should facilitate functional analyses that will be necessary to test whether ATRW is the primary sex-determining gene in axolotl or elucidate its role as a sexually antagonistic factor63,64. Methods for achieving targeted gene knockout and knock-ins have been developed in axolotl and could be adapted to better assess the functionality of ATRW in axolotls40,65,66.
Sex is an important biological variable in research, as it may contribute to variation in experimental studies. Because axolotl is an important model for many areas of research and has shown sex-specific effects, such as tail regeneration, it is important for investigators to differentiate sex effects from other experimental variables28. Until now it was necessary to visualize the sex organs, utilize axolotls that had produced gametes, or perform experiments in hybrid crosses that segregate markers at the linked locus E24C3 in order to accurately determine sex in axolotls29. However, many experiments utilize juvenile animals that may not have completed gonadal differentiation or maturation. With several robust markers for W-specific sequences in hand, it is now possible to precisely differentiate sex of an axolotl with a simple PCR67. These markers will also positively impact axolotl husbandry, as individuals may be housed and utilized in experiments accordingly.
Methods
Laser capture microdissection and amplification
Preparation of cells for metaphase spreads and laser capture were performed as previously described30. Briefly, fixed cells were spread on UV-treated 1.0 mm polyethylene naphthalate (PEN) membrane slides. Slides were inverted (membrane side down) over a steam bath of distilled water for 7 seconds. Immediately after steaming, 100 µl of the fixed cells were dropped across the middle of the slide lengthwise. Each slide was subsequently placed in a steam chamber at ~35 °C for 1 minute, then set on the hot plate for 5 minutes. After slides dried, chromosomes were stained via immersion in freshly made Giemsa stain (Sigma-Aldrich GS500-500 ML: 0.4% Giemsa, 0.7 g/L KH2PO4, 1.0 g/L Na2HPO4) for 2 minutes, rinsed in 95% ethanol, rinsed in distilled water, then allowed to dry in a desiccator until used.
The sex chromosome was captured using a Zeiss PALM Laser Microbeam Microscope at 40X magnification as previously described30. The sex chromosome was dissected individually using a Zeiss PALM Laser Microbeam Microscope at 40X magnification and catapulted into a Zeiss adhesive cap tube (Zeiss 415190-9191-000). 10 µl of a chromatin digestion buffer was pipetted into the cap30 and the tube was kept inverted overnight at 55 °C. After incubation, the sample was centrifuged briefly and incubated at 75 °C for 10 minutes and 95 °C for 4 minutes to inactivate the Proteinase K. Along with 23 other samples, the sex chromosome sample was immediately carried through full amplification using the Rubicon PicoPlex DNAseq Whole Genome Amplification (WGA) kit (R30050). Amplification followed the standard manufacturer protocol, with one exception: a chromatin digestion step replaced the cell extraction step. After amplification, an Agilent 2100 Bioanalyzer and accompanying DNA 12000 kit (Agilent DNA 12000 Kit 5067-1508) was used to estimate concentration and size distribution. The sex chromosome sample had a concentration >9 ng/µl and was sequenced on an Illumina HiSeq2500 (Hudson Alpha Institute for Biotechnology, Huntsville, Al). After initial sequencing, the same sample was further sequenced to generate paired-end 150 bp reads on a full lane of HiSeq2500.
Sequence analyses and assembly
Because amplified sequences contain a non-complex leader sequence corresponding to the pseudorandom primers that are used for whole chromosome amplification, reads were trimmed prior to further processing. Trimmomatic was used to remove leader sequences derived from phiX and to trim any window of 40 nucleotides with quality score lower than Q3068. Reads were then aligned to 945 model transcripts from the Ambystoma linkage map35 using the Burrows Wheeler Aligner with the single-end mapping option and BWA-MEM algorithm69. They were also aligned to several bacterial genomes as well as the human reference genome using the paired-end mapping option to identify exact matches for Bowtie 270. Paired reads that mapped concordantly to the human and bacterial genomes were considered potential contaminants and removed. After trimming and removal of potential contaminants, the reads were corrected with Blue71 using female A. mexicanum whole genome shotgun data30 and assembled with SOAPdenovo272.
To assign scaffolds from the whole genome assembly of a male axolotl genome to the Z chromosome, error-corrected laser capture reads were aligned as paired-end reads to the assembly with BWA-MEM and filtered to preserve only pairs with concordant reads that map to the reference with no mismatches69. For each scaffold we calculated physical coverage (i.e. coverage by paired-end fragments: bedtools v. 2.27, genomeCoverageBed, option pc73) and assigned scaffolds to the Z chromosome if at least 5% of their bases were covered by reads from laser capture sequencing.
FISH of sex-associated BAC E24C3
Fluorescent in situ hybridization of BACs to metaphase chromosome spreads were performed as previously described74,75. A Qiagen Large Construct kit (Qiagen Science, 12462) was used to extract bacterial artificial chromosome (BAC) DNA for E24C3 and E12A6, previously associated with sex29. Probes for in situ hybridization were labeled by nick-translation using direct fluorophores Cyanine 3-dUTP (Enzo Life Sciences, ENZ-42501) or Fluorescein-12-dUTP (Thermo Scientific, R0101) as described previously74 and hybridization of BAC probes was performed as previously described for axolotl chromosomes40.
Phenol-chloroform extraction in 1.2X SSC was used to isolate repetitive DNA fractions from female salamander tissue76. DNA was denatured for 5 minutes at 120 °C, re-associated at 60 °C for 1 hour to obtain Cot DNA. Microtubes containing the DNA were placed on ice for 2 minutes, then transferred to a bead bath at 42 °C for 1 hour with 5X S1 nuclease buffer and S1 nuclease for a concentration of 100 units per 1 mg DNA. DNA was precipitated with 0.1 volume of 3M sodium acetate and 1 volume isopropanol at room temperature, tubes were inverted several times and centrifuged at 14,000 rpm for 20 minutes at 4 °C. DNA was washed with 70% ethanol, centrifuged at 14,000 rpm for 10 minutes at 4 °C, air dried and solubilized in TE buffer.
Conservation and evolution of salamander chromosomes
To evaluate the sex chromosome assembly, we performed alignments between the sex chromosome assembly and reference transcripts (V4: Sal-Site)32 using megablast77 to identify genes that occur on the sex chromosome. These genes were then aligned (tblastx)78 to annotated protein coding genes from the chicken genome assembly (Gallus_gallus-4.0). Annotated genes from scaffolds assigned on the basis of read mapping were aligned (blastp)78 to this set of annotated chicken genes. Those with an alignment length of at least 50 amino acids and at least 60% identity were considered potential homologs.
A similar approach was taken to identify the homologous newt linkage group to assess potential sex candidate genes. Ambystoma reference transcripts from LG9 (V4) were aligned (tblastx)78 to the chicken genome assembly41. Using the same minimum thresholds as above, the potential homologs were then used to blast (tblastx)78 to the newt, Notophthalmus viridescens, reference transcripts36.
Identification of female-specific regions
We applied read depth of coverage analysis to identify single-copy regions in the assembly that have approximately half of the modal coverage in females and underrepresented/absent coverage in males. Reads were generated on an Illumina HiSeq2000 (Hudson Alpha Institute for Biotechnology, Huntsville, Al.) from DNA that was isolated via phenol-chloroform extraction76 from 48 individuals that were drawn from a previously described backcross mapping panel42. The resulting reads were aligned to the axolotl draft genome assembly using BWA-MEM (using default parameters) followed by filtering of secondary alignments (samtools view–F2308) and alignments clipped on both sides of the read. Merging of female and male bam files was performed using Samtools merge69,79.
We used DifCover (https://github.com/timnat/DifCover)80 to identify candidate female-specific regions. The method works by computing the ratio of female:male average read depth of coverage across continuous intervals containing approximately V, valid bases. Valid bases are defined by lower and upper limits on read depth of coverage for females (f) and males (m), respectively designated as minf, minm, maxf and maxm. If Cf and Cm define female and male coverage for a given valid base, then (1) Cf < maxf and Cm < maxm; and (2) Cf > minf or Cm > minm. The upper limits identify and allow skipping of fragments that contain repeats, while the lower limits serve to exclude underrepresented fragments with small numbers of reads in both males and females.
After testing, we chose V = 1000 and assigned lower limits equal to one third of modal coverage, (8 for females and 9 for males) and upper limits 3X of modal coverage, (75 for females and 87 for males). The enrichment scores [log2(standardized sperm coverage/blood coverage)] were computed for each interval. If the average coverage in males for an interval was zero, we replaced the coverage estimate with a non-zero positive value corresponding to alignment of half of one read. Some intervals were shorter than 1Kb and contained fewer than 1000 valid bases (short scaffolds or intervals that fall on the scaffold ends). These shorter intervals were filtered to exclude intervals with fewer than 500 bases or fewer than 200 valid bases.
Scaffolds that were validated through PCR in a panel of 6 females and 6 males were aligned to the V4 and V5 Ambystoma transcriptome assemblies in order to identify the genes present on the W-specific portion of the sex chromosome. If a transcript aligned to the scaffold with a percent identity higher than 95%, that transcript was blasted (blastx)78 to the NCBI nonredundant protein database to search for homologous genes.
Primer design and PCR
Primers were designed within the sex candidate regions identified using Primer381. Each primer was 25–28 bp in length, with a target melting temperature of 60 °C, 20–80% GC content and 150–400 bp product sizes depending on the size of the region and location of repeats (avoiding inclusion of repetitive sequence in primer and product). Fragments were amplified using standard PCR conditions (150 ng DNA, 50 ng of each primer, 200 mM each dATP, dCTP, dGTP, dTTP; thermal cycling at 94 °C for 4 minutes; 34 cycles of 94 °C for 45 seconds, 55 °C for 45 seconds, 72 °C for 30 seconds; and 72 °C for 7 minutes). Reactions were tested on a panel of six males and six females to validate sex specificity. Gel electrophoresis was performed and presence/absence was recorded for each set of primers (Supplementary Fig. 3). The scaffolds from which primers were designed were considered female-specific if the primers yielded specific amplicons in all six females and in no males.
Results from these data were used to develop a PCR based assay for determining sex in axolotls at any stage of development. This method uses a primer pair that amplifies a 219 bp DNA fragment in females and an internal control that yields a 486 bp DNA fragment in both sexes. This biplex PCR results in two bands (219 bp and 486 bp) for females and only the control band (486 bp) in males67.
Phylogenetic Reconstruction
Homologene was used to collect putative homology groups from the ATRX genes in a variety of eukaryotes82. Sequence for axolotl ATRX was obtained from Ambystoma reference transcripts, and the newt ATRX gene was obtained by aligning human ATRX to the newt reference transcriptome83. All sequences were aligned using MEGA784 via MUSCLE85. Sequences were trimmed to compare a conserved subregion of the sequence that was present in all species, a string of 251 codons (Fig. 5). Divergence time estimates were drawn from the TimeTree webserver49.
Accession Codes
Sequence data (48 sequenced axolotl genomes) are deposited at the NCBI short read archives (http://www.ncbi.nlm.nih.gov/sra) under study number PRJNA478224.
Electronic supplementary material
Acknowledgements
This study was supported by the National Institutes of Health through their support of this project (R24OD010435), the Ambystoma Genetic Stock Center (P40OD019794), (R01GM104123), and by the Army Research Office (W911NF1110475). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of National Institute of Health or the Army Research Office.
Author Contributions
M.C.K., J.J.S. and S.R.V. conceived of the study. M.C.K., N.T. and J.J.S. performed computational analyses. M.C.K. performed chromosome amplification and sequencing experiment, DNA extraction and sequencing experiment, and P.C.R. validation. V.T. performed in situ hybridizations.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Melissa C. Keinath, Email: keinath@carnegiescience.edu
Jeramiah J. Smith, Email: jjsmit2@uky.edu
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-36209-2.
References
- 1.Bull, J. J. Evolution of Sex Determining Mechanisms, (Benjamin/Cummings Publ. Co., 1983).
- 2.Bachtrog D. A dynamic view of sex chromosome evolution. Current opinion in genetics & development. 2006;16:578–585. doi: 10.1016/j.gde.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Cortez D, et al. Origins and functional evolution of Y chromosomes across mammals. Nature. 2014;508:488–493. doi: 10.1038/nature13151. [DOI] [PubMed] [Google Scholar]
- 4.Rice WR. Sex Chromosomes and the Evolution of Sexual Dimorphism. Evolution. 1984;38:735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- 5.Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- 6.Beukeboom, L. W. & Perrin, N. The Evolution of Sex Determination (2014).
- 7.Charlesworth B. The evolution of chromosomal sex determination and dosage compensation. Curr. Biol. 1996;6:149–162. doi: 10.1016/S0960-9822(02)00448-7. [DOI] [PubMed] [Google Scholar]
- 8.Connallon T, Clark AG. Sex linkage, sex-specific selection, and the role of recombination in the evolution of sexually dimorphic gene expression. Evolution. 2010;64:3417–3442. doi: 10.1111/j.1558-5646.2010.01136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillis DM, Green DM. Evolutionary changes of heterogametic sex in the phlogenetic history of amphibians. Journal of evolutionary biology. 1990;3:49–64. doi: 10.1046/j.1420-9101.1990.3010049.x. [DOI] [Google Scholar]
- 11.Schmid, M. et al. In Amphibian Cytogenetics and Evolution (eds Green, D. M. & Sessions, S. K.) 393–430 (Academic Press, 1991).
- 12.Ogata M, et al. Change of the heterogametic sex from male to female in the frog. Genetics. 2003;164:613–620. doi: 10.1093/genetics/164.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ezaz T, Stiglec R, Veyrunes F, Marshall Graves JA. Relationships between vertebrate ZW and XY sex chromosome systems. Curr. Biol. 2006;16:R736–R743. doi: 10.1016/j.cub.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Kamiya T, et al. A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu) PLoS Genet. 2012;8:e1002798. doi: 10.1371/journal.pgen.1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicoso B, Kaiser VB, Bachtrog D. Sex-biased gene expression at homomorphic sex chromosomes in emus and its implication for sex chromosome evolution. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6453–6458. doi: 10.1073/pnas.1217027110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vicoso B, Emerson JJ, Zektser Y, Mahajan S, Bachtrog D. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 2013;11:e1001643. doi: 10.1371/journal.pbio.1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stock M, et al. Ever-young sex chromosomes in European tree frogs. PLoS Biol. 2011;9:e1001062. doi: 10.1371/journal.pbio.1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DM, Sessions SK. Amphibian Cytogenetics and Evolution. Journal of evolutionary biology. 1991;6:300–302. doi: 10.1046/j.1420-9101.1993.6020300.x. [DOI] [Google Scholar]
- 19.Schmid, M. & Steinlein, C. Sex chromosomes, sex-linked genes, and sex determination in the vertebrate class amphibia. EXS, 143–176 (2001). [DOI] [PubMed]
- 20.Sessions SK, Bizjak Mali L, Green DM, Trifonov V, Ferguson-Smith M. Evidence for Sex Chromosome Turnover in Proteid Salamanders. Cytogenet Genome Res. 2016;148:305–313. doi: 10.1159/000446882. [DOI] [PubMed] [Google Scholar]
- 21.Humphrey RR. Reversal of sex in females of genotype WW in the axolotl (Siredon or Ambystoma mexicanum) and its bearing upon the role of the Z chromosomes in the development of the testis. J Exp Zool. 1948;109:171–185. doi: 10.1002/jez.1401090202. [DOI] [PubMed] [Google Scholar]
- 22.Humphrey RR, Frankhauser G. The origin of spontaneous and experimental haploids in the Mexican axolotl (Siredonor Ambystoma-mexicanum) J Exp Zool. 1957;134:427–447. doi: 10.1002/jez.1401340303. [DOI] [PubMed] [Google Scholar]
- 23.Armstrong JB. Genetic mapping in the Mexican axolotl, Ambystoma mexicanum. Can J Genet Cytol. 1984;26:1–6. doi: 10.1139/g84-001. [DOI] [PubMed] [Google Scholar]
- 24.Humphrey RR. Sex determination in the Ambystomatid salamanders: a study of the progeny of females experimentally converted into males. Am J Anat. 1945;76:33–66. doi: 10.1002/aja.1000760103. [DOI] [Google Scholar]
- 25.Lindsley DL, Fankhauser G, Humphrey RR. Mapping Centromeres in the Axolotl. Genetics. 1956;41:58–64. doi: 10.1093/genetics/41.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuny R, Malacinski GM. Banding differences between tiger salamander and axolotl chromosomes. Can J Genet Cytol. 1985;27:510–514. doi: 10.1139/g85-076. [DOI] [PubMed] [Google Scholar]
- 27.Sessions SK. Cytogenetics of diploid and triploid salamanders of the Ambystoma jeffersonianum complex. Chromosoma. 1982;77:599–621. doi: 10.1007/BF00286329. [DOI] [Google Scholar]
- 28.Voss GJ, Kump DK, Walker JA, Voss SR. Variation in salamander tail regeneration is associated with genetic factors that determine tail morphology. PloS one. 2013;8:e67274. doi: 10.1371/journal.pone.0067274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith JJ, Voss SR. Amphibian sex determination: segregation and linkage analysis using members of the tiger salamander species complex (Ambystoma mexicanum and A. t. tigrinum) Heredity. 2009;102:542–548. doi: 10.1038/hdy.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keinath MC, et al. Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci Rep. 2015;5:16413. doi: 10.1038/srep16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Callan HG. Chromosomes and nucleoli of the axolotl, Ambystoma mexicanum. J Cell Sci. 1966;1:85–108. doi: 10.1242/jcs.1.1.85. [DOI] [PubMed] [Google Scholar]
- 32.Smith JJ, et al. Sal-Site: integrating new and existing ambystomatid salamander research and informational resources. BMC.Genomics. 2005;6:181. doi: 10.1186/1471-2164-6-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whiteman HH. Evolution of Facultative Paedomorphosis in Salamanders. The Quarterly review of biology. 1994;69:205–221. doi: 10.1086/418540. [DOI] [Google Scholar]
- 34.Keinath, M. C. Characterization of a large vertebrate geome and homomorphic sex chromosomes in the axolotl, Ambystoma mexicanum Ph.D. thesis, University of Kentucky (2017).
- 35.Voss, S. R. et al. Origin of amphibian and avian chromosomes by fission, fusion, and retention of ancestral chromosomes. Genome Res, 10.1101/gr.116491.110 (2011). [DOI] [PMC free article] [PubMed]
- 36.Keinath MC, Voss SR, Tsonis PA, Smith JJ. A linkage map for the Newt Notophthalmus viridescens: Insights in vertebrate genome and chromosome evolution. Dev Biol. 2017;426:211–218. doi: 10.1016/j.ydbio.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.National Research Council (US) Subcommittee on Amphibian Standards. In Classification and Description of Amphibians Commonly Used for Laboratory Research Vol. 2 (National Academies Press, 1974).
- 38.Ye C, Ma ZS, Cannon CH, Pop M, Yu DW. Exploiting sparseness in de novo genome assembly. BMC Bioinformatics. 2012;13(Suppl 6):S1. doi: 10.1186/1471-2105-13-S6-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowoshilow S, et al. The axolotl genome and the evolution of key tissue formation regulators. Nature. 2018;554:50–55. doi: 10.1038/nature25458. [DOI] [PubMed] [Google Scholar]
- 40.Woodcock MR, et al. Identification of Mutant Genes and Introgressed Tiger Salamander DNA in the Laboratory Axolotl, Ambystoma mexicanum. Sci Rep. 2017;7:6. doi: 10.1038/s41598-017-00059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 42.Smith JJ, Kump DK, Walker JA, Parichy DM, Voss SR. A comprehensive expressed sequence tag linkage map for tiger salamander and Mexican axolotl: enabling gene mapping and comparative genomics in Ambystoma. Genetics. 2005;171:1161–1171. doi: 10.1534/genetics.105.046433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith JJ, Voss SR. Gene order data from a model amphibian (Ambystoma): new perspectives on vertebrate genome structure and evolution. BMC. Genomics. 2006;7:219. doi: 10.1186/1471-2164-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JJ, Voss SR. Bird and mammal sex-chromosome orthologs map to the same autosomal region in a salamander (ambystoma) Genetics. 2007;177:607–613. doi: 10.1534/genetics.107.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McElreavey K, Fellous M. Sex-determining genes. Trends Endocrinol. Metab. 1997;8:342–346. doi: 10.1016/S1043-2760(97)00135-5. [DOI] [PubMed] [Google Scholar]
- 46.Neri G, Opitz J. Syndromal (and nonsyndromal) forms of male pseudohermaphroditism. Am. J. Med. Genet. 1999;89:201–209. doi: 10.1002/(SICI)1096-8628(19991229)89:4<201::AID-AJMG4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 47.Pask A, Renfree MB, Marshall Graves JA. The human sex-reversing ATRX gene has a homologue on the marsupial Y chromosome, ATRY: implications for the evolution of mammalian sex determination. Proc. Natl. Acad. Sci. USA. 2000;97:13198–13202. doi: 10.1073/pnas.230424497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huyhn K, Renfree MB, Graves JA, Pask AJ. ATRX has a critical and conserved role in mammalian sexual differentiation. BMC Dev Biol. 2011;11:39. doi: 10.1186/1471-213X-11-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedges SB, Marin J, Suleski M, Paymer M, Kumar S. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. 2015;32:835–845. doi: 10.1093/molbev/msv037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaffer HB. Evolution in a Paedomorphic Lineage. II. Allometry and Form in the Mexican Ambystomatid Salamanders. Evolution. 1984;38:1207–1218. doi: 10.1111/j.1558-5646.1984.tb05644.x. [DOI] [PubMed] [Google Scholar]
- 51.Shaffer HB. Evolution in a Paedomorphic Lineage. I. An Electrophoretic Analysis of the Mexican Ambystomatid Salamanders. Evolution. 1984;38:1194–1206. doi: 10.1111/j.1558-5646.1984.tb05643.x. [DOI] [PubMed] [Google Scholar]
- 52.Stevenson, R. E. In GeneReviews(R) (eds Pagon, R. A. et al.) (1993).
- 53.Lee JS, et al. Alpha-thalassemia X-linked intellectual disability syndrome identified by whole exome sequencing in two boys with white matter changes and developmental retardation. Gene. 2015;569:318–322. doi: 10.1016/j.gene.2015.04.075. [DOI] [PubMed] [Google Scholar]
- 54.Bryant DM, et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017;18:762–776. doi: 10.1016/j.celrep.2016.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ayers KL, et al. RNA sequencing reveals sexually dimorphic gene expression before gonadal differentiation in chicken and allows comprehensive annotation of the W-chromosome. Genome Biol. 2013;14:R26. doi: 10.1186/gb-2013-14-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaffer HB, McKnight ML. The polytypic species revisited: genetic differentiation and molecular phylogenetics of the tiger salamander (Ambystoma tigrinum) (Amphibia: Caudata) complex. Evolution. 1996;50:417–433. doi: 10.1111/j.1558-5646.1996.tb04503.x. [DOI] [PubMed] [Google Scholar]
- 57.White, M. J. Animal cytology and evolution (Cambridge, 1973).
- 58.Schartl M. Sex chromosome evolution in non-mammalian vertebrates. Curr. Opin. Genet. Dev. 2004;14:634–641. doi: 10.1016/j.gde.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 59.Stock M, et al. Low rates of X-Y recombination, not turnovers, account for homomorphic sex chromosomes in several diploid species of Palearctic green toads (Bufo viridis subgroup) J Evol Biol. 2013;26:674–682. doi: 10.1111/jeb.12086. [DOI] [PubMed] [Google Scholar]
- 60.Perrin N. Sex reversal: a fountain of youth for sex chromosomes? Evolution. 2009;63:3043–3049. doi: 10.1111/j.1558-5646.2009.00837.x. [DOI] [PubMed] [Google Scholar]
- 61.Guerrero RF, Kirkpatrick M, Perrin N. Cryptic recombination in the ever-young sex chromosomes of Hylid frogs. J Evol Biol. 2012;25:1947–1954. doi: 10.1111/j.1420-9101.2012.02591.x. [DOI] [PubMed] [Google Scholar]
- 62.Bachtrog D, et al. Sex determination: why so many ways of doing it? PLoS Biol. 2014;12:e1001899. doi: 10.1371/journal.pbio.1001899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parker, G. A. In Sexual selection and reproductive competition in insects (eds Blum, M. S. & Blum, N. A.) 123–166 (Academic Press, 1979).
- 64.Holland B, Rice WR. Perspective: Chase-Away Sexual Selection: Antagonistic Seduction Versus Resistance. Evolution. 1998;52:1–7. doi: 10.1111/j.1558-5646.1998.tb05132.x. [DOI] [PubMed] [Google Scholar]
- 65.Fei JF, et al. CRISPR-mediated genomic deletion of Sox2 in the axolotl shows a requirement in spinal cord neural stem cell amplification during tail regeneration. Stem Cell Reports. 2014;3:444–459. doi: 10.1016/j.stemcr.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flowers GP, Timberlake AT, McLean KC, Monaghan JR, Crews CM. Highly efficient targeted mutagenesis in axolotl using Cas9 RNA-guided nuclease. Development. 2014;141:2165–2171. doi: 10.1242/dev.105072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Keinath MC, et al. A PCR based assay to efficiently determine the sex of axolotls. Axolotl. 2018;2:5–7. [Google Scholar]
- 68.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greenfield P, Duesing K, Papanicolaou A, Bauer DC. Blue: correcting sequencing errors using consensus and context. Bioinformatics. 2014;30:2723–2732. doi: 10.1093/bioinformatics/btu368. [DOI] [PubMed] [Google Scholar]
- 72.Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Timoshevskiy, V. A., Sharma, A., Sharakhov, I. V. & Sharakhova, M. V. Fluorescent in situ hybridization on mitotic chromosomes of mosquitoes. Journal of visualized experiments: JoVE, e4215, 10.3791/4215 (2012). [DOI] [PMC free article] [PubMed]
- 75.Timoshevskiy VA, Lampman RT, Hess JE, Porter LL, Smith JJ. Deep ancestry of programmed genome rearrangement in lampreys. Dev Biol. 2017;429:31–34. doi: 10.1016/j.ydbio.2017.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sambrook, J. & Russell, D. W. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc2006, 10.1101/pdb.prot4455 (2006). [DOI] [PubMed]
- 77.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 78.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith JJ, et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat Genet. 2018;50:270–277. doi: 10.1038/s41588-017-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Untergasser A, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wheeler DL, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abdullayev I, Kirkham M, Bjorklund AK, Simon A, Sandberg R. A reference transcriptome and inferred proteome for the salamander Notophthalmus viridescens. Exp Cell Res. 2013;319:1187–1197. doi: 10.1016/j.yexcr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 84.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cunningham F, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 88.Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 89.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tamura K, et al. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci USA. 2012;109:19333–19338. doi: 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shaffer HB, Pauly GB, Oliver JC, Trenham PC. The molecular phylogenetics of endangerment: cryptic variation and historical phylogeography of the California tiger salamander, Ambystoma californiense. Mol Ecol. 2004;13:3033–3049. doi: 10.1111/j.1365-294X.2004.02317.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.