

Abstract
Background
To assess the feasibility of using automated capture of Electronic Medical Record (EMR) data to build predictive models for amyotrophic lateral sclerosis (ALS) outcomes.
Methods
We used an Informatics for Integrating Biology and the Bedside search discovery tool to identify and extract data from 354 ALS patients from the University of Kansas Medical Center EMR. The completeness and integrity of the data extraction were verified by manual chart review. A linear mixed model was used to model disease progression. Cox proportional hazards models were used to investigate the effects of BMI, gender, and age on survival.
Results
Data extracted from the EMR was sufficient to create simple models of disease progression and survival. Several key variables of interest were unavailable without including a manual chart review. The average ALS Functional Rating Scale – Revised (ALSFRS-R) baseline score at first clinical visit was 34.08, and average decline was − 0.64 per month. Median survival was 27 months after first visit. Higher baseline ALSFRS-R score and BMI were associated with improved survival, higher baseline age was associated with decreased survival.
Conclusions
This study serves to show that EMR-captured data can be extracted and used to track outcomes in an ALS clinic setting, potentially important for post-marketing research of new drugs, or as historical controls for future studies. However, as automated EMR-based data extraction becomes more widely used there will be a need to standardize ALS data elements and clinical forms for data capture so data can be pooled across academic centers.
Electronic supplementary material
The online version of this article (10.1186/s12883-018-1208-z) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis, Motor neuron disease, Disease progression, Electronic medical record
Background
Amyotrophic Lateral Sclerosis (ALS) is a fatal neuro-degenerative disease. While over 50 clinical trials have been conducted over the last two decades, none have been successful save riluzole and edaravone [1], which at best offer modest improvements in survival or function [2]. While many studies may have failed because the drugs were ineffective, a recurring theme in ALS are trials which do not meet their primary outcome but yield indeterminate results [3]. Two major hurdles to conducting ALS trials are the rarity of ALS (3.9 in every 100,000 people in the US [4]) and the disease’s heterogeneity [5], which is a barrier to properly powered studies.
Methodology for rare-disease clinical trials is an important area of study for ALS researchers [6]. Enriching trials with historic controls has become possible due to the creation of large pooled placebo data sets [7] and is an approach used for selection of drugs for larger studies, such as in the lithium and rasagiline study [8–10]. Other benefits to large databases of ALS patients include constructing predictive models for screening particular subgroups of patients, which could reduce the heterogeneity of disease progression observed in the trial, or making interim decisions during the conduct of a clinical trial based on predicted and observed disease progression.
The wide implementation of Electronic Medical Record (EMR) systems across the United States, using one of two commercial systems, and the development automated abstraction and de-identification of data, create opportunities to: 1) better understand ALS disease progression and determinants of survival in the clinical setting; 2) use clinical data to enrich existing placebo-arm data sets to improve the power of trials; and 3) leverage this electronic infrastructure to run clinical trials – including EMR-based screening, randomization, and data collection. For these approaches to be worthwhile, we need to be able to demonstrate the feasibility of automatically extracting the data required for modelling ALS disease progression and survival directly from the EMR.
We consider the feasibility of constructing statistical models built with automatically captured EMR patient data from our ALS clinic at the University of Kansas Medical Center (KUMC). This is a key first-step in utilizing the EMR to augment clinical trials.
Methods
Study design
We first determined what specific data was necessary to build models for ALS disease progression and survival. Variables of interest for such models include, at a minimum, demographic information (age, race, and gender), survival information (vital status and date of death), ages of disease onset and diagnosis [5, 11], site of disease onset (typically bulbar or limb) [5, 12–14], riluzole use, BMI [5, 15], FVC [15, 16], and ALS Functional Rating Scale – Revised (ALSFRS-R) score [13, 17–19]. The ALSFRS-R, which is the gold-standard for measuring ALS disease progression, is a clinician-administered series of twelve questions which concern the ability to perform basic functional activities such as eating, walking, dressing, and breathing. Each question is rated on a 0–4 scale, with the overall score of 48 representing normal function [20].
To determine if these variables could be automatically extracted from the EMR, we conducted a retrospective chart review of patients seen at the KUMC ALS Clinic between summer 2013 and summer 2016. We obtained this data directly from the EMR using the KUMC Healthcare Enterprise Repository for Ontological Narration (HERON), powered by Informatics for Integrating Biology and the Bedside (i2b2), a discovery tool that allows searches of de-identified EMR data [21–23]. KUMC’s EMR is provided by Epic (EPIC EMR system, Epic Systems Corporation, Verona, USA, 2015. Using patient’s medical record numbers, this dataset was then verified for completeness and accuracy by manual review of the EMR records. Because we were interested in considering the efficiency of using automated tools versus manual review, the number of hours spent performing the automated review and manual review were tracked.
Study population
The ALS clinic at the University of Kansas Medical Center (KUMC) serves roughly 4 state regions across the Midwest (Kansas, Missouri, Oklahoma, Arkansas). At each visit, patient data collected by the clinician is entered in the EMR. Using HERON, we first performed a search using the ICD10 code for motor neuron disease and at least one visit. This would represent the full pool of patients seen in clinic over this time frame. Next we reduced this to patients having at least one ALSFRS-R score entered into the EMR (Fig. 1). Only patients seen in the ALS specialty clinic with a known diagnosis of motor neuron disease have ALSFRS-R scores in the EMR.
Fig. 1.
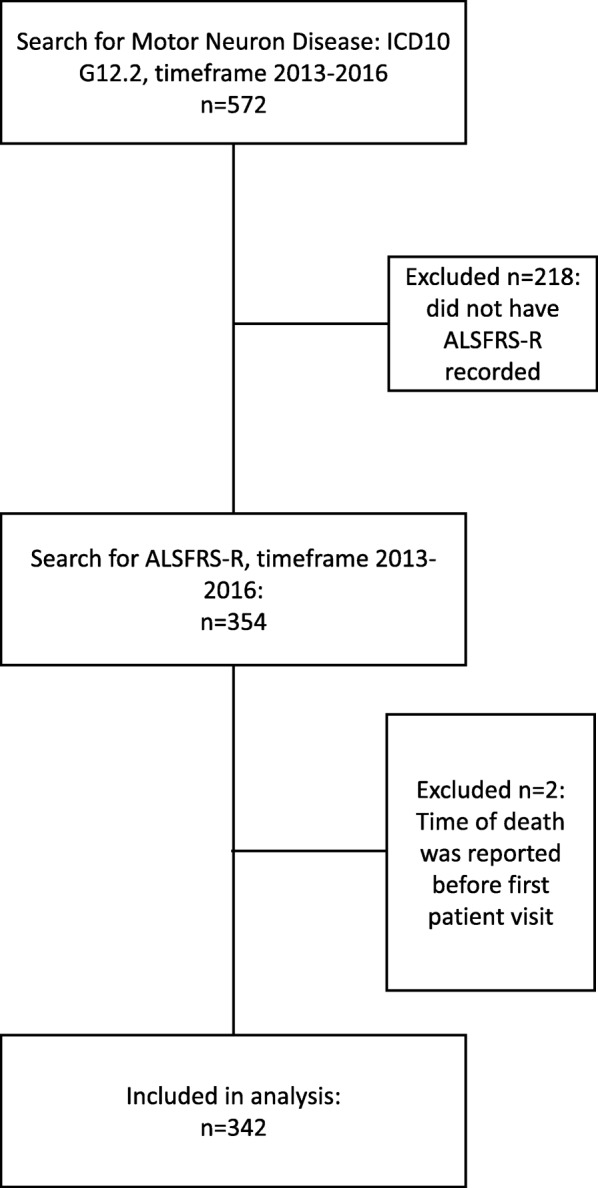
Patient inclusion / exclusion in study
Statistical methods
Analysis of disease progression
Disease progression is measured by patients’ average change per month in ALSFRS-R score. Each patient’s disease progression vs. time (as months since first clinical visit, where the first clinical visit is time 0) was modelled via a linear mixed model which included random slopes and random intercepts (these were allowed to correlate); the fixed effects for the intercept and slope of this model represent the average baseline ALSFRS-R score and average change in ALSFRS-R per month for the clinic. Individual estimates of patient baseline ALSFRS-R score can be obtained by adding the estimated fixed intercept effect to the patient’s estimated random intercept effect; similarly the individual estimate of a patient’s change in ALSFRS-R per month can be estimated by adding the fixed slope effect to the patient’s estimated random slope effect. Linearity was assumed from the literature [14, 24, 25] and verified via diagnostic plots (Additional file 1). These models were fit using the nlme package in R [26].
Analysis of survival
Our survival model analyzed time from patients’ first clinical visit to death (or censoring). Survival data captured by HERON includes both data from the EMR and from the Social Security Death Index [27]. Median survival was estimated by via a Kaplan-Meier approach with interval given by the log-log transformation. A Cox Proportional Hazards model was employed to assess the simultaneous effects of available predictors: BMI, age, and ALSFRS-R score at first visit, and gender. 72 patients were missing baseline BMI scores and were excluded from the Cox model. All analyses were done using R (version 3.2.4) [28].
Results
Accuracy of EMR data
A general search based on ICD10 code identified 572 subjects; 354 patients had at least one ALSFRS-R recorded in the EMR (62.4%), 352 of which were deemed eligible for analysis (two were excluded due to nonsensical death dates) (Fig. 1). Manual review verified ALSFRS-R and sub-scores as accurate.
Many variables of interest for modeling progression and survival (time of disease onset, time of disease diagnosis, and site of disease onset) were only available by manual chart review, because the EMR did not yet have a dedicated field to capture such information (Table 1). Other variables, though extracted via HERON, were not useful for analysis due to extreme sparsity (for example, raw FVC was missing from 59% of records).
Table 1.
Specific data automatically extracted from KUMC EMR by HERON, and data that required a manual chart review
| Information automatically extracted from the EMR using HERON | Information requiring manual chart review | |
|---|---|---|
| Demographic data | Subject age, race, gender, ethnicity | Date of disease onset, date of diagnosis, site of disease onset |
| Longitudinal data | ALSFRS-R and its sub scores, BMI, FVC (raw and percent-predicted) | |
| Medication history | Riluzole use | |
| Survival data | Death status, date of death |
The time spent coordinating with the team at HERON to properly identify and extract variables of interest took roughly 3 h. The manual review took over 30 h. Once the variables of interest were properly identified within the EMR, obtaining the data through HERON became a matter of minutes rather than hours.
Patient characteristics
Table 2 reports patient characteristics: participants at KUMC were predominantly male (57%), had an average age at first clinical visit of 64.1 years, 65% with limb onset, 63% taking riluzole, with an average ALSFRS-R at first visit of 34.5.
Table 2.
Demographic information of KUMC ALS clinical patients. Baseline is defined as the time of a patient’s first recorded ALSFRS-R score at KUMC
| KUMC | |
|---|---|
| Number of patients | 352 |
| Percent female / Male | 43/ 57 |
| Percent Caucasian / Non-Caucasian | 89/ 11 |
| Percent limb onset / Bulbar / Other | 65/ 27/ 8 |
| Percent using riluzole Yes / No / NA | 63/ 35 / 2 |
| Percent survived during follow up | 69 |
| Median time from baseline to last record, months (IQR) | 7.1 (17.7) |
| Median age at baseline (IQR) | 65.3 (11.2) |
| Median number of months from onset to baseline (IQR) | 19.0 (28.1) |
| Median baseline FVC percent predicted (IQR) | 70.0 (32.5) |
| Median BMI at baseline (IQR) | 26.4 (7.23) |
| Median number of ALSFRS-R assessments (IQR) | 3 (4) |
| Median baseline ALSFRS-R (SD) | 35.5 (10.0) |
Statistical results
Analysis of disease progression
The fixed-effect of clinic-level baseline ALSFRS-R score was 34.08 with 95% interval (33.28, 34.88), with random-effect standard deviation of 7.08 with 95% interval (6.49, 7.28). The fixed-effect of clinic-level disease progression (in terms of loss of ALSFRS-R per month) was 0.64 with 95% interval (0.56, 0.73), with random-effect standard deviation of 0.56 with 95% interval (0.48, 0.65). See Fig. 2 for graphical representation of the estimates of disease progression and baseline ALSFRS-R score by patient.
Fig. 2.
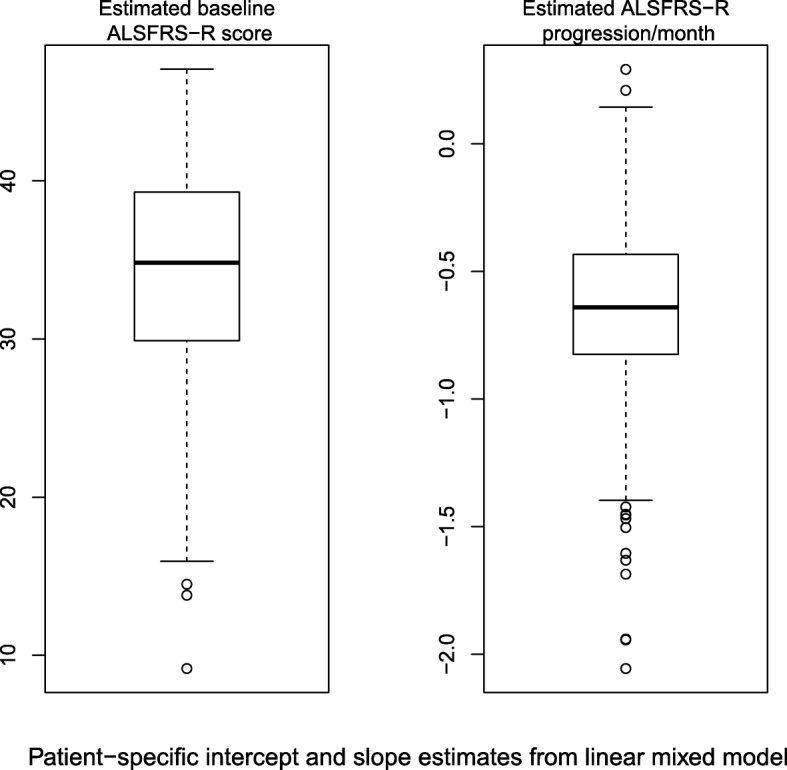
The distributions of baseline scores and monthly disease progression as estimated by the linear mixed effects model. These were calculated from the model by adding the fixed effect of intercept (or slope) to each patient’s estimated random effect of intercept (or slope)
Analysis of survival
Median survival time from first visit was 27 months (95% interval (22.7, 33.7)) for KUMC patients, as per Kaplan-Meier model. The Kaplan-Meier survival curve (unadjusted for other covariates) is given in Fig. 3. We observed a large number of censored observations (69% censored).
Fig. 3.
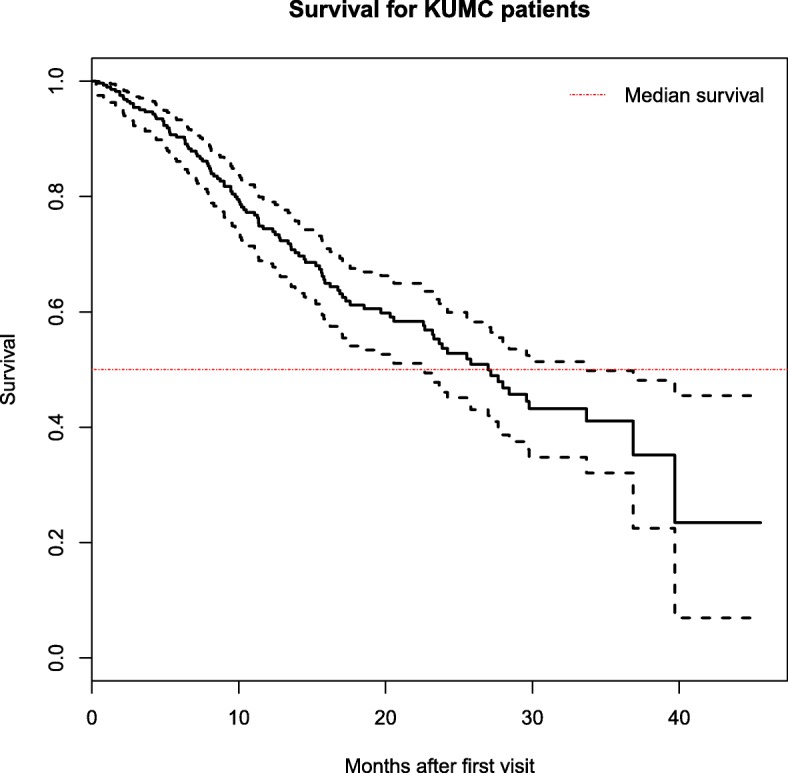
Kaplan-Meier survival curves and 95% confidence interval for KUMC patients. The horizontal line represents the median survival time, equivalent to where the y-axis is 0.5
Our Cox proportional hazards model found baseline ALSFRS-R score, baseline age, and baseline BMI as significant (p < 0.05) predictors of survival when α = 0.05. Higher baseline ALSFRS-R and BMI were related with improved survival, while higher baseline age was associated with decreased survival (Table 3).
Table 3.
Hazard ratios from Cox model
| Survival Model | |||
|---|---|---|---|
| Covariate | Hazard Ratio | 95% CI | P-value |
| Age (at onset) | 1.025 | (1.004, 1.047) | 0.019 |
| Sex (male) | 0.732 | (0.473, 1.133) | 0.161 |
| BMI (baseline) | 0.950 | (0.909, 0.993) | 0.022 |
| ALSFRS-R total (baseline) | 0.939 | (0.909, 0.970) | <0.001 |
Hazard ratios below one result in improved survival
Discussion
Here we demonstrate the feasibility of using an automated extraction tool (HERON) to obtain ALS patient data directly from the KUMC EMR which could be used for analysis of ALS disease progression and survival. While data pertaining to demographic, ALSFRS-R, and survival information was both readily obtainable and accurate, some key variables (especially disease onset time and riluzole use) were only available via manual EMR review and/or suffered from large amounts of missing data.
The main advantages to using automatic tools such as HERON includes that they can drastically reduce the amount of time needed to accurately capture EMR data when compared to a manual review of the EMR. This methodology is generalizable across other research sites: EPIC is one of the two major EMR record systems in the US, serving over 50% of patients in the US [29], and represents a large number of academic centers with ALS clinics. The automatic extraction tool HERON is powered by i2b2, which is used by dozens of research institutions within the US and abroad [30].
Looking towards the future, as EMR data becomes more complete, other advantages of using this approach will emerge. Advantages to complete and comprehensive ALS records in the EMR include allowing clinicians to track the performance of their patients clinic-wide and compare these to other ALS clinics, for both research and quality control purposes. For example, the average ALSFRS-R decline per month in the KUMC clinic of 0.64 is somewhat high compared to reports from other clinics, which report monthly ALSFRS-R declines of between 0.36 to 0.65 [14, 31–33]. Note that this may be because we were unable to adjust for how long patients’ have had the disease.
Other future advantages include the ability to perform retrospective studies quickly and efficiently, which could create support for new therapeutics or improvements to standards of care. This depends heavily on tracking of patients’ use of therapeutics in a way that is accessible in the EMR. EMR data could also be used to augment clinical trial data, being used as either a placebo/ standard of care arm or as historical controls [34]. This has become a vital issue for the broader ALS community. For example, approval of edaravone in the US has raised many questions about which patients will benefit from this therapy and for how long. This could be answered by pooling ALS clinic data. In addition, edaravone has put a limit on how broadly existing placebo data sets like PRO-ACT can be used for historical controls in clinical trials. Contemporary controls captured through automated EMR data abstraction could be one solution to this problem [1, 35, 36].
One current criticism of ALS clinical trials is that the ALS patients who serve in these trials are not representative of the general population [37], which is likely due to the rigorous inclusion/exclusion criteria for these trials. One simple solution to make ALS trials more representative is to simply modify the inclusion/exclusion criteria – however the resulting increased patient variability would require very large studies. Again we see the potential utility of EMR data: with a more general trial population, we would be free to use the EMR to augment the control population for these trials. Networks such as the Northeast or Western ALS Study Groups [38] could provide placebo or standard-of-care arms in a variety of designs, and could make such large-scale studies possible.
The main disadvantage of this approach is the current lack of completeness of the EMR with respect to critical ALS data, resulting in incomplete statistical models. To use the EMR as we propose across multiple academic centers, the ALS community would need to agree on a set of common data elements or ALS-related forms to capture in the EMR. Such agreement could allow common data dictionaries to be used to allow for automated data capture not just across academic centers, but across different EMR platforms (i.e. Epic and Cerner). Furthermore, physicians and their clinic personnel would need to adhere to these data dictionaries, and then rigorously enter all the required data for each patient at each visit. Many efforts have already been made toward developing these common data sets for ALS: much of the field already captures the ALSFRS-R, the FVC, and details about the diagnosis at each visit. In addition several initiatives are underway to standardize forms across institutions, with a suite of ALS clinic forms available for download through Epic Central.
One example of critical information that needs to be collected in a standardized way is disease onset time. Because disease duration (which is derived from disease onset time) is critical for both survival and disease progression modelling [5, 12, 24, 25, 39], it is necessary that ALS clinics dedicate a data-capture form for this, as opposed to entering it as free-text notes/comments where it is difficult to find systematically. Other critical variables include usage of approved therapeutics (such as riluzole or ederavone), time of diagnosis, and location of symptom onset.
Conclusions
We were able to use automated extraction tools to accurately obtain necessary variables from the EMR with which to create simple statistical models of both ALS disease progression and survival time. Key variables that might offer large improvements to these models (such as disease onset time or riluzole use) were unavailable via automatic extraction. In the future, as automated EMR data abstraction becomes increasingly important for post-marketing surveillance of FDA approved drugs, or for use as concurrent controls, the ALS community will need to adopt common data elements for the EMR. Optimal use of the EMR requires disease-specific key variables, such as disease-onset time for ALS, to be identifiable and obtainable by data extraction tools as well as rigorous data entry by clinical staff.
Additional file
Linearity of 16 randomly selected patients who had > 3 visits. For 16 randomly selected patients with more than three recorded visits, we show their ALSFRS-R score versus time in months, along with the fit regression line. This gives the reader a general idea of the linear decline of the ALSFRS-R seen in patients. (PDF 8 kb)
Acknowledgements
Not applicable
Funding
The Mabel A. Woodyard Fellowship in Neurodegenerative Disorders and the Roofe Fellowship in Neuroscience Research funded the writing of the manuscript, the statistical programming, and the analyses. All other funding bodies supported the authors’ time to work on this project during design, analysis, and manuscript preparation. This work was supported by a CTSA grant from NCRR and NCATS awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research # UL1TR000001 (formerly #UL1RR033179). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, NCRR, or NCATS. J.S. work on this project supported by a fellowship grant from the NCATS / Clinical Research in ALS and Related Disorders for Therapeutic Development Consortium awarded to the University of Miami (U54NS092091). HERON is supported by a CTSA grant from NCRR and NCATS awarded to the University of Kansas Medical Center for Frontiers: University of Kansas Clinical and Translational Science Institute # UL1TR002366 (formerly # UL1TR000001 and #UL1RR033179).
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due containing identifying medical information but de-identified data are available from the corresponding author on reasonable request.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- ALSFRS-R
Amyotrophic lateral sclerosis functional rating scale – revised
- EMR
Electronic medical record
- FVC
Forced vital capacity
- HERON
Healthcare Enterprise Repository for Ontological Narration
- I2b2
Informatics for Integrating Biology and the Bedside
- KUMC
University of Kansas Medical Center
Authors’ contributions
AK managed the data, performed statistical analyses, drafted and revised the manuscript. JS proposed the study, and assisted in the analysis and interpretation of data, drafting/revising the manuscript for content, and also provided study supervision. LW, OJ, and RB assisted with drafting/revising the manuscript and the acquisition of the data. BG and JH assisted with analysis and interpretation of data, verification of statistical methods, and drafting/revising the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This retrospective chart review was IRB-approved and received a waiver of consent by University of Kansas Medical Center Human Research Protection Program Institutional Review Board (IRB# STUDY00004291). Patient data was managed on secure servers at KUMC.
Consent for publication
Not applicable
Competing interests
JS is a consultant for aTyr, Acceleron, Fulcrum, Regeneron, and Strongbridge. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alex G. Karanevich, Phone: 816-813-8876, Email: akaranevich@embstats.com
Luke J. Weisbrod, Email: lweisbrod@kumc.edu
Omar Jawdat, Email: ojawdat@kumc.edu.
Richard J. Barohn, Email: rbarohn@kumc.edu
Byron J. Gajewski, Email: bgajewski@kumc.edu
Jianghua He, Email: jhe@kumc.edu.
Jeffrey M. Statland, Email: jstatland@kumc.edu
References
- 1.WRITING GROUP ON BEHALF OF THE EDARAVONE (MCI-186) ALS 18 STUDY GROUP Exploratory double-blind, parallel-group, placebo-controlled study of edaravone (MCI-186) in amyotrophic lateral sclerosis (Japan ALS severity classification: grade 3, requiring assistance for eating, excretion or ambulation) Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):40–48. doi: 10.1080/21678421.2017.1361441. [DOI] [PubMed] [Google Scholar]
- 2.Goyal NA, Mozaffar T. Experimental trials in amyotrophic lateral sclerosis: a review of recently completed, ongoing and planned trials using existing and novel drugs. Expert Opin Investig Drugs. 2014;23(11):1541–1551. doi: 10.1517/13543784.2014.933807. [DOI] [PubMed] [Google Scholar]
- 3.Katyal N, Govindarajan R. Shortcomings in the Current Amyotrophic Lateral Sclerosis Trials and Potential Solutions for Improvement. Front Neurol. 2017;8 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5626834/ [cited 17 Sep 2018]. [DOI] [PMC free article] [PubMed]
- 4.Paul Mehta M. Prevalence of Amyotrophic Lateral Sclerosis — United States, 2012–2013. MMWR Surveill Summ. 2016;65 Available from: https://www.cdc.gov/mmwr/volumes/65/ss/ss6508a1.htm [cited 17 Sep 2018]. [DOI] [PubMed]
- 5.Zach N, Ennist DL, Taylor AA, Alon H, Sherman A, Kueffner R, et al. Being PRO-ACTive: what can a clinical trial database reveal about ALS? Neurotherapeutics. 2015;12(2):417–423. doi: 10.1007/s13311-015-0336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilgers R-D, König F, Molenberghs G, Senn S. Design and analysis of clinical trials for small rare disease populations. J Rare Dis Res Treat. 2016:53–60.
- 7.Atassi N, Berry J, Shui A, Zach N, Sherman A, Sinani E, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology. 2014;83(19):1719–1725. doi: 10.1212/WNL.0000000000000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Statland JM, Moore D, Wang Y, Walsh M, Mozaffar T, Elman L, et al. Rasagiline for amyotrophic lateral sclerosis: a randomized controlled trial. Muscle Nerve. 2018. https://www.ncbi.nlm.nih.gov/pubmed/30192007. [DOI] [PMC free article] [PubMed]
- 9.Miller RG, Moore DH, Forshew DA, Katz JS, Barohn RJ, Valan M, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis. Neurology. 2011;77(10):973–979. doi: 10.1212/WNL.0b013e31822dc7a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordon PH, Cheung Y-K, Levin B, Andrews H, Doorish C, Macarthur RB, et al. A novel, efficient, randomized selection trial comparing combinations of drug therapy for ALS. Amyotroph Lateral Scler. 2008;9(4):212–222. doi: 10.1080/17482960802195632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Testa D, Lovati R, Ferrarini M, Salmoiraghi F, Filippini G. Survival of 793 patients with amyotrophic lateral sclerosis diagnosed over a 28-year period. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(4):208–212. doi: 10.1080/14660820410021311. [DOI] [PubMed] [Google Scholar]
- 12.Pastula DM, Coffman CJ, Allen KD, Oddone EZ, Kasarskis EJ, Lindquist JH, et al. Factors associated with survival in the National Registry of veterans with ALS. Amyotroph Lateral Scler. 2009;10(5–6):332–338. doi: 10.3109/17482960802320545. [DOI] [PubMed] [Google Scholar]
- 13.Elamin M, Bede P, Montuschi A, Pender N, Chio A, Hardiman O. Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol. 2015;262(6):1447–1454. doi: 10.1007/s00415-015-7731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnus T, Beck M, Giess R, Puls I, Naumann M, Toyka KV. Disease progression in amyotrophic lateral sclerosis: predictors of survival. Muscle Nerve. 2002;25(5):709–714. doi: 10.1002/mus.10090. [DOI] [PubMed] [Google Scholar]
- 15.Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills A-M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44(1):20–24. doi: 10.1002/mus.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006;77(3):390–392. doi: 10.1136/jnnp.2005.072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon PH, Cheng B, Salachas F, Pradat P-F, Bruneteau G, Corcia P, et al. Progression in ALS is not linear but is curvilinear. J Neurol. 2010;257(10):1713–1717. doi: 10.1007/s00415-010-5609-1. [DOI] [PubMed] [Google Scholar]
- 18.Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66(2):265–267. doi: 10.1212/01.wnl.0000194316.91908.8a. [DOI] [PubMed] [Google Scholar]
- 19.Kollewe K, Mauss U, Krampfl K, Petri S, Dengler R, Mohammadi B. ALSFRS-R score and its ratio: a useful predictor for ALS-progression. J Neurol Sci. 2008;275(1–2):69–73. doi: 10.1016/j.jns.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 20.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (phase III) J Neurol Sci. 1999;169(1–2):13–21. doi: 10.1016/S0022-510X(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 21.Murphy SN, Weber G, Mendis M, Gainer V, Chueh HC, Churchill S, et al. Serving the enterprise and beyond with informatics for integrating biology and the bedside (i2b2) J Am Med Inform Assoc. 2010;17(2):124–130. doi: 10.1136/jamia.2009.000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waitman LR, Warren JJ, Manos EL, Connolly DW. Expressing observations from electronic medical record flowsheets in an i2b2 based clinical data repository to support research and quality improvement. AMIA Annu Symp Proc. 2011;2011:1454–1463. [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy SN, Mendis ME, Berkowitz DA, Kohane I, Chueh HC. Integration of clinical and genetic data in the i2b2 architecture. AMIA Annu Symp Proc. 2006;1040. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1839291/. [PMC free article] [PubMed]
- 24.Armon C, Graves MC, Moses D, Forté DK, Sepulveda L, Darby SM, et al. Linear estimates of disease progression predict survival in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2000;23(6):874–882. doi: 10.1002/(SICI)1097-4598(200006)23:6<874::AID-MUS5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 25.Karanevich AG, Statland JM, Gajewski BJ, He J. Using an onset-anchored Bayesian hierarchical model to improve predictions for amyotrophic lateral sclerosis disease progression. BMC Med Res Methodol. 2018;18(1):19. doi: 10.1186/s12874-018-0479-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinheiro J, Bates D, DebRoy S, Sarkar D, Heisterkamp S, R-core. nlme: Linear and Nonlinear Mixed Effects Models. 2018. Available from: https://CRAN.R-project.org/package=nlme [cited 17 Sep 2018]
- 27.Social Security Death Master File -> Home. Available from: https://ladmf.ntis.gov/ [cited 17 Sep 2018]
- 28.R Core Team. R: a language and environment for statistical computing. [internet]. Vienna, Austria: R Foundation for Statistical Computing; 2017. Available from: https://www.r-project.org/.
- 29.jglaze@madison.com, 608-252-6138 JG. Epic Systems draws on literature greats for its next expansion. madison.com. Available from: https://madison.com/news/local/govt-and-politics/epic-systems-draws-on-literature-greats-for-its-next-expansion/article_4d1cf67c-2abf-5cfd-8ce1-2da60ed84194.html [cited 17 Sep 2018]
- 30.i2b2: Informatics for Integrating Biology & the Bedside. Available from: https://www.i2b2.org/work/i2b2_installations.html [cited 17 Sep 2018]
- 31.Shamshiri H, Fatehi F, Davoudi F, Mir E, Pourmirza B, Abolfazli R, et al. Amyotrophic lateral sclerosis progression: Iran-ALS clinical registry, a multicentre study. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7–8):506–511. doi: 10.3109/21678421.2015.1074698. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe H, Atsuta N, Nakamura R, Hirakawa A, Watanabe H, Ito M, et al. Factors affecting longitudinal functional decline and survival in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(3–4):230–236. doi: 10.3109/21678421.2014.990036. [DOI] [PubMed] [Google Scholar]
- 33.Mandrioli J, Biguzzi S, Guidi C, Sette E, Terlizzi E, Ravasio A, et al. Heterogeneity in ALSFRS-R decline and survival: a population-based study in Italy. Neurol Sci. 2015;36(12):2243–2252. doi: 10.1007/s10072-015-2343-6. [DOI] [PubMed] [Google Scholar]
- 34.Cowie MR, Blomster JI, Curtis LH, Duclaux S, Ford I, Fritz F, et al. Electronic health records to facilitate clinical research. Clin Res Cardiol. 2017;106(1):1–9. doi: 10.1007/s00392-016-1025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalin A, Medina-Paraiso E, Ishizaki K, Kim A, Zhang Y, Saita T, et al. A safety analysis of edaravone (MCI-186) during the first six cycles (24 weeks) of amyotrophic lateral sclerosis (ALS) therapy from the double-blind period in three randomized, placebo-controlled studies. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):71–79. doi: 10.1080/21678421.2017.1362440. [DOI] [PubMed] [Google Scholar]
- 36.Al-Chalabi A, Andersen PM, Chandran S, Chio A, Corcia P, Couratier P, et al. July 2017 ENCALS statement on edaravone. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(7–8):471–474. doi: 10.1080/21678421.2017.1369125. [DOI] [PubMed] [Google Scholar]
- 37.Chiò A, Canosa A, Gallo S, Cammarosano S, Moglia C, Fuda G, et al. ALS clinical trials: do enrolled patients accurately represent the ALS population? Neurology. 2011;77(15):1432–1437. doi: 10.1212/WNL.0b013e318232ab9b. [DOI] [PubMed] [Google Scholar]
- 38.Miller RG, Moore DH, Jackson CE. WALS study group. Western ALS Study Group Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(Suppl 1):121–124. doi: 10.1080/17434470410019988. [DOI] [PubMed] [Google Scholar]
- 39.Hothorn T, Jung HH. RandomForest4Life: a random Forest for predicting ALS disease progression. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(5–6):444–452. doi: 10.3109/21678421.2014.893361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Linearity of 16 randomly selected patients who had > 3 visits. For 16 randomly selected patients with more than three recorded visits, we show their ALSFRS-R score versus time in months, along with the fit regression line. This gives the reader a general idea of the linear decline of the ALSFRS-R seen in patients. (PDF 8 kb)
Data Availability Statement
The datasets generated and/or analysed during the current study are not publicly available due containing identifying medical information but de-identified data are available from the corresponding author on reasonable request.