

Abstract
Programmed cell death (PCD) plays critical roles in development, homeostasis, and both control and progression of a plethora of diseases, including cancer and neurodegenerative pathologies. Besides classical apoptosis, several different forms of PCD have now been recognized, including necroptosis. The way a cell dies determines the reaction of the surrounding environment, and immune activation in response to cell death proceeds in a manner dependent on which death pathways are activated. Apoptosis and necroptosis are major mechanisms of cell death that typically result in opposing immune responses. Apoptotic death usually leads to immunologically silent responses whereas necroptotic death releases molecules that promote inflammation, a process referred to as necroinflammation. Diseases of the nervous system, in particular neurodegenerative diseases, are characterized by neuronal death and progressive neuroinflammation. The mechanisms of neuronal death are not well defined and significant cross-talk between pathways has been suggested. Moreover, it has been proposed that the dying of neurons is a catalyst for activating immune cells in the brain and sustaining inflammatory output. In the current review we discuss the effects of apoptotis and necroptosis on inflammatory immune activation, and evaluate the roles of each cell death pathway in a variety of pathologies with specific focus on neurodegeneration. A putative model is proposed for the regulation of neuronal death and neuroinflammation that features a role for both the apoptotic and necroptotic pathways in disease establishment and progression.
Subject terms: Inflammation, Neuroscience
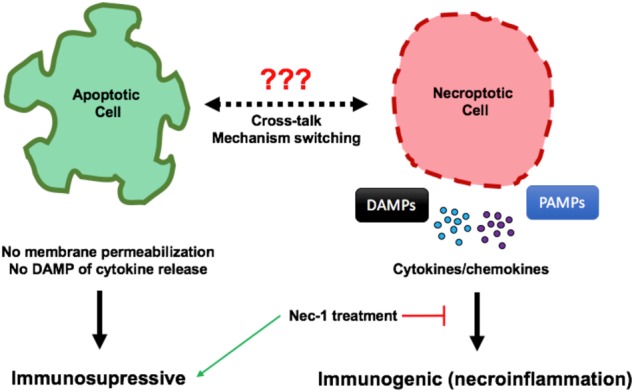
Facts
Apoptotic cell death is often immunosuppressive whereas necroptotic death is inflammatory.
Cross-talk between necroptosis and apoptosis exists in a variety of pathologies including neurodegenerative diseases such as Alzheimer’s.
Necroptosis may be activated in the hippocampus and cortex of human Alzheimer’s patients.
DAMPs released during necroptosis can activate microglial cells leading to neuroinflammation and promote further polymerization of amyloid beta.
Necrostatin-1 treatment is protective in a variety of diseases including ischemic pathologies and neurodegenerative conditions.
Open questions
Caspase-8 activates apoptosis and inhibits necroptosis, does it function as a molecular switch between death pathways in vivo?
What is the contribution of necroptosis to neuronal loss in neurodegenerative conditions?
Does engagement of the necroptotic mediators RIPK3 and MLKL have effects independent of cell death in the inflammatory response?
Can necroptosis and necroinflammation be specifically targeted as a therapeutic avenue for ameliorating inflammatory diseases?
Introduction
It has long been asserted that programmed cell death (PCD) plays critical roles in development and homeostatic maintenance of virtually every tissue in the body. Apoptosis is the most well-characterized PCD to date, but recently mechanisms such as necroptosis, pyroptosis, and ferroptosis have been described as pathways to the death of the cell. The roles of such pathways in development, homeostasis, and disease are emerging as an exciting research focus.
For the past few decades diseases characterized by aberrant cell death have simply been attributed to apoptosis. It is now becoming clear that the way a cell dies is of importance for the progression of diseases. Apoptosis can be described as a “clean” way for a cell to die. Apoptotic cells reduce to apoptotic bodies that are removed by resident phagocytic cells. Under most circumstances, as discussed below, apoptosis is immunologically silent and its activation does not promote a significant inflammatory or auto-immune response [1–7]. Apoptotic cell death is thus ideal for clearing non-functioning or malfunctioning cells without triggering an immune response, thereby preserving homeostatic integrity. Other forms of PCD, including necroptosis and pyroptosis, are a much “dirtier” means to an end. Both necroptosis and pyroptosis result in the “bursting” of the plasma membrane and subsequent release of the intracellular content into the surrounding microenvironment. The intracellular content consists of a variety of molecules that promote pro-inflammatory responses from the immune system. Thus, whereas apoptosis is generally anti-inflammatory, necroptosis and pyroptosis induce inflammation [8–10].
Diseases including atherosclerosis and ischemia–reperfusion injury following solid organ transplant are characterized by cell death and inflammatory activation. Similarly, neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are also characterized by neuronal death and excessive levels of neuroinflammation. In such neurodegenerative conditions, lines between apoptosis and other forms of cell death such as necroptosis are not clearly defined. Since the exact mechanisms that govern death of neurons in these diseases remain elusive, it provides an intriguing system for studying the interplay between various forms of cell death and associated immune consequences. Many studies have implicated neuroinflammation as not only a consequence of cellular death but also as a feedback mechanism promoting further abnormal neuron clearance [11–13]. In this review, we focus on the interplay between the apoptotic and necroptotic mechanisms of cell death and the impact they have on immune activation in both the physiological and pathological settings with specific focus on neurodegenerative conditions. Furthermore, we profile the therapeutic potential that exists from the modulation of these cell death pathways in the realm of both acute disease and chronic pathologies.
Apoptosis vs. necroptosis
Apoptosis is an evolutionarily conserved process that is morphologically characterized by a variety of features, including pyknosis, membrane blebbing, nuclear fragmentation, nuclear condensation, and apoptotic body formation [14]. The pathway of apoptosis has been well reviewed in detail, but in brief, activation of the apoptotic pathway is executed by the caspase family of cysteine proteases that have the ability to cleave a variety of key cellular substrates [15–20]. Two primary pathways of apoptotic activation exist [19], the extrinsic and intrinsic pathways. The intrinsic apoptotic pathway involves the release of cytochrome c following mitochondrial outer membrane permeabilization (MOMP), which in turn leads to the activation of caspase-9 [20, 21]. As the name suggests, this intrinsic pathway can be engaged by an assortment of intracellular signals generated by endoplasmic reticulum stress, reactive oxygen species (ROS), DNA damage, and growth factor deprivation. In contrast, extrinsic apoptotic activation usually occurs following the ligation of the death receptor subset of the tumor necrosis factor (TNF) receptor family, residing on the plasma membrane. Receptor activation induces a cascade that leads to the intracellular recruitment and activation of the initiator caspase-8, followed by downstream effector caspase activation [22, 23]. A classical example of the extrinsic apoptosis death pathway is the ligation of the death receptor, Fas (CD95), by Fas ligand (FasL, CD95L) as illustrated on the left-hand side of Fig. 1. Receptor ligation induces the formation of the death-inducing signaling complex (DISC) consisting of the Fas death domain (DD), the adaptor molecule FADD, and caspase-8/10 [24, 25]. Caspase-8 then homodimerizes, is stabilized by autoproteolytic cleavage, and initiates the downstream apoptotic pathway. Activation of either the extrinsic or intrinsic pathways ultimately leads to the downstream activation of caspases-3 and -7, the executioner caspases in apoptosis. In some cases the activity of the effector caspases is inhibited by X-linked Inhibitor of Apoptosis Protein (XIAP). However, caspase-8 can prevent this by cleaving the protein BID, which then activates the effector proteins BAX and BAK to mediate MOMP. Upon MOMP, proteins that antagonize XIAP (such as SMAC) are released from the mitochondrial intermembrane space, allowing executioner caspases to promote apoptosis [26, 27].
Fig. 1.

Ligated CD95 and TRAILR recruit FADD via homotypic death domain (DD) interactions. FADD then recruits caspase-8 via death effector domain (DED) interactions, leading to homodimerization and activation of caspase-8 to initiate apoptosis. TNFR1 ligation leads to the recruitment of TRADD, TRAF2, cIAP1/2, and RIPK1. RIPK1 is ubiquitylated by cIAP1/2 and retained in the complex to mediate activation of NF-κB. RIPK1 is deubiquitylated by CYLD and leaves the complex to recruit FADD via homotypic DD interactions and RIPK3 through RIP homotypic interaction motif (RHIM) domain interactions. RIPK1-FADD recruits caspase-8 and apoptosis ensues. RIPK1–RIPK3 interaction leads to RIPK3 ameloid formation, MLKL activation, and engagement of necroptosis. NF-κB expresses the pro-survival gene cFLIPL which heterodimerizes with caspase-8, resulting in inhibition of caspase-8 activation and apoptosis. Caspase-8 retains its catalytic activity and is able to cleave RIPK1 and RIPK3, thereby blocking necroptosis
Cell survival vs. cell death by apoptosis involving MOMP in mammals is governed by the Bcl-2 family of proteins. The Bcl-2 family consists of both pro-apoptotic and anti-apoptotic members that have the ability to heterodimerize, establishing an antagonistic balance between pro- and anti-apoptotic regulators that control the decision of a cell to live or die [28, 29]. Most upstream signals that stimulate apoptotic activation function to alter the ratio of these regulators, shifting the balance towards MOMP. Bcl-2 family-mediated control of mitochondrial cytochrome c release has been well characterized [30–32].
Necroptosis is a form of PCD that, like extrinsic apoptosis, can be executed upon death receptor activation as well as upon activation of some Toll-like receptors (TLRs) and the intracellular sensor DAI/ZBP1. Necroptosis is both morphologically and mechanistically distinct from apoptosis. Whereas extrinsic apoptosis depends on initiator and effector caspase activation, necroptosis is a kinase-mediated pathway. Necroptosis induced by activation of the death receptor tumor necrosis factor receptor 1 (TNFR1) has been most extensively characterized [33, 34]. TNFR1 ligation-induced necroptosis depends on the recruitment and deubiquitylation of the receptor interacting serine/threonine kinase (RIPK) 1 (Fig. 1). RIPK1 then autophosphorylates and recruits RIPK3 through homotypic RHIM domain interactions of both proteins [35, 36]. This leads to RIPK3 oligomerization, activation by autophosphorylation and the subsequent engagement and phosphorylation of the pseudokinase, Mixed Lineage Kinase-Like (MLKL), the effector protein that once activated translocates to the plasma membrane, where it induces rupture and subsequent cell death [37]. Engagement of the receptors TLR3 and TLR4, IFNAR, and ZBP1 can also result in necroptosis [38–43]. Although the exact mechanism of IFNAR-induced necroptosis remains unclear, ligation of TLR3, TLR4, and ZBP1 can result in RIPK3 activation through RHIM domain interactions. Whereas ZBP1 has at least three internal RHIM domains, TLR3 and TLR4 ligation activates the RHIM domain-containing adaptor protein TRIF. RIPK3 can bind to and oligomerize on these RHIM domain-containing proteins and recruit and activate MLKL to execute necroptosis. However, engagement of these necroptosis-inducing receptors also rapidly leads to the activation of transcription factors, such as NF-κB and p38, and subsequent expression of pro-inflammatory and pro-survival genes such as CFLAR [38, 44]. cFLIPL (encoded by CFLAR) is a protein that closely resembles caspase-8, but lacks a catalytic domain. Caspase-8 preferentially heterodimerizes with cFLIPL, and thereby alters its ability to engage apoptosis. The proteolytic activity of caspase-8–cFLIPL specifically cleaves certain substrates, among which are RIPK1 and RIPK3. The FADD–caspase-8–cFLIPL complex thus inhibits necroptosis initiated by TNFR1 or the TLRs. As mentioned, necroptosis induced by TNFR1 ligation requires RIPK1. FADD can bind RIPK1 through death domain interactions and thereby brings the caspase-8–cFLIPL heterodimer in close enough proximity to cleave RIPK1 and RIPK3. TLR- and ZBP1-induced necroptosis does not rely on RIPK1 to activate RIPK3 [41]. Instead, RIPK1 has a protective role in TLR ligation, since it is recruited to activated RIPK3, bringing with it the inhibitory caspase-8–cFLIPL heterodimer through the FADD adaptor protein. Thus, only in settings where the catalytic activity of caspase-8 is inhibited or disrupted can necroptosis generally ensue.
In contrast to the relatively “clean” death associated with apoptosis, cells that are necroptotic release their intracellular contents en masse into the surrounding milieu, due to the plasma membrane rupture mediated by MLKL. The highly inflammatory response that occurs following intracellular content release into the microenvironment (necroinflammation) after necroptosis may be a consequence of several of these intracellular components, since the cytosol can contain various pro-inflammatory cytokines (induced by NF-κB and p38), as well as constitutive damage-associated molecular patterns (DAMPs) [45–47].
The in vivo role of necroptosis has been proposed to contribute to a variety of conditions, including atherosclerosis, pancreatitis, inflammatory bowel disease, and some cancers. In regard to the role of necroptosis in neurodegeneration, it has been implicated in amyotrophic lateral sclerosis and perhaps multiple sclerosis [48–50]. More recently, necroptotic activation has been suggested in the brains of Alzheimer’s patients [51]. These studies establish a pathological role for necroptosis that has often been overshadowed by its cousin, apoptosis.
Consequences of apoptotic cell death for immune activation
As described above, apoptosis typically leads to an immunologically silent or immune-suppressive outcome. The immunologically silent feature of apoptosis is often attributed to its capacity to contain intracellular components of the dying cell, preventing the release of inflammatory mediators. However, in some settings apoptotic cells can inhibit an inflammatory response in a more indirect way. When a cell dies by apoptosis, a hallmark of death is the reallocation of the membrane lipid phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane [52, 53]. The exposure of PS allows for apoptotic cells to be identified by professional phagocytic cells for clearance [54–56]. Apoptotic cells are preferentially recognized by T-cell immunoglobulin and mucin domain-containing molecule 4 (Tim4), a PS-sensing receptor expressed on macrophages, among other receptors [57, 58]. Binding of PS on apoptotic cells to Tim4 on macrophages promotes the phagocytic engulfment of the dying/dead cell and subsequent degradation in a process known as efferocytosis. Once the cell has been internalized, it is now subject to degradation in the lysosomal compartment. This process culminates with the production of immunoregulatory cytokines, among which is the anti-inflammatory cytokine IL-10. In the absence of IL-10 production by the macrophage, the expressed pro-inflammatory cytokines can induce an inflammatory response [59]. This suggests that without IL-10, apoptotic cells have the potential to induce an inflammatory response.
The process of LC3-associated phagocytosis (LAP) has been implicated as an important mediator of immune silence during efferocytosis. LAP is a non-canonical function of autophagy proteins, where components of the autophagy pathway conjugate LC3 to phagosome membranes in a receptor- and cargo-dependent fashion [59–64]. Our laboratory has recently shown that abrogation of LAP in macrophages is sufficient to abolish the immunologically silent response to apoptotic cells [62, 63]. Following engulfment of a dying cell, a switch from IL-10 production to pro-inflammatory IL-1β and IL-6 cytokine expression occurs in the absence of LAP. This molecular switch in cytokine production reduces the pool of anti-inflammatory mediators and increases inflammatory molecules, effectively causing apoptotic cells to become pro-inflammatory as illustrated in Fig. 2. This demonstrates that even though apoptotic cells have the ability to mask their intracellular immunogenic contents during the cell death process, once cells are consumed by macrophages, the ability to maintain a non-immunogenic setting is dependent on the phagocyte and its associated pathways.
Fig. 2.
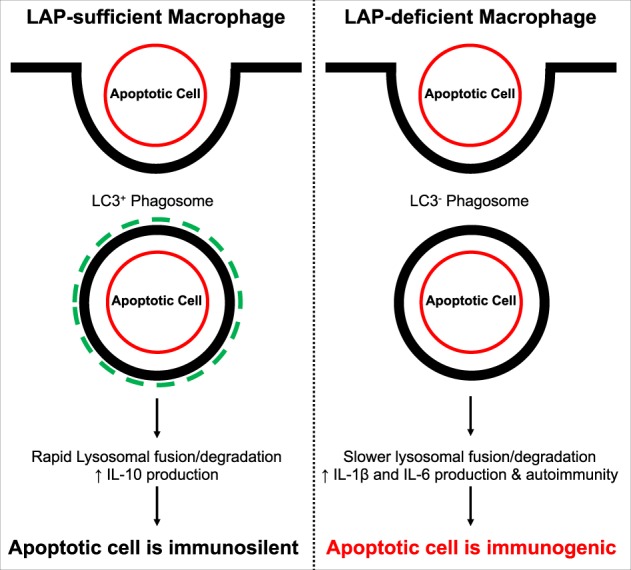
The immunogenic state of apoptotic cells is in part dependent on macrophage efferocytosis. LC3-associated phagocytosis (LAP) plays a critical role in maintaining immunosuppressive activation following apoptotic cell engulfment. As shown in the left panel, macrophages that are LAP-sufficient are able to conjugate LC3 to phagosomal membranes which leads to rapid lysosomal fusion and degradation. Furthermore, it promotes the production of anti-inflammatory cytokines including IL-10 by mechanisms that are still under investigation. In contrast, LAP-deficient macrophages that phagocytose apoptotic cells have a slow rate of lysosomal fusion and cargo degradation that leads to the polarization of the cell towards a pro-inflammatory state, including production and release of IL-1β and IL-6. Moreover, LAP-deficiency leads to an accumulation of un-cleared apoptotic cells that can further induce autoimmunity
Moreover, it is likely that it is not only the clearance mechanism following apoptotic death that controls or dampens immune activation. Caspase cascade activation has been proposed to contribute to the immune silence of apoptotic cells [65]. Intriguingly, we should keep in mind that while caspase activation is dispensable for cell death, caspase-dependent apoptosis appears to be the only form of cell death that does not elicit an immune response. Interestingly, studies have demonstrated a role for caspase activation in the inhibition of type I interferon (IFN) production in response to mitochondrial DNA that is released during apoptotic death, suggesting a critical role of the apoptotic caspases in the silencing of cell-intrinsic immune responses (such as that to mitochondrial DNA) [66, 67]. Therefore, the ability to promote immune silence is likely to be a combination of cell-intrinsic processes within the apoptotic cell as well as dampened activation of immune cells during efferocytosis.
DAMPing out immune silence
In contrast to apoptotic cells, necroptotic cells are unable to restrict the release of their intracellular contents. Following MLKL-mediated membrane permeabilization, a variety of pro-inflammatory molecules are directly released from the dying cell. Pro-inflammatory cytokines and chemokines are released from necroptotic corpses. Cytokines such as TNF can lead to further activation of cell death pathways including necroptosis in surrounding cells [46, 47]. Moreover, these pro-inflammatory mediators can directly influence cells of the immune system such as macrophages, leading to further inflammatory output in what could be described as a positive feedback loop for inflammation.
In addition to the direct release of inflammatory cytokines during necroptosis, there is likewise a release of DAMPs. Compared to PAMPs which are pathogen-derived molecules recognized by the innate immune system, DAMPs are endogenous host biomolecules such as uric acid, proteins, DNA and RNA, and purines such as adenosine and ATP [68]. When released during necroptosis, DAMPs can engage pattern-recognition receptors such as TLRs, nucleotide-binding domain-containing molecules, and C-type lectin receptors on bystander (immune) cells. Ligation of these surface immune receptors promotes inflammatory cytokine production via activation of NF-κB and other inflammatory transcriptional programs [69].
An example of a DAMP released upon necroptosis is the prototypic alarmin high-mobility group box-1 protein (HMGB1). HMGB1 is a non-histone nuclear protein that has been shown to stimulate the innate immune response and drive the pathogenesis of multiple inflammatory diseases when aberrantly released from cells [70]. Under physiological conditions HMGB1 is an important modulator of DNA organization and transcriptional regulation. While HMGB1 can be expelled from both active and dying cells, an acute, highly reactive pool is released during necroptosis following cell lysis. Liberated HMGB1 appears to bind immune receptors such as C-X-C motif chemokine 12 receptor (CXCR12), TLR4, and the receptor for advanced glycation endproducts (RAGE) upon which the inflammatory loop cascades [71–73].
Although necroptosis is generally considered to be a pro-inflammatory stimulus, in certain circumstances necroptosis may function in an anti-inflammatory role. Convincing evidence has been reported that triggering necroptosis can attenuate TNF- and lipopolysaccharide-induced cytokine production and consequent inflammation [74]. In this study, TNF was able to induce RIPK3-dependent necroptosis upon inhibition of caspase activation, and this suppressed inflammatory cytokine production. Upon ablation of either RIPK3 or MLKL, cytokine production under these conditions was restored. This suggests that termination of cell viability by activation of necroptosis is responsible for decreased cytokine synthesis vs. exacerbation of inflammatory cytokine production by the host immune system, as is frequently speculated. These results were further supported by another study that used the TNF/zVAD-fmk model of shock and lethality, where the pancaspase inhibitor zVAD-fmk is used to force TNF-mediated necroptosis. In this study they showed inhibition of necroptosis using necrostatin-1 (Nec-1) failed to provide protection and mice died at an accelerated rate compared to treatment without Nec-1 [75]. While evidence clearly defining an anti-inflammatory role of necroptosis remains controversial, it is important to remain congnizant of this possibility, at least in certain settings.
The role of necroinflammation in vivo
The most well-characterized models of necroinflammation are focused primarily on pathologies that are defined by acute injury, such as those induced by ischemia–reperfusion. Other settings being actively investigated include pathogenic infection, inflammatory bowel disease (IBD), acute kidney injury, non-alcoholic steatohepatitis, and central nervous system (CNS) pathologies such as stroke, traumatic brain injury (TBI), and neurodegenerative and neuroinflammatory diseases. While this list is by no means complete, most of these pathological settings are acute in nature and/or have a significant combination of a cell death component coupled with a severe inflammatory state.
For convenience, we can loosely group diseases and conditions into two basic categories for the purpose of examining necroptosis and necroinflammation in vivo. The first are pathologies that are characterized by ischemia followed by reperfusion. Ischemia–reperfusion injury during transplant, acute myocardial infarction, and stroke share similar outcomes with regards to tissue damage. The second group are diseases that include neurodegenerative conditions, TBI, pathogenic infections, acute kidney disease, and IBD. This group does not have the underlying theme of ischemia–reperfusion, but are all characterized by high levels of cellular death and progressive inflammation.
In many of the studies implicating necroptosis in disease, Nec-1 has been employed to inhibit necroptosis. Care must be taken to interpret these results, however, as Nec-1 is not a specific inhibitor of RIPK1; Nec-1 inhibits indoleamine 2,3-dioxygenase [76] as well as some conditions of ferroptosis. More specific inhibitors of RIPK1 are described and some have shown beneficial effects in disease models [77–79]. In addition to specificity, the pharmaco-kinetics of Nec-1 should be taken into consideration and the varying effects of each Nec-1 analog [80]. It is important to note that the half-life of Nec-1 in vivo varies depending on which analog is used, ranging from 15 to 60 min [79]. Thus, studies that only rely on Nec-1 should be considered suggestive, rather than definitive. Similarly, while RIPK3 is required for necroptosis, it also has other signaling roles, and in at least one case, a role for RIPK3 in pathogenesis was shown to be independent of necroptosis [81]. Again, care must be used in concluding a role for necroptosis in a disease process based only on ablation of RIPK3 [82].
Nec-1 treatment can decrease ischemia–reperfusion injury in murine models, and mice that are deficient in RIPK3 are protected from ischemia–reperfusion injury [83–85]. More recent work has established that abrogation of the necroptotic pathway whether genetically or by chemical inhibition is sufficient to decrease necroinflammation in addition to abolishing cell death [86]. One study found that expression of RIPK3 was increased by over 50% in heart following myocardial ischemia, and RIPK3-deficient mice were protected from myocardial damage [87]. Perhaps more importantly, this study showed that RIPK3 had a direct detrimental role in the remodeling of the heart following injury, and high levels of inflammatory mediators, increased infiltration of inflammatory T cells, and high level of ROS production were observed in RIPK3-sufficient animals compared to those that were RIPK3-deficient.
Necroptosis in neurological disorders including TBI is less well investigated. It has recently been shown that administration of Nec-1 attenuates cell death and brain damage following a TBI [88–90]. Inhibition of necroptosis was also able to improve post-injury spatial memory and behavior. However, it must be noted that another study suggested that improvement of TBI symptoms following Nec-1 administration may in part be due to secondary effects of the compound on alternative death pathways, and evidence was provided that both the apoptotic and autophagic pathways were attenuated following Nec-1 treatment during TBI with a likewise decrease in inflammation [88]. A suppression of both Beclin-1 and autophagic LC3-II activation as well as a concomitant reduction in pro-apoptotic Bcl-2 and caspase-3 cleavage were observed, suggesting that the effect of Nec-1 in reducing TBI-induced tissue damage in this model could be due to its coaction on multiple pathways and not only on necroptosis.
In regard to neurodegenerative conditions, fewer studies probing the role of necroptosis exist. One piece of evidence comes from genetic studies in a murine model of amyotrophic lateral sclerosis (ALS). Genetic mutations in the optineurin gene, which have been described in ALS patients, result in increased turnover of the necroptotic regulator RIPK1 leading to downregulation of necroptosis and development of ALS-like symptoms [50, 91]. More recently and to our knowledge there are two studies demonstrating a putative role for necroptosis in AD. The first observed increased expression of RIPK1 and MLKL in the temporal gyrus of brains from AD patients [51]. Necrosome formation and phosphorylation of MLKL was also observed in patient samples. Strikingly over 60% of phosphorylated MLKL was identified in neurons within the hippocampus. The second study identified a death-independent role of RIPK1 where it may modulate the activation of microglia in AD and moreover may mediate the transition to the disease-associated microglial state [92]. These findings provide evidence that not only is necroptosis engaged in AD, but death-independent functions of the necroptotic regulators likely exist. It is probable that necroptotic induction also occurs in other diseases characterized by neuronal cell death, especially under conditions that have a high correlative level of inflammatory immune activation.
Modes of cell death in neurodegeneration
In the introduction of this review we initially proposed the idea that the nervous system, and more specifically neurodegenerative conditions provide a model for studying not only necroinflammation, but the cross-talk and balance between the necroptotic and apoptotic pathways of cell death. In the vast majority of neurodegenerative conditions, we are cognizant of many of the death-inducing stimuli, nonetheless the definition of the mechanism by which death proceeds remains elusive. The apoptotic pathway has been described to contribute to neuronal death in many models of neurodegeneration, including AD, PD, ALS, and multiple sclerosis (MS). Most studies examining neuronal death in AD and other conditions typically evaluate traditional markers of apoptotic activation, including caspase cleavage and PS redistribution, with mixed results. The ability to identify apoptotic cells morphologically is also hindered in most neurodegenerative conditions, as dying neural cells, even when experimentally induced to undergo apoptosis, have varying morphological profiles that are atypical to most apoptotic cells. Multiple studies evaluating dying neurons in the substantia nigra in models of PD show very low levels of dopaminergic neurons that stain positive for DNA fragmentation using TUNEL [93]. Moreover, these neurons have only a marginal increase in the expression of pro-apoptotic proteins and have moderate levels of cleaved caspase-3, suggesting apoptosis is only one potential death pathway activated in this disease [94–98].
On the other hand, mutations in the presenilin 1 (PS1) and amyloid precursor protein (APP) genes associated with familial early onset AD have been shown to participate in the sensitization of neurons to apoptosis [99, 100]. APP mutations associated with human disease have been shown to induce neuronal DNA fragmentation and caspase-3 cleavage [101, 102], favoring a role for apoptosis. Additionally, recent studies have shown that the accumulation of amyloid beta (Aβ), the aberrant cleavage product of APP that is a causative factor of AD, suppresses Bcl-2 expression and promotes pro-apoptotic BAX upregulation [103]. In contrast, correlative evidence between necroptotic protein expression and brain weight in human AD patients exists [51]. Patients with higher levels of RIPK1 and MLKL had significantly lower brain weights, suggestive of increased neuronal death. Moreover, in murine models of early onset AD, approximately a 10% reduction in neuronal death was observed in animals treated with Nec-1 [92, 104]. The opposite effect was observed in AD mice that carried a constitutively active MLKL transgene [51].
Necroinflammation as a contributor to neuroinflammation
Whether neurons are prone to apoptosis or necroptosis still leaves the fact that in the end they die, and their death further promotes the establishment and progression of neuroinflammation. In this section of the review, we speculate on the contributions of both apoptosis and necroptosis in regulating an inflammatory immune reaction in neuroinflammatory environments.
There is currently a lack of evidence supporting either pathway fully. However, there are suggestions that demonstrate both pathways likely have a role in regulating neuron demise and inflammation. We propose that both pathways indeed contribute to neuron loss in diseases such as AD, and that each pathway impacts the immune response, albeit in opposing manners and likely at differing times during disease pathology. Using AD as a model to illustrate this hypothesis, we can imagine that the aberrant accumulation of Aβ and subsequent hyperphosphorylation of the microtubule protein Tau in neurons, both instrumental factors in AD pathology, are the beginnings of significant stress to the neuronal population in the hippocampus and certain regions of the cerebral cortex as reviewed previously [105]. In an attempt to mitigate tissue damage from increasing inflammation stemming from the processing of Aβ and soluble Aβ peptides by microglial cells (macrophage-like CNS immune cells) and the accumulation of non-functional Tau in neurons, cells activate the apoptotic pathway in an effort to clear “beyond-repair” neurons and maintain homeostatic integrity.
As neurons begin to die via apoptosis, the dying cells and corpses must be scavenged and cleared by microglial cells to prevent further immune activation [106, 107]. It has been well established that Aβ plaque-associated microglia often have an impairment in either phagocytic uptake or intracellular processing of internalized cargo [108–111]. These deficiencies have been shown to lead to a state of reactive microgliosis, characterized by microglial activation and expansion coupled with pro-inflammatory cytokine production and release [112–114]. These events lead to the establishment of an autoreactive environment that is prime for fueling inflammatory responses. One of the primary pro-inflammatory cytokines that is elevated in the AD brain is TNF. We can speculate that increased levels of TNF lead to the activation of further rounds of cell death through ligation of TNFR1. A recent study demonstrated two caspase-8 variants that are found in human AD and that lead to a reduction in caspase-8 activity and consequently, apoptosis [115]. It is wholly feasible that even a slight alteration in caspase-8 activity in even a subset of neurons may shift the mechanism of death from apoptosis towards necroptosis.
If neurons begin to die via the necroptotic pathway, the massive release of intracellular contents into the immediate milieu of the brain would be further detrimental. As shown in Fig. 3, the release of DAMPs including HMGB1, which was recently shown to drive AD pathology by promoting Aβ polymerization, would exacerbate disease progression [109, 116–119]. This model of cooperative death mechanisms is consistent with the time frame of disease advancement. In the early stages of disease, low levels of apoptotic activity occur with a gradual switch to necroptosis over time, leading to an increase in the rate of cell death. This timeline is fairly consistent with the typical profile of AD progression and is also correlated to the levels of neuroinflammation found in patient samples. This can be observed in mouse models that slowly develop neurobehavioral disorders that progress and worsen with time, in regard to both inflammation and behavioral impairment [120].
Fig. 3.

Here we present a putative model for necroptosis and necroinflammation in conditions of neurodegeneration using AD as an example. The lytic nature of necroptosis results in the release of DAMPs from the intracellular environment. DAMPs including HMGB1 have been shown to have detrimental effects in the contexts of both neuroinflammation and AD. Left—HMGB1 promotes the polymerization and of Aβ and subsequent Aβ plaque formation. Middle—HMGB1 also has a negative impact on efferocytosis by macrophages and microglial cells by decreasing activity and phagocytic capacity. Right—HMGB1 and other DAMPs can activate pro-inflammatory transcriptional programs on target cells such as microglia. Sustained activation and further release of DAMPs from dying cells will promote a phenotype of reactive microgliosis, leading to persistent inflammation
Therapeutic potential of targeting necroptosis in neurodegeneration
We have seen how necroptotic inhibition, whether genetic or pharmaceutical, can impact an extensive group of pathologies. While shared features including cell death and inflammation are common among them, they are distinct in their locations and causative factors. The contribution of necroptosis and necroinflammation in the majority of these settings is becoming more appreciated and as such, the therapeutic potential of targeting the necroptotic pathway as a treatment avenue is of broad interest.
While genetic manipulation would provide ultimate control of regulating a particular cell death pathway and cross-talk with other mechanisms, the ability to perform in vivo modifications in a cell-specific manner is still in its infancy. There are studies actively using Nec-1 in an effort to treat neurodegenerative diseases such as AD and PD as shown in Table 1, in addition to other peripheral diseases. Many results from these studies show that Nec-1 treatment provides not only a reduction in disease symptoms such as behavior and memory in the case of AD, but also alters the biochemical and physiological architecture associated with these pathologies. While the results from these studies and others are still preliminary, they provide encouraging evidence demonstrating the necessity to further pursue employing necroptotic inhibition as a clinical modality.
Table 1.
Studies evaluating the use of necroptosis inhibition for treating neurodegenerative conditions
| Condition | Mode of inhibition | Outcome | References |
|---|---|---|---|
| Alzheimer’s disease (AD) | Necrostatin-1 | Improved behavior and reduction in AD-induced neuronal death | [51, 104] |
| Reduced Aβ and Tau deposition | |||
| Amelioration of cognitive impairment | |||
| Amyotrophic lateral sclerosis (ALS) | RIPK1/3 Mutation | Increased neuron viability in ALS-causing optineurin (OPTN)−/− mice | [91, 121] |
| Necrostatin-1 | Improved neuron survival in ALS astrocyte co-cultures in vitro | ||
| Huntington’s disease (HD) | Necrostatin-1 | Improved behavior and delayed symptom onset in HD murine model | [122] |
| Parkinson’s disease | Necrostatin-1 | Necroptotic inhibition protected dopaminergic neurons in culture | [123] |
Conclusions
With the development of new murine models of both apoptosis and necroptosis, our understanding of the roles of each cell death pathway in vivo is rapidly expanding. Limitations remain with respect to modulating individual pathways due to the high levels of redundancy and compensation between cell death mechanisms, particularly in vivo. However, the studies presented herein have demonstrated an intricate relationship between apoptosis and necroptosis. Moreover, the immune consequences of cell death activation are becoming clearer. It is evident that the necroptotic pathway and associated necroinflammation is present in a variety of healthcare-related disparities including ischemic injury during organ transplantation, as well as debilitating neurodegenerative conditions, stroke, and myocardical infarction. The latter conditions are all highly correlative to an aging population and as such will likely become even more prevelant over the next few decades. The ability to modulate necroptosis using specific inhibitors will provide a viable means of targeting the necroptotic pathway for clinical therapy. As we have evaluated in this review, numerous studies are utilizing Nec-1 administration in murine models of neurodegeneration as well as ischemia–reperfusion and TBI to evaluate the protective effects of necroptotic inhibition in pathology, and this will be improved by the use of more specific inhibitors. This capability to specifically target a singular cell death pathway that is involved in promoting immune responses has significant therapeutic promise that will likely be of substantial importance as a broad treatment for inflammatory disease.
Acknowledgements
D.R.G. is supported by funding from the National Institutues of Health and the Lupus Research Alliance. B.L.H. is generously supported by the John H. Sununu Endowed Fellowship and National Institutes of Health grants AI138492 and JTWS6172 . B.T. is supported by the Paul Barrett Endowed Fellowship.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by F. Pentimalli
References
- 1.Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–25. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson TA, Herndon J, Elzey B, Griffith TS, Schoenberger S, Green DR. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8(+) T cells produce active immune unresponsiveness. J Immunol. 2002;168:5589–95. doi: 10.4049/jimmunol.168.11.5589. [DOI] [PubMed] [Google Scholar]
- 3.Green DR. The cell’s dilemma, or the story of cell death: an entertainment in three acts. FEBS J. 2016;283:2568–76. doi: 10.1111/febs.13658. [DOI] [PubMed] [Google Scholar]
- 4.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–63. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffith TS, Kazama H, VanOosten RL, Earle JK, Jr, Herndon JM, Green DR, et al. Apoptotic cells induce tolerance by generating helpless CD8+T cells that produce TRAIL. J Immunol. 2007;178:2679–87. doi: 10.4049/jimmunol.178.5.2679. [DOI] [PubMed] [Google Scholar]
- 6.Griffith TS, Yu X, Herndon JM, Green DR, Ferguson TA. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity. 1996;5:7–16. doi: 10.1016/s1074-7613(00)80305-2. [DOI] [PubMed] [Google Scholar]
- 7.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 8.Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75. doi: 10.1111/imr.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang W, Lin J, Dong J, Li D. Pyroptosis: an inflammatory cell death implicates in atherosclerosis. Med Hypotheses. 2013;81:484–6. doi: 10.1016/j.mehy.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 11.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorman AM. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med. 2008;12:2263–80. doi: 10.1111/j.1582-4934.2008.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1):1003. [PMC free article] [PubMed]
- 14.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Julien O, Wells JA. Caspases and their substrates. Cell Death Differ. 2017;24:1380–9. doi: 10.1038/cdd.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 2004;13:1979–87. doi: 10.1110/ps.04789804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994;8:1613–26. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- 18.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–52. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 19.Strasser A, Harris AW, Huang DC, Krammer PH. Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–47. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 21.Spierings D, McStay G, Saleh M, Bender C, Chipuk J, Maurer U, et al. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science. 2005;310:66–67. doi: 10.1126/science.1117105. [DOI] [PubMed] [Google Scholar]
- 22.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–4. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 23.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–27. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 24.Fu TM, Li Y, Lu A, Li Z, Vajjhala PR, Cruz AC, et al. Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol Cell. 2016;64:236–50. doi: 10.1016/j.molcel.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61:834–49. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 27.Munoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA. 2006;103:11573–8. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25:46–55. doi: 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 30.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–6. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 32.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 33.Belizario J, Vieira-Cordeiro L, Enns S. Necroptotic cell death signaling and execution pathway: lessons from knockout mice. Mediat Inflamm. 2015;2015:128076. doi: 10.1155/2015/128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Almagro MC, Vucic D. Necroptosis: pathway diversity and characteristics. Semin Cell Dev Biol. 2015;39:56–62. doi: 10.1016/j.semcdb.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–21. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277:76–89. doi: 10.1111/imr.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. 2016;20:674–81. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33. doi: 10.1038/nature20559. [DOI] [PubMed] [Google Scholar]
- 41.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–8. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–42. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 45.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 46.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson B. Necroptosis: a new way of dying? Cancer Biol Ther. 2016;17:899–910. doi: 10.1080/15384047.2016.1210732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao L, Yu S, Ji W, Li H, Gao Y. The contribution of necroptosis in neurodegenerative diseases. Neurochem Res. 2017;42:2117–26. doi: 10.1007/s11064-017-2249-1. [DOI] [PubMed] [Google Scholar]
- 49.Morrice JR, Gregory-Evans CY, Shaw CA. Necroptosis in amyotrophic lateral sclerosis and other neurological disorders. Biochim Biophys Acta. 2017;1863:347–53. doi: 10.1016/j.bbadis.2016.11.025. [DOI] [PubMed] [Google Scholar]
- 50.Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol. 2017;154:1–20. doi: 10.1016/j.pneurobio.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, et al. Necroptosis activation in Alzheimer’s disease. Nat Neurosci. 2017;20:1236–46. doi: 10.1038/nn.4608. [DOI] [PubMed] [Google Scholar]
- 52.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16. [PubMed] [Google Scholar]
- 54.Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79:896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- 55.Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, et al. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med. 2004;200:459–67. doi: 10.1084/jem.20040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krahling S, Callahan MK, Williamson P, Schlegel RA. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999;6:183–9. doi: 10.1038/sj.cdd.4400473. [DOI] [PubMed] [Google Scholar]
- 57.Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, et al. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc Natl Acad Sci USA. 2014;111:E1463–72. doi: 10.1073/pnas.1320174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–9. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 59.Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844–54. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
- 60.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 61.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–9. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA. 2011;108:17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-associated phagocytosis and inflammation. J Mol Biol. 2017;429:3561–76. doi: 10.1016/j.jmb.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–77. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pisetsky DS. The expression of HMGB1 on microparticles released during cell activation and cell death in vitro and in vivo. Mol Med. 2014;20:158–63. doi: 10.2119/molmed.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209:551–63. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–73. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–88. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 73.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–7. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22:1313–27. doi: 10.1038/cdd.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Linkermann A, Brasen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol Med. 2012;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013;20:185–7. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.von Massenhausen A, Tonnus W, Himmerkus N, Parmentier S, Saleh D, Rodriguez D, et al. Phenytoin inhibits necroptosis. Cell Death Dis. 2018;9:359. doi: 10.1038/s41419-018-0394-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harris PA, Bandyopadhyay D, Berger SB, Campobasso N, Capriotti CA, Cox JA, et al. Discovery of small molecule RIP1 kinase inhibitors for the treatment of pathologies associated with necroptosis. ACS Med Chem Lett. 2013;4:1238–43. doi: 10.1021/ml400382p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Degterev A, Maki JL, Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, III, Tait SWG, et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell. 2017;169:301–13. doi: 10.1016/j.cell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science. 2016;352:aaf2154. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 83.Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–9. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–68. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol. 2016;27:27–39. doi: 10.1681/ASN.2015040405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103:206–16. doi: 10.1093/cvr/cvu146. [DOI] [PubMed] [Google Scholar]
- 88.Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ, Liu WL, et al. Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem Res. 2012;37:1849–58. doi: 10.1007/s11064-012-0791-4. [DOI] [PubMed] [Google Scholar]
- 89.Chen F, Su X, Lin Z, Lin Y, Yu L, Cai J, et al. Necrostatin-1 attenuates early brain injury after subarachnoid hemorrhage in rats by inhibiting necroptosis. Neuropsychiatr Dis Treat. 2017;13:1771–82. doi: 10.2147/NDT.S140801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28:1564–73. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–8. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci USA. 2017;114:E8788–E8797. doi: 10.1073/pnas.1714175114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jellinger KA. Cell death mechanisms in neurodegeneration. J Cell Mol Med. 2001;5:1–17. doi: 10.1111/j.1582-4934.2001.tb00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wullner U, Kornhuber J, Weller M, Schulz JB, Loschmann PA, Riederer P, et al. Cell death and apoptosis regulating proteins in Parkinson’s disease—a cautionary note. Acta Neuropathol. 1999;97:408–12. doi: 10.1007/s004010051005. [DOI] [PubMed] [Google Scholar]
- 95.Kingsbury AE, Mardsen CD, Foster OJ. DNA fragmentation in human substantia nigra: apoptosis or perimortem effect? Mov Disord. 1998;13:877–84. doi: 10.1002/mds.870130604. [DOI] [PubMed] [Google Scholar]
- 96.Jellinger KA, Stadelmann CH. The enigma of cell death in neurodegenerative disorders. J Neural Transm Suppl. 2000;60:21–36. [DOI] [PubMed]
- 97.Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13:221–7. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- 98.Kosel S, Egensperger R, von Eitzen U, Mehraein P, Graeber MB. On the question of apoptosis in the parkinsonian substantia nigra. Acta Neuropathol. 1997;93:105–8. doi: 10.1007/s004010050590. [DOI] [PubMed] [Google Scholar]
- 99.Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J Neurochem. 1999;72:1019–29. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]
- 100.Lucassen PJ. Presenilins and cellular damage; a link through amyloid? J Alzheimers Dis. 2000;2:61–7. doi: 10.3233/jad-2000-2108. [DOI] [PubMed] [Google Scholar]
- 101.Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, et al. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci. 1999;19:6955–64. doi: 10.1523/JNEUROSCI.19-16-06955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 103.Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J Neurosci. 1996;16:7533–9. doi: 10.1523/JNEUROSCI.16-23-07533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang SH, Lee DK, Shin J, Lee S, Baek S, Kim J, et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med. 2017;9:61–77. doi: 10.15252/emmm.201606566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 106.Janda E, Boi L, Carta AR. Microglial phagocytosis and its regulation: a therapeutic target in Parkinson’s disease? Front Mol Neurosci. 2018;11:144. doi: 10.3389/fnmol.2018.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li C, Zhao B, Lin C, Gong Z, An X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-kappaB signaling. Cell Biol Int. 2018. 1–13. [DOI] [PMC free article] [PubMed]
- 108.Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takata K, Takada T, Ito A, Asai M, Tawa M, Saito Y, et al. Microglial amyloid-beta1-40 phagocytosis dysfunction is caused by high-mobility group box protein-1: implications for the pathological progression of Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:685739. doi: 10.1155/2012/685739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, et al. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol Neurodegener. 2011;6:45. doi: 10.1186/1750-1326-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sokolowski JD, Mandell JW. Phagocytic clearance in neurodegeneration. Am J Pathol. 2011;178:1416–28. doi: 10.1016/j.ajpath.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13:24. doi: 10.1186/s13024-018-0254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging. 2014;35:1012–23. doi: 10.1016/j.neurobiolaging.2013.10.089. [DOI] [PubMed] [Google Scholar]
- 115.Rehker J, Rodhe J, Nesbitt RR, Boyle EA, Martin BK, Lord J, et al. Caspase-8, association with Alzheimer’s disease and functional analysis of rare variants. PLoS ONE. 2017;12:e0185777. doi: 10.1371/journal.pone.0185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meneghini V, Bortolotto V, Francese MT, Dellarole A, Carraro L, Terzieva S, et al. High-mobility group box-1 protein and beta-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-kappaB axis: relevance for Alzheimer’s disease. J Neurosci. 2013;33:6047–59. doi: 10.1523/JNEUROSCI.2052-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fujita K, Motoki K, Tagawa K, Chen X, Hama H, Nakajima K, et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci Rep. 2016;6:31895. doi: 10.1038/srep31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kong ZH, Chen X, Hua HP, Liang L, Liu LJ. The oral pretreatment of glycyrrhizin prevents surgery-induced cognitive impairment in aged mice by reducing neuroinflammation and Alzheimer’s-related pathology via HMGB1 inhibition. J Mol Neurosci. 2017;63:385–95. doi: 10.1007/s12031-017-0989-7. [DOI] [PubMed] [Google Scholar]
- 119.Jang A, Liew H, Kim YM, Choi H, Kim S, Lee SH, et al. p35 deficiency accelerates HMGB-1-mediated neuronal death in the early stages of an Alzheimer’s disease mouse model. Curr Alzheimer Res. 2013;10:829–43. doi: 10.2174/15672050113109990135. [DOI] [PubMed] [Google Scholar]
- 120.Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002;5:633–9. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- 121.Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–8. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhu S, Zhang Y, Bai G, Li H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2011;2:e115. doi: 10.1038/cddis.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu JR, Wang J, Zhou SK, Yang L, Yin JL, Cao JP, et al. Necrostatin-1 protection of dopaminergic neurons. Neural Regen Res. 2015;10:1120–4. doi: 10.4103/1673-5374.160108. [DOI] [PMC free article] [PubMed] [Google Scholar]