

Abstract
Background
Spinal cord injury (SCI) is a serious disease with high disability and mortality rates, with no effective therapeutic strategies available. In SCI, abnormal DNA methylation is considered to be associated with axonal regeneration and cell proliferation. However, the roles of key genes in potential molecular mechanisms of SCI are not clear.
Material/Methods
Subacute spinal cord injury models were established in Wistar rats. Histological observations and motor function assessments were performed separately. Whole-genome bisulfite sequencing (WGBS) was used to detect the methylation of genes. Gene ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed using the DAVID database. Protein–protein interaction (PPI) networks were analyzed by Cytoscape software.
Results
After SCI, many cavities, areas of necrotic tissue, and many inflammatory cells were observed, and motor function scores were low. After the whole-genome bisulfite sequencing, approximately 96 DMGs were screened, of which 50 were hypermethylated genes and 46 were hypomethylated genes. KEGG pathway analysis highlighted the Axon Guidance pathway, Endocytosis pathway, T cell receptor signaling pathway, and Hippo signaling pathway. Expression patterns of hypermethylated genes and hypomethylated genes detected by qRT-PCR were the opposite of WGBS data, and the difference was significant.
Conclusions
Abnormal methylated genes and key signaling pathways involved in spinal cord injury were identified through histological observation, behavioral assessment, and bioinformatics analysis. This research can serve as a source of additional information to expand understanding of spinal cord-induced epigenetic changes.
MeSH Keywords: DNA Methylation, Epigenomics, Nerve Regeneration, Spinal Cord Injuries
Background
Spinal cord injury (SCI) can lead to severe autonomic, sensory, and motor dysfunction [1]. It is estimated that more than 3 million people live with spinal cord injury and its worldwide incidence is 23 to 70 individuals per million [2]. Due to the high incidence and high disability rate of spinal cord injury, a serious burden has been placed on society and families. Spinal cord injury can be divided into primary mechanical injury and secondary injury [3]. According to the pathogenesis and the time after injury, the secondary injury process can be divided into acute, subacute, and chronic phases [4]. In addition, ‘microenvironment imbalance’ is considered to be the main cause of the poor regeneration and recovery of SCI [5]. Microenvironmental imbalances are often accompanied by increased inhibitory factors, loss of neurons, filling of glial cells and reduction of promoting factors at different times and spaces [6]. Multiple cells combined with different nutritional factors or scaffolds have become the focus of spinal cord injury repair [7]. However, treatment with cells, surgery, or medication have been unable to completely cure spinal cord injuries [8].
Conrad Waddington defined the term “epigenetics” to describe inherited changes in phenotypes without genotypic changes [9,10]. At present, epigenetics usually refers to a stable genetic phenotype resulting from a chromosome change without variations in the DNA sequence [11]. Due to the role of transcriptional and epigenetic regulations, even though mature cells start off with the same genotype, their phenotypes may quite different [12]. In the brain of adult vertebrates, the formation of new neurons occurs in a specific population of cells. Nerve regeneration is extremely difficult under normal physiological conditions, and it is usually described as being induced after spinal cord injury [13]. Although the exact mechanism of neural repair is not yet clear, previous studies have shown that specific cytoplasmatic factors (exosome), transcriptional factor network, and epigenetic regulators play key roles in nerve regeneration [14].
DNA methylation is one of the most thoroughly studied epigenetic modifications [15]. The characteristic of DNA methylation is adding a methyl group to cytosine nucleotide without changing the properties of base pairs. Due to the influence of environment or age, different DNA methylation patterns affect the expression of genes involved in crosstalk between neural activity and inflammatory pathways, further contributing to various diseases [16]. Previous studies have confirmed that DNA methylation is associated with a variety of diseases such as cancer [17], Alzheimer’s disease [18], and hematological diseases [19]. DNA methyltransferases are key enzymes in the process of DNA methylation. More and more studies have shown that DNA methyltransferase plays a critical role in the early development of the central nervous system (CNS), including cognition, learning, and memory [20]. However, the effect of DNA methylation on spinal cord injury has been unclear.
In the present study, whole-genome bisulfite sequencing (WGBS) technology was used to assess tissue before and after spinal cord transection in rats. The discovery of abnormal DNA methylation in the thoracic spinal cord might provide a new repair approach for epigenetic therapies of spinal cord injury.
Material and Methods
Animals
Adult female Wistar rats (approximately 230–250 g, provided by Radiation Study Institute-Animal Center, Tianjin, China, License Key: SCXK2012-0004) were used in this study. Two experimental groups were established: a sham group (n=9) and a SCI group (n=9). All animal experiments were performed according to the guidelines for laboratory animal safety and care as issued by the Ethics Committee of Tianjin Medical University General Hospital and the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). All procedures performed in the study involving animals were consistent with the ethical standards set by the above-mentioned institutions.
Spinal cord transection
Adult female Wistar rats were used for spinal cord transection as described earlier [21–23]. In brief, all rats subjected to SCI were deeply anesthetized with isoflurane to minimize suffering. Following laminectomy at the T10–11 vertebral level, a 2-mm segment of spinal cord with associated spinal roots was completely removed at the T10 spinal cord level. Sham control rats also underwent laminectomy without contusion. For postoperative care, the bladder was emptied manually twice a day for a month. All rats received an intramuscular injection of penicillin (40 000 U/kg/day) for 5 days to prevent infection.
DNA methylation analysis
DNA was extracted from the spinal cord using a DNA extraction kit (TIANamp Genomic DNA Kit, China) according to the manufacturer’s instructions. Five hundred nanograms of bisulfite-converted DNA per sample were analyzed by Illumina Infinium Human Methylation 450 BeadChip array (Illumina, China). Raw data analysis and preliminary data quality control were performed with GenomeStudio software 2011.1 (Illumina, China). Specific experimental procedures for DNA methylation sequencing are shown in Figure 1. For further gene expression analysis, all data were imported into Cytoscape software (v3.6.1) and GraphPad Prism software (Graph Pad v6.01) for functional analysis and statistical analysis [24]. Differentially methylated genes (DMGs) were identified (mean methylation difference ≥20, P<0.001) as described earlier [25]. Using the bioinformatics resources of DAVID 6.7 (https://david.ncifcrf.gov/), the Gene Ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially methylated genes were performed [26].
Figure 1.
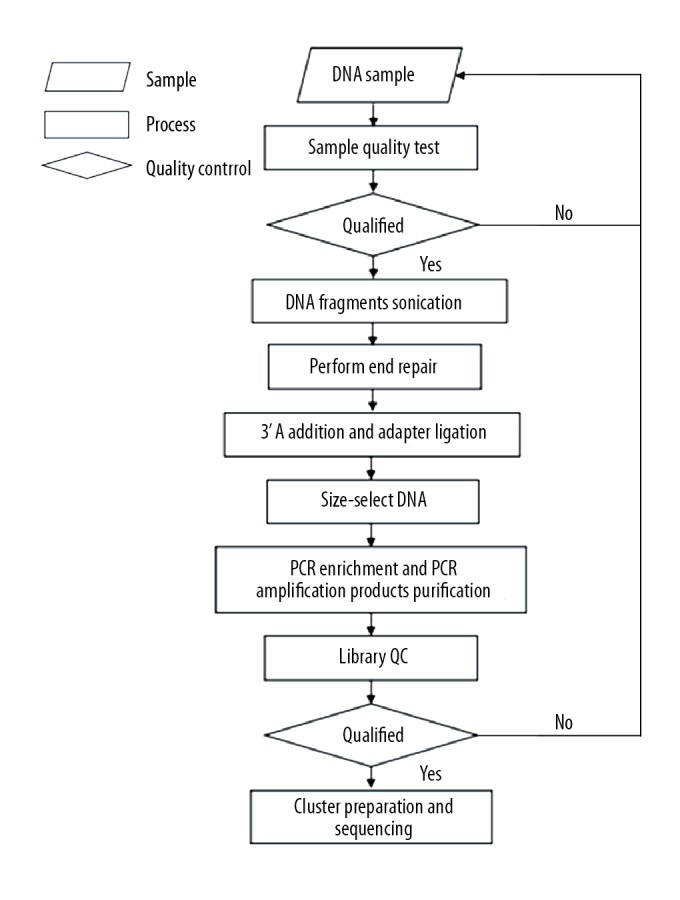
Concise experimental procedure for the whole-genome bisulfite sequencing.
Histology and immunohistochemistry
The histological evaluation was performed at 4 weeks post surgeries. The rats were anesthetized with isoflurane and transcardially perfused with 4% paraformaldehyde in PBS. Spinal cord tissue was cut into paraffin sagittal sections of 7 μm thickness. After the paraffin sections were prepared, the paraffin sections were stained with hematoxylin-eosin (Solarbio, China), as described previously [27]. Finally, the stained sections were observed under a microscope (Nikon, Japan).
Quantitative real-time PCR
Total RNA was extracted from spinal cord tissues using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions [28]. One microgram of total RNA per sample was reverse-transcribed using a Reverse Transcription Kit (Applied Biosystems, USA). Quantitative real-time RT-PCR was performed on a LightCycler® 480 Real-Time PCR System (Roche, Germany) using SYBR-Green (Thermal, USA). GAPDH acted as internal control. The primers are listed in Table 1. All samples were analyzed in duplicate, then the average value of the duplicates was used for quantification.
Table 1.
Information on primer sequences.
| Gene | Forward primer 5′ to 3′ | Reverse primer 5′ to 3′ | Annealing temperature (°C) |
|---|---|---|---|
| Csf2 | AATGACATGCGTGCTCTGGAGAAC | TCGTCTGGTAGTGGCTGGCTATC | 54 |
| Fars2 | CCACCTGGCAGAACTTCGATAGC | GTCACGCCGATACACATCACCTAC | 54 |
| Synj2 | TCCATGTCTCGTACCATCCAGTCC | CCGTGTTGTCCAGCAGCATCC | 53 |
| Ppp3cc | TCCGAGGCTGCTCCTACTTCTTC | AGCCAGTTGCTTGGTTCTTCCTG | 54 |
| Stat4 | CAGGACTGGAAGAAGCGGCAAC | AAGCAGTTCTGAAGCTGGTCCAAC | 53 |
| Pcsk2 | CACAGTCAACGCAACCAGGAGAG | ACCTTGGAGTCGTCGTCTCTTGG | 54 |
| Dnm3 | GTCACACCAGCCAACACCGATC | GGTGATAACGCCAATGGTCCTCAG | 54 |
| Hmgcll1 | ACTCCAGGCAGCATGAAGACAATG | TCATGGCAGTGAACAGCAAGAGC | 54 |
Behavioral analysis
After surgery, hindlimb function of the rats was evaluated with the Basso, Beattie, and Bresnahan (BBB) open field locomotor test [29]. BBB scores were taken 3 days prior to injury, and once each week following SCI, for 8 weeks. BBB scores of each animal were calculated as the average of movement scores between the 2 hind limbs. Two independent researchers blind of the different experimental treatments determined the BBB scores.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism software. Data are reported as mean ± standard deviations. The BBB scores data were evaluated using 2-way analysis of variance (ANOVA). P<0.05 was considered a statistically significant difference.
Results
Histological and behavioral evaluation after spinal cord injury
After spinal cord injury, the loss of neuronal cell was noticeable and axons were severed. Cells and tissue morphology of the sham group were relatively complete (Figure 2A). Inflammatory cell infiltration, bleeding, and glial scars were observed in the SCI group (Figure 2B). Motor function recovery was evaluated using the BBB open field locomotor test. The BBB scores ranged from 0 (no hindlimb movement) to 21 (normal hindlimb move) according to the rating scale. After successful spinal cord injury, the BBB score of all rats in the SCI group was 0. After 8 weeks, the score of motor function of some rats in the SCI group reached 5 (Figure 2C).
Figure 2.

Histological observation and motor function assessment after spinal cord injury. (A) Hematoxylin-eosin staining of spinal cord sections in the sham group at 8 weeks after reperfusion. (B) Hematoxylin-eosin staining of spinal cord sections in the SCI group at 8 weeks after reperfusion. (C) BBB scores of Wistar rats. Values are means ±SE (* P<0.0001).
Identification of DMGs in SCI
After the whole-genome bisulfite sequencing, a total of 623 487 210 clean reads in the sham group and 623 545 728 clean reads in the SCI group were obtained, respectively. There were 1158 differentially methylated genes identified (Table 2). Among differentially methylated genes, 50.95% were hypermethylated and 49.05% were hypomethylated. A total of 370 differentially methylated genes between the sham group and SCI group were selected (P<0.05). According to screening criteria (mean methylation difference ≥20, P<0.001), 96 methylated genes were selected. Among them, 50 genes were hypermethylated and 46 genes were hypomethylated (Tables 3, 4). All of the aberrantly expressed genes are shown in a heat map in Figure 3.
Table 2.
Differential methylation in the spinal cord.
| Class | Hypermethylated | Hypomethylated | Total |
|---|---|---|---|
| Differentially methylated genes | 590 | 568 | 1158 (100%) |
| DMG, P<0.05, mean.meth.diff=20 | 189 | 181 | 370 (31.95%) |
| DMG, P<0.001, mean.meth.diff=20 (remove repetition) | 50 | 46 | 96 (8.29%) |
Table 3.
Complete list of the 50 hypermethylated genes.
| Chr | Symbol | ID | Length | Num. CpGs | DMR. p value | DMR. q value | Mean. meth. diff |
|---|---|---|---|---|---|---|---|
| chr10 | Fbxw11 | NM_001106993_I2_introns | 13 | 3 | 3.97E-13 | 3.71E-11 | 41.39596773 |
| chr1 | Gna14 | NM_001013151_I1_introns | 44 | 3 | 4.18E-10 | 1.02E-08 | 39.78573567 |
| chr2 | Chi3l3 | NM_001191712_I5_introns | 289 | 3 | 6.72E-07 | 5.21E-06 | 39.31869094 |
| chr1 | Vps13a | NM_001100975_E54_exon | 51 | 3 | 4.76E-10 | 1.07E-08 | 38.36766934 |
| chr15 | Adra1a | NM_017191_I1_introns | 203 | 4 | 7.06E-13 | 2.54E-11 | 38.16288829 |
| chr6 | Frmd6 | NM_001271054_I1_introns | 171 | 3 | 7.03E-12 | 2.11E-10 | 37.4625921 |
| chr7 | Palm | NM_130829_I8_introns | 170 | 4 | 0.000442034 | 0.001304001 | 36.40773047 |
| chr6 | Nubpl | NM_001185025_I4_introns | 121 | 3 | 4.52E-07 | 6.02E-06 | 35.86094377 |
| chr17 | Crem | NM_001110860_I3_introns | 105 | 3 | 8.12E-05 | 0.000517023 | 35.7757685 |
| chr7 | Fam227a | NM_001130581_I20_introns | 61 | 3 | 0.000174069 | 0.000641881 | 35.64110942 |
| chr10 | Fstl4 | NM_001107000_I4_introns | 15 | 3 | 1.34E-05 | 0.000156783 | 34.86382548 |
| chr2 | Col11a1 | NM_013117_I49_introns | 226 | 3 | 1.04E-05 | 5.08E-05 | 34.8266253 |
| chr10 | Neurl1b | NM_001142652_I4_introns | 80 | 7 | 9.05E-06 | 0.000120934 | 34.13727909 |
| chr13 | Plxna2 | NM_001105988_I3_introns | 219 | 3 | 3.78E-05 | 0.000184147 | 33.8264037 |
| chr12 | Fry | NM_001170398_I63_introns | 55 | 4 | 8.19E-08 | 3.44E-06 | 33.49560871 |
| chr10 | Snx29 | NM_001109526_I7_introns | 6 | 4 | 2.56E-05 | 0.000217367 | 33.28848981 |
| chr10 | Litaf | NM_001105735_I1_introns | 54 | 5 | 6.91E-10 | 2.59E-08 | 33.03121583 |
| chr1 | Slc22a3 | NM_019230_I1_introns | 28 | 3 | 3.77E-09 | 7.37E-08 | 33.00571733 |
| chr18 | Fbn2 | NM_031826_I10_introns | 202 | 4 | 1.04E-13 | 5.10E-12 | 32.59773088 |
| chr17 | Susd3 | NM_001107341_I1_introns | 228 | 4 | 5.87E-05 | 0.000435361 | 31.38181808 |
| chr15 | Cysltr2 | NR_131894_I4_introns | 390 | 4 | 0.000284111 | 0.000601647 | 30.97831867 |
| chr15 | Fndc3a | NM_001107278_I21_introns | 516 | 5 | 0.000898606 | 0.001586244 | 30.17566608 |
| chr2 | Fat4 | NM_001191705_I1_introns | 449 | 5 | 2.40E-05 | 0.000101298 | 30.11072466 |
| chr1 | Syt3 | NM_019122_I5_introns | 71 | 3 | 2.07E-07 | 2.64E-06 | 29.85324558 |
| chr18 | Ldlrad4 | NM_001271365_I1_introns | 85 | 3 | 0.000175281 | 0.00071573 | 29.56178745 |
| chr19 | Cdh13 | NM_138889_I4_introns | 116 | 10 | 7.40E-08 | 3.11E-06 | 29.18274327 |
| chr10 | Asic2 | NM_001034014_I6_introns | 11 | 4 | 0.000718251 | 0.002857722 | 28.85755303 |
| chr1 | Il4r | NM_133380_I1_introns | 111 | 3 | 0.000835518 | 0.002523782 | 26.43736728 |
| chr8 | Kirrel3 | NM_001048215_I1_introns | 146 | 3 | 0.000935696 | 0.00519023 | 26.2257812 |
| chr1 | Ezr | NM_019357_I12_introns | 86 | 13 | 3.17E-07 | 3.31E-06 | 25.69879184 |
| chr2 | Bank1 | NM_001047918_I10_introns | 696 | 6 | 0.000248188 | 0.000607409 | 25.27410866 |
| chr15 | Ppp3cc | NM_134367_I8_introns | 628 | 3 | 1.49E-09 | 1.79E-08 | 25.22411799 |
| chr10 | Cpped1 | NM_001013963_I3_introns | 45 | 4 | 0.000373298 | 0.001745166 | 25.13311283 |
| chr1 | Ust | NM_001108458_I5_introns | 138 | 3 | 2.52E-11 | 1.23E-09 | 25.06445554 |
| chr9 | Stat4 | NM_001012226_I10_introns | 125 | 3 | 6.22E-06 | 4.71E-05 | 24.88762551 |
| chr10 | Rab11fip4 | NM_001107023_I3_introns | 29 | 3 | 0.000195025 | 0.001022057 | 24.47716696 |
| chr10 | Dexi | NM_001109026_I1_introns | 51 | 4 | 2.70E-09 | 6.48E-08 | 24.31882587 |
| chr17 | Mpp7 | NM_001100575_I12_introns | 128 | 3 | 0.000163608 | 0.000880968 | 24.29724306 |
| chr10 | Zc3h7a | NM_001108262_E23_exon | 50 | 3 | 0.00033137 | 0.001621921 | 23.86498374 |
| chr6 | Slc8a1 | NM_001270773_I6_introns | 151 | 5 | 0.000633381 | 0.001794639 | 23.70245762 |
| chr3 | Tspan18 | NM_001107750_I8_introns | 41 | 3 | 0.000144543 | 0.00100661 | 23.25712602 |
| chr1 | Arntl | NM_024362_I2_introns | 43 | 3 | 3.93E-14 | 2.88E-12 | 22.86236695 |
| chr15 | Ppp2r2a | NM_053999_I7_introns | 205 | 15 | 7.78E-09 | 5.60E-08 | 22.86049685 |
| chr2 | Arsb | NM_033443_I4_introns | 1282 | 21 | 5.69E-14 | 2.65E-12 | 22.42210761 |
| chr5 | Slco5a1 | NM_001107898_I8_introns | 154 | 4 | 0.000156566 | 0.0007552 | 22.36790713 |
| chr8 | Arhgap20 | NM_213629_I9_introns | 59 | 5 | 0.000990502 | 0.00519023 | 21.72558987 |
| chr6 | Arid4a | NM_001108029_I5_introns | 100 | 4 | 0.00047549 | 0.00142647 | 21.4264141 |
| chr1 | Syt17 | NM_138849_I3_introns | 56 | 3 | 1.50E-05 | 6.87E-05 | 21.27406378 |
| chr1 | Synm | NM_001134858_I2_introns | 104 | 5 | 9.91E-07 | 8.06E-06 | 21.19753403 |
| chr6 | Rtn1 | NM_053865_I49_introns | 141 | 3 | 4.75E-05 | 0.000195013 | 21.15397951 |
Table 4.
Complete list of the 46 hypomethylated genes.
| Chr | Symbol | ID | Length | Num. CpGs | DMR. p value | DMR. q value | Mean. meth. diff |
|---|---|---|---|---|---|---|---|
| chr8 | Snx1 | NM_053411_I8_introns | 16 | 3 | 0.000178924 | 0.001674221 | 39.23422802 |
| chr10 | Nubp1 | NM_001009619_I9_introns | 51 | 3 | 2.77E-09 | 6.48E-08 | 38.19511889 |
| chr19 | Atp6v0d1 | NM_001011927_I7_introns | 27 | 3 | 8.97E-07 | 1.88E-05 | 37.45568608 |
| chr8 | Bckdhb | NM_019267_I3_introns | 91 | 6 | 4.56E-11 | 1.99E-09 | 36.09637024 |
| chr6 | Prkch | NM_031085_I10_introns | 130 | 4 | 5.03E-06 | 3.02E-05 | 35.65709264 |
| chr13 | Hmcn1 | NM_001271292_I106_introns | 168 | 3 | 4.14E-08 | 4.04E-07 | 35.19645258 |
| chr1 | Zp2 | NM_031150_E10_exon | 79 | 4 | 0.00049086 | 0.001580462 | 33.48819301 |
| chr2 | Ptpn22 | NM_001106460_I13_introns | 70 | 3 | 5.71E-05 | 0.000202829 | 32.53343885 |
| chr2 | Skiv2l2 | NM_001034093_I2_introns | 95 | 4 | 3.00E-08 | 3.49E-07 | 31.5071179 |
| chr1 | Tulp2 | NM_001012168_I1_introns | 110 | 6 | 9.46E-07 | 7.92E-06 | 31.18947695 |
| chr12 | Fry | NM_001170398_I14_introns | 19 | 3 | 2.19E-05 | 0.000288601 | 30.41340002 |
| chr1 | Slco3a1 | NM_177481_I8_introns | 85 | 3 | 0.000528807 | 0.001684136 | 30.13666411 |
| chr1 | Atp10a | NM_001141935_I3_introns | 27 | 6 | 1.03E-06 | 8.14E-06 | 30.06814676 |
| chr1 | Prkg1 | NM_001105731_I15_introns | 70 | 5 | 0.000356284 | 0.001186265 | 28.94112061 |
| chr4 | Grm7 | NM_031040_I7_introns | 192 | 3 | 0.000245364 | 0.001003763 | 28.53745911 |
| chr10 | Carhsp1 | NM_152790_I2_introns | 102 | 5 | 2.26E-05 | 0.000211468 | 28.25784027 |
| chr2 | Noct | NM_138526_I1_introns | 17 | 3 | 0.000373085 | 0.000889665 | 28.10593157 |
| chr15 | Gpc5 | NM_001107285_I2_introns | 205 | 4 | 2.72E-09 | 2.44E-08 | 27.84298084 |
| chr16 | Nrg1 | NM_001271130_I1_introns | 272 | 4 | 0.000226123 | 0.000621838 | 27.46776641 |
| chr7 | Dmc1 | NM_001130567_I6_introns | 134 | 3 | 0.000623225 | 0.001671375 | 27.18046865 |
| chr8 | Tex264 | NM_001007665_I3_introns | 13 | 4 | 4.67E-12 | 3.06E-10 | 26.94493645 |
| chr1 | Oprm1 | NR_027877_I3_introns | 167 | 4 | 5.98E-05 | 0.000236705 | 26.39509712 |
| chr1 | Ntrk3 | NM_001270655_I14_introns | 19 | 4 | 0.000915163 | 0.00273615 | 26.34513213 |
| chr6 | Psma3l | NM_001004094_I5_introns | 128 | 3 | 1.84E-06 | 1.57E-05 | 26.13483738 |
| chr6 | Psma3 | NM_017280_I5_introns | 128 | 3 | 1.84E-06 | 1.57E-05 | 26.13483738 |
| chr7 | Cpq | NM_031640_I8_introns | 174 | 5 | 0.000464492 | 0.001305002 | 25.03471249 |
| chr1 | Tpd52l1 | NM_001044295_I1_introns | 43 | 4 | 2.22E-06 | 1.41E-05 | 24.99670494 |
| chr1 | RGD1307603 | NM_001134508_E3_exon | 35 | 6 | 6.18E-05 | 0.000241568 | 24.98945466 |
| chr19 | Gfod2 | NM_001107421_I2_introns | 100 | 9 | 4.56E-06 | 6.38E-05 | 24.49436882 |
| chr10 | Cyth1 | NM_053910_E12_exon | 56 | 6 | 2.55E-05 | 0.000217367 | 24.45542274 |
| chr13 | Gpatch2 | NM_001011909_I1_introns | 258 | 3 | 9.48E-05 | 0.000369638 | 23.92339964 |
| chr9 | Myo1b | NM_053986_I3_introns | 136 | 6 | 8.64E-05 | 0.000351935 | 23.4475614 |
| chr18 | Ldlrad4 | NM_001271365_I5_introns | 494 | 14 | 5.50E-07 | 5.39E-06 | 23.43565697 |
| chr6 | Frmd6 | NM_001271054_I1_introns | 304 | 4 | 0.000701646 | 0.001871057 | 23.28083922 |
| chr1 | Plpp4 | NM_001191631_I5_introns | 86 | 3 | 1.05E-05 | 5.31E-05 | 23.22492261 |
| chr1 | Gpr139 | NM_001024241_I1_introns | 38 | 3 | 0.00021821 | 0.000752181 | 22.95290692 |
| chr10 | Ccdc40 | NM_001134688_I1_introns | 38 | 4 | 4.16E-05 | 0.000338144 | 22.84818656 |
| chr13 | Cntnap5b | NM_001047873_I1_introns | 329 | 3 | 1.10E-05 | 6.16E-05 | 22.56555396 |
| chr19 | Zfp612 | NM_001107428_I3_introns | 169 | 5 | 0.000905479 | 0.004444048 | 22.51841655 |
| chr8 | Dpp8 | NM_001108159_I13_introns | 120 | 13 | 0.000326038 | 0.002512407 | 22.4358468 |
| chr9 | Sphkap | NM_001127492_I13_introns | 199 | 4 | 0.000143173 | 0.000508033 | 21.12261507 |
| chr1 | Ipcef1 | NM_001170799_I1_introns | 88 | 4 | 0.000961996 | 0.002847121 | 20.82969003 |
| chr5 | Slco5a1 | NM_001107898_I1_introns | 62 | 3 | 8.67E-05 | 0.000473983 | 20.47392161 |
| chr4 | Prickle2 | NM_001107876_I1_introns | 181 | 7 | 0.000537784 | 0.001861562 | 20.45460701 |
| chr1 | Hddc2 | NM_001108460_I2_introns | 61 | 4 | 2.61E-05 | 0.000112648 | 20.32430339 |
| chr9 | Kcnh8 | NM_145095_I9_introns | 302 | 5 | 3.28E-05 | 0.000150472 | 20.01509207 |
Figure 3.

Representative heat map of the top 100 differentially methylated genes. Red indicates hypermethylated genes and blue indicates hypomethylated genes.
GO enrichment analysis and KEGG pathway analysis
The results of GO enrichment analysis are presented in Table 5. In the biological processes (BP), the hypermethylated genes were significantly enriched in spermatogenesis, regulation of cell shape, and neurogenesis. Regarding the molecular function (MF), the hypermethylated genes were mainly enriched in plasma protein binding and metal ion binding. In the cellular component (CC), the hypermethylated genes were significantly enriched in membrane, extracellular exosome, and membrane. The biological processes enriched by the hypomethylated genes included brain development, protein phosphorylation, and response to ethanol. In molecular function, the hypomethylated genes were mainly enriched in protein binding and calcium ion binding. In the cellular component, the hypomethylated genes were enriched in cytoplasm and extracellular exosome.
Table 5.
Gene ontology analysis of aberrantly methylated-differentially expressed genes in spinal cord injury.
| Category | Term | Count | % | P value |
|---|---|---|---|---|
| GOTERM_BP_DIRECT | GO:0007283 spermatogenesis | 6 | 5 | 0.083899175 |
| GOTERM_BP_DIRECT | GO:0008360 regulation of cell shape | 5 | 4.1 | 0.007915904 |
| GOTERM_BP_DIRECT | GO:0022008 neurogenesis | 4 | 3.3 | 0.00873703 |
| GOTERM_BP_DIRECT | GO:0006470 protein dephosphorylation | 4 | 3.3 | 0.032688039 |
| GOTERM_BP_DIRECT | GO:0006897 endocytosis | 4 | 3.3 | 0.046516317 |
| GOTERM_CC_DIRECT | GO:0005886~plasma membrane | 33 | 0.2 | 0.035034848 |
| GOTERM_CC_DIRECT | GO:0070062~extracellular exosome | 27 | 0.1 | 0.005859253 |
| GOTERM_CC_DIRECT | GO:0016020~membrane | 22 | 0.1 | 0.018591495 |
| GOTERM_CC_DIRECT | GO:0005887~integral component of plasma membrane | 14 | 0.1 | 0.003864181 |
| GOTERM_CC_DIRECT | GO:0048471~perinuclear region of cytoplasm | 13 | 0.1 | 0.000430891 |
| GOTERM_MF_DIRECT | GO:0005515~protein binding | 19 | 0.1 | 0.005508607 |
| GOTERM_MF_DIRECT | GO:0046872~metal ion binding | 15 | 0.1 | 0.032965821 |
| GOTERM_MF_DIRECT | GO:0005509~calcium ion binding | 12 | 0.1 | 0.002804787 |
| GOTERM_BP_DIRECT | GO:0007420~brain development | 8 | 6.1 | 0.00348475 |
| GOTERM_BP_DIRECT | GO:0006468~protein phosphorylation | 8 | 6.1 | 0.043342319 |
| GOTERM_BP_DIRECT | GO:0045471~response to ethanol | 6 | 4.6 | 0.008263832 |
| GOTERM_BP_DIRECT | GO:0007399~nervous system development | 6 | 4.6 | 0.009359172 |
| GOTERM_BP_DIRECT | GO:0007613~memory | 5 | 3.8 | 0.002955411 |
| GOTERM_CC_DIRECT | GO:0005737~cytoplasm | 46 | 35.1 | 0.023089945 |
| GOTERM_CC_DIRECT | GO:0070062~extracellular exosome | 34 | 26 | 0.00016196 |
| GOTERM_CC_DIRECT | GO:0016020~membrane | 21 | 16 | 0.084368826 |
| GOTERM_CC_DIRECT | GO:0005829~cytosol | 18 | 13.7 | 0.031630643 |
| GOTERM_CC_DIRECT | GO:0005887~integral component of plasma membrane | 15 | 11.5 | 0.003637411 |
| GOTERM_MF_DIRECT | GO:0005515~protein binding | 22 | 16.8 | 0.001727352 |
| GOTERM_MF_DIRECT | GO:0005509~calcium ion binding | 10 | 7.6 | 0.042887314 |
| GOTERM_MF_DIRECT | GO:0042803~protein homodimerization activity | 10 | 7.6 | 0.088758008 |
| GOTERM_MF_DIRECT | GO:0030165~PDZ domain binding | 4 | 3.1 | 0.043421165 |
| GOTERM_MF_DIRECT | GO:0005516~calmodulin binding | 4 | 3.1 | 0.08759155 |
KEGG pathway analysis results are shown in Table 6. According to KEGG pathway analysis, the DMGs were significantly enriched in the Axon guidance pathway, Endocytosis pathway, T cell receptor signaling pathway, and Hippo signaling pathway.
Table 6.
KEGG pathway analysis of aberrantly methylated-differentially expressed genes in spinal cord injury.
| Pathway name | Gene num | P-value | Genes |
|---|---|---|---|
| Hypermethylation | |||
| Calcium signaling pathway | 7 | 0.001367652 | Htr7, Gna14, Adra1a, Cysltr2, Itpr3, Ppp3cc, Slc8a1 |
| Endocytosis pathway | 6 | 0.041783996 | Acap1, Ehd4, Rab11fip4, Sh3kbp1, Smurf1, Dnm3 |
| T cell receptor signaling pathway | 4 | 0.020396716 | Csf2, Ctla4, Ppp3cc, Vav2 |
| Axon guidance | 4 | 0.053653812 | Dpysl2, Plxna2, Ppp3cc, Robo2 |
| Dopaminergic synapse | 4 | 0.054679627 | Arntl, Itpr3, Ppp2r2a, Ppp3cc |
| Taste transduction | 3 | 0.053663995 | Asic2, Itpr3, Trpm5 |
| Hypomethylation | |||
| Endocytosis pathway | 6 | 0.041783996 | Arfgef1, Flt1, Cyth1, Dnm3, Snx1, Spg21 |
| Hippo signaling pathway | 4 | 0.082043834 | Frmd6, Bmp5, Ctnna2, Dlg2 |
PPI network analysis
Protein–protein interaction (PPI) networks analysis was performed using Cytoscape software. The PPI network of hypermethylated/hypomethylated genes is shown in Figure 4. According to Figure 4A, a total of 48 nodes and 41 interaction pairs were included in this network. Some proteins involved in the Calcium signaling pathway, such as Htr7, Gna14, Adra1a, Cysltr2, Itpr3, Ppp3cc, and Slc8a1, are central nodes in this network. The top core genes were chosen: Csf2, Fars2, Synj2, Ppp3cc, Stat4, Casp8, Cysltr2, and Tiel. These genes and the number of gene cords are shown in Figure 4B. A total of 43 nodes and 33 interaction pairs were included in this network (Figure 4C). Some proteins involved in the Endocytosis pathway, such as Arfgef1, Flt1, Cyth1, Dnm3, Snx1, and Spg21, are central nodes in this network. The top core genes were chosen: Pcsk2, Dnm3, Hmgcll1, Flt1, Plcg2, RT1-Db1, Ntrk3, and Atp10a. These genes and the number of gene cords are shown in Figure 4D.
Figure 4.

PPI network and the Core genes. (A) PPI network of hypermethylated genes. (B) Core genes of hypermethylated genes. (C) PPI network of hypomethylated genes. (D) Core genes of hypomethylated genes.
Genes expression validation by qRT-PCR
In addition to validating the WGBS analysis results, qRT-PCR was used to quantify parts of mRNAs of corresponding methylated genes in the SCI group compared with the sham group. Among these core genes, there were 5 differentially hypermethylated genes (Csf2, Fars2, Synj2, Ppp3cc, and Stat4) and 3 differentially hypomethylated genes (Pcsk2, Dnm3, and Hmgcll1). A search of PubMed revealed that all of these genes are involved in central nervous system repair. Figure 5 shows that 5 mRNAs of differentially hypermethylated genes were downregulated in the SCI group compared with the sham group (P<0.05), and 3 mRNAs of differentially hypomethylated genes were upregulated in the SCI group compared with the sham group (P<0.05).
Figure 5.
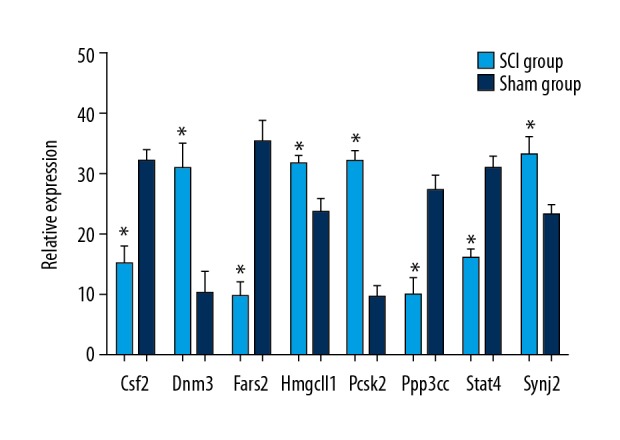
Validation of the differential expression of 8 mRNAs of corresponding methylated genes identified in the WGBS in the SCI group compared with the sham group by qRT-PCR. Values are means ±SE (* P<0.05).
Discussion
During the past decade, few studies have revealed the epigenetic changes that accompany the formation and development of the central nervous system [30,31]. In the early stages of the development of the central nervous system, DNA methylation may play an essential role. It is reported that DNA methylation regulates the differentiation of neurons, which is closely related to adult learning, memory, and cognition [32]. During the process of DNA methylation changes, it is easy to cause more epigenetic diseases due to other factors such as the environment. Therefore, whole-genome bisulfite sequencing of DNA methylation helps reveal epigenetic modifications underlying a variety of complex diseases.
In this study, we first established a transection model of spinal cord injury. Then, histological and motor function scores of Wistar rats before and after spinal cord injury were assessed. We further used whole-genome bisulfite sequencing to analyze epigenetic changes in rat spinal cords before and after injury. In bioinformatics analysis, approximately 96 differential DNA methylation genes were identified, including 50 hypermethylation genes and 46 hypomethylated genes. After GO enrichment analysis, KEGG signaling pathway analysis, and PPI network analysis of these significantly different DNA methylated genes, several core genes in this epigenetic change were screened out, such as Csf2, Fars2, Synj2, Ppp3cc, Stat4, Pcsk2, Dnm3, and Hmgcll1. In addition, we used qRT-PCR to verify the expression of these genes.
Bioinformatics analysis was performed on the selected hypomethylated genes. In GO analysis of biological processes, these hypomethylated genes were significantly enriched in brain development, protein phosphorylation, and response to ethanol, while in the cellular component, the hypomethylated genes were enriched in cytoplasm and extracellular exosome. These hypomethylated genes may be closely related to the formation and regeneration of the central nervous system, and epigenetic changes in these genes may lead to the proliferation and migration of nerve cells (e.g., neurons, oligodendrocytes, and astrocytes) after nerve injury. Among these genes, Dnm3 attracted our attention. Previous reports have shown that Dnm3 is an important epigenetic marker for early detection of breast cancer [33]. There is increasing evidence that Dnm3 plays a critical role in primordial short stature, neurodevelopmental impairments, and microcephaly [34]. In addition, Dnm3 is expressed in the brain and contributes to myelin formation, which promotes axon maturation and myelination [35–36]. This is consistent with changes in hypomethylated genes affecting axonal remodeling after spinal cord injury in rats in the present study. In KEGG analysis, these hypomethylated genes were significantly enriched in the Endocytosis pathway and Hippo signaling pathway. This may be related to the influence of Dnm3 on the coupling between the post-synaptic density scaffold and the endocytic zones [37]. Previous studies have shown that the use of a proliferation-inducing medium containing Y-27632 (Rho/Rho-kinase pathway inhibitor) to culture neural stem cells (NSCs) can activate the Hippo signaling pathway and enhance axon regeneration of NSCs [38], and found that inhibition of ROCK mediates neurite outgrowth in NSCs by activating the Hippo signaling pathway. This is consistent with the results of the present study. After PPI analysis of the hypomethylated genes, the top 5 core genes were Pcsk2, Dnm3, Hmgcll1, Flt1, and Plcg2.
In GO analysis, we found that the hypermethylated genes enriched in biological processes included spermatogenesis, neurogenesis, and regulation of cell shape. In molecular function, the hypermethylated genes were mainly enriched in metal ion binding and plasma protein binding. Regarding the cellular component, we found the hypermethylated genes were significantly enriched in membrane, extracellular exosome, and membrane. Csf2 is a member of the colony-stimulating factors (CSFs) family. This cytokine family includes widely known hematopoietic growth factors [39,40]. Initially, colony-stimulating factor 2 was reported to play a key role in embryonic and early nervous system development [41[. A previous study focused on long noncoding RNAs and messenger RNAs indicated that Csf2 contributes to pathogenesis in the immediate phase of spinal cord injury in adult SD rats [42]. These results all suggest that Csf2 genes may be potential biomarkers in the central nervous system. KEGG analysis showed that these hypermethylated genes were significantly enriched in the T cell receptor signaling pathway, Axon guidance pathway, Calcium signaling pathway, Dopaminergic synapse pathway, and Taste transduction pathway. Previous research has indicated that T cell receptor signaling pathways are crucial in development of neuropathic pain following spinal cord injury [43]. Another study demonstrated that changes in the Axon guidance pathway along with an upregulation of voltage-dependent calcium channel alpha (2) delta-1 subunit Cacna2d1, could contribute to increased mechanical sensitivity [44]. These pathway changes are consistent with the results of our study. In addition, we performed PPI network analysis on hypermethylated genes; the top 5 core genes were Csf2, Fars2, Synj2, Ppp3cc, and Stat4.
Although this study is the first to reveal epigenetic changes after spinal cord injury in Wistar rats, there are still some limitations that need to be addressed. First, we used rodent models, and primate models and even human studies are needed. Second, the central nervous system contains the spinal cord and brain, and the structure and function of the brain are more complex than in the spinal cord, but we did not perform epigenetic studies of the brain. Third, DNA methylation is an important part of epigenetics, and after spinal cord injury, histone modification, gene silencing, and changes in genomic imprinting need to be further explored. In addition, the type of spinal cord injury should be assessed and the screening of more core genes is needed in future work. Despite these limitations, this study furthers understanding of epigenetic changes in spinal cord injury.
Conclusions
The present study performed a comprehensive bioinformatics analysis; 96 differential DNA methylation genes were identified in the thoracic spinal cord tissue following transection injury compared with sham group samples. Among them, 50 genes were hypermethylated and 46 genes were hypomethylated. Moreover, the Axon guidance pathway, Endocytosis pathway, T cell receptor signaling pathway, and Hippo signaling pathway were identified and may be significant mechanisms involved. Core genes such as Csf2, Fars2, Synj2, Ppp3cc, Stat4, Pcsk2, Dnm3, and Hmgcll1 are potential new markers for more accurate diagnosis and effective therapy of spinal cord injury. These markers can be used in drug therapy to alleviate the development of neuropathic pain caused by spinal cord injury and to promote axon maturation, myelin formation, and nerve repair.
Footnotes
Source of support: This study was supported by grants from the State Key Program of National Natural Science Foundation of China (81330042); Special Program for Sino-Russian Joint Research Sponsored by the Ministry of Science and Technology (2014DFR31210); International Cooperation Program of National Natural Science Foundation of China (81620108018); State General Program National Natural Science Foundation of China (81371957); and the Key Program Sponsored by the Tianjin Science and Technology Committee, China (14ZCZDSY00044, 13RCGFSY19000)
Conflicts of Interest
None.
References
- 1.Anderson MA, Burda JE, Ren Y, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532(7598):195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assinck P, Duncan GJ, Hilton BJ, et al. Cell transplantation therapy for spinal cord injury. Nat Neurosci. 2017;20(5):637–47. doi: 10.1038/nn.4541. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues LF, Moura-Neto V, E Spohr TCLS. Biomarkers in spinal cord injury: From prognosis to treatment. Mol Neurobiol. 2018;55(8):6436–48. doi: 10.1007/s12035-017-0858-y. [DOI] [PubMed] [Google Scholar]
- 4.Nees TA, Finnerup NB, Blesch A, Weidner N. Neuropathic pain after spinal cord injury: The impact of sensorimotor activity. Pain. 2017;158(3):371–76. doi: 10.1097/j.pain.0000000000000783. [DOI] [PubMed] [Google Scholar]
- 5.Fan B, Wei Z, Yao X, et al. Microenvironment imbalance of spinal cord injury. Cell Transplant. 2018;27(6):853–66. doi: 10.1177/0963689718755778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker CG, Becker T, Hugnot JP. The spinal ependymal zone as a source of endogenous repair cells across vertebrates. Prog Neurobiol. 2018;170:67–80. doi: 10.1016/j.pneurobio.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Shi G, Fan B, et al. Polycaprolactone electrospun fiber scaffold loaded with iPSCs-NSCs and ASCs as a novel tissue engineering scaffold for the treatment of spinal cord injury. Int J Nanomedicine. 2018;13:6265–77. doi: 10.2147/IJN.S175914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niclis JC, Turner C, Durnall J, et al. Long-distance axonal growth and protracted functional maturation of neurons derived from human induced pluripotent stem cells after intracerebral transplantation. Stem Cells Transl Med. 2017;6(6):1547–56. doi: 10.1002/sctm.16-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183(4676):1654–55. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- 10.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 11.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 12.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15(2):163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Gould E. How widespread is adult neurogenesis in mammals? Na Rev Neurosci. 2007;8(6):481–88. doi: 10.1038/nrn2147. [DOI] [PubMed] [Google Scholar]
- 14.Ming GL, Song H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron. 2011;70(4):687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nwaobi SE, Lin E, Peramsetty SR, Olsen ML. DNA methylation functions as a critical regulator of Kir4.1 expression during CNS development. Glia. 2014;62(3):411–27. doi: 10.1002/glia.22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaze J, Wang J, Ho L, et al. Polyphenolic compounds alter stress-induced patterns of global DNA methylation in brain and blood. Mol Nutr Food Res. 2018;62(8):e1700722. doi: 10.1002/mnfr.201700722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Losi L, Fonda S, Saponaro S, et al. Distinct DNA methylation profiles in ovarian tumors: Opportunities for novel biomarkers. Int J Mol Sci. 2018;19(6) doi: 10.3390/ijms19061559. pii: E1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicolia V, Cavallaro RA, Lopez-Gonzalez I, et al. DNA methylation profiles of selected pro-inflammatory cytokines in Alzheimer disease. J Neuropathol Exp Neurol. 2017;76(1):27–31. doi: 10.1093/jnen/nlw099. [DOI] [PubMed] [Google Scholar]
- 19.Miyake K, Kawaguchi A, Miura R, et al. Association between DNA methylation in cord blood and maternal smoking: The Hokkaido Study on Environment and Children’s Health. Sci Rep. 2018;8(1):5654. doi: 10.1038/s41598-018-23772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–74. doi: 10.1038/nature26000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du BL, Zeng X, Ma YH, et al. Graft of the gelatin sponge scaffold containing genetically-modified neural stem cells promotes cell differentiation, axon regeneration, and functional recovery in rat with spinal cord transection. J Biomed Mater Res A. 2015;103(4):1533–45. doi: 10.1002/jbm.a.35290. [DOI] [PubMed] [Google Scholar]
- 22.Lai BQ, Wang JM, Ling EA, et al. Graft of a tissue-engineered neural scaffold serves as a promising strategy to restore myelination after rat spinal cord transection. Stem Cells Dev. 2014;23(8):910–21. doi: 10.1089/scd.2013.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu P, Woodruff G, Wang Y, et al. Long-distance axonal growth from human induced pluripotent stem cells after spinal cord injury. Neuron. 2014;83(4):789–96. doi: 10.1016/j.neuron.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shannon P, Markiel A, Ozier O, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Garrett-Bakelman FE, Akalin A, et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013;14(Suppl 5):S10. doi: 10.1186/1471-2105-14-S5-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- 27.Li G, Cao Y, Shen F, et al. Mdivi-1 inhibits astrocyte activation and astroglial scar formation and enhances axonal regeneration after spinal cord injury in rats. Front Cell Neurosci. 2016;10:241. doi: 10.3389/fncel.2016.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao L, Wang Q, Zhang C, Huang C. Genome-wide DNA methylation analysis of articular chondrocytes identifies TRAF1, CTGF, and CX3CL1 genes as hypomethylated in osteoarthritis. Clin Rheumatol. 2017;36(10):2335–42. doi: 10.1007/s10067-017-3667-9. [DOI] [PubMed] [Google Scholar]
- 29.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12(1):1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 30.Aiba T, Saito T, Hayashi A, et al. Does the prenatal bisphenol A exposure alter DNA methylation levels in the mouse hippocampus?: An analysis using a high-sensitivity methylome technique. Genes Environ. 2018;40:12. doi: 10.1186/s41021-018-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Xiao B, Chen XS. DNA methylation: A new player in multiple sclerosis. Mol Neurobiol. 2017;54(6):4049–59. doi: 10.1007/s12035-016-9966-3. [DOI] [PubMed] [Google Scholar]
- 32.Huang WQ, Yi KH, Li Z, et al. DNA methylation profiling reveals the change of inflammation-associated ZC3H12D in leukoaraiosis. Front Aging Neurosci. 2018;10:143. doi: 10.3389/fnagi.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uehiro N, Sato F, Pu F, et al. Circulating cell-free DNA-based epigenetic assay can detect early breast cancer. Breast Cancer Res. 2016;18(1):129. doi: 10.1186/s13058-016-0788-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burkardt DD, Rosenfeld JA, Helgeson ML, et al. Distinctive phenotype in 9 patients with deletion of chromosome 1q24-q25. Am J Med Genet A. 2011;155A(6):1336–51. doi: 10.1002/ajmg.a.34049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sala C, Piëch V, Wilson NR, et al. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–30. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 36.Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40(1):55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu J, Helton TD, Blanpied TA, et al. Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to Homer. Neuron. 2007;55(6):874–89. doi: 10.1016/j.neuron.2007.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia XF, Ye F, Wang YB, Feng DX. ROCK inhibition enhances neurite outgrowth in neural stem cells by upregulating YAP expression in vitro. Neural Regen Res. 2016;11(6):983–87. doi: 10.4103/1673-5374.184499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson SA. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor Rev. 2007;18(3–4):287–98. doi: 10.1016/j.cytogfr.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 40.Sferruzzi-Perri AN, Macpherson AM, Roberts CT, Robertson SA. Csf2 null mutation alters placental gene expression and trophoblast glycogen cell and giant cell abundance in mice. Biol Reprod. 2009;81(1):207–21. doi: 10.1095/biolreprod.108.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith A, Witte E, McGee D, et al. Cortisol inhibits CSF2 and CSF3 via DNA methylation and inhibits invasion in first-trimester trophoblast cells. Am J Reprod Immunol. 2017;78(5) doi: 10.1111/aji.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou H, Shi Z, Kang Y, et al. Investigation of candidate long noncoding RNAs and messenger RNAs in the immediate phase of spinal cord injury based on gene expression profiles. Gene. 2018;661:119–25. doi: 10.1016/j.gene.2018.03.074. [DOI] [PubMed] [Google Scholar]
- 43.He X, Fan L, Wu Z, et al. Gene expression profiles reveal key pathways and genes associated with neuropathic pain in patients with spinal cord injury. Mol Med Rep. 2017;15(4):2120–28. doi: 10.3892/mmr.2017.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capasso KE, Manners MT, Quershi RA, et al. Effect of histone deacetylase inhibitor jnj-26481585 in pain. J Mol Neurosci. 2014;55(3):570–78. doi: 10.1007/s12031-014-0391-7. [DOI] [PubMed] [Google Scholar]