

Abstract
Introduction:
There are currently no effective therapeutics for Alzheimer disease (AD). Clinical trials targeting amyloid beta thus far have shown very little benefit and only in the earliest stages of disease. These limitations have driven research to identify alternative therapeutic targets, one of the most promising is the triggering receptor expressed on myeloid cells 2 (TREM2).
Areas covered:
Here, we review the literature to-date and discuss the potentials and pitfalls for targeting TREM2 as a potential therapeutic for AD. We focus on research in animal and cell models for AD and central nervous system injury models which may help understand the role of TREM2 in disease.
Expert opinion:
Studies suggest TREM2 plays a key role in AD pathology; however, results have been conflicting about whether TREM2 is beneficial or harmful. More research is necessary before designing TREM2-targeting therapies. Successful therapeutics will most likely be administered early in disease.
Keywords: TREM2, Alzheimer disease, cerebrospinal fluid, genetic association, microglia
1. Introduction
Approximately 5.5 million individuals have been currently diagnosed with Alzheimer disease (AD) in the United States (US) and as the population 65 years and older increases, that number is estimated to almost triple by the year 2050 [1]. AD is reported to be the sixth-leading cause of death in the US; however, after three decades of research leading to tremendous breakthroughs in identifying much of the underlying neuropathology of AD, there are currently no treatments to prevent or slow progression of the disease. The defining neuropathological hallmarks of AD are extracellular plaques primarily consisting of aggregated β-amyloid (Aβ) deposits and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein (ptau). Mutations in amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) cause rare (< 1% of cases) Mendelian early-onset AD and variants in these genes are also associated with non-Mendelian late-onset AD. Although late-onset AD is highly heritable, estimated as high as 79% in twin studies [2], it is also genetically complex; with more than 25 genetic loci associated with risk identified thus far only about half of the heritability is explained by these associations [3, 4]. The strongest genetic risk factor for AD is a variant in the gene encoding apolipoprotein E (APOE) known as the ε4 allele; individuals carrying one ε4 allele have a threefold increase in AD risk and two ε4 alleles confer 8- to 12-fold increased risk [5, 6].
Although promising treatments have been tested in clinical trials none have met with much success. Most of the therapeutic approaches thus far have targeted Aβ, and although immunization against Aβ has reduced plaques and showed some cognitive improvement in transgenic mouse models for AD [7], human clinical trials have shown, at best, very small benefits in patients with mild cognitive symptoms [8]. Phase III trials of anti-Aβ antibodies have not shown any improvement in primary outcomes for patients with mild to moderate AD [9–11] and the field continues to search for effective treatment for this devastating disease. Research has revealed that AD pathology appears at least 10–20 years before the onset of clinical symptoms, and many experts in the field believe that once symptoms of memory decline appear the pathological damage may be irreversible [12]. This suggests that treatment would likely be most effective in the preclinical stage of AD; however, currently there are no biomarkers with the sensitivity and specificity necessary to identify preclinical individuals, presenting another obstacle for clinical research.
Cerebrospinal fluid (CSF) levels of tau, ptau, and Aβ are considered the gold standard for AD biomarkers. In 2013, an extensive review that included a total of 1,172 individuals with MCI at baseline (430 of whom converted to AD within 1–4 years) estimated that CSF tau had a median specificity of 72% and sensitivity of 75% and CSF ptau had a median specificity of 47.5% and sensitivity of 81% [13]. The ratio of CSF tau (or ptau) and Aβ42 has been reported to be the most accurate thus far [13–18]. However, inter-individual variability, overlap between preclinical cases and controls, and cross-study measurement variability reduce diagnostic specificity of these biomarkers, particularly for prodromal AD [19–23]. One promising biomarker of recent interest is CSF soluble triggering receptor expressed on myeloid cells 2 protein (sTREM2) mostly because there is a strong genetic association between TREM2 and AD; additionally, sTREM2 levels in CSF have been found elevated in individuals with AD and changes were observed in preclinical AD [24–27]. The genetic associations between TREM2 and disease and potential for sTREM2 as a disease biomarker are discussed further in the next section.
2. TREM2 associated with disease
2.1. Nasu-Hakola disease causal variants
Nasu-Hakola disease (NHD), also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), is a rare recessively-inherited disease characterized by the presence of bone cysts and progressive presenile dementia that usually begins around the ages of 25 – 40 years and leads to death by the age of 50 [28]. The first identified causal gene for NHD was the gene encoding TYRO protein tyrosine kinase binding protein (TYROBP also known as DAP12) which is a transmembrane protein important in innate immune signaling [29]. TYROBP is expressed in microglia in the brain and is an adapter protein for the triggering receptor expressed on myeloid cells 2 protein (TREM2). In 2002, researchers identified variants in TREM2 that were associated with NHD in families who did not carry previously identified TYROBP mutations [30]. In addition to a splice-site mutation, researchers identified four other TREM2 mutations in two families and four individuals who did not carry TYROBP mutations: p.W78X, p.W44X, p.K186N, and p.D134G [30]. Dendritic cells taken from three NHD patients carrying a shared ancestral deletion within TYROBP that was identified in a Finnish population, referred to as the PLOSLFin DAP12 mutation, and from two NHD patients carrying TREM2 (p.Q33X or p.V126G) mutations showed differential expression of several genes associated with inflammatory and innate immune response when compared to cells taken from non-carriers [31]. Interestingly, TREM2 expression was significantly down-regulated in dendritic cells from patients with TYROBP mutations similarly to the TREM2 mutation carriers, but the TREM2 mutations did not appear to influence the expression levels of TYROBP [31]. This suggests that disrupted expression of TREM2, not TYROBP, may be responsible for NHD symptoms and that TREM2 may be the best therapeutic target for NHD.
2.2. TREM2 risk variants for Human acquired colesteatoma and dementia
TREM2 gene expression was recently reported to be associated with a disease that is classified by bone destruction, but no cognitive symptoms. Human acquired cholesteatoma causes hearing loss and vestibular neuritis, usually in response to bacterial infection, and is characterized by proliferation of keratinized epithelia inside the ear as well as erosion of ossicles and temporal bone [32]. Inflammatory response is considered the most important factor in the bone destruction observed in acquired cholesteatoma [32]. RT-PCR of mRNA extracted from skin taken from patients and healthy controls revealed that TREM2 gene expression was significantly upregulated in patients with acquired cholesteatoma. Analyses of TREM2-deficient (Trem2−/−) mouse models of experimentally acquired cholesteatoma revealed that TREM2 appears to act through the TLR4 signaling pathway [32].
A number of studies have identified TREM2 variants associated with dementia and FTD-like disease without the presence of bone cysts. Functional neuroimaging (99mTc-ECD SPECT) and neuropsychological analyses of unaffected individuals from an Italian family with two homozygous p.Q33X carriers who were diagnosed with NHD, revealed that heterozygous p.Q33X carriers had visuo-spatial memory deficits associated with hypoperfusion in the basal ganglia [33]. A linkage study of a Lebanese family identified a novel splice-site deletion within the first intron of TREM2 in three individuals who had presented with early-onset dementia beginning around the ages 30–35 but had no signs of bone cysts [34]. This study also determined that although the TREM2 transcripts appeared normal in lymphocytes and cultured fibroblasts taken from family members, the expression of TREM2 was significantly down-regulated and the expression of other genes were disrupted as well (BCL3, FSCN, NFKBIA, NEDD9, and SPP1) [34]. More recently a few studies have reported associations between TREM2 missense variants and FTD or FTD-like dementia [35–38].
2.3. Alzheimer disease risk variants
Several studies have demonstrated that TREM2 variants are associated with AD risk [39–43]. In 2013, two groups independently identified, for the first time, a rare variant in TREM2 (p.R47H) that increased risk for AD almost three-fold, the greatest genetic risk for AD identified since APOE ε4 [40, 43]. Guerreiro et al. also observed other TREM2 variants in AD cases, but not controls, in their discovery set: p.H157Y, p.R98W, p.D87N, p.T66M, p.Y38C, and p.Q33X [40]. Pooled sequencing of TREM2 allowed researchers to identify additional AD risk variants, and revealed that disparities in minor allele frequency resulted in a different set of risk variants in African Americans compared to Europeans [41, 42]. Other studies have demonstrated that different populations have different TREM2 risk variants [44, 45].
Further evidence for the association of TREM2 in AD is that TREM2 protein levels were found elevated in post-mortem temporal brain tissue taken from AD patients [46]. The increased TREM2 protein levels were positively correlated with ptau and caspase 3 and negatively correlated with SNAP25, suggesting an association between TREM2 levels, apoptosis, and synapse loss in AD [46]. Additionally, microglia associated with neuropathology stained positively for TREM2, supporting previous research that associated TREM2 with microglia [46]. The role of TREM2 in microglia is discussed further in section 3.
2.4. Soluble TREM2 as a disease biomarker
There is evidence that TREM2 may be involved in multiple sclerosis (MS) although no TREM2 variants have been reported to be associated with MS. Soluble TREM2 (sTREM2) can be measured in CSF and was found elevated in patients with MS in the relapsing-remitting phase (RRMS), the secondary progressive phase (SPMS), and the primary progressive phase (PPMS) [47, 48]. RRMS patients treated with natalizumab (a humanized monoclonal antibody against the cell adhesion molecule α4-integrin) showed CSF sTREM2 levels were reduced to levels similar to controls after 12 months of treatment and patients showed improvement of clinical symptoms [48].
Studies have also demonstrated that CSF sTREM2 levels are elevated in early stages of AD [24–26] and changes in CSF sTREM2 can be observed in preclinical individuals after amyloid deposition, increasing approximately five years before the onset of clinical symptoms and remaining significantly elevated five years after onset [27]. CSF levels of sTREM2 were positively correlated with tau and ptau levels, but were not correlated with CSF Aβ [24–26]. CSF levels of sTREM2 have also been associated with increased gray matter volume suggestive f brain swelling during neuroinflammation [49].
There is evidence that CSF levels of sTREM2 are influenced by previously identified TREM2 risk variants, and that the risk variants for different diseases have very different effects on sTREM2 levels [25, 50]. The TREM2 p.R47H AD risk variant was associated with higher levels of CSF sTREM2, whereas carriers of variants associated with NHD had lower CSF sTREM2 levels compared to controls [25]. Levels of sTREM2 were virtually undetectable in CSF from individuals who were homozygous for the p.T66M mutation, which has been primarily associated with FTD risk, and a heterozygous p.T66M carrier was reported to have significantly lower levels similar to NHD-risk variant carriers [25, 50].
There is a great deal of interest in the field to identify biomarkers that are less invasive than CSF biomarkers, so researchers have tested sTREM2 levels in blood as well. One of the earlier studies that found higher sTREM2 in CSF from MS patients did not find a difference in sTREM2 levels measured in blood from the same individuals [47]. TREM2 protein levels and mRNA were measured in blood extracted from 116 individuals with “probable” AD and 116 sex- and age-matched healthy controls and were reported to be significantly higher in AD patients; however, the reported sensitivity (68%) and specificity (72%) were too low for blood levels of TREM2 to be a reliable biomarker [51]. Another study in 2014, which had reported higher sTREM2 levels in CSF taken from AD patients, did not find a significant difference in plasma levels of sTREM2 between AD cases (n = 51) and controls (n = 86) [50]. This finding was replicated in a more recent study that analyzed sTREM2 levels in CSF and plasma taken from a well-characterized AD cohort (73 cases and 107 healthy controls) which showed significantly elevated CSF sTREM2 in AD cases but no difference in sTREM2 plasma levels [25]. Based on research thus far, it appears that although CSF levels of sTREM2 may be a promising AD biomarker, plasma sTREM2 levels probably are not going to be as informative.
3. TREM2 in microglia
With this strong association between TREM2, dementia, and Alzheimer disease, it became important to understand what the normal function of TREM2 is in the brain and how dysfunction of TREM2 may contribute to disease. To identify the most important functions of TREM2 in microglia that is known to-date, a careful literature review was conducted by first using the search terms ‘TITLE-ABS-KEY (trem2 AND microglia) AND (LIMIT-TO (DOCTYPE, “ar”))’ in Scopus in February 2018. Figure 1 illustrates the most cited research of TREM2 and microglia, with the primary findings depicted in a Sankey plot generated using the googleVis version 0.6.2 package in R version 3.4 [52]. We evaluated the 20 most cited research articles in depth and selected 14 of the 20 most cited articles. Figure 1 describes 1. the main conclusion, 2. related TREM2 variants or disturbance, 3. disease background, 4. research model, and 5. reference.
Figure 1.
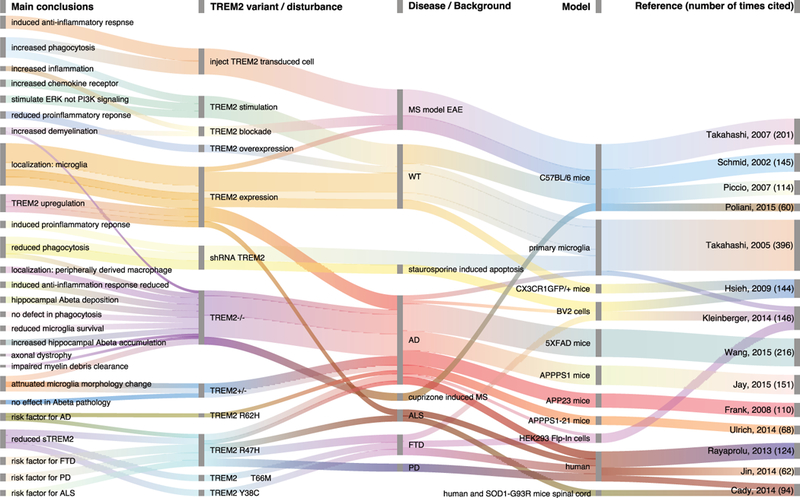
Sankey plot depicting the 14 most cited articles in a literature review of TREM2 and microglia research. Columns show the main conclusion, related TREM2 variants or disturbance, disease background, research model, and reference (the number of times the reference was cited at time of literature review is in parentheses). Wavy lines depict the main conclusions that were reported in different studies and the different conclusions reported in one reference.
TREM2 is part of the triggering receptor expressed on myeloid cells family which regulate innate immune response and are expressed on a variety of myeloid cells including microglia, macrophages, monocytes, osteoclasts, dendritic cells, and neutrophils. TREM1 was the first TREM identified and is the best characterized. Both TREM1 and TREM2 encode proteins that form transmembrane receptor-signaling complexes with the TYRO binding protein (aka DNAX-activating protein of 12 kDa, or DAP12). TREM1 is highly expressed on monocytes, macrophages, and neutrophils; whereas TREM2 is highly expressed on macrophages, dendritic cells, osteoclasts, and microglia[53]. Studies suggest that TREM2 is necessary for maturation of dendritic cells and osteoclasts [54, 55]. In the central nervous system, direct RNA sequencing of murine microglia demonstrated that TREM2 is one of the highest expressed receptors in microglia and is specifically enriched on microglia compared to astrocytes and neurons [56]. TREM2 appears to be necessary for microglial survival, inflammatory response, and induced phagocytosis (Figure 2). Studies of cuprizone-induced demyelination in Trem2−/− mice showed reduced clearance of myelin debris, increased axonal pathology, and worse clinical symptoms than WT mice [57]. There was also evidence that microglial activation was reduced in the Trem2−/− mice when measuring major histocompatibility complex class II (MHC II) and inducible nitric oxide synthase (iNOS) expression, and differential expression analyses suggested that deficient lipid metabolism may be responsible [57].
Figure 2.
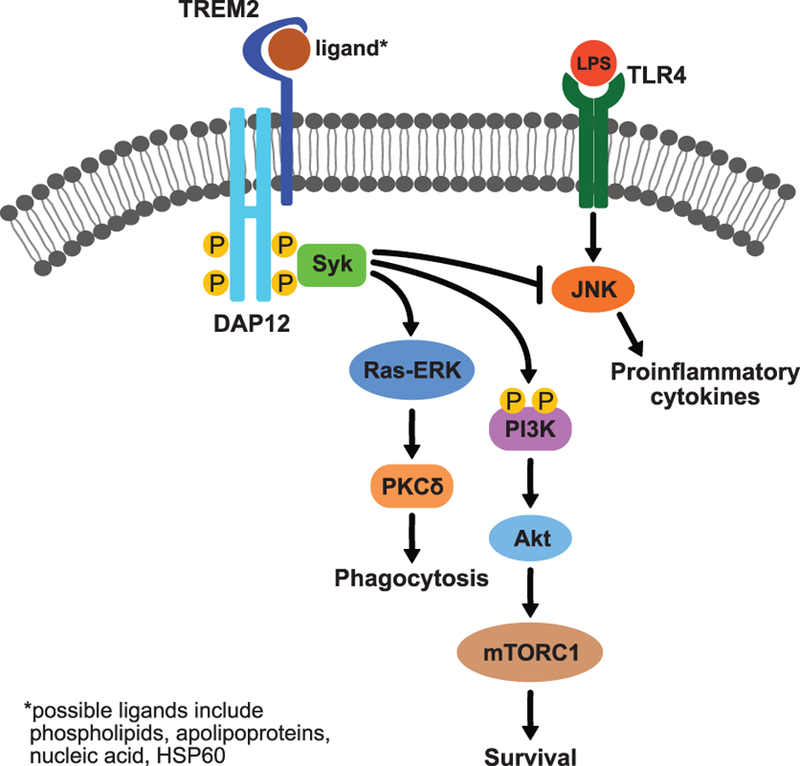
TREM2 molecular signaling in microglia, initiating phagocytosis and microglial survival or modulating proinflammatory response, is not well characterized so only validated components are displayed. In vitro studies have suggested possible ligands for TREM2, which bind TREM2 to initiate signaling. TREM2 associates with DAP12, which is phosphorylated to activate signaling through recruitment of spleen tyrosine kinase (Syk). Syk activates the Ras/extracellular signal-regulated kinase (ERK)/protein kinase C (PKC) pathway to initiate phagocytosis. Microglial survival is initiated by the activation of the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin complex 1 (mTORC1) pathway. TREM2 is thought to influence inflammatory response by inhibiting the c-Jun N-terminal kinase (JNK) pathway which can be activated when ligands like lipopolysaccharide (LPS) bind toll-like receptor-4 (TLR4), initiating the production of proinflammatory cytokines.
Animal model studies have suggested that TREM2 plays a key role in modulating microglial response in the presence of AD pathology [50, 58–61]. There have been conflicting conclusions from these studies about whether this role is beneficial or harmful, key evidence from these studies is summarized in Figure 3. Trem2 knockdown in microglia from APPswe/PS1dE9 (APP/PS1) mice showed reduced microglial phagocytosis of Aβ and increased pathology, suggesting that TREM2 is necessary for keeping amyloid pathology in check by phagocytosis [62]. Analyses of Trem2−/− 5XFAD crossbred mice showed significant Aβ deposition in the hippocampus and reduced survival of microglia due to apoptosis [60]. However, other studies suggest that inhibiting Trem2 may be beneficial instead [63, 64]. Four-month-old Trem2−/− APP/PS1 mice had fewer plaque-associated macrophages and reduced neuroinflammation, amyloid accumulation in the hippocampus, and tau pathology [63]. Knockout of Trem2 in a PS19 mouse model for tauopathy (expressing human tau (1N4R) with the FTD-linked P301S mutation) showed a reduction of neurodegeneration and inflammation [64]. One possible reason for the apparently conflicting evidence for TREM2 having a protective or harmful role in disease could be that these studies represent different stages of AD and the presence of different disease pathology. The 5XFAD transgenic mice carry mutations in both PSEN1 and APP, resulting in early and rapid Aβ accumulation (by 6 months) and cognitive deficits (by 5 months) [65]. The APP/PS1 transgenic mice carry fewer mutations in both APP and PSEN1, resulting in slower accumulation of Aβ (beginning at 6 months and abundant by 9 months) and show a later cognitive decline (by 12 months) [66]. The PS19 mice carry a human tau transgene with the p.P301S mutation which causes FTD in humans, resulting in extensive tangle pathology and gliosis by 9 months as well as severe neurodegeneration, all in the absence of amyloid pathology [67].
Figure 3.
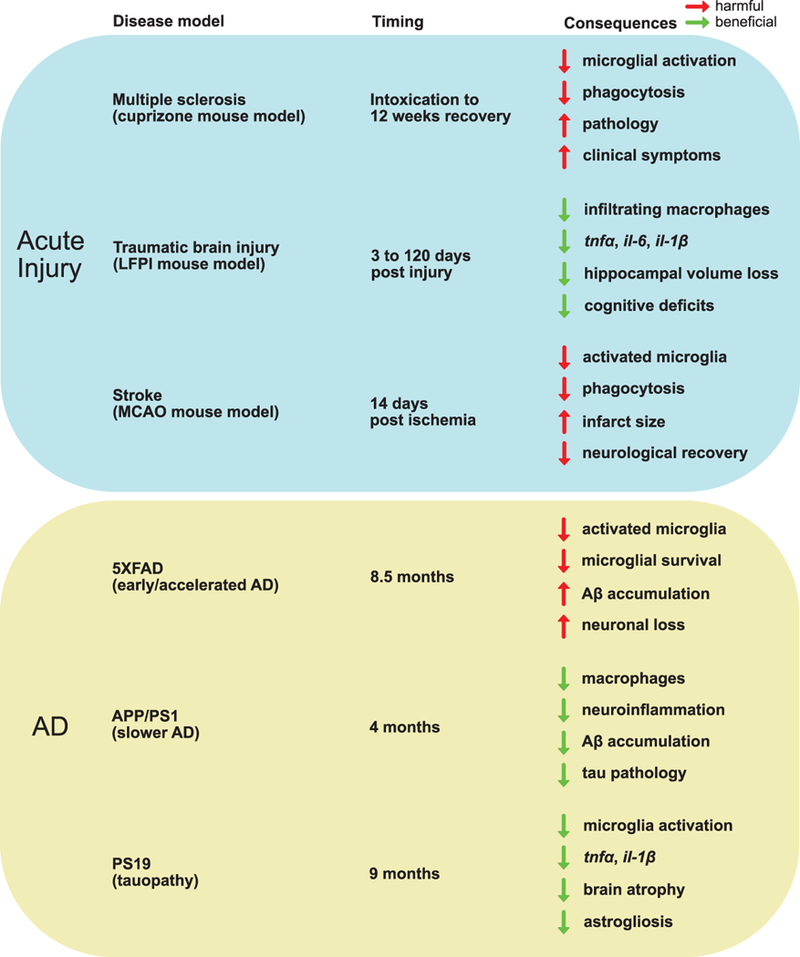
Summary of key findings in TREM2 knockout models. The top box (blue) shows three studies representing acute injury: a cuprizone intoxication mouse model for demyelination observed in multiple sclerosis (Cantoni et al, 2005), a lateral fluid percussion model for TBI (Saber et al, 2017), and an experimental stroke mouse model using permanent MCAO (Kawabori et al, 2015). The lower box (yellow) shows three studies of AD mouse models crossed with Trem2−/− representing different disease pathology: the 5XFAD mouse model which develops cognitive deficits and amyloid accumulation by 6 months (Wang et al, 2015), the APP/PS1 mouse model which develops amyloid accumulation 6–9 months and cognitive decline by 12 months (Jay et al, 2015), and the PS19 mouse model which is a model for tauopathy and has no amyloid pathology (Leyns et al, 2017). The timing column represents when the researchers observed the consequences/effects shown in the right-hand column, which represents the difference in effect of Trem2−/− compared to WT. The arrows represent whether the effect on the described observation to the right of the arrow is increased (pointing up) or decreased (pointing down) and the arrow color represents whether the effect is considered harmful (red) or beneficial (green).
Research of acquired brain injury models, in combination with the AD mouse model research, further suggests that some of the conflicting results from studies of TREM2 function are context specific: differences in acute vs chronic injury and time course. Traumatic brain injury (TBI) may increase risk for AD and it is interesting to note that Trem2 gene expression was found elevated for at least seven days post injury in a lateral fluid percussion injury mouse model for TBI [68]. Trem2−/− mice had fewer infiltrating peripheral macrophages and reduced gene expression of tnfα, il-6, and il-1β compared to WT three days post injury [68]. After 120 days post injury, Trem2−/− mice showed reduced loss of hippocampal volume and attenuation of cognitive deficits compared to WT, which led the authors to conclude that Trem2 deficiency is neuroprotective by reducing infiltrating macrophages in response to injury [68]. However, in an experimental stroke mouse model using permanent middle cerebral artery occlusion (MCAO), Trem2 deficiency resulted in fewer activated microglia, reduced phagocytosis, larger infarct size, and worse neurological recovery fourteen days post ischemia, suggesting TREM2 is necessary for stroke recovery [69]. These disparate results, also summarized in Figure 3, suggest that the role of TREM2 in neuroinflammation, neurodegeneration, and neuroprotection is highly dynamic and depends on cell type (peripheral macrophages or microglia), type of injury, and timing.
TREM2 makes up part of a receptor complex which binds ligands and initiates signaling; therefore, determining which ligands bind this receptor may help understand the function of TREM2 in microglia. Identification of endogenous TREM2 ligands has not been comprehensive, but some in vitro studies provide compelling evidence for several ligands including molecules that are important in AD. Several studies have suggested that TREM2 binds anionic ligands on cells, both bacterial and mammalian, such as phospholipids, nucleic acids, and proteoglycans [70]. Other studies have reported other ligands such as heat shock protein 60 (HSP60) and apolipoproteins [70]. The first reported specific TREM2 agonist was HSP60, which was found to induce phagocytosis [71]. Researchers studying an ischemia cell model reported that although they did not find TREM2 signaling in response to HSP60, they did find evidence that high-molecular-weight nucleic acids may act as ligands to initiate TREM2 signaling in response to apoptotic cells [69]. One putative ligand for TREM2 which is of particular interest in AD is APOE and reduced binding affinity for APOE was observed for the TREM2 p.R47H mutation [72]. Another group identified apolipoproteins, including clusterin and APOE, as TREM2 ligands and also reported less binding affinity for not only the p.R47H mutations but p.D87N and p.R62H variants as well [73]. They also found that the loss of function TREM2 mutations associated with NHD (p.Y38C and p.T66M) showed no binding affinity for apolipoproteins [73]. Recent research suggests that APOE signaling, induced by TREM2, drives post-transcriptional changes that cause microglia to promote neurodegeneration and that targeting Apoe in mice restored the neuroprotective milieu [58]. APOE influences both amyloid- and tau-mediated AD pathogenesis through multiple mechanisms including metabolism and aggregation of Aβ, lipid transport, synaptic plasticity, neuroinflammation, and many others [74]. Since both TREM2 and APOE are associated with AD risk, this TREM2-APOE pathway in microglia most likely plays a key role in AD pathology.
4. TREM2 biological pathways and activity
4.1. TREM2 Cleavage
As mentioned in section 2.4, sTREM2 can be measured in CSF and plasma. In 2014, Jin et al. reported three distinct transcripts for TREM2 were present in human brain tissue, including a short transcript which was alternatively spliced to exclude exon 4 (which contains the transmembrane domain) encoding for sTREM2 [41]. Other studies reported evidence for sequential proteolytic cleavage of the transmembrane protein also resulting in sTREM2 [75] and recently two groups independently reported a consensus of the exact cleavage site on TREM2 that is targeted by ADAM10 and other proteases to generate sTREM2; though there was some disagreement about which proteases, other than ADAM10, also cleave TREM2 [76, 77]. Through mass spectrometry analyses and targeted in vivo experiments, they determined that ADAM-mediated cleavage occurs between histidine 157 and serine 158, which is interestingly where an AD risk variant has been identified in various studies (p.H157Y) [45, 76–79]. Both groups found increased shedding of TREM2 in cells expressing the p.H157Y mutant, which would result in increased levels of sTREM2 and less cell surface expression of TREM2 [76, 77]. Mutations such as p.T66M and p.Y38C, which are in the Ig-like domain and prevent the protein from leaving the endoplasmic reticulum, resulted in both reduced cell surface expression of TREM2 and levels of sTREM2 [50, 79, 80]. The p.R47H variant did not appear to affect glycosylation or normal trafficking of TREM2 to the same extent as p.T66M and p.Y38C, but the glycosylation pattern was different from WT, suggesting some impairment that could affect normal TREM2 function [80]. Together, these studies suggest that the role of TREM2 mutations in disease may not be simply a gain or loss of function of the receptor.
4.2. Effect of TREM2 variants on trafficking, shedding, and sTREM2 levels
TREM2 localization can greatly impact TREM2 signaling. TREM2 trafficking appears to be a highly dynamic process involving endocytosis and recycling. Under baseline conditions, TREM2 is localized in the trans-Golgi network [81, 82], endosomes [83], and in exocytic vesicles [81]. TREM2 is internalized in a clathrin-dependent manner and recycled back to the plasma membrane through vacuolar protein sorting 35 (Vps35) and beclin-1 [84, 85]. In the absence of Vps35, TREM2 is degraded by the lysosome [85].
Heterologous expression of TREM2 variants (p.T66M, p.Y38C, and p.R47H) in various cell lines (Hek293, HeLa, and murine microglial BV2) significantly reduced TREM2 surface expression and the amount of sTREM2 in conditioned media [50, 80]. The p.T66M and p.Y38C variants increased localization of TREM2 to the endoplasmic reticulum [50, 80], affecting proper folding and stability of the protein [86] and consequently degrading TREM2 by the ERAD pathway [80]. However, the p.R47H variant did not appear to alter surface expression of TREM2 [80] or it reduced expression to a lesser extent than the other variants [50]. Also unlike the other variants, p.R47H TREM2 was primarily localized to the trans-Golgi network [50, 80]. Overall, these studies suggest that TREM2 variants associated with NHD, FTD, or FTD-like disease affect TREM2 maturation, cell surface transport, and proteolytic processing whereas the AD-associated p.R47H mutation has a milder effect on protein maturation and secretion. However, researchers have focused on the cleaved form of sTREM2 and very little attention has been paid to the alternatively spliced form of sTREM2. Further research will be necessary to determine how the different TREM2 variants may influence alternative splicing.
5. Current research targeting TREM2
The extensive genetic and biomarker evidence for the potential role of TREM2 in AD risk has prompted several groups to determine whether TREM2 may be a viable therapeutic target for AD. TREM2 variants that are causal for NHD appear to be loss-of-function and many researchers have hypothesized that the AD risk variants result in some loss-of-function as well. As detailed in Section 3, a great deal of research has focused on this loss-of-function hypothesis and reported conflicting conclusions about the beneficial or harmful effect of TREM2 deficiency. Analyses of Trem2−/− 5XFAD crossbred mice showed significant Aβ deposition in the hippocampus and reduced survival of microglia due to apoptosis, suggesting the loss of TREM2 function is harmful [60]. Trem2 knockdown in microglia cultured from the APPswe/PS1dE9 mice reduced microglial phagocytosis of Aβ, also suggesting TREM2 deficiency is harmful [62]. However, a mouse model for earlier stages of AD showed that Trem2 depletion resulted in reduced amyloid deposition in the hippocampus, suggesting that loss-of-function of TREM2 is beneficial [87]. In the context of tauopathy without amyloidosis, PS19/Trem2−/− mice showed reduced brain atrophy, suggesting a beneficial effect of TREM2 deficiency [64]. These results demonstrate that the role of TREM2 in AD is unlikely to be attributable to a simple partial loss-of-function.
TREM2 gene transcript and protein levels appear to be elevated in human AD brains [88, 89]. AD patients carrying the p.R47H variant showed a trend toward increased TREM2 protein levels, suggesting that the p.R47H mutation does not appear to have a negative influence on protein levels but actually the opposite [89]. The authors suggested that the p.R47H variant may affect TREM2 binding function instead of expression levels [89]. Some research suggests that increasing TREM2 levels or activity may be beneficial in AD. TREM2 protein levels were upregulated in a transgenic mouse model for AD (APPswe/PS1dE9) at 7-months of age when amyloid deposition and cognitive impairment is present, and TREM2 levels were positively correlated with levels of Aβ42 [62]. Lentiviral overexpression of Trem2 in vivo significantly reduced neuropathology and improved cognitive symptoms, suggesting that upregulation of TREM2 is a protective response to AD pathology [62]. However, overexpression of TREM2 by lentivirus did not improve cognitive performance or reduce neuropathology in older (18 months old) APPswe/PS1dE9 mice, suggesting that TREM2 may only be a viable therapeutic target early in disease [90].
It is unclear how different TREM2 variants affect protein levels or activity, so it is useful to study specific variants rather than manipulating gene expression. The AD-associated p.R47H TREM2 mutation has been shown to impair binding of phospholipids and lipoproteins in TREM2 reporter cells [60]. Human brain tissue from p.R47H carriers showed less microglia enveloping amyloid plaques resulting in larger plaques with branched amyloid fibrils [91]. Based on these (and similar) findings, TREM2 activating antibodies have been proposed as a potential therapeutic tool. TREM2 activating antibodies can activate ERK and calcium signaling and appear to improve maturation and migration of osteoclasts and dendritic cells [54, 92]. However, there are challenges with this approach. TREM2 appears to influence phagocytosis and anti-TREM2 antibodies may appear as a substrate for cellular uptake [81]. Alternatively, anti-TREM2 antibodies may inhibit binding and internalization of TREM2 ligands [60]. In considering anti-TREM2 antibodies as a potential therapeutic, it is also important to note that the role of sTREM2 in health and disease is not yet understood [93] and most anti-TREM2 antibodies against the extracellular domain target both membranous and soluble forms of TREM2. Chronic treatment with TREM2-activating antibodies could also have toxic effects through increased activation of microglia and extra-CNS macrophages, so the timing of this type of therapeutic would be absolutely crucial to reduce treatment duration. Although there are currently no in vivo examples of TREM2 antibody-stimulated agonism successfully reducing AD-related pathology, a patent application search suggests this is the most active pharmacological interest thus far. As shown in Figure 4, the number of TREM2-related patent applications sharply increased after the first reported TREM2 agonist (Hsp60) in 2009 [71] and increased dramatically again after the first reported association between TREM2 and AD at the beginning of 2013 [40, 43].
Figure 4.
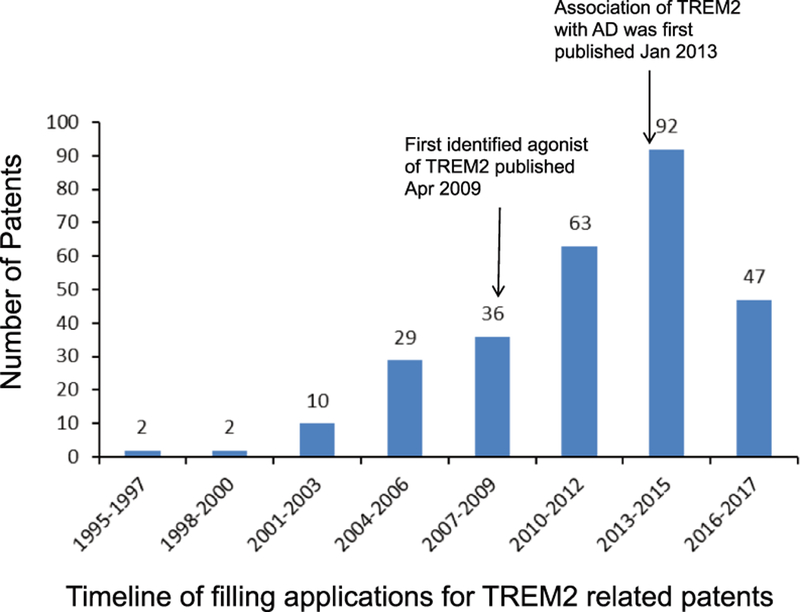
The number of TREM2-related patent applications every 2 years starting from 1995, which was the oldest record found, to near the end of 2017. Found in a Google Patents (https://patents.google.com) search performed on 15 November 2017. Numbers at the top of each bar are the number of patents reported within the 2-year span.
An alternative strategy to using activating antibodies may be to increase TREM2 gene expression or protein levels, making more TREM2 available. Analyses of murine primary microglial cultures have shown that Trem2 knockdown increased protein levels of TNFα and inhibited phagocytosis while overexpression of Trem2 not only increased phagocytosis but also reduced proinflammatory response [94]. Follow-up studies in experimental autoimmune encephalomyelitis (EAE) mouse models for MS supported these findings [95]. Ectopic gene expression of Trem2 in myeloid precursor cells injected intravenously improved MS-like clinical symptoms via increased phagocytosis by Trem2-transduced myeloid cells, decreased expression of proinflammatory genes (TNFα, IFNγ, and IL-1β), and increased expression of anti-inflammatory IL-10 [95]. It is important to note that the Trem2-transduced myeloid cells injected into healthy mice showed no signs of activation, and activated cells in the EAE-diseased mice were localized to the inflammatory lesions in the spinal cord [95]. A more recent study of APPswe/PS1dE9 mice demonstrated that lentiviral overexpression of Trem2 in the brain ameliorated AD-associated neuropathology and cognitive function [62].
Of some concern are the seemingly conflicting results of different studies that suggest TREM2 may enhance inflammatory response or TREM2 reduces inflammatory response. Some researchers suggest that the different cell types expressing TREM2 may influence these different responses [32]. Studies of TREM2 expressed on macrophages or microglia suggest that TREM2 ameliorates the inflammatory response [95–98], whereas studies of dendritic cells suggest TREM2 exacerbates inflammatory response [32, 54]. This hypothesis is plausible considering the roles of these cell types in the innate immune system (macrophages) and the adaptive immune system (dendritic cells). Additionally, microglia clearly play a bigger role in the CNS than that of simple immune cells; they are important for homeostasis, synaptic plasticity, and learning and memory [99–103]. Recent studies have demonstrated that TREM2 is important for microglial metabolism [104]; inhibition of TREM2 would disrupt all of the various microglial roles in the brain.
6. Expert Opinion
There are several factors to consider when developing TREM2-directed therapeutics. First, some argue that TREM2 may not be a broadly applicable therapeutic target for AD since the minor allele frequency of TREM2 variants which confer a strong risk for developing AD are present in less than 1% of AD patients. However, extensive evidence has shown that neuroinflammation and immune response play a significant role in AD pathology and TREM2 is important in innate immunity. Thus far research has focused on overall TREM2 expression levels or TREM2 variants associated with AD risk; however, it is probable that variants in other genes influence TREM2 protein levels or activity, or disrupt other processes within the immune pathway. For instance, it has been shown that TREM2 expression is disrupted by TYROBP mutations in the absence of TREM2 mutations, suggesting that TREM2 may still be involved in disease in TREM2 mutation non-carriers [31]. Therefore, although TREM2 variants are present in a small number of AD patients, targeting TREM2 or other molecules in the same biological pathway will most likely be applicable for many AD cases whether they carry a TREM2 mutation or not.
The timing of intervention will always be important when designing AD treatment. Cellular and animal models demonstrate that TREM2 deficiency has a changing role throughout the progression of AD, by reducing amyloid pathology early in disease but increasing amyloid accumulation later in disease [60, 63, 87]. These studies, combined with studies that show CSF levels of sTREM2 are significantly elevated in preclinical and early AD [24–26], suggest that trials using TREM2-directed therapies would probably be most successful in earlier stages of disease. However, this brings us to the question about what these TREM2-directed therapeutics would be designed to do.
It is important to understand how the identified TREM2 variants increase AD risk. Most research has focused on TREM2 expression, with many groups studying Trem2 knockouts. This approach may not be the best recapitulation of what happens in the presence of these TREM2 mutations. Some researchers concluded that their studies demonstrated there should be less TREM2 expression and some researchers concluded the opposite. Recent research suggests the answer isn’t that simple. It is important to remember a few things here: 1. protein expression level does not represent protein activity; 2. there are at least two forms of TREM2 that may be important, the membrane-bound part of the receptor and the soluble form (which can result from cleavage or alternative splicing); and 3. just as microglia can be protective or damaging depending on the situation, TREM2 probably also has the potential to be protective or damaging. Therapeutics aimed at simply reducing or increasing TREM2 expression or activity are unlikely to be successful; it is more likely that effective treatment will focus within the pathways involving TREM2, targeting disrupted processes instead. Researchers very recently reported their findings from BAC transgenic Trem−/− mice expressing humanized common variant TREM2 or p.R47H mutant TREM2 crossed with 5XFAD mice [105]. They demonstrated that although shedding of sTREM2 was similar in vitro, sTREM2 was found on plaques and neurons in mice expressing WT Trem2 but not in the mice expressing p.R47H [99]. They also reported that plaque-induced microgliosis was impaired in the p.R47H mice, suggesting that sTREM2 interacts with plaques and may be important for microglial response to amyloidosis [99]. More research is necessary to determine what role, if any, sTREM2 has in AD pathology and experiments need to be carefully designed to truly model the TREM2 variants associated with AD risk to better understand their influence on disease.
Therapeutics targeting TREM2, are unlikely to be simple drugs administered orally or injected. If the hypothesis that TREM2 acts differently on the different cell types is correct, not only will interventions need to cross the blood brain barrier, but they will have to be highly specific. Results from the earlier study by Takahashi et al., where they injected myeloid precursor cells expressing wild-type Trem2 into the EAE mouse model for MS, suggest that cell-based therapeutics may be a viable option [95]. Still in its infancy, cell-based therapies for AD have been discussed by researchers over the past few years, primarily focusing on the potential of using induced pluripotent stem cells (iPSCs) to replace lost neurons and the numerous pitfalls and limitations for these methods [106, 107]; however, other researchers have discussed the potentials for glial cell-based therapies as potentially safer alternatives [108]. We think that utilizing gene therapies to introduce properly functioning microglia early in disease is more likely to be successful than attempting to replace lost neurons using iPSCs after the damage has been done. Although a few potential treatments for AD have reached human clinical trials, more work will be necessary to identify therapies to effectively halt or slow disease progression. Extensive evidence suggests that successful AD treatment will need to be administered early in disease, and research has been underway to identify preclinical or prodromal AD. Studies thus far indicate that a combination of biomarkers and genetic risk scoring will provide the best predictive power to find presymptomatic patients who may benefit from treatment. Identification of these individuals will be invaluable for developing effective AD therapeutics.
AD research has progressively shown an important role of immune response and inflammation in disease pathology, so it is not surprising that TREM2 is such a promising target for early AD therapeutics. As we discuss in this manuscript, more research will be necessary to determine how TREM2 influences disease before we will know how to target TREM2 to effectively treat AD. Recent research has started to move in the right direction by studying specific AD-associated TREM2 mutations as well as the potential role of sTREM2. Results from these studies will prove valuable not only for developing TREM2-targeted therapeutics but for further understanding the underlying biology of disease and possibly revealing additional treatments.
Article Highlights.
Vaccines targeting amyloid have reached phase 3 clinical trials but have currently shown little or no improvement in cognitive symptoms, leading researchers to search for alternative AD therapeutics.
Since pathology appears years before cognitive symptoms, treatments for AD most likely need to be administered early in disease to be affective.
TREM2 has been strongly associated with AD and plays a key role in innate immune response; however, the exact mechanisms are unclear. There is conflicting evidence about whether TREM2-targeted therapeutics should enhance or downregulate expression of TREM2.
Context is important for understanding the role of TREM2 in AD; microglial activation state, presence of amyloid-mediated or tau-mediated pathology, and acute vs chronic injury can all have different effects that can be beneficial or harmful.
Most research to-date has explored Trem2 knockout/knockdown; however, it has been demonstrated that different TREM2 mutations associated with disease influence sTREM2 levels differently. More research using transgenic models studying identified TREM2 mutations is necessary.
Acknowledgments
Funding
This manuscript is supported by grants from the National Institutes of Health (NIH) (R01-AG044546, P01-AG003991, U01-AG05241102, and RF1-AG053303), the Alzheimer Association (NIRG-11–200110, BAND-14–338165, and BFG-15–362540). Y Deming is supported by an NIMH training grant (T32MH014877). BA Benitez is supported by 2018 pilot funding from the Hope Center for Neurological Disorders and the Danforth Foundation Challenge at Washington University.
Footnotes
Declaration of Interests
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewers Disclosures
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Papers of special note have been highlighted as:
* of interest
** of considerable interest
References
- 1.Alzheimer’s Association, 2017 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 2017. 13(4):325:373. [Google Scholar]
- 2.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, et al. , Role of genes and environments for explaining Alzheimer disease. Archives of General Psychiatry, 2006. 63(2):168–174. [DOI] [PubMed] [Google Scholar]
- 3.Escott-Price V, Shoai M, Pither R, Williams J, et al. , Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging, 2017. 49:214 e7–214 e11. [DOI] [PubMed] [Google Scholar]
- 4.Ridge PG, Hoyt KB, Boehme K, Mukherjee S, et al. , Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging, 2016. 41:200 e13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, et al. , Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 1993. 261(5123):921–3. [DOI] [PubMed] [Google Scholar]
- 6.Holtzman DM, Herz J, and Bu G, Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med, 2012. 2(3):a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenk D, Barbour R, Dunn W, Gordon G, et al. , Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature, 1999. 400(6740):173–7. [DOI] [PubMed] [Google Scholar]
- 8.Panza F, Solfrizzi V, Imbimbo BP, and Logroscino G, Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: the point of no return? Expert Opin Biol Ther, 2014. 14(10):1465–76. [DOI] [PubMed] [Google Scholar]
- 9.Doody RS, Thomas RG, Farlow M, Iwatsubo T, et al. , Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med, 2014. 370(4):311–21. [DOI] [PubMed] [Google Scholar]
- 10.Salloway S, Sperling R, Fox NC, Blennow K, et al. , Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med, 2014. 370(4):322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vandenberghe R, Rinne JO, Boada M, Katayama S, et al. , Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther, 2016. 8(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDade E and Bateman RJ, Stop Alzheimer’s before it starts. Nature, 2017. 547(7662):153–155. [DOI] [PubMed] [Google Scholar]
- 13.Ritchie C, Smailagic N, Noel-Storr AH, Ukoumunne O, et al. , CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev, 2017. 3:CD010803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blennow K, Dubois B, Fagan AM, Lewczuk P, et al. , Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement, 2015. 11(1):58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Jong D, Jansen RW, Kremer BP, and Verbeek MM, Cerebrospinal fluid amyloid beta42/phosphorylated tau ratio discriminates between Alzheimer’s disease and vascular dementia. J Gerontol A Biol Sci Med Sci, 2006. 61(7):755–8. [DOI] [PubMed] [Google Scholar]
- 16.Doecke JD, Rembach A, Villemagne VL, Varghese S, et al. , Concordance Between Cerebrospinal Fluid Biomarkers with Alzheimer’s Disease Pathology Between Three Independent Assay Platforms. J Alzheimers Dis, 2018. 61(1):169–183. [DOI] [PubMed] [Google Scholar]
- 17.Fagan AM, Roe CM, Xiong C, Mintun MA, et al. , Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol, 2007. 64(3):343–9. [DOI] [PubMed] [Google Scholar]
- 18.Harari O, Cruchaga C, Kauwe JS, Ainscough BJ, et al. , Phosphorylated tau-Abeta42 ratio as a continuous trait for biomarker discovery for early-stage Alzheimer’s disease in multiplex immunoassay panels of cerebrospinal fluid. Biol Psychiatry, 2014. 75(9):723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonell A, Mansilla A, Rami L, Llado A, et al. , Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J Alzheimers Dis, 2014. 42(3):901–8. [DOI] [PubMed] [Google Scholar]
- 20.Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, et al. , YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry, 2010. 68(10):903–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellwig K, Kvartsberg H, Portelius E, Andreasson U, et al. , Neurogranin and YKL-40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimers Res Ther, 2015. 7(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrin RJ, Craig-Schapiro R, Malone JP, Shah AR, et al. , Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer’s disease. PLoS One, 2011. 6(1):e16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutphen CL, Jasielec MS, Shah AR, Macy EM, et al. , Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol, 2015. 72(9):1029–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heslegrave A, Heywood W, Paterson R, Magdalinou N, et al. , Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol Neurodegener, 2016. 11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, et al. , Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol, 2016. 131(6):925–33.* Initial report of the influence of disease-associated TREM2 mutations on CSF sTREM2 levels, demonstrating that different variants have opposite affects on sTREM2 levels.
- 26.Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, et al. , sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med, 2016. 8(5):466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, et al. , Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med, 2016. 8(369):369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paloneva J, Autti T, Raininko R, Partanen J, et al. , CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology, 2001. 56(11):1552–8. [DOI] [PubMed] [Google Scholar]
- 29.Paloneva J, Kestila M, Wu J, Salminen A, et al. , Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet, 2000. 25(3):357–61. [DOI] [PubMed] [Google Scholar]
- 30.Paloneva J, Manninen T, Christman G, Hovanes K, et al. , Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet, 2002. 71(3):656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiialainen A, Veckman V, Saharinen J, Paloneva J, et al. , Transcript profiles of dendritic cells of PLOSL patients link demyelinating CNS disorders with abnormalities in pathways of actin bundling and immune response. J Mol Med (Berl), 2007. 85(9):971–83. [DOI] [PubMed] [Google Scholar]
- 32.Jiang H, Si Y, Li Z, Huang X, et al. , TREM-2 promotes acquired cholesteatoma-induced bone destruction by modulating TLR4 signaling pathway and osteoclasts activation. Sci Rep, 2016. 6:38761.* Authors suggest the different roles of TREM2 in inflammatory response may be related to the cell type on which it is expressed.
- 33.Montalbetti L, Ratti MT, Greco B, Aprile C, et al. , Neuropsychological tests and functional nuclear neuroimaging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct Neurol, 2005. 20(2):71–5. [PubMed] [Google Scholar]
- 34.Chouery E, Delague V, Bergougnoux A, Koussa S, et al. , Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat, 2008. 29(9):E194–204. [DOI] [PubMed] [Google Scholar]
- 35.Borroni B, Ferrari F, Galimberti D, Nacmias B, et al. , Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging, 2014. 35(4):934 e7–10. [DOI] [PubMed] [Google Scholar]
- 36.Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, et al. , Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging, 2014. 35(3):726 e11–9. [DOI] [PubMed] [Google Scholar]
- 37.Guerreiro R, Bilgic B, Guven G, Bras J, et al. , A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiology of Aging, 2013. 34(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rayaprolu S, Mullen B, Baker M, Lynch T, et al. , TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener, 2013. 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benitez BA, Cooper B, Pastor P, Jin SC, et al. , TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging, 2013. 34(6):1711 e15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, et al. , TREM2 Variants in Alzheimer’s Disease. New England Journal of Medicine, 2013. 368(2):117–127.** First repoted (at the same time as Jonsson et al) genetic association of TREM2 and AD risk.
- 41.Jin SC, Benitez BA, Karch CM, Cooper B, et al. , Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet, 2014. 23(21):5838–46.* Identified for the first time an alternative transcript in the brain that encodes the soluble form of TREM2.
- 42.Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, et al. , TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener, 2015. 10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, et al. , Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. New England Journal of Medicine, 2013. 368(2):107–116.** First repoted (at the same time as Guerreiro et al) genetic association of TREM2 and AD risk.
- 44.Bonham LW, Sirkis DW, Fan J, Aparicio RE, et al. , Identification of a rare coding variant in TREM2 in a Chinese individual with Alzheimer’s disease. Neurocase, 2017. 23(1):65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang T, Tan L, Chen Q, Tan MS, et al. , A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol Aging, 2016. 42:217 e1–3. [DOI] [PubMed] [Google Scholar]
- 46.Lue LF, Schmitz CT, Serrano G, Sue LI, et al. , TREM2 Protein Expression Changes Correlate with Alzheimer’s Disease Neurodegenerative Pathologies in Post-Mortem Temporal Cortices. Brain Pathol, 2015. 25(4):469–80.** This study demonstrated TREM2 protein expression is associated with AD neuropathology.
- 47.Piccio L, Buonsanti C, Cella M, Tassi I, et al. , Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain, 2008. 131(Pt 11):3081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohrfelt A, Axelsson M, Malmestrom C, Novakova L, et al. , Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult Scler, 2016. 22(12):1587–1595. [DOI] [PubMed] [Google Scholar]
- 49.Gispert JD, Suarez-Calvet M, Monte GC, Tucholka A, et al. , Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimers Dement, 2016. 10.1016/j.jalz.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, et al. , TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med, 2014. 6(243):243ra86. [DOI] [PubMed] [Google Scholar]
- 51.Hu N, Tan MS, Yu JT, Sun L, et al. , Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J Alzheimers Dis, 2014. 38(3):497–501. [DOI] [PubMed] [Google Scholar]
- 52.Gesmann M and de Castillo D, Using the Google Visualisation API with R. R Journal, 2011. 3(2):40–44. [Google Scholar]
- 53.Pelham CJ and Agrawal DK, Emerging roles for triggering receptor expressed on myeloid cells receptor family signaling in inflammatory diseases. Expert Rev Clin Immunol, 2014. 10(2):243–56. [DOI] [PubMed] [Google Scholar]
- 54.Bouchon A, Hernandez-Munain C, Cella M, and Colonna M, A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med, 2001. 194(8):1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cella M, Buonsanti C, Strader C, Kondo T, et al. , Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med, 2003. 198(4):645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hickman SE and El Khoury J, TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol, 2014. 88(4):495–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantoni C, Bollman B, Licastro D, Xie M, et al. , TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol, 2015. 129(3):429–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krasemann S, Madore C, Cialic R, Baufeld C, et al. , The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 2017. 47(3):566–581 e9.* Showed evidence for a specific TREM2-APOE pathway that may be important in neurodegenerative disease.
- 59.Ulrich JD, Finn MB, Wang Y, Shen A, et al. , Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener, 2014. 9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Cella M, Mallinson K, Ulrich JD, et al. , TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell, 2015. 160(6):1061–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Ulland TK, Ulrich JD, Song W, et al. , TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med, 2016. 213(5):667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang T, Tan L, Zhu XC, Zhang QQ, et al. , Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology, 2014. 39(13):2949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jay TR, Miller CM, Cheng PJ, Graham LC, et al. , TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med, 2015. 212(3):287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leyns CEG, Ulrich JD, Finn MB, Stewart FR, et al. , TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A, 2017. 114(43):11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oakley H, Cole SL, Logan S, Maus E, et al. , Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci, 2006. 26(40):10129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, et al. , Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep, 2006. 7(9):940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, et al. , Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron, 2007. 53(3):337–51. [DOI] [PubMed] [Google Scholar]
- 68.Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, et al. , Triggering Receptor Expressed on Myeloid Cells 2 Deficiency Alters Acute Macrophage Distribution and Improves Recovery after Traumatic Brain Injury. J Neurotrauma, 2017. 34(2):423–435. [DOI] [PubMed] [Google Scholar]
- 69.Kawabori M, Kacimi R, Kauppinen T, Calosing C, et al. , Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci, 2015. 35(8):3384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kober DL and Brett TJ, TREM2-Ligand Interactions in Health and Disease. J Mol Biol, 2017. 429(11):1607–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stefano L, Racchetti G, Bianco F, Passini N, et al. , The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem, 2009. 110(1):284–94. [DOI] [PubMed] [Google Scholar]
- 72.Atagi Y, Liu CC, Painter MM, Chen XF, et al. , Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem, 2015. 290(43):26043–50.* First study to report ApoE as a ligand for TREM2
- 73.Yeh FL, Wang Y, Tom I, Gonzalez LC, et al. , TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron, 2016. 91(2):328–40. [DOI] [PubMed] [Google Scholar]
- 74.Zhao N, Liu CC, Qiao W, and Bu G, Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol Psychiatry, 2018. 83(4):347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wunderlich P, Glebov K, Kemmerling N, Tien NT, et al. , Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem, 2013. 288(46):33027–36.* Demonstrated for the first time that soluble TREM2 could be shed from the cell membrane by sequential proteolytic cleavage.
- 76.Schlepckow K, Kleinberger G, Fukumori A, Feederle R, et al. , An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med, 2017. 9(10):1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thornton P, Sevalle J, Deery MJ, Fraser G, et al. , TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med, 2017. 9(10):1366–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang T, Hou JK, Gao Q, Yu JT, et al. , TREM2 p.H157Y Variant and the Risk of Alzheimer’s Disease: A Meta-Analysis Involving 14,510 Subjects. Curr Neurovasc Res, 2016. 13(4):318–320. [DOI] [PubMed] [Google Scholar]
- 79.Song W, Hooli B, Mullin K, Jin SC, et al. , Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement, 2017. 13(4):381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park JS, Ji IJ, An HJ, Kang MJ, et al. , Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic, 2015. 16(5):510–8. [DOI] [PubMed] [Google Scholar]
- 81.Prada I, Ongania GN, Buonsanti C, Panina-Bordignon P, et al. , Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience, 2006. 140(4):1139–48. [DOI] [PubMed] [Google Scholar]
- 82.Varnum MM, Clayton KA, Yoshii-Kitahara A, Yonemoto G, et al. , A split-luciferase complementation, real-time reporting assay enables monitoring of the disease-associated transmembrane protein TREM2 in live cells. J Biol Chem, 2017. 292(25):10651–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raha AA, Henderson JW, Stott SR, Vuono R, et al. , Neuroprotective Effect of TREM-2 in Aging and Alzheimer’s Disease Model. J Alzheimers Dis, 2017. 55(1):199–217. [DOI] [PubMed] [Google Scholar]
- 84.Lucin KM, O’Brien CE, Bieri G, Czirr E, et al. , Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron, 2013. 79(5):873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yin J, Liu X, He Q, Zhou L, et al. , Vps35-dependent recycling of Trem2 regulates microglial function. Traffic, 2016. 17(12):1286–1296. [DOI] [PubMed] [Google Scholar]
- 86.Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, et al. , Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jay TR, Hirsch AM, Broihier ML, Miller CM, et al. , Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci, 2017. 37(3):637–647.** This study demonstrated the changing role of TREM2 through disease progression; TREM2 deficiency in an AD mouse model was protective early in disease but enhanced amyloid pathology later in disease.
- 88.Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, et al. , Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol Aging, 2013. 34(8):2077 e11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma L, Allen M, Sakae N, Ertekin-Taner N, et al. , Expression and processing analyses of wild type and p.R47H TREM2 variant in Alzheimer’s disease brains. Mol Neurodegener, 2016. 11(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang T, Wan Y, Zhang YD, Zhou JS, et al. , TREM2 Overexpression has No Improvement on Neuropathology and Cognitive Impairment in Aging APPswe/PS1dE9 Mice. Mol Neurobiol, 2017. 54(2):855–865. [DOI] [PubMed] [Google Scholar]
- 91.Yuan P, Condello C, Keene CD, Wang Y, et al. , TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron, 2016. 92(1):252–264. [DOI] [PubMed] [Google Scholar]
- 92.Humphrey MB, Daws MR, Spusta SC, Niemi EC, et al. , TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res, 2006. 21(2):237–45. [DOI] [PubMed] [Google Scholar]
- 93.Zhong L, Chen XF, Wang T, Wang Z, et al. , Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med, 2017. 214(3):597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takahashi K, Rochford CD, and Neumann H, Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med, 2005. 201(4):647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi K, Prinz M, Stagi M, Chechneva O, et al. , TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med, 2007. 4(4):e124.** This study demonstrated potential for cell-based gene therapy targeting TREM2 to treat multiple sclerosis, by enhanced phagocytosis of cell debris and attenuated proinflammatory response after intravenous injection of TREM2-transduced myeloid precursor cells into EAE-disease mice.
- 96.Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, et al. , Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol, 2006. 177(4):2051–5. [DOI] [PubMed] [Google Scholar]
- 97.Piccio L, Buonsanti C, Mariani M, Cella M, et al. , Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol, 2007. 37(5):1290–301. [DOI] [PubMed] [Google Scholar]
- 98.Turnbull IR, Gilfillan S, Cella M, Aoshi T, et al. , Cutting edge: TREM-2 attenuates macrophage activation. J Immunol, 2006. 177(6):3520–4. [DOI] [PubMed] [Google Scholar]
- 99.Parkhurst CN, Yang G, Ninan I, Savas JN, et al. , Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 2013. 155(7):1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sierra A, Tremblay ME, and Wake H, Never-resting microglia: physiological roles in the healthy brain and pathological implications. Front Cell Neurosci, 2014. 8:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wake H, Moorhouse AJ, Miyamoto A, and Nabekura J, Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci, 2013. 36(4):209–17. [DOI] [PubMed] [Google Scholar]
- 102.Wu Y, Dissing-Olesen L, MacVicar BA, and Stevens B, Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol, 2015. 36(10):605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang J, Malik A, Choi HB, Ko RW, et al. , Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron, 2014. 82(1):195–207. [DOI] [PubMed] [Google Scholar]
- 104.Ulland TK, Song WM, Huang SC, Ulrich JD, et al. , TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell, 2017. 170(4):649–663 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Song WM, Joshita S, Zhou Y, Ulland TK, et al. , Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med, 2018. 10.1084/jem.20171529.** First study to use BAC transgenic mouse model to investigate a disease-specific TREM2 mutation (p.R47H) and demonstrated that soluble TREM2 localized differently in mutant mice.
- 106.Ross CA and Akimov SS, Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum Mol Genet, 2014. 23(R1):R17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pen AE and Jensen UB, Current status of treating neurodegenerative disease with induced pluripotent stem cells. Acta Neurol Scand, 2017. 135(1):57–72. [DOI] [PubMed] [Google Scholar]
- 108.Hunsberger JG, Rao M, Kurtzberg J, Bulte JWM, et al. , Accelerating stem cell trials for Alzheimer’s disease. Lancet Neurol, 2016. 15(2):219–230. [DOI] [PubMed] [Google Scholar]