

Abstract
Pyroptosis and necroptosis represent two pathways of genetically encoded necrotic cell death. Although these cell death programmes can protect the host against microbial pathogens, their dysregulation has been implicated in a variety of autoimmune and auto-inflammatory conditions. The disease-promoting potential of necroptosis and pyroptosis is likely a consequence of their ability to induce a lytic cell death. This cell suicide mechanism, distinct from apoptosis, allows the release of immunogenic cellular content, including damage-associated molecular patterns (DAMPs), and inflammatory cytokines such as interleukin-1β (IL-1β), to trigger inflammation. In this Review, we discuss recent discoveries that have advanced our understanding on the primary functions of pyroptosis and necroptosis, including evidence for the specific cytokines and DAMPs responsible for driving inflammation. We compare the similar and unique aspects of pyroptotic- and necroptotic-induced membrane damage, and explore how these may functionally impact distinct intracellular organelles and signalling pathways. We also examine studies highlighting the crosstalk that can occur between necroptosis and pyroptosis signalling, and evidence supporting the physiological significance of this convergence. Ultimately, a better understanding of the similarities, unique aspects and crosstalk of pyroptosis and necroptosis will inform as to how these cell death pathways might be manipulated for therapeutic benefit.
Subject terms: Interleukins, Cell death and immune response

Facts
Pyroptosis and necroptosis are lytic, inflammatory types of programmed cell death that require the membrane damaging GSDMD and MLKL proteins, respectively.
Pyroptosis and necroptosis protect against infections and can be triggered by pathogen and host molecules.
A number of DAMPs are released by pyroptotic and necroptotic cell death, which have the capacity to trigger inflammatory responses.
GSDMD and MLKL signalling both cause potassium efflux to trigger the NLRP3 inflammasome, resulting in IL-1β-driven inflammation.
Open Questions
Are there DAMPs that are released that are unique to pyroptosis and necroptosis?
What are the key necroptotic and pyroptotic DAMPs that contribute to inflammatory responses?
Does MLKL-induced NLRP3 inflammasome activity contribute to necroptotic-induced inflammatory disease?
How critical are GSDMD membrane pores for IL-1β release and inflammatory responses in different diseases?
Introduction
The idea of cells equipped with an internal instruction manual to die was first proposed back in 1965 from the study of developing insect cells [1]. Since then, extensive research has been carried out and has shed light on the significance and the mechanism of programmed cell death. It is now established that cells can commit suicide, either during development, as a consequence of pathogen invasion, or following cellular stress and metabolic disturbances. The three genetically defined cell death pathways that have been intensively investigated in recent years are apoptosis, necroptosis, and pyroptosis.
Apoptosis was the first programmed cell death described [2], and in most cases apoptotic effectors such as caspase-3 and -7 ensure that it remains immunologically silent [3–6]. In contrast, necroptosis and pyroptosis are lytic cell death modalities that allow the release of potential immunostimulatory molecules. Genetic evidence supports the idea that, when activated, these cell death pathways can induce potent inflammatory responses in vivo, and thereby may contribute to various inflammatory disease pathologies [7–10]. However, it is worth considering that the act of cell death, regardless of its mechanism, will invariably abrogate cell-intrinsic inflammatory cytokine production. This may reflect cell-type and stimulus-specific pyroptotic and necroptotic signalling scenarios and therefore, in some circumstances, the bystander DAMPs released following pyroptosis and necroptosis may be less inflammatory than if the cell did not receive the cues for suicide [11–16]. In this Review, we focus on the immunogenic nature of necroptosis and pyroptosis. We compare the similarities and differences in how the necroptotic and pyroptotic effectors induce membrane damage, and the mounting evidence documenting how these modes of cell death allow the subsequent release of inflammatory cytokines and DAMPs. We also discuss emerging studies suggesting that necroptotic crosstalk with the pyroptotic machinery may significantly contribute to the necroptotic inflammatory response.
An overview of pyroptosis
The act of pyroptosis was first described in 1992 [17], but the term was coined in 2001 following the observation that bacteria-infected macrophages underwent a rapid lytic cell death dependent on caspase-1 activity [18]. Although traditionally defined as caspase-1-mediated cell death, studies have revealed other caspases, caspase-11 and its human orthologs caspase-4 and -5 [19, 20], and more recently the apoptotic effector caspase, caspase-3 [21, 22], as being capable of triggering pyroptosis (Fig. 1).
Fig. 1.

Gasdermins form membrane pores to cause pyroptosis. A wide array of extracellular stimuli can drive pyroptosis. In the canonical model of pyroptosis, inflammasome sensor proteins, such as NLRP3, recognize cellular stressors, including those from bacteria, viruses, toxins, ATP, uric acid crystals, silica, and DAMPs. These stressors activate NLRP3 indirectly through potassium efflux, which leads to NEK7 binding NLRP3 to trigger its oligomerization. NLRP3 subsequently activates caspase-1 via the adaptor protein ASC. Caspase-1 processes and activates IL-1β and IL-18, and also cleaves GSDMD to release the membrane pore-forming GSDMD-N domain. GSDMD-N pores promote the release of activated IL-1β and IL-18 and, most likely, DAMPs that can be accommodated by the 10–20 nm pore diameter. Additional DAMPs will be released following the collapse of the plasma membrane. Cytosolic LPS binds Caspase-4/5/11 to trigger their cleavage of GSDMD, but not IL-1β and IL-18. In addition, recent research has revealed how the apoptotic effector caspase, caspase-3, can cleave GSDME to also cause pyroptotic death. DAMPs damage-associated molecular patterns, LPS lipopolysaccharide
The execution of pyroptotic cell death via these caspases is a result of their ability to cleave and activate specific members of the pore-forming gasdermin gene family, which incorporates six genes in humans and ten in mice. To date, caspase-1/4/5/11 have been reported to target gasdermin D (GSDMD) [23, 24], while caspase-3 can process gasdermin E (GSDME/DFNA5) [21, 22]. Gasdermin N- and C-terminal linker domain cleavage releases an activated N-terminal region from an inhibitory C-terminal fragment. As such, expression of the gasdermin-N domain alone from GSDMA, GSDMA3, GSDMB, GSDMAC, GSDMD and GSDME can signal cell death [23, 25], although proteases that might target GSDMA to GSDMC, and the physiological functions of these gasdermins, requires further study. Nevertheless, a body of evidence supports the idea that when released by proteolysis, the gasdermin-N domain binds to acidic phospholipids, such as phsophoinositides found on the inner leaflet of the mammalian plasma membrane, to form oligomeric death-inducing pores [25–28]. In vitro studies also suggest that gasdermins can target bacterial membranes to cause lysis, although in vivo reports indicate that bacteria survive the act of pyroptosis and are cleared by neutrophils [31, 32]. Regardless, we can now define mammalian cell pyroptosis as being gasdermin-dependent.
In the canonical model of caspase-1-mediated pyroptosis, recognition of inflammatory ligands leads to activation of intracellular multiprotein signalling complexes known as the inflammasomes (Fig. 1). Among the best studied inflammasome sensors are absent in melanoma 2 (AIM2), Pyrin, and the NOD-like receptor (NLR) family members, NLRP1, NLRP3, and NLRC4 [33]. The AIM2 inflammasome, for example, binds specifically to, and is activated by, cytosolic double-stranded DNA. On the other hand, the NLRP3 inflammasome responds to a diverse set of molecules, such as ATP, crystalline (e.g. cholesterol crystals) and viral components which, with few exceptions, trigger NLRP3 activity by causing potassium efflux and subsequent association of NEK7 (NIMA-related kinase 7) with NLRP3 [29, 34]. In many cases, including NLRP3, caspase-1 is recruited to the inflammasome sensor proteins via the CARD-domain containing adaptor protein, ASC, which triggers auto-processing of the inactive form of caspase-1 to its catalytically active species, p46 and p33/p10 subunits [35]. Activated caspase-1 cleaves and activates GSDMD, as well as the inflammatory cytokines, IL-1β and IL-18. In contrast to caspase-1, non-canonical inflammasomes are defined by their requirement for caspase-4/5/11, which have been reported to directly bind cytosolic LPS [20], resulting in their targeting and activation of GSDMD. Although caspase-4/5/11 do not process IL-1β and IL-18 directly [36], their activity does cause GSDMD-mediated potassium efflux, which suffices to induce canonical NLRP3 inflammasome formation and IL-1β activation [30, 37]. To date, caspase-1- and caspase-11-induced GSDMD pore formation appears requisite for the efficient release of activated IL-1β, although other membrane damaging pathways, such as those mediated by mixed lineage kinase domain like pseudokinase (MLKL) or monosodium urate crystals, have been documented to allow activated IL-1β release in the absence of GSDMD [24, 38, 39].
An overview of necroptosis
TNF superfamily receptors, such as TNF Receptor 1 (TNFR1), CD95, TRAIL-R1, and TRAIL-R2, can transmit a cell death signal upon ligand binding via a conserved cytosolic death domain, and are therefore known as death receptors (DRs). The first genetic determinant of DR-induced necroptosis, RIPK1, was discovered by Holler et al. [40] 18 years ago. However, the term used to describe this non-apoptotic cell death pathway, necroptosis, was not coined until 2005 by Degterev et al. [41]. In this study, a novel compound was identified, necrostatin-1, that blocks the ability of RIPK1 kinase to induce necroptosis in TNF-treated cell lines. This discovery sparked tremendous interest in the cell death community to further characterize the pathway and, in less than a decade, the two downstream core components of the necropototic machinery, RIPK3 and MLKL, have been identified [7] (Fig. 2). Nevertheless, the quest to acquire a complete picture of the pathway is an ongoing adventure, highlighted by the identification of post-translational regulators [42], and experiments showing that activated MLKL does not necessarily commit a cell to death [43].
Fig. 2.
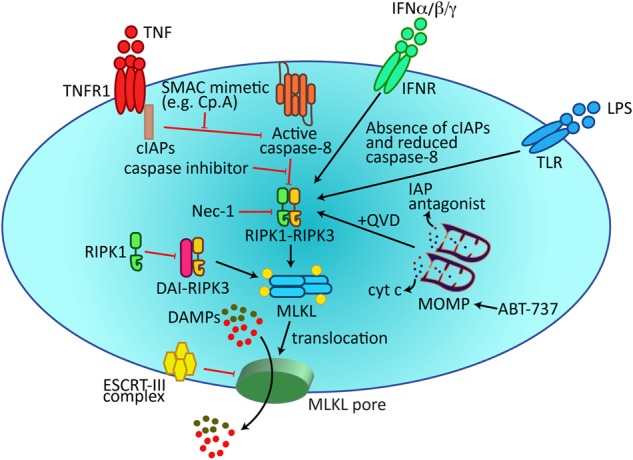
Membrane-associated MLKL induces necroptosis. Necroptosis can be triggered by death receptors (e.g. TNFR1), the IFNR, and TLR3/4, which promote the assembly of a RIPK1-RIPK3-MLKL signalling complex. RIPK3-mediated phosphorylation of MLKL results in MLKL translocation to the plasma membrane to induce membrane damage. Damaged membrane shedding, and cell death, is limited by the ESCRT-III complex. Recent findings have documented additional necroptotic triggers, such as BH3-mimetic (ABT-737)-induced MOMP or ZBP1/DAI-induced RIPK3 dimerization. cIAPs cellular inhibitor of apoptosis proteins, DAI DNA-dependent activator of IFN-regulatory factors, DAMPs damage-associated molecular patterns, ESCRT-III endosomal sorting complex required for transport (ESCRT) complex III, INFR interferon receptor, LPS lipopolysaccharide, MOMP mitochondrial outer membrane permeabilization, Nec-1 Necrostatin-1, Smac mimetic; an IAP antagonist, QVD; a pan-caspase inhibitor
Necroptosis is frequently observed when DR apoptotic signalling components are rendered non-functional. For example, most studies to date trigger DR necroptotic killing by genetic or chemicial inhibition of the cellular inhibitors of apoptosis proteins (cIAP1/2) and caspase-8, which normally act to ubiquitylate (cIAP1/2) or cleave (caspase-8) RIPK1/3 to limit necroptosis [44]. However, when caspase-8 and/or cIAPs are removed, the activation of RIPK1/3 by DRs (e.g. TNFR1, CD95, TRAIL-R), Toll-like receptors (i.e. TLR3 or TLR4), or the cytosolic Z-DNA/Z–RNA sensing receptor, Z-DNA binding protein 1 (ZBP1/DAI/DLM-1), can directly induce necroptosis. Activation of these receptors triggers the formation of a RIPK1-RIPK3 cell death platform, termed the necrosome, via RIPK1/3 RHIM-RHIM domain interactions that adopt a hetero-amyloid structure [45]. Subsequent RIPK3-mediated phosphorylation of the necroptotic executioner MLKL [46, 47] triggers MLKL oligomerization and membrane association, resulting in plasma membrane damage and the release of potential DAMPs [48]. Although RIPK1 and its kinase activity is essential for necroptosis in many conditions, recent studies have uncovered how a RIPK1 scaffolding function, in particularly the RHIM, acts as an essential inhibitor of lethal necroptotic killing by preventing ZBP1 binding and oligomerization of RIPK3 [49, 50]. It is also pertinent to note that both RIPK1 and RIPK3 have several non-necroptotic transcriptional (e.g. inflammatory cytokine induction) and post-translational (e.g. activation of apoptotic cell death) functions [51]. Therefore, mammalian necroptosis is best defined by its requirement for MLKL.
Pyroptosis and necroptosis: death with a difference?
A body of evidence shows that both pyroptosis and necroptosis represent lytic cell death pathways that are inflammatory in nature. However, as suggested by their different genetic determinants, pyroptotic and necroptotic killing may also serve distinct purposes. For example, necroptosis is mostly observed as a backup cell death defence mechanism that is triggered when apoptosis is hindered, such as during pathogen infection [52, 53], which is highlighted by caspase-8 inhibition of lethal necroptotic signalling [54, 55]. On the other hand, pyroptosis is a primary cellular response following the sensing of potentially damaging insults, which include pathogen ligands, DAMPs, altered levels of host metabolites and environmental irritants.
GSDMD versus MLKL: mechanism of membrane damage
Activation of GSDMD is mediated by proteolytic cleavage, whereas a phosphorylation event is essential for MLKL activation during necroptosis. RIPK3 phosphorylation of MLKL will shift the equilibrium from inert cytoplasmic monomeric MLKL to membrane-associated MLKL oligomers [56–58]. The monomer-to-oligomer transition as an essential step in the capacity of MLKL to induce membrane damage has also been observed in the GSDMD-N domain [25, 28, 59]. Whether GSDMD and MLKL-induced membrane damage are sufficient to allow the release of DAMPs and other small soluble cytosolic factors prior to complete cell lysis, and their physiological relevance, is a topic of ongoing interest. However, reports have suggested that pre-lytic GSDMD pores may allow ion flux, or even cytokine release, prior to plasma membrane rupture [60]. Several studies have further hinted on this possibility, for instance, with MLKL/GSDMD-mediated or GSDMD-independent IL-1β release being detected prior to cellular lysis, or even in the complete absence of cell death [38, 61–66].
The GSDMD-N domain oligomers form membrane pores with ring diameters of ~10–20 nm [25, 27], while cryo-electron microscopy indicates that the GSDMA-N domain forms a 26- to 28-fold single ring anti-parallel β-barrel 18 nm pore [67]. Unlike GSDMD, the mechanism of the membrane damage resulting from activated MLKL is less clear. RIPK3-mediated phosphorylation of the MLKL activation loop within the pseudokinase domain unleashes the killer, oligomeric and membrane associated, N-terminal 4-Helical Bundle (4HB) domain. The ability of the MLKL 4HB domain to permeabilize liposomes suggests it has direct pore forming, or membrane damaging capacity [68–70], and the size of MLKL-initiated membrane perforations have been estimated at ~4 nm [71]. However, other studies have suggested that activated MLKL binds ion channels and induces ion flux [56, 57], or can form cation channels [72]. Moreover, mutant MLKL constructs have been documented to oligomerize and associate with membranes, yet fail to induce cell death, implying that additional factors may be required for membrane perforation [58]. More recently, it was reported that activated MLKL formed disulphide bond-dependent amyloid-like fibres that were required for necroptotic killing [73]. Whether these MLKL fibres recruit additional necroptotic factors to cellular membranes, or directly engage membrane binding and damage, remains unclear.
MLKL and GSDMD killing can result in distinct morphologies [59, 60]. This may be explained by variation in MLKL and GSDMD membrane targeting and pore formation, or experimental differences in the kinetics of necroptotic and pyroptotic killing. The non-selective diffusion of ions triggered by GSDMD-N pore formation has been suggested to explain the reduced cell swelling and flattened cytoplasm during pyroptosis, and cells undergoing pyroptosis remain adherent until the plasma membrane is compromised [59, 60]. In contrast, necroptotic signalling results in cellular detachment, and ion-selective MLKL damage has been reported to impact intracellular osmolarity and cause cell swelling and subsequent osmolysis [59]. However, as both cell death mechanisms invariably rupture the plasma membrane, Annexin V staining of phosphatidylserine does not distinguish between apoptosis, pyroptosis, or necroptosis [43, 74, 75].
GSDMD versus MLKL: membrane association and subcellular localization
The killer GSDMD-N domain has been reported to bind phosphoinositides (PIPs), such as PI(4,5)P2 and PI(3, 4,5)P2, which are found on the inner leaflet of the plasma membrane, and also to cardiolipin [25, 28, 59]. Cardiolipin is present in bacterial membranes and may therefore also explain the direct in vitro bacterial killing capacity of the GSDMD-N domain [25, 28]. The presence of GSDMD binding PIPs on numerous organelles, the co-fractionation of the GSDMD-N domain to intracellular membranes, such as endosomes and lysosomes, and the observation that lysosome and mitochondrial damage occurs during pyroptotic signalling prior to plasma membrane rupture [25, 60], suggests that GSDMD might target and perforate multiple organelles, not just the plasma membrane.
The lipid binding profile of MLKL has been reported to be similar to GSDMD, with studies demonstrating that MLKL can bind to cardiolipin and negatively charged PIPs to enable its redistribution to the plasma membrane [57, 68, 69]. In particular, it has been suggested that MLKL activation first triggers MLKL 4HB domain low-affinity PIP binding to enable plasma membrane targeting, which subsequently exposes high-affinity MLKL 4HB domain PIP-binding sites, with a preference for PI(4,5)P2 interactions [70]. Other than the plasma membrane, localization studies have implicated MLKL in targeting a diverse number of cellular organelles, including lysosomes, mitochondria, the endoplasmic reticulum [68] and the nucleus [76]. Whether MLKL functionally impacts these other organelles has yet to be thoroughly assessed. However, emerging studies have implicated nuclear MLKL in limiting its necroptotic killing potential [77], mitochondrial MLKL in RIPK3-mediated induction of aerobic respiration [78, 79], while activated MLKL may disrupt lysosomal function [80] and also promote vesicle trafficking and extracellular release [43, 74, 81]. Notably, the latter event has been reported to allow shedding of activated MLKL, thereby removing damaged membranes and promoting cellular survival [43, 81]. Whether similar membrane release functions exist to limit gasdermin killing has yet to be documented, although like necroptosis, membrane blebbing has been observed during pyroptotic killing [32, 60].
What are the primary functions of GSDMD-mediated pyroptosis?
Gain-of-function mutations in inflammasome sensor proteins, such as NLRP3, Pyrin, NLRC4 and NLRP1, all result in auto-inflammatory disease that is typically treated by blocking IL-1 activity. Although the initial characterization of GSDMD documented its importance in IL-1β and IL-18 release from macrophages [23, 24], whether GSDMD mediated IL-1β release in vivo was a key determinant in inflammasome-driven disease remained unclear. Recently, mice expressing activating mutant Pyrin, responsible for Familial Mediterranean Fever (FMF) in humans that is successfully treated with anti-IL-1 biologics, were crossed to GSDMD deficient animals [82]. The complete amelioration of disease symptoms, such as elevated IL-1β, runting, tissue injury, anaemia, neutrophilia and splenomegaly, demonstrated that an important in vivo function of GSDMD, and by extension pyroptosis, is to allow the exit of inflammasome-activated cytokines such as IL-1β.
Other inflammasome-driven auto-inflammatory disease model mice have yet to be crossed to GSDMD deficient mice to define if disease pathology is rescued to the same extent as IL-1β and/or IL-18 loss. However, NLRP3 mutant mice resembling CAPS are better protected following the loss of caspase-1 compared to the combined deletion of the IL-1R and IL-18 [83]. This highlights that in some autoinflammatory disease models, pyroptosis and/or DAMP release may play a significant role in disease pathology. In line with this idea, haematopoietic progenitor cell pyroptosis is observed in NLRP1a auto-activating mice that is also independent of IL-1 [84].
The loss of inflammasome sensors, adaptors, caspases, and cytokines, have documented their importance for the control of many pathogen infections, including bacterial, fungal, and protozoan [10]. Distinguishing the functions of pyroptosis versus inflammasome-activated cytokines is challenging as both are initiated by caspase-1/4/5/11 signalling. However, recent evidence supports the idea that pyroptosis is responsible for HIV-induced CD4 T-cell death [85], and the clearance of intracellular infections. For example, elegant genetic experiments comparing IL-1β and IL-18 deficiency with caspase-1/11 loss have documented that pyroptosis is required for optimal control of bacterial infections, including Legionella pneumophila, Bulkholderia thailandensis, Chromobacterium violaceum, and flagellin-expressing Salmonella typhimurium [31, 86–88]. Macrophage pyroptotic structures termed pore-induced intracellular traps (PITS) have been described to capture intracellular bacteria to promote bacterial clearance, akin to neutrophil extracellular traps (NETS), and it has been suggested that necroptotic-induced structures may play a similar role [32]. Interestingly, chemical induction of macrophage apoptosis upon Legionella infection prevents lethal disease and also triggers bacterial clearance, implying that cell death of pathogen-containing cells, regardless of its modality, can act as a potent anti-pathogen immune response [89].
The role of the terminal effector of pyroptosis, GSDMD, in infection has yet to be examined in detail. However, one recent study reported that diminished neutrophil killing resulting from GSDMD deficiency enhances host responses against Escherichia coli infection [14]. Whether pyroptosis is beneficial or harmful to the host may be cell type and context-dependent, and therefore warrants further research.
What are the primary functions of MLKL-mediated necroptosis?
Evidence to date supports the hypothesis that mammalian necroptosis primarily exists to counteract pathogen infections, to destroy the microbial replicative niche and/or to trigger an inflammatory anti-microbial response through the release of DAMPs. First, necroptosis is triggered by PRRs, such as DAI and TLRs, which have evolved to sense pathogen molecules. Second, the physiological engagement of necroptosis can occur in vivo following infection, and can serve as an efficient back-up cell death mechanism when apoptosis is inhibited by pathogen molecules [53, 90]. Third, both viral and bacterial pathogens have been shown to directly target the necroptotic machinery to prevent cell death, suggesting that pathogen fitness is enhanced by limiting necroptotic killing and/or the ensuing host inflammatory response [91]. Fourth, unlike deletion of essential apoptotic effectors, genetic deletion of the core necroptotic machinery, RIPK3 and MLKL, has no significant role in animal development. This phenotype, or lack thereof, is similar to inflammasome-deficient animals, which display no abnormalities until challenged with infectious or damaging environmental agents.
The genetic deletion of inhibitors of necroptosis, such as RIPK1, FADD, and Caspase-8, has provided proof of MLKLs ability to trigger tissue damage and induce potent inflammatory responses [8]. This implies that the unwanted engagement of necroptosis may contribute to inflammatory-driven diseases, similar to the disease-inducing potential of excessive inflammasome signalling. In line with this idea, necroptotic MLKL signalling has been implicated in various pathological settings, such as TNF-induced shock [92–94] and ischaemia-reperfusion injuries involving the kidney and heart [95–97]. Several other inflammatory conditions, such as atherosclerosis, have also been linked to RIPK1 and/or RIPK3 signalling, although a role for MLKL has yet to be defined [8].
Necroptotic to pyroptotic crosstalk: is the NLRP3 inflammasome a driver of necroptotic-induced inflammation?
As documented above, pyroptosis and necroptosis appear to benefit mammalian health by inducing the death of infected or damaged cells, and by their capacity to activate inflammatory signalling pathways. Genetic evidence indicates that a primary function of GSDMD-mediated pyroptosis is to allow the release of the caspase-1 activated cytokines, IL-1β and IL-18. Therefore, the question arises as to whether necroptosis-induced inflammation might also be driven by the release of inflammasome-activated cytokines? Several lines of evidence lead credence to this hypothesis. Initial studies demonstrated that the deletion of genes linked to human inflammatory diseases, the inhibitor of apoptosis proteins (IAPs) [98–100], and the ubiquitin-editing enzyme A20 [101], induced RIPK3 to promote NLRP3-caspase-1-mediated IL-1β secretion. Subsequently, genetic experiments, including the use of MLKL and inflammasome knockout macrophages, confirmed that necroptosis signalling could trigger a RIPK3-MLKL-NLRP3-Caspase-1 axis that resulted in IL-1β maturation [38, 39, 65, 102] (Fig. 3). Notably, single cell analysis documented that activated MLKL triggered NLRP3 in a cell intrinsic manner, thereby eliminating the potential for NLRP3 to be activated as a consequence of DAMP release [38]. Mechanistically, membrane-associated MLKL caused potassium efflux to induce NLRP3 signalling, in line with how NLRP3 is activated by chemically and structurally diverse stimuli.
Fig. 3.

Necroptotic signalling activates the NLRP3 inflammasome to drive inflammation. Both the pyroptosis and the necroptosis pathways can activate the NLRP3-IL-1β signalling axis. Data support the hypothesis that both caspase-4/5/11 induction of GSDMD pores, or MLKL-induced membrane damage, cause potassium efflux. As a result, NEK7 binds and triggers NLRP3 inflammasome formation. Activated IL-1β is subsequently released from pre-lytic GSDMD and MLKL membrane perforations that may also accommodate appropriately sized DAMPs. Subsequent cellular rupture allows the release of larger DAMPs. DAMPs; damage-associated molecular patterns
The first in vivo evidence that necroptotic-induced IL-1 is important for necroptotic-driven inflammation resulted from the restricted deletion of the key RIPK3-MLKL necroptotic repressor, caspase-8, in dendritic cells (DCs) [103]. Unlike the RIPK3-MLKL-driven embryonic lethality resulting from whole animal caspase-8 loss, DC-restricted caspase-8 deletion was well tolerated. Strikingly, however, the loss of DC caspase-8 dramatically sensitized mice to endotoxic shock that was prevented by inhibition of RIPK1 kinase activity, co-deletion of RIPK3, or downstream inhibition of IL-1 and, in vitro, MLKL silencing. On the other hand, TNF neutralization had no impact. This study is important because it demonstrates that necroptotic-induced disease may, in some circumstances, be inhibited by targeting a single downstream inflammatory mediator. More recently, it was reported that MLKL activation in the intestinal mucosa was required for inflammasome-mediated protection against Salmonella infection [104], while Staphylococcus toxin-induced MLKLtriggered NLRP3 activation to promote damaging inflammation [105]. It will be interesting to define which other necroptotic disease models may also respond to NLRP3 inflammasome inhibition, or whether there are other specific DAMPS, such as HMGB1, whose targeting may be useful to limit damaging, necroptotic-induced, inflammation.
Sterile DAMPs associated with pyroptosis and necroptosis, and the evidence for their physiological significance
Pyroptotic and necroptotic death pathways both allow the release of immunogenic cellular content (DAMPs), which have been suggested as direct activators of PRRs and immune cells that can trigger inflammatory responses (Table 1). While this is certainly true for members of the IL-1 family, such as IL-33 and IL-1α, the in vivo evidence for direct inflammatory functions of other proposed DAMPs has been controversial. For example, heat shock proteins (HSPs) have been suggested to limit inflammation, with HSP70 expression conferring protection against the TNF shock model [106] that is mediated by necroptotic signalling. Similarly, HMGB1 has been suggested to act as an indirect amplifier of TLR-induced inflammation, not a direct ligand [107], and in some inflammatory models thought to be driven by HMGB1, its genetic deletion has little impact [108]. Perhaps surprisingly, it has been assumed that the types of DAMPs released following necroptosis or pyroptosis are similar, independent of the molecular mechanism that triggers membrane rupture. In this section, we examine DAMPs that activate caspase-1 and pyroptotic responses, the DAMPs released as a result of necroptotic or pyroptotic killing, and the evidence to support their physiological significance.
Table 1.
Sterile DAMPs associated with inflammasome, pyroptosis, and necroptosis signalling
| DAMPs | Experimental validation | References |
|---|---|---|
| HMGB1 | Observed in both pyroptosis and necroptosis, and has been reported to propagate inflammation in vitro and in vivo during both these modes of cell death. | [111, 114, 115, 118, 120, 125] |
| S100a9 | Not reported in pyroptosis. Released during necroptosis but its significance remains undetermined. | [129] |
| HSPs (e.g. HSP70 and HSP90) | HSP90 is required for both NLRP3 inflammasome and necroptotic signalling, while HSP70 expression can protect from TNF-induced shock, which is mediated by both necroptotic and apoptotic cell death. Potential DAMP role not determined. | [106, 130–132] |
| ATP | Released during necrotic cell death and chemotherapy-induced necroptosis. At high concentrations ATP is a potent activator of the NLRP3 inflammasome. | [109, 118, 134]. |
| Mitochondria/mtDNA | Released following TNF-induced apoptotic/necroptotic cell death in vivo in a RIPK3-dependent manner. Necroptotic disease relevance unclear. Cytochrome C released following pyroptosis, suggesting extracellular mtDNA will also be present. Cytosolic mtDNA preferentially activates the AIM2 inflammasome. | [60, 92, 138, 139, 143]. |
| ASC specks | ASC particles released during pyroptosis can be phagocytosed to propagate inflammasome activation in neighbouring cells. Detected in human and murine inflammatory disease serum in vivo, but physiological relevance yet to be clarified. Potential to occur following MLKL-induced inflammasome activation not yet reported. | [126, 127] |
| IL-1α | Can be released in a caspase-1 dependent manner during pyroptosis. Also released from macrophages during necroptosis, RIPK3/caspase-8 apoptotic signalling, or apoptotic bodies. Implicated in numerous cell death-associated pathologies. | [38, 92, 98, 100, 146, 153, 168] |
| IL-33 | Necroptosis increases serum levels of cleaved IL-33 in vivo, predicted to be biologically active. LPS lethality is reduced in IL-33 deficient animals. | [112, 120, 129] |
| Cholesterol and MSU crystals | Activate the NLRP3 inflammasome to promote atherosclerosis (cholesterol crystals) and gout (MSU crystals). Evidence accumulated using mouse models supported by clinical studies targeting IL-1. | [148, 164–166] |
DAMPs damage-associated molecular patterns, HMGB1 high-mobility group box 1 protein, HSP heat shock protein, mtDNA mitochondrial DNA, MSU monosodium urate
Pyroptosis and HMGB1
The injection of high-dose LPS into mice has been documented to cause lethality through caspase-11 and GSDMD pyroptotic activity [19, 24]. GSDMD-induced NLRP3 activity also likely impacts LPS responses in vivo, as loss of NLRP3 [109], or combined targeting of IL-1β and IL-18 [110] can also protect against LPS killing. However, evidence suggests that these inflammasome-associated cytokines, and NLRP3 itself, are dispensable at higher doses of LPS, or under different laboratory/environmental conditions [19, 111], implicating pyroptotic cell death and/or the combined actions of other inflammatory mediators, such as IL-33 [112] and TNF [113], in murine endotoxic shock lethality.
Inflammasome-mediated pyroptosis promotes the release of HMGB1, and the neutralization of HMGB1 has also been reported to protect against LPS lethality [114, 115], even when killing occurs independent of IL-1β and IL-18 [111]. Similarly, the activation of NLRC4-driven pyroptosis triggers HMGB1 release, with the inflammatory functions of released HMGB1 dependent on HMGB1 acetylation and disulphide bonding [116]. Interestingly, extracellular HMGB1 may further propagate inflammatory responses through bystander macrophage endocytosis, resulting in lysosomal rupture and caspase-1 activation [117]. Perplexingly, however, the genetic deletion of HMGB1 in interferon responsive cells, as opposed to the use of neutralizing antibodies, did not alter LPS-induced lethality despite significantly reduced serum HMGB1 levels [108]. It is possible that unknown intracellular functions of HMGB1 are important for protecting against target cell LPS responses, similar to HSP70 [106]. Regardless, the genetic loss of HMGB1 has confirmed it as a bona fide DAMP, as its deletion significantly reduces neutrophil recruitment following necrotic tissue damage, and confers protection from hepatic ischaemia reperfusion- and acetaminophen-induced liver injury.
Necroptosis and HMGB1
Unlike pyroptosis, a role or HMGB1 in necroptotic-induced inflammation is less clear. While HMGB1 can clearly contribute to detrimental inflammation in necrotic mouse models [48, 118, 119], the levels of plasma HMGB1 in neonate RIPK1-deficient mice was reported as similar to wild-type animals, despite other inflammatory markers, such as IL-33, being elevated in a RIPK3- and MLKL-dependent manner [120]. In fact, it has been suggested that HMGB1-RAGE-induced transcriptional responses may suppress LPS-induced necroptosis by limiting TLR4 receptor expression levels [121]. Nevertheless, the activation of necroptosis signalling in cancer cell lines has been reported to trigger immunogenic cell death that can enhance tumour immunity. In this context, it has been documented that, in some cancer cell types, RIPK3 and MLKL are required for optimal release of HMGB1 and ATP from tumour cells killed by chemotherapy, and that these DAMPs generate an efficient anti-cancer immune response [118]. In other cancer cell types, RIPK1-RIPK3 activation of NF-κB and cytokine production, not necroptotic DAMP release, has been suggested as the key determinant for efficient anti-cancer immune responses [122]. It will, therefore, be informative to define if, in cancer models treated with necroptotic stimuli [123, 124], DAMPs such as HMGB1 and ATP contribute to cancer immunity.
Two inflammatory disease animal models that have implicated MLKL and necroptosis in disease pathology are TNF-induced systemic inflammatory response syndrome (SIRS) and kidney-ischaemia reperfusion injury [97]. Although in vitro TNF-induced necroptosis can result in HMGB1 release, its role in TNF SIRS severity remains to be addressed. Interestingly, however, the neutralization of HMGB1 can significantly protect from kidney-ischaemia reperfusion injury, while the administration of recombinant HMGB1 can exacerbate kidney damage in this model [125]. Therefore, necroptotic-mediated HMGB1 release may contribute to ischaemia-reperfusion pathology.
ASC specks
Following pyroptosis, released ASC specks have been reported to act in a DAMP-like manner. For example, they have the capacity to activate extracellular caspase-1, as well as be engulfed by bystander macrophages, resulting in lysosomal rupture and subsequent engagement of cytosolic caspase-1 [126, 127]. The injection of ASC specks into mice is also sufficient to generate inflammatory responses. Notably, ASC specks have been detected in broncoalveolar lavages from inflamed lungs and bacterially infected mice, as well as the serum of CAPS patients, who express gain-of-function NLRP3 mutations and are successfully treated with anti-IL-1 biologics [126, 127]. Whether extracellular ASC specks act as a bona fide DAMP to influence inflammatory responses in these diseases, however, requires further research.
S100
The S100 protein consists of 24 members that exhibit diverse functions, including in inflammation by acting as proinflammatory DAMPs during accidental necrosis [128]. Although the expression level of one of the members, S100a9, was upregulated in caspase-8-deficient epidermal keratinocytes [129], it is not clear at this stage whether the increase was related to necroptosis. There is currently no report regarding the presence of this DAMP during pyroptosis.
HSPs
Currently there is a lack of data available examining the release of these immunogenic proteins during pyroptosis and necroptosis, even though these chaperones have been frequently associated with cell death. HSP70 release, for instance, was only reported after necrotic cell death [130], and its expression actually confers protection against the TNF shock model [106] that is mediated by necroptotic killing [97]. Intracellular roles of HSP90 include essential functions in activating the NLRP3 inflammasome [131] and necroptotic cell death [132]. However, whether this chaperone is released in the extracellular milieu and retains DAMP-like functions in both scenarios has not been determined.
ATP
Cell death and tissue damage results in high extracellular levels of ATP which can act as an immunomodulatory DAMP, including a potent ability to trigger activation of the NLRP3 inflammasome and pyroptosis in innate immune cells [133]. In murine cells, TLR stimulation by ligands, such as LPS, primes cells for inflammasome activation by inducing NLRP3 and IL-1β expression, but is insufficient for inflammasome activation. However, high levels of extracellular ATP act on the purinergic receptor, P2X7R, to trigger potassium ion efflux and NLRP3 inflammasome activity [109]. On the other hand, in human monocytes, LPS alone can stimulate both IL-1β synthesis (priming) and inflammasome activation. It has been suggested that LPS stimulates ATP release, which then acts in an autocrine manner to activate the P2X7R and NLRP3 signalling [134]. However, more recent reports indicate that LPS-induced IL-1β activation in human monocytes results from a TLR-caspase-8-NLRP3 signalling axis that occurs independent of ATP release and cell death [135]. It has also been suggested that chemotherapeutic-induced tumour cell death and ATP release triggers P2X7R-induced NLRP3 activation in DCs to promote anti-tumour immune responses [118, 136]. Similarly, pressure-disrupted necrotic cells can trigger bystander cell NLRP3 inflammasome activation in vitro and in vivo, and this appears to result from the ability of NLRP3 to respond to released mitochondrial ATP [137].
Mitochondrial DNA
Another DAMP that has the ability to trigger innate immune responses is mitochondrial DNA (mtDNA). It was suggested that blockade of mitophagy and the accumulation of damaged mitochondria, or canonical NLRP3 inflammasome activators themselves, trigger mitochondrial apoptosis to promote oxidized mtDNA release, which subsequently binds and activates NLRP3 [138, 139]. However, experiments revealed that mtDNA, if present in the cytosol, is most efficient in activating the DNA-sensing AIM2 inflammasome [138–140]. Moreover, deletion of the essential mitochondrial apoptotic effectors BAX and BAK had no influence on the capacity of canonical NLRP3 activators to trigger NLRP3 signalling [141]. On the other hand, it has been reported that downstream of NLRP3 and AIM2 inflammasome formation, caspase-1 mediates mitochondrial damage and inhibits mitophagy [142]. The block in autophagy-mediated mitochondrial degradation suggests that mitochondrial components, such as mtDNA, are likely to accumulate and be released into the extracellular milieu during pyroptosis, albeit this has yet to be experimentally tested. However, cytochrome C has been detected in the media following pyroptotic death in vitro, implying that mtDNA will also be released [60].
Whether mtDNA acts as a bona fide DAMP following necroptotic killing has yet to be determined. It was recently documented that TNF-induced necroptosis results in release of extracellular intact mitochondria, and that these mitochondria can be phagocytosed by macrophages and DCs to induce cytokine production and cellular maturation, respectively [143]. This observation contrasts findings suggesting that necrotic cell death triggers the release of mtDNA to signal inflammation [144], and results documenting the detection of mtDNA in the plasma of TNF-challenged wildtype, but not RIPK3-deficient, mice [92]. Whether necroptotic-induced mitochondria or mtDNA release significantly impacts necroptotic inflammatory responses in vivo is unclear.
IL-1α
The spectrum of immune consequences resulting from IL-1R signalling is not only IL-1β-dependent. IL-1α activates the same cell surface receptor as IL-1β, IL-1R, and is frequently released from dying cells, including downstream of inflammasome activation [145]. However, caspase-1 does not target IL-1α for proteolysis. Instead, caspase-1 degrades the IL-1R2 that is bound to IL-1α, thereby releasing IL-1α and allowing its processing by macrophage calpain-like proteases to a p17 fragment that has increased biological activity [146, 147]. However, it has been reported that full-length IL-1α also possesses biological activity and its release is often observed in cell death-associated conditions, such as atherosclerosis and ischaemia-reperfusion injury, for which both necroptotic and pyroptotic signalling components have been implicated [97, 148–152]. Notably, elevated serum and peritoneal lavage fluid IL-1α (and IL-1β) is also observed following sensitization to apoptotic, LPS-induced, caspase-8 activation, both in vitro and in vivo, implying that IL-1α has the potential to trigger an inflammatory response regardless of the cell death mechanism [98, 100]. This idea is consistent with findings documenting the inflammatory potential of IL-1α containing apoptotic bodies [153].
Macrophages undergoing TNF-induced necroptotic death also release IL-1α [38]. In the TNF-induced shock model, IL-1α serum levels are elevated in wildtype, RIPK3- and MLKL-deficient animals, but do not increase with TNF injection upon loss of both MLKL and Caspase-8, consistent with this model triggering both necroptotic and apoptotic cell death [97]. Similarly, the plasma of RIPK1-deficient animals contained greatly elevated levels of IL-1α that is a likely consequence of MLKL and caspase-8 killing [154]. RIPK3 loss was reported to protect from cecal ligation and puncture (CLP)-induced sepsis, and this also correlated with reduced blood cytokine levels, including IL-1α [92]. Of note, loss of the inflammasome activated cytokines, IL-1β and IL-18 protect against CLP lethality, but in a caspase-1 and -11 independent manner [110], implicating other pathways, such as caspase-8, in their cleavage-induced activation [98, 155]. Although the contribution of IL-1α to CLP animal mortality was not examined in this study [110], combined therapeutic targeting of IL-18, and anakinra to inhibit both IL-1α and IL-1β, afforded greater protection from CLP-induced killing than genetic loss of just IL-1β and IL-18. Whether IL-1α, IL-1β and IL-18 activation and release in this model reflects necroptotic and/or apoptotic signalling remains unclear [156].
IL-33
IL-33, like IL-1α, is a chromatin-associated member of the IL-1 family implicated in innate and adaptive immune responses. During apoptosis, IL-33 is cleaved by apoptotic caspases to destabilize the protein and limit its biological activity [157]. Although IL-33 was suggested to be activated by caspase-1 processing [158], subsequent studies have reported that IL-33 is not a substrate for caspase-1[157, 159], or that caspase-1 can inactivate it [160]. Regardless, upon necrosis induction, through agents like hydrogen peroxide or sodium azide treatment, full-length IL-33 is readily detected in the cell supernatant, and recombinant full-length IL-33 is an efficient activator of the interleukin-1 receptor family member, ST2 [157]. Notably, an IL-33 fragment, with a molecular weight likely reflecting increased biological activity, was detected in the plasma of RIPK1-deficient mice that was dependent on the presence of RIPK3 and MLKL, but not caspase-8 [154]. Together with an earlier observation highlighting increased IL-33 expression in necroptotic epidermal keratinocytes [129], these findings implicate IL-33 as a necroptotic DAMP. It remains feasible that in the above genetic models studied, IL-33 release reflects the indirect actions of necroptotic killing. The potential for direct pyroptotic-induced IL-33 release also remains to be clarified. However, LPS-induced lethality is reduced in IL-33-deficient mice [112] implying that IL-33 may contribute to the pyroptotic-generated inflammatory response. Nonetheless, in other inflammatory models, such as allergic asthma, caspase-1 has been reported to either limit [161], or increase [162], IL-33 levels.
Cholesterol crystals and MSU
Cholesterol crystals are a feature of atherosclerotic plaques and can be phagocytosed by macrophages to activate the NLRP3 inflammasome. The loss of NLRP3 and IL-1 signalling significantly protects against atherosclerosis in low-density lipoprotein (LDL)-receptor-deficient mouse models [148]. Additional DAMP molecules, including fatty acids and oxidized LDL, have also been implicated in NLRP3 activation in the context of cardiovascular disease [163]. In line with sterile DAMPs triggering inflammasome-driven inflammation that can contribute to coronary heart disease, the recent CANTOS trial results demonstrated the clinical efficacy of inhibiting IL-1β to protect from cardiovascular events [164].
MSU crystallization in joints acts as a potent DAMP that causes the inflammatory arthritis termed gout. Like cholesterol crystals, MSU crystals are phagocytosed by macrophages to induce lysosomal rupture and subsequent NLRP3 inflammasome activity [165]. Mouse models suggested that targeting NLRP3 and IL-1 would be beneficial in the treatment of gout, with subsequent clinical trials using anti-IL-1 biologics proving this to be correct [166]. Therefore, there is significant evidence implicating mammalian particulate DAMPs as inducers of IL-1-driven disease. Whether GSDMD contributes to these pathologies will be of significant interest to determine [167].
Conclusions
A significant body of evidence implicates both necroptotic and pyroptotic killing in host protection from infectious pathogens. As demonstrated by the apoptotic clearance of intracellular pathogens [89], any mode of cell death has the capacity to destroy a pathogens replicative niche and expose it to the immune system. The potential advantage conferred by pyroptosis and necroptosis is the release of immunogenic molecules that can contribute to anti-microbial activity, or even anti-cancer immune responses. At face value, therefore, both modes of lytic cell death appear remarkably similar. However, while it has been established that a principle function of the GSDMD pores are to ensure the efficient release of activated IL-1β and IL-18, it remains unclear whether MLKL-induced membrane perforations are similar in nature and can also target the release of specific disease-causing inflammatory mediators. Although initial studies suggest that necroptosis can drive pathological NLRP3-caspase-1 and IL-1β activation in innate immune cells, the key inflammatory DAMPs and cytokines released following necroptotic killing of cell types that do not express the inflammasome machinery will be important to investigate. Moreover, the physiological roles of pyroptotic gasdermins other than GSDMD remain very much unclear. Whether sub-lethal gasdermin activation and targeting to distinct intracellular membranes can occur, as reported for MLKL, will be important to investigate. Clearly, the uncovering of these new cell death effectors, MLKL and gasdermins, has sparked renewed interest into how distinct cell death modalities sculpt developmental and immune responses. A greater understanding of their mechanisms and physiological roles will inform as to how these new cell death effectors might be targeted for the treatment of inflammatory conditions, or triggering of cancer cell death and anti-cancer immune responses.
Acknowledgements
D.F. is supported by a Melbourne Research Scholarship from The University of Melbourne (Australia). J.E.V. is supported by a NHMRC Career Development Fellowship (1141466). Due to space constraints, we apologize to those whose work we could not cite.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by F. Pentimalli
References
- 1.Lockshin RA, Williams CM. Programmed cell death--I. Cytology of degeneration in the intersegmental muscles of the Pernyi silkmoth. J Insect Physiol. 1965;11:123–33. doi: 10.1016/0022-1910(65)90099-5. [DOI] [PubMed] [Google Scholar]
- 2.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindqvist LM, Frank D, McArthur K, Dite TA, Lazarou M, Oakhill JS, et al. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ 2017;25:782–794. [DOI] [PMC free article] [PubMed]
- 4.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–62. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–77. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segawa K, Nagata S. An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25:639–50. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–36. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 8.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689–97. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv Immunol. 2017;135:81–117. doi: 10.1016/bs.ai.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166:1–15. doi: 10.1111/j.1365-2249.2011.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity. 2016;45:513–26. doi: 10.1016/j.immuni.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22:1313–27. doi: 10.1038/cdd.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014;8:570–82. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 2018;22:2924–36. doi: 10.1016/j.celrep.2018.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang TB, Jeong JS, Yang SH, Kovalenko A, Wallach D. Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. 2018;25:1107–17. doi: 10.1038/s41418-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Penaloza HF, et al. Necroptosis Promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016;16:2219–30. doi: 10.1016/j.celrep.2016.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–9. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- 18.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 19.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 20.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 22.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 24.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 25.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 26.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–63. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. 2015;45:2918–26. doi: 10.1002/eji.201545655. [DOI] [PubMed] [Google Scholar]
- 31.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213:2113–28. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 34.He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215:827–40. doi: 10.1084/jem.20172222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramirez MLG, Poreba M, Snipas SJ, Groborz K, Drag M, Salvesen GS. Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem. 2018;293:7058–67. doi: 10.1074/jbc.RA117.001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. 2015;45:2927–36. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- 38.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA. 2017;114:E961–E969. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol. 2017;198:2156–64. doi: 10.4049/jimmunol.1601757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–95. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 41.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 42.Murphy JM, Vince JE. Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Res 2015, 4. [DOI] [PMC free article] [PubMed]
- 43.Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell. 2017;169:286–300.e16. doi: 10.1016/j.cell.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24:1184–95. doi: 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, et al. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell. 2018;173:1244–53.e10. doi: 10.1016/j.cell.2018.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 47.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33. doi: 10.1038/nature20559. [DOI] [PubMed] [Google Scholar]
- 50.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature. 2016;540:124–8. doi: 10.1038/nature20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan N, Lawlor KE, Murphy JM, Vince JE. More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr Opin Immunol. 2014;26:76–89. doi: 10.1016/j.coi.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 52.Brault M, Oberst A. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol. 2017;95:131–6. doi: 10.1038/icb.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naderer T, Fulcher MC. Targeting apoptosis pathways in infections. J Leukoc Biol. 2018;103:275–85. doi: 10.1189/JLB.4MR0717-286R. [DOI] [PubMed] [Google Scholar]
- 54.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Li W, Ren J, Huang D, He WT, Song Y, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. 2014;111:15072–7. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26:1007–20. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 2018 10.1038/s41418-018-0106-7. [DOI] [PMC free article] [PubMed]
- 61.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. 2018;48:584–92. doi: 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 63.Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol. 2016;197:1353–67. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conos SA, Lawlor KE, Vaux DL, Vince JE, Lindqvist LM. Cell death is not essential for caspase-1-mediated interleukin-1beta activation and secretion. Cell Death Differ. 2016;23:1827–38. doi: 10.1038/cdd.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–46. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 66.Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1beta maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 2018;24:1425–33. doi: 10.1016/j.celrep.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 67.Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62–67. doi: 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 69.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 70.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. 2016;61:589–601. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ros U, Pena-Blanco A, Hanggi K, Kunzendorf U, Krautwald S, Wong WW, et al. Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep. 2017;19:175–87. doi: 10.1016/j.celrep.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. MLKL forms cation channels. Cell Res. 2016;26:517–28. doi: 10.1038/cr.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci USA. 2017;114:E7450–9. doi: 10.1073/pnas.1707531114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15:e2002711. doi: 10.1371/journal.pbio.2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol. 2013;25:363–72. doi: 10.1093/intimm/dxs161. [DOI] [PubMed] [Google Scholar]
- 76.Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ 2015: advance online publication 17 July 2015: 2010.1038/cdd.2015.2092. [DOI] [PMC free article] [PubMed]
- 77.Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol. 2018;1:6. doi: 10.1038/s42003-017-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Z, Wang Y, Zhang Y, He X, Zhong CQ, Ni H, et al. RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat Cell Biol. 2018;20:186–97. doi: 10.1038/s41556-017-0022-y. [DOI] [PubMed] [Google Scholar]
- 79.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 80.Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 81.Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65.e7. doi: 10.1016/j.immuni.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med. 2018;215:1519–29. doi: 10.1084/jem.20172060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest. 2013;123:4695–705. doi: 10.1172/JCI71543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012;37:1009–23. doi: 10.1016/j.immuni.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–14. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JP, Coers J, et al. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. 2015;18:320–32. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, et al. Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity. 2015;43:987–97. doi: 10.1016/j.immuni.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–8. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Speir M, Lawlor KE, Glaser SP, Abraham G, Chow S, Vogrin A, et al. Eliminating Legionella by inhibiting BCL-XL to induce macrophage apoptosis. Nat Microbiol. 2016;1:15034. doi: 10.1038/nmicrobiol.2015.34. [DOI] [PubMed] [Google Scholar]
- 90.Silke J, Hartland EL. Masters, marionettes and modulators: intersection of pathogen virulence factors and mammalian death receptor signaling. Curr Opin Immunol. 2013;25:436–40. doi: 10.1016/j.coi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 91.Orzalli MH, Kagan JC. Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 2017;27:800–9. doi: 10.1016/j.tcb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 93.Linkermann A, Brasen JH, De Zen F, Weinlich R, Schwendener RA, Green DR, et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol Med. 2012;18:577–86. doi: 10.2119/molmed.2011.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 95.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–61. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 96.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–18. doi: 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 97.Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–27. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 99.Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014;7:1796–808. doi: 10.1016/j.celrep.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 100.Lawlor KE, Feltham R, Yabal M, Conos SA, Chen KW, Ziehe S, et al. XIAP loss triggers RIPK3- and caspase-8-driven IL-1beta activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 2017;20:668–82. doi: 10.1016/j.celrep.2017.06.073. [DOI] [PubMed] [Google Scholar]
- 101.Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, et al. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015;42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 104.Yu SX, Chen W, Liu ZZ, Zhou FH, Yan SQ, Hu GQ, et al. Non-hematopoietic MLKL protects against salmonella mucosal infection by enhancing inflammasome activation. Front Immunol. 2018;9:119. doi: 10.3389/fimmu.2018.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K, et al. HSP70 protects against TNF-induced lethal inflammatory shock. Immunity. 2002;16:685–95. doi: 10.1016/s1074-7613(02)00310-2. [DOI] [PubMed] [Google Scholar]
- 107.Tsan MF. Heat shock proteins and high mobility group box 1 protein lack cytokine function. J Leukoc Biol. 2011;89:847–53. doi: 10.1189/jlb.0810471. [DOI] [PubMed] [Google Scholar]
- 108.Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125:539–50. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 110.Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014;189:282–91. doi: 10.1164/rccm.201308-1535OC. [DOI] [PubMed] [Google Scholar]
- 111.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–92. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA. 2010;107:18581–6. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 115.Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. 2017;7:5850. doi: 10.1038/s41598-017-06205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nystrom S, Antoine DJ, Lundback P, Lock JG, Nita AF, Hogstrand K, et al. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013;32:86–99. doi: 10.1038/emboj.2012.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39. doi: 10.1038/cdd.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology. 2016;5:e1149673. doi: 10.1080/2162402X.2016.1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 120.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–88. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 121.Li Z, Scott MJ, Fan EK, Li Y, Liu J, Xiao G, et al. Tissue damage negatively regulates LPS-induced macrophage necroptosis. Cell Death Differ. 2016;23:1428–47. doi: 10.1038/cdd.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–34. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med. 2016;8:339ra369. doi: 10.1126/scitranslmed.aad3099. [DOI] [PubMed] [Google Scholar]
- 124.McComb S, Aguade-Gorgorio J, Harder L, Marovca B, Cario G, Eckert C, et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med. 2016;8:339ra370. doi: 10.1126/scitranslmed.aad2986. [DOI] [PubMed] [Google Scholar]
- 125.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21:1878–90. doi: 10.1681/ASN.2009101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–37. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–48. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 128.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med. 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 129.Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O, et al. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med. 2009;206:2161–77. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.El Mezayen R, El Gazzar M, Seeds MC, McCall CE, Dreskin SC, Nicolls MR. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 2007;111:36–44. doi: 10.1016/j.imlet.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol. 2007;8:497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- 132.Jacobsen AV, Lowes KN, Tanzer MC, Lucet IS, Hildebrand JM, Petrie EJ, et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016;7:e2051. doi: 10.1038/cddis.2015.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31. doi: 10.1016/j.immuni.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 134.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci USA. 2008;105:8067–72. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gaidt MM, Hornung V. Alternative inflammasome activation enables IL-1beta release from living cells. Curr Opin Immunol. 2016;44:7–13. doi: 10.1016/j.coi.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 137.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–93. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171:1057–71.e11. doi: 10.1016/j.cell.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014;15:982–90. doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111:15514–9. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Maeda A, Fadeel B. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis. 2014;5:e1312. doi: 10.1038/cddis.2014.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 146.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 147.Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity. 2013;38:285–95. doi: 10.1016/j.immuni.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Di Paolo NC, Shayakhmetov DM. Interleukin 1alpha and the inflammatory process. Nat Immunol. 2016;17:906–13. doi: 10.1038/ni.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lin J, Li H, Yang M, Ren J, Huang Z, Han F, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–10. doi: 10.1016/j.celrep.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 151.Bakker PJ, Butter LM, Claessen N, Teske GJ, Sutterwala FS, Florquin S, et al. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol. 2014;184:2013–22. doi: 10.1016/j.ajpath.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. 2010;185:6277–85. doi: 10.4049/jimmunol.1002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Berda-Haddad Y, Robert S, Salers P, Zekraoui L, Farnarier C, Dinarello CA, et al. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc Natl Acad Sci USA. 2011;108:20684–9. doi: 10.1073/pnas.1116848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers TW, et al. RIPK1 regulates RIPK3-MLKL driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157:1175–88. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 155.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, et al. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–73. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 158.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 159.Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284:19420–6. doi: 10.1074/jbc.M901744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. 2009;106:9021–6. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol. 2015;7:351–65. doi: 10.1093/jmcb/mjv012. [DOI] [PubMed] [Google Scholar]
- 162.Menzel M, Akbarshahi H, Mahmutovic Persson I, Puthia M, Bjermer L, Uller L. Caspase-1 deficiency reduces eosinophilia and interleukin-33 in an asthma exacerbation model. ERJ Open Res 2017, 3. pii: 00047-2017 [DOI] [PMC free article] [PubMed]
- 163.Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb. 2017;24:443–51. doi: 10.5551/jat.RV17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 165.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 166.de Lautour H, Dalbeth N, Taylor WJ. Outcome measures for gout clinical trials: a summary of progress. Curr Treat Options Rheumatol. 2015;1:156–66. [Google Scholar]
- 167.Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL-1beta secretion by triggering necrosis. Cell Rep. 2015;11:1535–48. doi: 10.1016/j.celrep.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 168.Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 2017;21:3846–59. doi: 10.1016/j.celrep.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]