

Abstract
The Working Group of the International Agency for Research on Cancer classified the consumption of processed meat as carcinogenic to humans (Group 1), and classified red meat as probably carcinogenic to humans (Group 2A); consumption of both meat types is associated with an increased risk of colorectal cancer. These classifications are based on a compilation of epidemiology data and mechanistic evidence from animal and human studies. The curing of meats with nitrite can produce carcinogenic N-nitroso compounds (NOCs), and the smoking of meat produces polycyclic aromatic hydrocarbons (PAHs). The high-temperature cooking of meat also produces carcinogenic heterocyclic aromatic amines (HAAs). The ingestion of heme from meat can catalyze the formation of NOC and lipid peroxidation products (LPOs) in the digestive tract. Many of these chemicals form DNA adducts, some of which can induce mutations and initiate carcinogenesis. Another recent hypothesis is that N-glycolylneuraminic acid, a non-human sialic acid sugar present in red meat, becomes incorporated in the cell membrane, triggering the immune response with associated inflammation and reactive oxygen species, which can contribute to DNA damage, tumor promotion, and cancer. The mechanisms by which these chemicals in meat induce DNA damage, and the impact of dietary and host factors that influence the biological potency of these chemicals are highlighted in this updated report.
Keywords: DNA damage, genotoxicant, colorectal cancer
1. Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and the third leading cause of cancer death in both men and women in the United States.[1] The risks for developing CRC are thought to be attributed to lifestyle factors such as the diet, alcohol and tobacco usage, physical exercise, and obesity.[2] The differences in rates of CRC in different geographic locations and temporal changes in risk among immigrant populations suggest that diet and lifestyle strongly influence the occurrence of CRC, and that inherited genetic mutations account for a minor percentage of CRC incidence.[1, 3] Single nucleotide polymorphisms in genes that encode for carcinogen metabolism enzymes that influence the biological activity of procarcinogens,[4–6] epigenetic factors, aberrant microRNA activity, and chronic inflammation contribute to the development of CRC.[7, 8] Recently, the role of the intestinal bacterial flora has emerged as an increasingly important factor in CRC.[9] However, the definitive biochemical mechanisms and causative factors contributing to DNA damage, mutations, and development of CRC remain unclear.
In 2015, a Working Group convened at the International Agency for Research on Cancer (IARC) in Lyon, France, to evaluate the carcinogenicity of the consumption of red meat and processed meat. In a report published in Lancet Oncol.,[10] the Working Group reported that “On the basis of the large amount of data and the consistent associations of colorectal cancer with consumption of processed meat across studies in different populations, which make chance, bias, and confounding unlikely as explanations, a majority of the Working Group concluded that there is sufficient evidence in human beings for the carcinogenicity of the consumption of processed meat.” A large body of epidemiological studies have reported that the consumption of processed meats and red meats are risk factors for colorectal cancer (CRC).[11–13] Processed meat was classified as carcinogenic to humans (Group 1), and red meat was classified as probably carcinogenic to humans (Group 2A). The consumption of red meat was also reported to be positively associated with pancreatic and prostate cancer and processed meat with gastric cancer. However, the Working Group noted that there was inadequate evidence in experimental animals for the carcinogenicity of consumption of red meat and of processed meat. The mechanistic evidence was based on genotoxic effects of certain processed meats or red meats, or some of their components in experimental rodent models.[12, 14–16]
The Working Group defined red meat as unprocessed mammalian muscle meats, including beef, veal, pork, lamb, mutton, horse, or goat meat. Red meat is usually consumed cooked. Processed meat was defined as meat treated through salting, curing, fermentation, smoking, or other processes to enhance flavor or improve preservation. Most processed meats contain pork or beef, but might also contain other red meats, offal (for example liver), or meat byproducts such as blood.[10]
Several classes of carcinogens formed in processed and cooked red meats are proposed to contribute to CRC: N-nitroso compounds (NOCs) in cured meats;[17, 18] heterocyclic aromatic amines (HAAs) formed in well-done cooked meats and poultry;[19, 20] and polycyclic aromatic hydrocarbons (PAHs) formed in smoked meats and meats cooked under flame.[21] In addition, ingested heme can catalyze the nitrosation of endogenous secondary amines[14] and exert pro-oxidative effects by catalysis of lipid peroxidation in the gastrointestinal tract.[16] All of these chemicals can form DNA adducts, and if not repaired by enzyme systems,[22] some of the DNA adducts can induce mutations during cell division and lead to the development of cancer.[23] IARC has classified several NOCs and PAHs as Group 1 carcinogens, and several HAAs are designated as Group 2A or 2B carcinogens. The identification of the causative agents involved in the development of CRC is important since the modification of diets or changes in the methods of preparing meats can mitigate some chemical exposures that contribute to the cancer burden.[24] Some epidemiologic studies have reported an elevated risk for CRC and other cancers with consumption of meats cooked well-done at high temperatures, whereas other studies have not found this association.[25–31] Thus, the dietary data have been suggestive but inconsistent. A main limiting factor in epidemiological studies is the uncertainty in the quantitative estimates of chronic exposure to different types of carcinogens in meat. The concentrations of genotoxicants in meat can range over 100-fold, depending on the processing and methods of cooking meat. There is a critical need to develop and employ specific and quantitative measurements of stable, long-lived biomarkers for reliable assessments of exposures, estimation of the biologically effective dose, and the DNA damage induced by chemicals in processed and red meats for human risk assessment.[32–34] Some of these prototypical chemical carcinogens in meat and their mechanisms of DNA damage, and the impact of dietary and host factors that influence the biological potency of these chemicals are presented in this updated report.
1.1. Mechanisms of genomic damage and DNA adduct formation by components in processed and cooked meat.
The proposed biochemical mechanisms by which genotoxicants and components in processed and red meat induce DNA damage in the colorectum are depicted in Figure 1.
Figure 1.
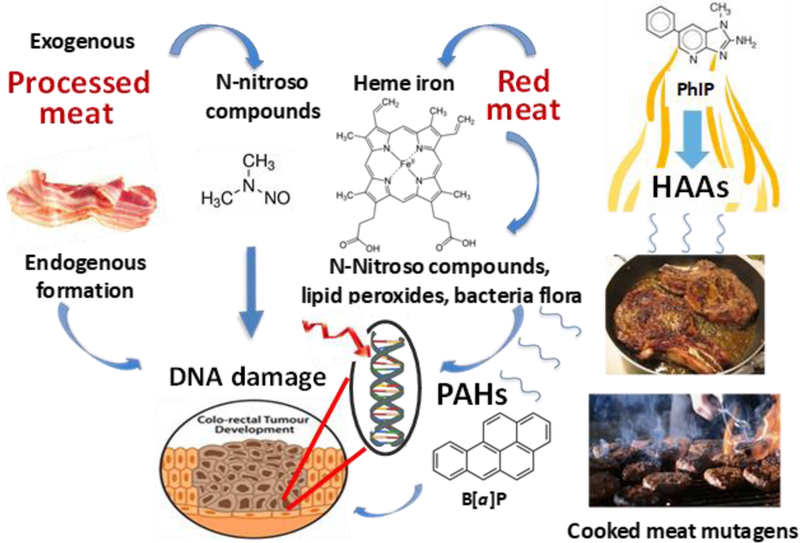
Mechanisms of DNA damage in colorectum by genotoxicants and components in meat and modulating effects of the bacterial flora. Mechanisms are adapted from references [35, 36] and citations within.
N-Nitroso compounds.
Carcinogenic NOCs formed during the curing of meats include N-nitrosodimethylamine (NDMA), N-nitrosodiethylamine, N-nitrosodibutylamine, N-nitrosopyrrolidine, and N-nitrospiperidine. The levels of NOCs in cured meats range from less than one part-per-billion (ppb) up to 130 ppb.[17, 18] NOCs undergo metabolic activation by cytochrome P450 2E1 expressed in the GI tract.[37] For example, the reactive methylating intermediate of NMDA forms N7-methyl-2´-deoxyguanosine (N7-MedG), which leads to abasic site formation, DNA strand breaks, and cytotoxicity.[38] Another adduct occurs through O6-methylation of deoxyguanosine (dG) to form O6-methyl-2´-deoxyguanosine (O6-MedG) (Figure 2).[39]
Figure 2.
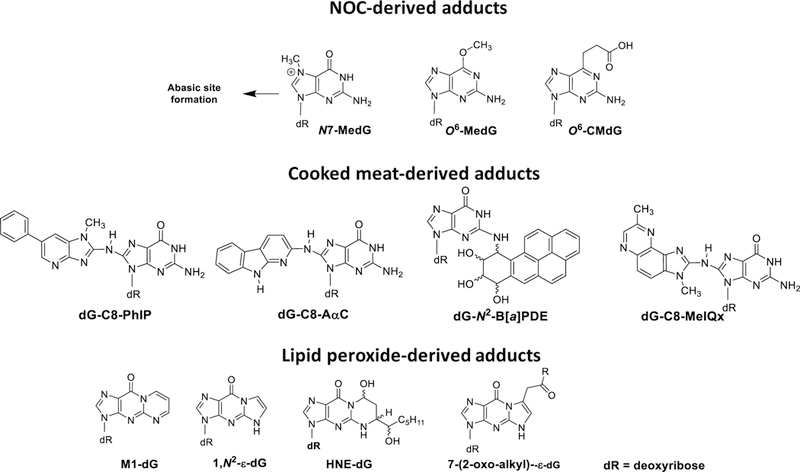
Chemical structures of DNA adducts derived from prototypical NOCs, HAAs and PAHs, and lipid peroxides.
Recent attention has focused on endogenously formed NOCs, which become elevated in the gastrointestinal tract following consumption of processed or red meats.[14] Nitrosated glycine, dipeptides, or N-nitroso bile acid conjugates, such as N-nitrosoglycocholic acid, form O6-MedG and O6-carboxymethyl-2´-deoxyguanosine (O6-CMdG) (Figure 2),[39] which induce G-A transitions and G-T transversions that contribute to the mutations in cancer driver genes, including H-ras and K-ras oncogenes and the p53 tumor suppressor gene, in the gastrointestinal tract of omnivores.[40, 41] The levels of endogenously produced NOCs in feces of healthy human subjects on a fresh red meat- or processed meat diet were ten-fold or greater than those levels formed in feces of volunteers on a vegetarian diet, and the percentage of colonic exfoliated cells staining positive for O6-CMdG was significantly (P < 0.001) higher in feces of subjects on the high red meat diet than those consuming a vegetarian diet.[14] However, mutations were not detected in the K-ras gene of exfoliated colonocytes.[42]
Heme iron – a pro-oxidant.
The ingestion of heme iron from hemoglobin or myoglobin mediates the formation of lipid peroxidation and apparent total NOCs in the colon.[13, 16, 36, 42] The feeding of hemin (ferriheme), but not protoporphyrin IX, ferric citrate or bilirubin leads to the formation of cytotoxic and potentially DNA-damaging agents, and cell proliferation of the colonic mucosa of rodents.[43] Malondialdehyde (MDA) is one product of lipid peroxidation that reacts with dG to form the cyclic adduct 3-(2-deoxy-β-D-erythropenta-furano-syl)pyrimido[1,2-α]purin-10(3H)-one 2′-deoxyguanosine (M1-dG).[44] MDA and other genotoxic lipid oxidation products, such as those formed from 4-oxo-2-alkenals (Figure 2),[45] are hypothesized to be elevated in humans who eat processed or red meats.[16, 46]
Heterocyclic aromatic amines and polycyclic aromatic hydrocarbons.
HAAs, including 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) and 2-amino-9H-pyrido[2,3-b]indole (AαC) are abundant HAAs formed in well-done cooked meats. HAAs undergo metabolism, by cytochrome P450 enzymes, to form genotoxic N-hydroxylated metabolites.[19] These metabolites undergo further metabolism with conjugation enzymes, such N-acetyltransferases or sulfotransferases, to generate reactive intermediates that bind to DNA (Figure 2).[32] PAHs, such as benzo[a]pyrene (B[a]P), also undergo bioactivation by human cytochrome P450 enzymes to form genotoxic species.[47] The reactive intermediates are anti-diol-epoxides of the bay region of PAH molecules, although trans-dihydrodiols of some PAHs can contribute to DNA damage and oxidative stress through their oxidation to o-quinones.[48] PAHs that arise in tobacco smoke are also believed contribute to lung cancer in smokers.[49]
N-glycolylneuraminic acid (Neu5Gc).
Red meats are rich with glycans containing a non-human variant of sialic acid called N-glycolylneuraminic acid (Neu5Gc).[50] Neu5Gc is not biosynthesized in humans, but it is bioavailable and becomes incorporated in tissues of omnivores.[50] Interactions of this antigen with circulating anti-Neu5Gc antibodies in a murine model have been shown promote chronic inflammation, leading to reactive oxygen species that can contribute to carcinogenesis and tumor progression (Figure 3).
Figure 3.
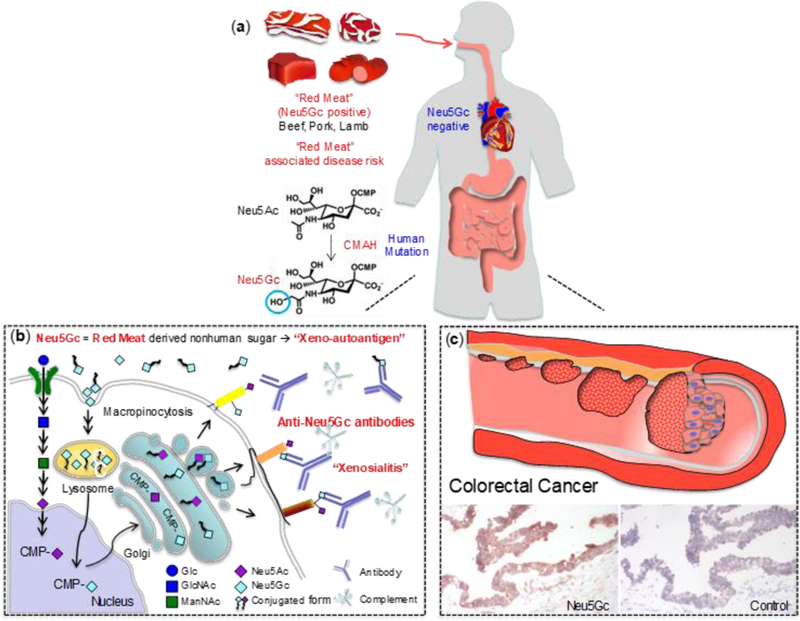
Potential CRC risk associated with metabolic incorporation of non-human sialic acid N-glycolylneuraminic acid (Neu5Gc) from red meat. (a) Neu5Gc is present in beef, pork, lamb, but humans cannot synthesize Neu5Gc. (b) Neu5Gc can be incorporated into human cells through the same pathway used for Neu5Ac recycling. (c) Endocytosed Neu5Gc is used as a substrate for the synthesis of sialylated glycans in the Golgi. Cell surface glycans containing Neu5Gc may be targeted by circulating anti-Neu5Gc antibodies and complement, leading to a human specific inflammation, termed xenosialitis.[50] The figure was kindly provided by Dr. Kunio Kawanishi and adapted with permission from reference 50.
2. Methods to detect DNA adducts in humans.
Despite the large body of epidemiology data on the risk of processed or red meat diet in the development of CRC,[11, 13] there is a paucity of physico-chemical data on the DNA adducts formed in the colorectum. The absence of specific biomarkers for distinguishing between DNA adducts occurring through dietary intake or from endogenous processes has hindered our ability to substantiate any of the proposed chemicals and biochemical mechanisms involved in CRC. 32P-postlabeling, immunohistochemistry (IHC), gas chromatography/mass spectrometry (GC/MS) and liquid chromatography mass spectrometry (LC/MS) have served as the major methods to measure DNA adducts in humans. The advantages and limitations of these technologies have been reviewed,[51–54] and the technologies are briefly highlighted here in context to the characterization of DNA adducts in colorectum.
2.1. 32P-Postlabeling
32P-Postlabeling remains the most commonly used method to screen for DNA adducts because it is a highly sensitive technique.[51] The DNA is enzymatically digested to 3′-phospho-2′-deoxyribonucleotides, and 32P-orthophosphate from [γ−32P] ATP is transferred to the 5′-OH position of the 2′-deoxyribonucleotide adduct, by polynucleotide kinase. The 5′−32P-labeled nucleotides are usually resolved by multi-dimensional thin-layer chromatography using autoradiography for detection or by HPLC with radiometric detection. The technique has revealed that human DNA is modified with many environmental and dietary chemicals, and endogenous electrophiles to form numerous putative DNA adducts some of which may occur in the colorectum.[55, 56] However, the identification and quantification of DNA adducts cannot be achieved by 32P-postlabeling. Thus, the conclusions drawn from epidemiological studies employing 32P-postlabeling have often provided ambiguous results about the role of diet, chemical exposures and their relationships to DNA adducts, and the impact of genetic polymorphisms in genes that encode for carcinogen metabolism enzymes on cancer risk.[57–59]
2.2. Immunochemical methods
Several DNA adducts have been screened, by immunochemical methods, in human colorectal or other tissues.[38, 60–63] These methods employ ELISA (enzyme-linked immunosorbent assay) plate-based assays, radio immunoassays, or slot-blot methods to screen for DNA adducts, using antibodies raised against carcinogen-treated DNA or DNA adducts coupled to carrier proteins. Immunohistochemical (IHC) detection of DNA adducts in tissue section-cuts mounted on slides is another screening method and allows for the visualization of the DNA adduct within specific cell types of a tissue.[52] IHC is especially suitable for archived formalin-fixed paraffin-embedded (FFPE) tissues for which there is a clinical diagnosis of disease. An important drawback of immuno-based detection methods is that the specificity of antibodies, even monoclonal antibodies, for DNA adducts is uncertain as they may cross-react with other DNA lesions or cellular components, leading to errors in identification and quantification. Also, immunodetection methods can only be performed on DNA lesions for which antibodies are available. Signals for putative colorectal adducts of O6-MedG,[60, 62] O6-CMdG,[42] and M1-dG[61] have been detected by IHC or slot blot methods. A high red meat diet increased rectal O6-MedG adduct levels in healthy subjects by 21% relative to baseline levels, based on immuno detection.[62] In another study, the mean percentage of exfoliated colonocytes staining positive for O6-CMdG in feces of subjects on a red meat diet was increased by 2.5-fold compared to the levels for the same subjects on a vegetarian diet.[42] M1-dG was detected, by means of a slot blot method, in more than 90% of the colorectal specimens of men and women from European Prospective Investigation on Cancer.[61]
2.3. Mass spectrometry methods
GC/MS with electron impact ionization and more recently negative ion chemical ionization have been employed to measure DNA adducts (primarily used for oxidized DNA bases) where adduct structures can be corroborated from the MS fragmentation spectra. The DNA is usually hydrolyzed with acid or base to produce the aglycone adducts.[54] DNA adducts require chemical derivatization to increase the volatility required for GC analysis. The derivatization process is often conducted at elevated temperature (> 100 °C). Thus, the DNA adducts must be stable to the harsh conditions of DNA hydrolysis and to elevated temperatures employed in GC/MS. The base hydrolysis of DNA was employed to recover PhIP from human colorectal DNA, followed by electron capture MS, which is also known as GC-negative ion chemical ionization (NICI)-MS.[64] Presumably, the liberated PhIP was derived from the N-(2′-deoxyguanosine-8-yl)-PhIP (dG-C8-PhIP). PhIP was detected at levels of several adducts per 108 DNA bases in two out of six human colon samples, when assayed by this method. To the author’s knowledge, there are no other reports on the analysis of DNA adducts in human colorectal tissue by GC/MS.
The online coupling of LC to electrospray ionization (ESI) MS is the most commonly used technology to measure many DNA adducts which would otherwise undergo thermal decomposition by GC-MS.[65–67] Most DNA adducts are detected following nuclease digestion of the DNA to produce the modified 2′-deoxyribonucleoside adducts.[67] Because of the basic properties of the nitrogenous nucleobase moieties, DNA adducts are usually analyzed in the positive ionization mode. A common feature of most DNA adducts is their propensity to lose the 2′-deoxyribose (dR) moiety (116 Da or 116.0473 Da in high-resolution accurate mass (HRAMS)), when subjected to collision-induced dissociation (CID).[68] This transition is commonly used to measure DNA adducts by targeted MS2 methods employing triple quadrupole MS, and more recently by ion trap (IT)/Orbitrap MSn scanning methods.[67, 69–72] However, there are no reports in the literature employing LC/MS2 to measure 2′-deoxyribonucleoside DNA adducts in the colorectum from chemicals derived from processed red meat or cooked meats. Another approach is to analyze for the DNA adducts as the modified DNA base. Following formic acid hydrolysis of DNA, Orbitrap-HRAMS was employed using wide-selected ion monitoring or HRAMS/MS2 to screen for the aglycones of 30 putative DNA adducts in human colon; O6-CMdG was tentatively identified as one lesion.[73]
2.4. Other methods to detect DNA damage
There is one report on the detection of a putative B[a]P DNA adduct in colorectum, following acid hydrolysis of DNA. The liberated tetraol r-7,c-10,t-8,t-9-tetrahydroxy-7,8,9,10-tetrahydro-B[a]P was detected, by HPLC/fluorescence, in four out of seven colon mucosa samples at levels ranging between 0.2 and 1.0 adducts per 108 DNA bases.[74] These findings were not confirmed by specific MS-based methods.
DNA damage has also been assessed by the Comet Assay. The alkaline comet assay detects DNA strand breaks (SBs) and alkali-labile sites at frequencies from a few hundred to several thousand breaks per eukaryote cell and can visualize DNA damage introduced by endogenous electrophiles or exposures to exogenous genotoxicants.[75] The treatment of the genome with DNA glycosylases allows for measurement of damage other than SBs, where damaged bases in DNA are removed, resulting in the formation of apurinic/apyrimidinic (AP) sites, which are readily detected by the comet assay. The assay is sensitive and can screen for a broad range of types of DNA damage. However, the Comet Assay, like 32P-postlabeling and immunodetection methods, fails to identify the specific chemicals involved in DNA adduct formation. DNA damage in human colon of volunteers who consumed cooked meat was characterized by the Comet Assay. When volunteers ate a diet of high-temperature cooked red meat containing elevated levels of HAAs, rectal biopsy cell DNA damage increased, when measured by the Comet assay.[76] The DNA damage was reduced by nearly two-fold when the cooked meat was consumed with cruciferous vegetables, yogurt, and chlorophyllin tablets.[76] These food components reduce the genotoxicity of HAAs in rodent studies,[77, 78] suggesting a plausible role for HAAs in cooked meat in DNA damage of the rectum.[76]
3. Factors that modulate carcinogenicity
There are many dietary and host factors that can influence the biological potency of genotoxicants. Chemoprotective agents in the diet and beverages and the bacterial flora are important factors that impact the carcinogenic potential of genotoxicants in cooked meat.
3.1. Chemoprevention
Numerous studies have reported protective beneficial health effects of naturally occurring phytochemicals, including polyphenolics and thiocyanates, found in cruciferous vegetables, foods, and beverages against carcinogens.[79, 80] Beneficial effects have also been reported on short chain fatty acid microbial metabolites, which are formed in fermented dairy products and regulate epithelial cell homeostasis.[81] These biochemicals have been reported to exert protection towards dietary, tobacco-associated, and mycotoxin carcinogens.[80, 82–85] Wattenberg classified chemopreventive agents into three broad categories, with markedly different functions: 1) agents that can prevent the formation of carcinogenic compounds from their precursors or diminish carcinogen bioavailability; 2) agents that can block the metabolic activation of carcinogens, scavenge reactive intermediates, or alter metabolism by changes in the expression of enzymes of metabolism involved in bioactivation or detoxification; and 3) agents that can interfere in the process by which initiated cells progress to neoplasia.[82] Recently certain chemicals have been shown to act on the repair and replication processes of damaged DNA, resulting in decreases in mutation frequency.[80] It is also now recognized that dietary factors contribute to chemoprevention by modulating epigenetic events, including the DNA methylation status, that alters expression of genes involved carcinogenesis.[86]
3.2. The role of bacterial flora in colorectal cancer
Bacteria constitute about 90% of all cells in the human body, and it has been estimated that these bacteria are comprised of over 1000 different species.[9] Trillions of commensal bacteria, termed “the microbiota,” are in close proximity to a single layer of intestinal epithelial cells. Commensal bacteria are involved in the intestinal architecture and possess important homeostatic immune and metabolic functions. Commensal bacteria and their metabolites affect the proliferation and survival of epithelial cells and also provide protection against pathogens.[87] The majority of microbes reside in the gastrointestinal tract in communities defined as the microbiome. The microbiome is dynamic and its composition and functionality is influenced by the diet, environment, and physiological changes, such as diseases. An unbalanced change in the microbiota ecosystem leads to dysbiosis and can result in the development of inflammatory bowel disease, metabolic syndrome, and initiate CRC.[87] A model of the microbial ecology involved in the onset of CRC and progression was proposed by Tjalsma,[9] and termed the bacterial driver-passenger model. In this model, CRC development is initiated by over colonization of the colorectum with indigenous bacteria with pro-carcinogenic features – these microbes are termed bacterial drivers, and damage epithelial DNA and contribute to CRC initiation. Several candidate bacterial drivers have been identified and include superoxide-producing strains of Enterococcus faecalis, and colibactin-producing Escherichia coli strains. Colibactin is a potent genotoxin that is can form DNA adducts and crosslinks, induces DNA strand breaks and cell cycle arrest, and also promotes tumor growth.[88–91] Other pro-inflammatory members of Bacteroides fragilis Enterobacteriaceae, such as Shigella, Citrobacter and Salmonella have been associated with early stages of CRC as possible bacterial drivers.[92]
There are also a number of bacteria that exert antimutagenic/anticarcinogenic effects. For example, certain strains of lactobacillus and bifidobacterium species inhibit DNA damage and decrease the incidence of colon carcinogenesis of 1,2-dimethylhydrazine and HAAs in rodents by either binding to the procarcinogens, catalyzing the detoxication of the chemicals, or by induction of the immune response.[93–95]
Bacterial enzymes can metabolize procarcinogens. The enzymes include: β-glucuronidase, β-glycosidase, azoreductase, nitroreductase, and nitrate reductase.[96] For example, the glucuronide conjugates of HAAs and their N-hydroxylated HAAs, and those of hydroxylated PAHs can undergo enzymatic hydrolysis by β-glucuronidases of the bacterial flora, which liberate the aglycones that may exert genotoxic effects within the GI tract, or undergo enterohepatic circulation and modulate carcinogenic risk (Figure 4).[97, 98]
Figure 4.
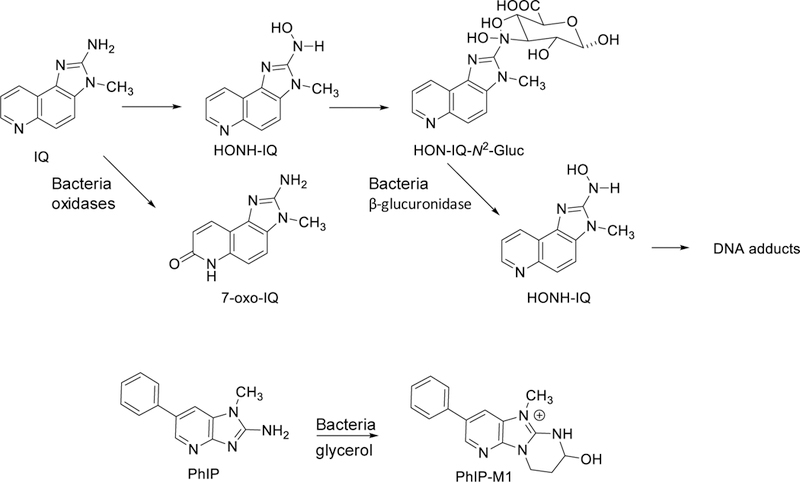
Metabolism of HAAs by mammalian P450s (CYP) and UDP-glucuronosyltransferases (UGT), and metabolism of HAAs or their metabolites by bacterial gut flora.
Eubacterium and Clostridium oxidize IQ-type compounds to form 7-oxo species, such as 2-amino-3-methyl-3H-imidazo[4,5-f]quinolone (7-oxo-IQ) (Figure 4).[99] These compounds are directly mutagenic in the Ames test strain Salmonella; however, they are not carcinogenic in rodents.[100] The human gut microbiota also can contribute to the detoxification of PhIP by forming 7-hydroxy-5-methyl-3-phenyl-6,7,8,9-7-tetrahydropyrido[3′,2′:4,5]imidazo[1,2-a]pyrimidin-5-ium chloride (PhIP-M1), a conjugate formed by Lactobacillus reuteri, Eubacterium hallii, and strains of Enterococcus in the presence of glycerol.[101–103] PhIP-M1 has been detected as a minor metabolite in human urine and feces.[104] The human colon microbiota also catalyze the formation of 7-hydroxybenzo[a]pyrene in vitro.[105] At the elevated doses employed in carcinogenesis studies, the oral exposure to B[a]P significantly altered the composition and the abundance of the gut microbiota and led to moderate inflammation in the ileal and colonic mucosa of C57BL/6 mice.[106] Collectively, these findings highlight the complex effects of the diet, and the interplay of commensal and pathogenic bacteria microbiome on microbial composition and activity in the colon that impact human health and disease states.
Conclusions.
Epidemiological studies have often linked the frequent consumption of processed meats and cooked red meats with an elevated risk for CRC.[11, 13] Multiple hypotheses have been proposed to explain the increased risk of CRC associated with meat consumption.[107] Many different classes of genotoxicants present in processed and cooked meats are capable of forming pro-mutagenic DNA adducts in humans, which can contribute to CRC.[13, 31, 46] In addition, endogenous nitrosation processes produce reactive intermediates that can induce DNA damage, and possibly lead to mutations in the colonocytes.[14] Pro-oxidants in cooked red and processed meats, including heme, ingested fats and lipid peroxidation products,[108] and Neu5Gc[50] may contribute to inflammation and tumor promotion, leading to the development of CRC. It is also important to consider the role of the bacterial flora of the gut in the development of CRC. Gut bacteria have critical homeostatic and immune functions, and are capable of metabolizing endogenous and xenobiotic chemicals, including the bioactivation and detoxification of carcinogens.[78, 107] The increased risk for CRC may not be associated with one single chemical, but due to the presence of a complex mixture of chemicals and bacterial flora acting on multiple stages of CRC development.[18] Clearly, more human studies with controlled meat diets and the identification and quantification of colorectal biomarkers of DNA damage, such as DNA adducts, by specific mass spectrometric methods, and linking these adducts to mutations[109] can advance our understanding of the chemicals in the diet and those produced endogenously that damage DNA and may contribute CRC risk.[32, 110]
The IARC working group concluded that for every 50 grams of processed meat or 100 grams of red meat eaten, the relative risk of colon cancer was increased by about 18% compared to those individuals who ate the least meats.[10] This relative risk is modest compared to the relative risk of developing lung cancer from smoking cigarettes, which ranges between 1000–3000%.[111] Nevertheless, exposure to genotoxicants in the diet should be avoided. It should be recognized that consumption of red meat does have beneficial effects. Red meat is a nutritious food and an important source of protein with all essential amino acids, highly bioavailable iron, zinc, selenium, and B vitamins, especially vitamin B12 in the diet.[112] There are ways to eat healthier meat products by avoiding the consumption of processed meats treated with nitrite or by not over-cooking or charring of red meat. The consumption of lean red meats in moderation,[2] combined with poultry, fish, whole grains, vegetables, and fruits can provide a well-balanced and healthy diet.
Acknowledgment.
The research conducted in the author’s lab on cooked meat mutagens and colorectal cancer has been supported by National Institutes of Health Grants R01CA122320 and R01CA134700.
Biography

Dr. Robert Turesky is a Professor in the Department of Medicinal Chemistry, and Director of the Masonic Cancer Center's Analytical Biochemistry shared resource, a mass spectrometry facility devoted to the cancer and chemoprevention programs at the University of Minnesota. Dr. Turesky received his PhD in nutrition and food science from M.I.T. Prior to his current position, Dr. Turesky served as Group Leader of the Biomarkers Unit, Nestlé Research Center, Lausanne, Switzerland (1986 – 2000); Division Director of Chemistry, National Center for Toxicological Research, U.S. Food and Drug Administration, Jefferson, AR, (2000 – 2004); and Principal Investigator, Wadsworth Center, New York State Department of Health (2004 – 2013). He investigates the biochemical toxicology of dietary and environmental genotoxicants and applies mass spectrometry methods to identify and measure biomarkers of these chemicals in molecular epidemiology studies that seek to understand the role of chemical exposures in the etiology of cancer.
References
- [1].American Cancer Society. Cancer Facts & Figures 2018 Atlanta: American Cancer Society, Atlanta, Georgia, 2018. [Google Scholar]
- [2].World Cancer Research Fund / American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective, 2007.
- [3].Power DG, Gloglowski E, Lipkin SM, Hematol Oncol Clin North Am 2010, 24, 837. [DOI] [PubMed] [Google Scholar]
- [4].Le Marchand L, Hankin JH, Wilkens LR, Pierce LM, Franke A, et al. , Cancer Epidemiol Biomarkers Prev 2001, 10, 1259. [PubMed] [Google Scholar]
- [5].Le Marchand L, Donlon T, Seifried A, Wilkens LR, Cancer Epidemiol Biomarkers Prev 2002, 11, 1019. [PubMed] [Google Scholar]
- [6].Kury S, Buecher B, Robiou-du-Pont S, Scoul C, Sebille V, et al. , Cancer Epidemiol Biomarkers Prev 2007, 16, 1460. [DOI] [PubMed] [Google Scholar]
- [7].Fearon ER, Vogelstein B, Cell 1990, 61, 759. [DOI] [PubMed] [Google Scholar]
- [8].Colussi D, Brandi G, Bazzoli F, Ricciardiello L, Int J Mol Sci 2013, 14, 16365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tjalsma H, Boleij A, Marchesi JR, Dutilh BE, Nat Rev Microbiol 2012, 10, 575. [DOI] [PubMed] [Google Scholar]
- [10].Bouvard V, Loomis D, Guyton KZ, Grosse Y, Ghissassi FE, et al. , The Lancet Oncology 2015, 16, 1599.26514947 [Google Scholar]
- [11].Norat T, Bingham S, Ferrari P, Slimani N, Jenab M, et al. , J Natl Cancer Inst 2005, 97, 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kim D, Lee YJ, Ryu HY, Lee JH, Kim HK, et al. , J Appl Toxicol 2013, 33, 63. [DOI] [PubMed] [Google Scholar]
- [13].Corpet DE, De Smet S, Demeyer D, Meat Sci 2014, 98, 115. [DOI] [PubMed] [Google Scholar]
- [14].Joosen AM, Kuhnle GG, Aspinall SM, Barrow TM, Lecommandeur E, et al. , Carcinogenesis 2009, 30, 1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Santarelli RL, Vendeuvre JL, Naud N, Tache S, Gueraud F, et al. , Cancer Prev Res (Phila) 2010, 3, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bastide NM, Chenni F, Audebert M, Santarelli RL, Tache S, et al. , Cancer Res 2015, 75, 870. [DOI] [PubMed] [Google Scholar]
- [17].Lijinsky W, Mutat Res 1999, 443, 129. [DOI] [PubMed] [Google Scholar]
- [18].Demeyer D, Mertens B, De Smet S, Ulens M, Crit Rev Food Sci Nutr 2016, 56, 2747. [DOI] [PubMed] [Google Scholar]
- [19].Sugimura T, Wakabayashi K, Nakagama H, Nagao M, Cancer Sci 2004, 95, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Felton JS, Jagerstad M, Knize MG, Skog K, Wakabayashi K, in ‘Food Borne Carcinogens Heterocyclic Amines’, Eds. Nagao M, Sugimura T, John Wiley & Sons Ltd, Chichester, England, 2000, p. 31. [Google Scholar]
- [21].Phillips DH, Mutat Res 1999, 443, 139. [DOI] [PubMed] [Google Scholar]
- [22].Sancar A, Reardon JT, Adv Protein Chem 2004, 69, 43. [DOI] [PubMed] [Google Scholar]
- [23].Delaney JC, Essigmann JM, Chem Res Toxicol 2008, 21, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Doll R, Peto R, J Natl Cancer Inst 1981, 66, 1191. [PubMed] [Google Scholar]
- [25].Cross AJ, Sinha R, Environ Mol Mutagen 2004, 44, 44. [DOI] [PubMed] [Google Scholar]
- [26].Rohrmann S, Hermann S, Linseisen J, Am J Clin Nutr 2009, 89, 1418. [DOI] [PubMed] [Google Scholar]
- [27].Sander A, Linseisen J, Rohrmann S, Cancer Causes Control 2011, 22, 109. [DOI] [PubMed] [Google Scholar]
- [28].Nothlings U, Yamamoto JF, Wilkens LR, Murphy SP, Park SY, et al. , Cancer Epidemiol Biomarkers Prev 2009, 18, 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nowell S, Coles B, Sinha R, MacLeod S, Luke RD, et al. , Mutat Res 2002, 506–507, 175. [DOI] [PubMed]
- [30].Nowell S, Ratnasinghe DL, Ambrosone CB, Williams S, Teague-Ross T, et al. , Cancer Epidemiol Biomarkers Prev 2004, 13, 270. [DOI] [PubMed] [Google Scholar]
- [31].Zheng W, Lee SA, Nutr Cancer 2009, 61, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Turesky RJ, Le Marchand L, Chem Res Toxicol 2011, 24, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Himmelstein MW, Boogaard PJ, Cadet J, Farmer PB, Kim JH, et al. , Crit RevToxicol 2009, 39, 679. [DOI] [PubMed] [Google Scholar]
- [34].Jarabek AM, Pottenger LH, Andrews LS, Casciano D, Embry MR, et al. , Crit Rev Toxicol 2009, 39, 659. [DOI] [PubMed] [Google Scholar]
- [35].Kim S, Guo J, O'Sullivan MG, Gallaher DD, Turesky RJ, Environ Mol Mutagen 2016, 57, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bastide NM, Pierre FH, Corpet DE, Cancer Prev Res (Phila) 2011, 4, 177. [DOI] [PubMed] [Google Scholar]
- [37].Yang CS, Yoo J-SH, Ishizaki H, Hong J, Drug Metabolism Reviews 1990, 22, 147. [DOI] [PubMed] [Google Scholar]
- [38].Harrison KL, Wood M, Lees NP, Hall CN, Margison GP, et al. , Chemical research in toxicology 2001, 14, 295. [DOI] [PubMed] [Google Scholar]
- [39].Harrison KL, Jukes R, Cooper DP, Shuker DE, Chem Res Toxicol 1999, 12, 106. [DOI] [PubMed] [Google Scholar]
- [40].Gottschalg E, Scott GB, Burns PA, Shuker DE, Carcinogenesis 2007, 28, 356. [DOI] [PubMed] [Google Scholar]
- [41].Lees NP, Harrison KL, Hall CN, Margison GP, Povey AC, Carcinogenesis 2004, 25, 1243. [DOI] [PubMed] [Google Scholar]
- [42].Lewin MH, Bailey N, Bandaletova T, Bowman R, Cross AJ, et al. , Cancer Res 2006, 66, 1859. [DOI] [PubMed] [Google Scholar]
- [43].Sesink AL, Termont DS, Kleibeuker JH, Van der Meer R, Cancer Res 1999, 59, 5704. [PubMed] [Google Scholar]
- [44].Marnett LJ, Plastaras JP, Trends Genet 2001, 17, 214. [DOI] [PubMed] [Google Scholar]
- [45].Kawai Y, Nuka E, J Clin Biochem Nutr 2017, 1. [DOI] [PMC free article] [PubMed]
- [46].Oostindjer M, Alexander J, Amdam GV, Andersen G, Bryan NS, et al. , Meat Sci 2014, 97, 583. [DOI] [PubMed] [Google Scholar]
- [47].Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, et al. , Cancer Res 1996, 56, 2979. [PubMed] [Google Scholar]
- [48].Penning TM, Chemical research in toxicology 2014, 27, 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].IARC, ‘IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking’, Vol. 83, International Agency for Research on Cancer, Lyon, France, 2002. [PMC free article] [PubMed] [Google Scholar]
- [50].Alisson-Silva F, Kawanishi K, Varki A, Mol Aspects Med 2016, 51, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Phillips DH, Cancer Lett 2012, 334, 5. [DOI] [PubMed] [Google Scholar]
- [52].Poirier MC, Santella RM, Weston A, Carcinogenesis 2000, 21, 353. [DOI] [PubMed] [Google Scholar]
- [53].Klaene JJ, Sharma VK, Glick J, Vouros P, Cancer Lett 2013, 334, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dizdaroglu M, Mutat Res Rev Mutat Res 2015, 763, 212. [DOI] [PubMed] [Google Scholar]
- [55].Pfohl-Leszkowicz A, Grosse Y, Carriere V, Cugnenc PH, Berger A, et al. , Cancer Res 1995, 55, 5611. [PubMed] [Google Scholar]
- [56].Umemoto A, Kajikawa A, Tanaka M, Hamada K, Seraj MJ, et al. , Carcinogenesis 1994, 15, 901. [DOI] [PubMed] [Google Scholar]
- [57].Agudo A, Peluso M, Munnia A, Lujan-Barroso L, Sanchez MJ, et al. , Cancer Epidemiol Biomarkers Prev 2012, 21, 685. [DOI] [PubMed] [Google Scholar]
- [58].Ricceri F, Godschalk RW, Peluso M, Phillips DH, Agudo A, et al. , Cancer Epidemiol Biomarkers Prev 2010, 19, 3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ho V, Peacock S, Massey TE, Godschalk RW, van Schooten FJ, et al. , Environ Mol Mutagen 2015, 56, 609. [DOI] [PubMed] [Google Scholar]
- [60].Povey AC, Hall CN, Badawi AF, Cooper DP, O'Connor PJ, Gut 2000, 47, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Leuratti C, Watson MA, Deag EJ, Welch A, Singh R, et al. , Cancer Epidemiol Biomarkers Prev 2002, 11, 267. [PubMed] [Google Scholar]
- [62].Le Leu RK, Winter JM, Christophersen CT, Young GP, Humphreys KJ, et al. , Br J Nutr 2015, 114, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pratt MM, John K, MacLean AB, Afework S, Phillips DH, et al. , IntJEnvironResPublic Health 2011, 8, 2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Friesen MD, Kaderlik K, Lin D, Garren L, Bartsch H, et al. , Chem Res Toxicol 1994, 7, 733. [DOI] [PubMed] [Google Scholar]
- [65].Tretyakova N, Goggin M, Sangaraju D, Janis G, Chem Res Toxicol 2012, 25, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu S, Wang Y, Chem Soc Rev 2015, 44, 7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Guo J, Turesky RJ, Curr Protoc Nucleic Acid Chem 2016, 66, 7 24 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wolf SM, Vouros P, Chem Res Toxicol 1994, 7, 82. [DOI] [PubMed] [Google Scholar]
- [69].Bessette EE, Goodenough AK, Langouet S, Yasa I, Kozekov ID, et al. , Anal Chem 2009, 81, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gu D, Turesky RJ, Tao Y, Langouet SA, Nauwelaers GC, et al. , Carcinogenesis 2012, 33, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xiao S, Guo J, Yun BH, Villalta PW, Krishna S, et al. , Anal Chem 2016, 88, 12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yun BH, Rosenquist TA, Sidorenko V, Iden CR, Chen CH, et al. , Chem Res Toxicol 2012, 25, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hemeryck LY, Decloedt AI, Vanden Bussche J, Geboes KP, Vanhaecke L, Anal Chim Acta 2015, 892, 123. [DOI] [PubMed] [Google Scholar]
- [74].Alexandrov K, Rojas M, Kadlubar FF, Lang NP, Bartsch H, Carcinogenesis 1996, 17, 2081. [DOI] [PubMed] [Google Scholar]
- [75].Langie SAS, Azqueta A, Collins AR, Front Genet 2015, 6, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shaughnessy DT, Gangarosa LM, Schliebe B, Umbach DM, Xu Z, et al. , PLoS One 2011, 6, e18707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Higdon JV, Delage B, Williams DE, Dashwood RH, Pharmacol Res 2007, 55, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Walton GE, Gibson GR, in ‘Nutrition and Health: Bioactive Compounds and Cancer’, Eds. Milner JA, Romagnolo DF, Humana Press, New York: 2010, p. 181. [Google Scholar]
- [79].Talalay P, Fahey JW, JNutr 2001, 131, 3027S. [DOI] [PubMed] [Google Scholar]
- [80].Dashwood RH, Mutat Res 2002, 511, 89. [DOI] [PubMed] [Google Scholar]
- [81].Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, et al. , Science 2013, 341, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wattenberg LW, Cancer Res 1985, 45, 1. [PubMed] [Google Scholar]
- [83].Wattenberg LW, Cancer Res 1992, 52, 2085s. [PubMed] [Google Scholar]
- [84].Hecht SS, Kassie F, Hatsukami DK, Nat Rev Cancer 2009, 9, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kensler TW, Egner PA, Wang JB, Zhu YR, Zhang BC, et al. , Gastroenterology 2004, 127, S310. [DOI] [PubMed] [Google Scholar]
- [86].Ho E, Beaver LM, Williams DE, Dashwood RH, Adv Nutr 2011, 2, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Martin R, Miquel S, Ulmer J, Kechaou N, Langella P, et al. , Microb Cell Fact 2013, 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, et al. , Science 2006, 313, 848. [DOI] [PubMed] [Google Scholar]
- [89].Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, et al. , Proc Natl Acad Sci U S A 2010, 107, 11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R, Gut Microbes 2014, 5, 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Vizcaino MI, Crawford JM, Nat Chem 2015, 7, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Candela M, Turroni S, Biagi E, Carbonero F, Rampelli S, et al. , World journal of gastroenterology : WJG 2014, 20, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Goldin BR, Gorbach SL, J Natl Cancer Inst 1980, 64, 263. [DOI] [PubMed] [Google Scholar]
- [94].Reddy BS, Rivenson A, Cancer Res 1993, 53, 3914. [PubMed] [Google Scholar]
- [95].Zsivkovits M, Fekadu K, Sontag G, Nabinger U, Huber WW, et al. , Carcinogenesis 2003, 24, 1913. [DOI] [PubMed] [Google Scholar]
- [96].Rowland IR, Curr Pharm Des 2009, 15, 1524. [DOI] [PubMed] [Google Scholar]
- [97].Humblot C, Murkovic M, Rigottier-Gois L, Bensaada M, Bouclet A, et al. , Carcinogenesis 2007, 28, 2419. [DOI] [PubMed] [Google Scholar]
- [98].Alexander J, Wallin H, Rossland OJ, Solberg KE, Holme JA, et al. , Carcinogenesis 1991, 12, 2239. [DOI] [PubMed] [Google Scholar]
- [99].Van Tassell RL, Kingston DG, Wilkins TD, Mutat Res 1990, 238, 209. [DOI] [PubMed] [Google Scholar]
- [100].Weisburger JH, Rivenson A, Kingston DG, Wilkins TD, Van Tassell RL, et al. , Princess Takamatsu Symp 1995, 23, 240. [PubMed] [Google Scholar]
- [101].Vanhaecke L, Van Hoof N, Van Brabandt W, Soenen B, Heyerick A, et al. , J Agric Food Chem 2006, 54, 3454. [DOI] [PubMed] [Google Scholar]
- [102].Fekry MI, Engels C, Zhang J, Schwab C, Lacroix C, et al. , Environ Microbiol Rep 2016, 8, 201. [DOI] [PubMed] [Google Scholar]
- [103].Engels C, Schwab C, Zhang J, Stevens MJ, Bieri C, et al. , Sci Rep 2016, 6, 36246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Vanhaecke L, Knize MG, Noppe H, De Brabander H, Verstraete W, et al. , Food Chem Toxicol 2008, 46, 140. [DOI] [PubMed] [Google Scholar]
- [105].Van de Wiele T, Vanhaecke L, Boeckaert C, Peru K, Headley J, et al. , Environmental health perspectives 2005, 113, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ribiere C, Peyret P, Parisot N, Darcha C, Dechelotte PJ, et al. , Sci Rep 2016, 6, 31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Quigley EM, Gastroenterol Hepatol (N Y) 2013, 9, 560. [PMC free article] [PubMed] [Google Scholar]
- [108].Boyle P, Langman JS, BMJ 2000, 321, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, et al. , Nature 2013, 500, 415.23945592 [Google Scholar]
- [110].Hwang E-S, Bowen PE, Critical Reviews in Food Science and Nutrition 2007, 47, 27. [DOI] [PubMed] [Google Scholar]
- [111].Klurfeld DM, Animal Frontiers 2018, In Press. [DOI] [PMC free article] [PubMed]
- [112].Klurfeld DM, Meat Sci 2015, 109, 86. [DOI] [PubMed] [Google Scholar]