

Abstract
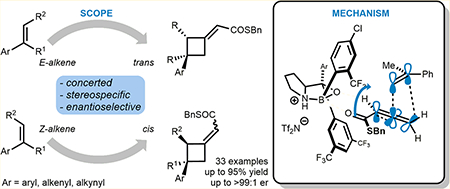
Identification of a novel catalyst–allenoate pair allows enantioselective [2+2] cycloaddition of α-methylstyrene. To understand the origin of selectivity, a detailed mechanistic investigation was conducted. Herein, two competing reaction pathways are proposed, which operate simultaneously and funnel the alkenes to the same axially chiral cyclobutanes. In agreement with the Woodward–Hoffmann rules, this mechanistic curiosity can be rationalized through a unique symmetry operation that was elucidated by deuteration experiments. In the case of 1,1-diarylalkenes, distal communication between the catalyst and alkene is achieved through subtle alteration of electronic properties and conformation. In this context, a Hammett study lends further credibility to a concerted mechanism. Thus, extended scope exploration, including β-substitution on the alkene to generate two adjacent stereocenters within the cyclobutane ring, is achieved in a highly stereospecific and enantioselective fashion (33 examples, up to >99:1 er).
Graphical Abstract
INTRODUCTION
Construction of multiple stereocenters in a single event has become an increasingly important strategy to build molecular complexity in an efficient and economical way. Arguably, one of the most powerful methods to achieve such a transformation is the Diels–Alder reaction.1 It is generally postulated that the aforementioned reaction involves a symmetry-allowed cyclic transition state as predicted by the Woodward–Hoffmann rules.2 Consequently, the reaction can be performed in a stereospecific fashion, allowing the generation of all possible isomers from the respective E- or Z-alkenes. Despite significant advances in the realm of enantioselective Diels–Alder reactions,3 the analogous, concerted [2+2] cycloaddition of alkenes has remained a challenge. Particularly, methods that utilize activated olefins are especially difficult. In contrast to the Diels–Alder reaction, recent methods to construct enantioenriched arylcyclobutanes by [2+2] cycloaddition often proceed through stepwise processes, resulting in decreased reaction selectivity (Scheme 1a).4,5 With respect to Lewis acid catalyzed examples, gold complexes have been utilized to activate alkynes6 or allenes7 to achieve enantioselective cycloaddition with highly activated styrene derivatives. In addition, copper and zinc have been used with electron rich arylalkenes to give cyclobutanes with good control of enantioselectivity.8 Alternatively, chiral amines can be used to assemble cyclobutanes via ionic intermediates.9 Photochemical methods10 often exhibit increased tolerance for electron-poor styrenes, but generally involve excited state biradical intermediates, which lead to either stereoconvergence10f or erosion of diastereoselectivity.10g Overall, only a few examples,9a report good stereospecificity through trapping of reactive intermediates at low temperatures.
Scheme 1.

Enantioselective Arylcyclobutane Synthesis
To address this problem, we recently focused our efforts on rendering alkene-allenoate cycloadditions enantioselective.11 Based on several reports in the literature,11b,12 alkene-allenoate cycloadditions appear to be concerted and therefore stereo-specific in nature, which provides a unique opportunity for an in-depth study (Scheme 1b). Herein, we report the enantioselective [2+2] cycloaddition of α-methylstyrene through identification of a novel alkene-allenoate pair. This method does not only enable catalytic enantioselective formation of quaternary carbon centers13 but also exemplifies how elucidation of reaction mechanism can go hand in hand with expansion of scope. Ultimately, we propose models that reliably explain the observed selectivity for a range of activated alkenes in this unique [2+2] cycloaddition.
RESULTS AND DISCUSSION
Optimization.
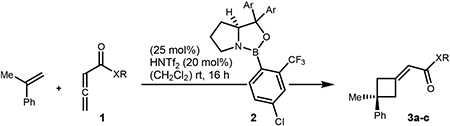
We initially envisioned accessing cyclobutanes bearing a quaternary center by using activated 1,1-disubstituted alkenes (e.g., α-methylstyrene). Preliminary data suggested that the identity of the allenoate ester has a significant influence on modulating reactivity as well as selectivity.12d As such, investigations of various allenoates 1 were undertaken (Table 1).
Table 1.
Reaction Optimization
| Entry | XR | Ar | Yieldb | erc |
|---|---|---|---|---|
| 1 | OBn (1a) | Ph (2a) | 95% | 78:22 |
| 2 | OCH2CF3 (1b) | Ph (2a) | 46% | 76:24 |
| 3 | SBn (1c) | Ph (2a) | 72% | 81:19 |
| 4 | SBn (1c) | 3,5-(CH3)2-C6H3 (2b) | 71% | 90:10 |
| 5 | SBn (1c) | 3,5-(tBu)2-C6H3 (2c) | 90% | 80:20 |
| 6 | SBn (1c) | 3,5-(OMe)2-C6H3 (2d) | 56% | 87:13 |
| 7 | SBn (1c) | 3,5-(CF3)2-C6H3 (2e) | 96% | 96:4 |
| 8 | OBn (1a) | 3.5-(CF3)2-C6H3 (2e) | 87% | 86:14 |
See the Supporting Information for experimental details.
Determi-nation by 1H NMR of the crude reaction mixture utilizing a calibrated standard.
Determined by HPLC analysis using a chiral column.
Changing from benzyl (1a) to the more reactive 2,2,2-trifluoroethyl ester (1b) under our previously optimized reaction conditions resulted in decreased yield, due to competitive polymerization of the starting materials under the reaction conditions (Table 1, compare entries 1 and 2). We next examined the use of thiobenzyl allenic ester 1c in the reaction and were pleased to find a slight increase in reaction selectivity, albeit with decreased overall yield (compare entries 1 and 3). Confident that reaction yield could be improved through catalyst control, we studied modifications of the diarylprolinol scaffold. Whereas increasing the steric size of the aryl groups from phenyl (2a) to xylyl (2b) provided a significant increase in reaction selectivity, further increase to sterically bulkier 3,5-(tBu)2-C6H3 catalyst 2c resulted in a substantially less selective reaction (compare entries 3–5). Investigation of more electron rich aryl groups (i.e., entry 6, 2d) resulted in a marked decrease in overall reaction yield while providing higher reaction selectivity. Significant improvements in both reaction yield and enantioselectivity was observed when more electron deficient 3,5-(CF3)2-C6H3 catalyst 2e was examined in the reaction (entry 7). Interestingly, utilizing the same catalyst with benzyl allenoate resulted in a smaller increase in enantioselectivity (compare entries 1 and 8).
Mechanism.
To account for the observed enantiomer obtained in the reaction we propose that upon binding of the Lewis basic carbonyl oxygen to the Lewis acidic boron atom,14 the orientation of the allenoate may be fixed by a putative C— H⋯O hydrogen bonding interaction (Scheme 2a).15 As the bottom face of the allenoate is effectively blocked by the large aryl groups of the catalyst, approach of the alkene may only occur from the top face. Additionally, the sterically large phenyl group of the alkene is oriented distal to the large catalyst–substrate complex. Conveniently, the planar character of the phenyl group thereby also minimizes steric interaction with the protruding C–H bond of the allene, resulting in two plausible transition states (Scheme 2a, TS-A1 and TS-B1) for alkene approach.
Scheme 2.

Model for Enantioselectivity
We propose a concerted, asynchronous [π2s+(π2s+π2s)] cycloaddition in which the direction of rotation of the electron deficient allenic π-bond is dictated by the Woodward–Hoffmann rules (indicated by the blue arrows).2,16 Because of the unusual symmetry of the system, both transition states lead to the same enantiomer. The absolute configuration of the cycloadduct 3c was proven through hydrolysis of the thioester and subsequent analysis of the corresponding carboxylic acid 4 via X-ray diffraction.17 To distinguish TS-A1 and TS-B1, cis-β-deutero-α-methylstyrene 5 was subjected to the reaction conditions (Scheme 2b). To our surprise, the cycloadduct 6 was obtained as an 83:17 Z:E mixture suggesting that both pathways are operating. Accordingly, trans-β-deutero-α-methylstyrene 7 furnished cyclobutane 8 as a 86:14 Z:E mixture. Assignment of the respective E- and Z-isomers was achieved through derivatization and subsequent NOE analysis of the respective tertiary alcohol 9 (see Supporting Information for details). Thus, we were able to deduce TS-A1 to be energetically favored, which can be explained by the alkene being distal to the bulky boroaryl group of the catalyst. In addition, the cycloaddition was highly stereospecific, as indicated by the two different pairs of products generated from the respective cis- and trans-deuteroalkene. This suggests a concerted mechanism, which was not necessarily to be expected considering the stabilization of a potential benzylic carbocation in a stepwise process.
To gain further insight into the reaction mechanism, we became interested in differentiating 1,1-biaryl alkenes based on their steric and/or electronic properties (Scheme 3a). Herein, only one aryl ring is in conjugation with the π-system, resulting in substantial steric differentiation of the two aryl groups (Scheme 3a, TS-A2). We propose preferential reaction occurs with the more reactive conformer of alkene (TS-A2, X = more electron donating than Y), providing an excellent setting to undertake a more detailed Hammett study. Electronically differentiated biaryl alkenes 10 were evaluated in the reaction. Moderate to good yields were obtained depending on the electronic properties of the biarylalkenes. In agreement with our model, increased disparity between the two aryl groups resulted in improved reaction enantioselectivity (products 11a–11f). We found a good correlation between log(er) and σ+ with a ρ value <1, suggesting the build-up of positive charge in the transition state in a less sensitive fashion than the parent SN1 reaction.18 This correlates with a concerted, highly asynchronous cycloaddition. According to the regression equation obtained from the small training set used for the Hammett study, an enantiomeric ratio of 92:8 was predicted for alkene 12 bearing two electronically altered rings19 (herein Δσ+ was obtained from the parent σ+ values for para-CF3 and para-OMe).18 When 12 was subjected to the reaction conditions, cycloadduct 13 was obtained in 58% yield and 92:8 er highlighting the potential of this type of enantiodiscrimination (Scheme 3b). Proof of absolute stereochemistry was achieved by hydrolysis of product 13 and X-ray analysis of the respective biarylcyclobutanecarboxylic acid 14.
Scheme 3.

Distal Differentiation of 1,1-Biarylalkenes
To further probe our hypothesis, differentially substituted 1,1-biaryl olefin 15 was synthesized and examined in the reaction (Scheme 3c). As the aryl groups of this alkene possess more similar electronic properties, the rotation of one aryl group out of conjugation is primarily driven by adverse intramolecular steric interactions. We hypothesized the ortho-tolyl group would preferentially rotate out of plane to minimize 1,3-allylic strain (TS-A3, Scheme 3c), thus providing a similar steric environment as proposed in TS-A2. Gratifyingly, 16 was obtained in 48% yield and 89:11 er, lending support to our mechanistic hypothesis.
Scope.
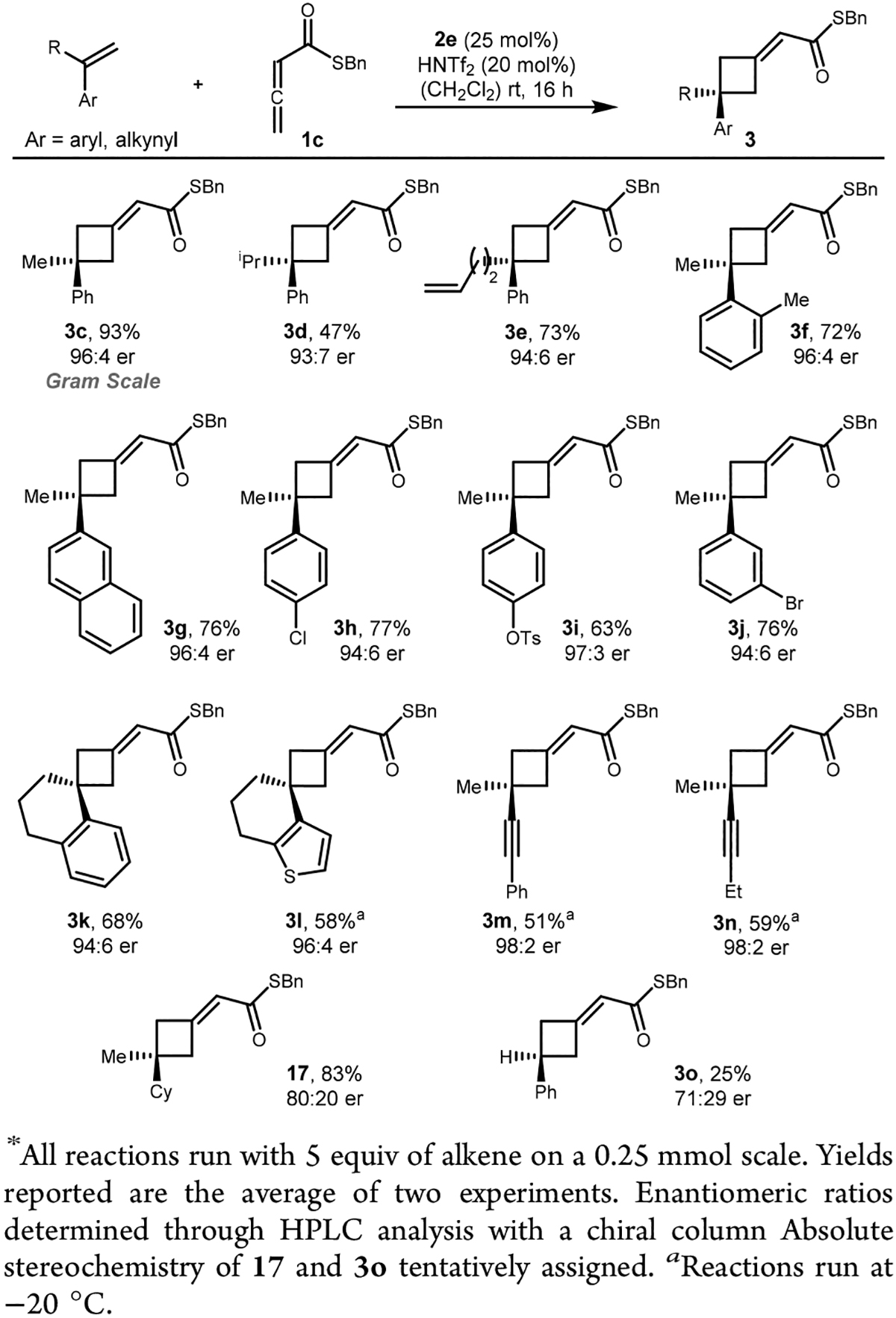
With a catalyst system that allowed for the cycloaddition of activated alkenes in hand, we examined the substrate scope of the reaction (Scheme 4). α-Methylstyrene underwent [2+2] cycloaddition in 93% yield and 96:4 er (product 3c) on gram scale (5.26 mmol) with no loss in reaction selectivity. Increasing the steric size of the α-substituent was investigated and proceeds with high enantioselectivity (products 3d and 3e). High chemoselectivity for the activated alkene in the presence of an unactivated alkene was observed to provide 3e in 72% yield and 96:4 er, with no trace of cycloadducts derived from the reaction of the unactivated olefin. Several steric and electronic perturbations of the aromatic ring have been investigated (products 3f–3l). The cycloaddition proceeded in good yield with sterically encumbered (product 3f), halogenated (products 3h and 3j), and electron-poor (products 3i and 3j) vinyl arenes. Spirocyclic cyclobutane derivatives can also be accessed from the requisite 1,1-disubstituted olefin in good yield and high enantioselectivity (product 3k and 3l). Heterocycles bearing weakly basic heteroatoms, such as thiophene, were also tolerated (product 3l). In some cases, dropping the temperature improved the yield by decreasing the rate of alkene polymerization. Interestingly, enynes underwent cycloaddition without any interference of the alkyne moiety yielding 3m and 3n in moderate yield and excellent enantioselectivity.
Scheme 4.
1,1-Disubstituted Alkene Substrate Scope*
Replacement of the aryl group with a cyclohexyl group resulted in substantial decrease in enantioselectivity, demonstrating that the aryl group is necessary to obtain highly enantioenriched products (Scheme 4, compare products 17 and 3c). Substitution at the α-position was also essential for successful reaction, as styrene itself performed poorly in terms of reactivity and selectivity under several reaction conditions (product 3o, see Supporting Information for further details). Partial polymerization of styrene presumably accounts for the low yield, whereas low enantioselectivities for 17 and 3o can be rationalized by lack of steric differentiation with the protruding C–H bond of the allene.
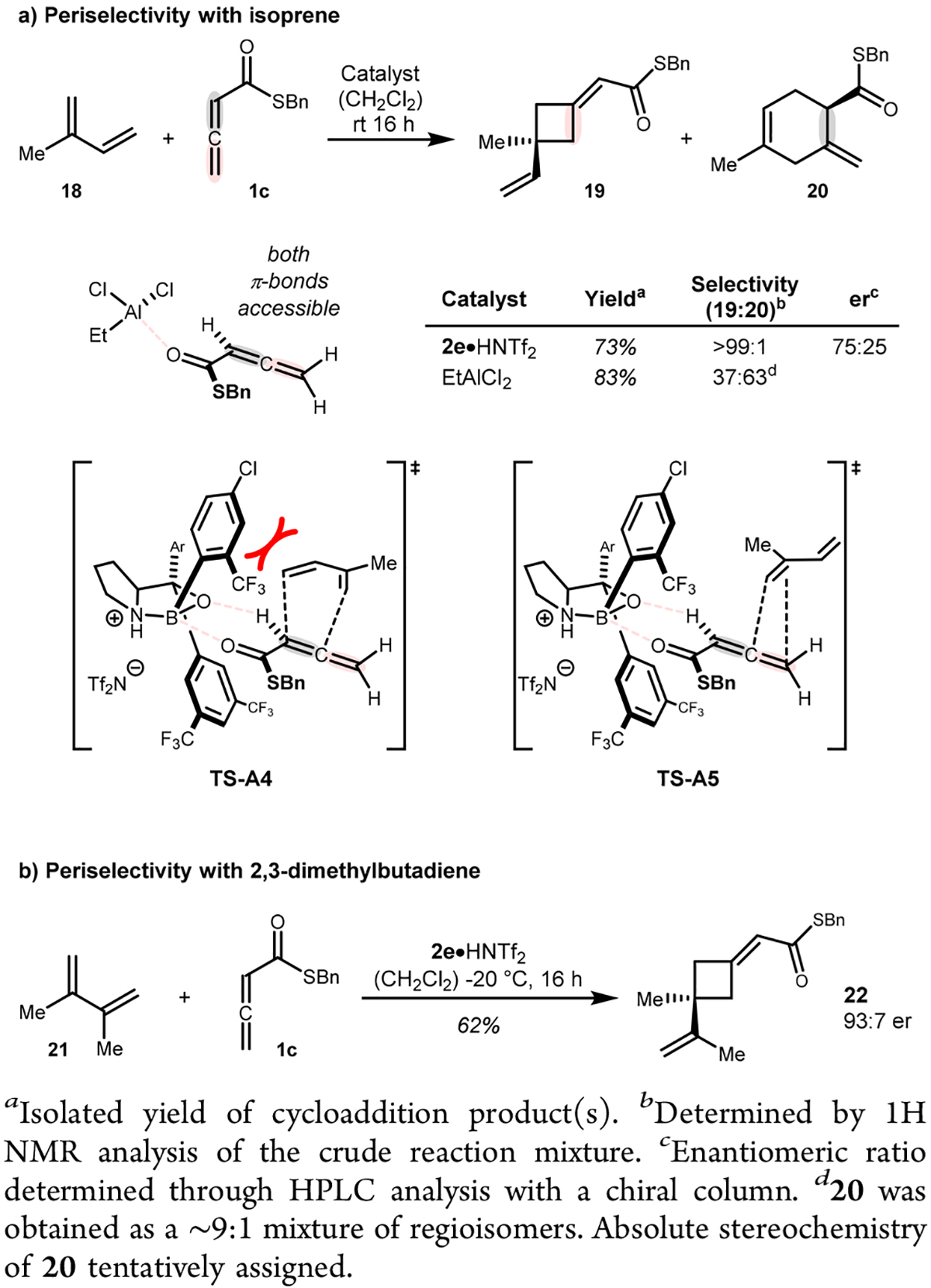
To further expand the reaction scope, we investigated commodity dienes, such as isoprene (18), in the cycloaddition reaction (Scheme 5). Initially, when EtAlCl2 was used as a Lewis acid, low periselectivity was observed, favoring Diels–Alder product 20. In stark contrast to EtAlCl2, catalyst 2e allowed for >99:1 selectivity favoring [2+2] cycloadduct 19. The reaction also occurred with high chemoselectivity, as only the more substituted alkene of isoprene underwent cyclo-addition; however, the observed enantioselectivity was only moderate for this reaction. We assume that upon binding of allenoate 1c to catalyst 2e, the internal π-bond (Scheme 5a, marked in gray in TS-A4) is sufficiently blocked by the large boroaryl group of the catalyst, leaving the distal π-bond (marked in red in TS-A5) more readily accessible for [2+2] cycloaddition.20 It should be noted that our system complements previous reports on Diels–Alder reactions between allenoates and cyclic dienes.14b,21,22 α-Substitution, as imposed by 2,3-dimethylbutadiene (21), significantly improved the enantioselectivity while preserving the high level of peri-selectivity (Scheme 5b, product 22).
Scheme 5.
Catalyst Induced Periselectivity
Application to β-Substitution.
The potential to generate two adjacent stereocenters piqued our curiosity to further study β-substitution of the alkene starting materials and test our proposed models. We initiated this survey with cyclic alkene 23. Gratifyingly, the reaction proceeded in good yield, regioselectivity, and with excellent enantioselectivity, but resulted in the formation of both alkene isomers (Scheme 6, Z-24 and ent-E-24) in a 69:31 ratio. Separation by column chromatography revealed Z-isomer Z-24 as the major product. In accordance with the deuteration experiment and as a consequence of β-substitution on the alkene, the two TS do not lead to the same absolute configuration within the cyclobutane ring (see Scheme 6a, TS-A6 and TS-B2). To verify this hypothesis, Z-24 and ent-E-24 were individually transformed to ketone 25 by oxidative cleavage using a modified Lemieux–Johnson oxidation.23 As expected, 25 revealed opposite absolute configuration indicated by opposite optical rotation.
Scheme 6.
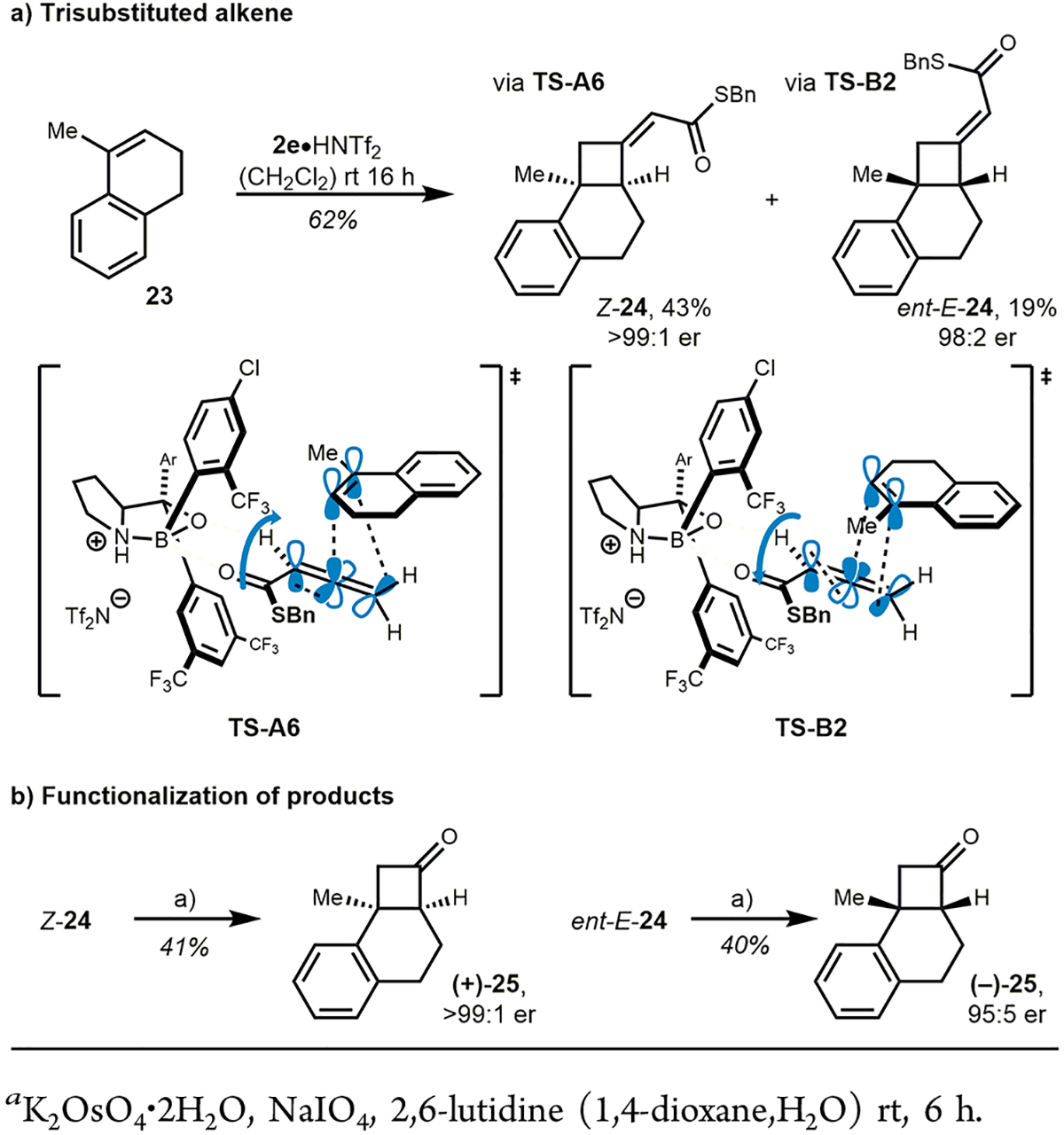
Initial Study on Trisubstituted Alkenes
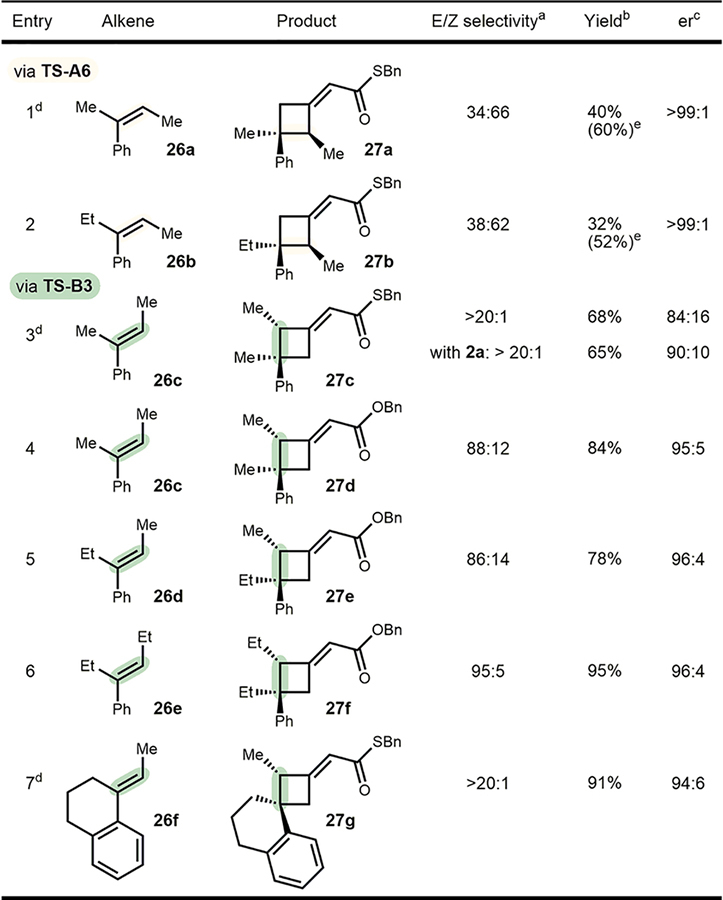
Acyclic trisubstituted alkenes should, based on our mechanistic study, undergo cycloaddition in a concerted, stereospecific fashion. To demonstrate the utility of such an attribute, different pairs of acyclic E- and Z-alkenes were subjected to the optimized conditions (Table 2). As seen for alkene 23, modest E/Z selectivity was observed for Z-alkenes; however, the respective cyclobutanes 27a and 27b were formed with very high enantioselectivity (entries 1 and 2, the respective TS-A6 and TS-B2 from Scheme 6 are likely operating and explain the observed selectivity for alkenes 26a and 26b). Gratifyingly, no detectable amounts of the diastereomeric product originating from a nonstereospecific process was observed (compare entry 1 and 3). Interestingly, E-alkenes (entries 3–7) performed significantly better in terms of E/Z selectivity of products. Considering the two transition states TS-A7 and TS-B3, this is not surprising because TS-A7 encounters a substantial steric interaction between the E-β-substituent and the boroaryl group, whereas TS-B3 is less affected by this substituent pointing away from the large boroaryl group (see Scheme 7a for details). Thus, E-isomeric cyclobutanes were formed almost exclusively from E-alkenes; however, product 27c (entry 3) was obtained with low enantioselectivity even when the temperature was decreased to −20 °C. A modest improvement was accomplished by exchanging catalyst 2c with 2a to provide the desired product in 90:10 er. Interestingly, when benzyl allenoate 1a was used instead of its thio-analog 1c, good enantioselectivity was achieved while maintaining the high level of E/Z selectivity (entry 4). Steric bulk, as imposed by ethyl groups on their respective positions (alkene 26d and 26e), was well tolerated giving products 27e and 27f in good yield and enantioselectivity. Finally, cyclic alkene 26f proceeded in 88% yield and 94:6 er with thiobenzyl allenic ester 1c. Its absolute stereochemistry was unambiguously determined by X-ray diffraction of the respective pentabromophenyl ester 28 and was found to be in agreement with the proposed models (Scheme 7b).
Table 2.
Stereospecific [2+2] Cycloadditions
|
Determination by 1H NMR of the crude reaction mixture utilizing a calibrated standard. Reactions run under optimized conditions using catalyst 2e.
Yields reported of pure major isomer as average of two experiments.
Enantiomeric ratio of the major isomer (see Supporting Information for er of minor isomers).
Reactions run at −20 °C.
Combined yield of both isomers in parentheses.
Scheme 7.

TS for E-Alkenes
CONCLUSION
In summary, a method for enantioselective [2+2] cyclo-additions of activated alkenes with allenoates has been developed. Supported by mechanistic evidence, this reaction resembles a rare example of a concerted, enantioselective [2+2] cycloaddition with activated alkenes. As such, its potential to generate molecular complexity, with precise control of stereochemistry, makes the reaction especially attractive toward the synthesis of cyclobutane containing targets.
Supplementary Material
ACKNOWLEDGMENTS
We thank Indiana University and the National Institutes of Health (R01GM110131) for financial support. The Deutsche Forschungsgemeinschaft (WI 4933/1–2) is acknowledged for postdoctoral fellowship support to J.M.W.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10008.
Crystallographic data for C13H14O2 (CIF)
Crystallographic data for C22H17Br5O2 (CIF)
Experimental procedures, analytical data for all new compounds (PDF)
Crystallographic data for C20H17F3O3 (CIF)
REFERENCES
- (1).(a) Diels O; Alder K Über die Ursachen der Azoesterreaktion. Justus Liebigs Ann. Chem 1926, 450, 237. [Google Scholar]; (b) Diels O; Alder K Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem 1928, 460, 98. [Google Scholar]; (c) Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed 2002, 41, 1668. [DOI] [PubMed] [Google Scholar]
- (2).(a) Woodward RB; Hoffmann R The Conservation of Orbital Symmetry. Angew. Chem., Int. Ed. Engl 1969, 8, 781. [Google Scholar]; (b) Fleming I Molecular Orbitals and Organic Chemical Reactions; Wiley: Chichester, 2009. [Google Scholar]
- (3).For reviews, see:Kagan HB; Riant O Catalytic asymmetric Diels Alder reactions. Chem. Rev 1992, 92, 1007.Corey EJ Catalytic Enantioselective Diels–Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications. Angew. Chem., Int. Ed 2002, 41, 1650.
- (4).For recent reviews about enantioselective arylcyclobutane syntheses, see:Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed 2015, 54, 3872.Xu Y; Conner ML; Brown MK Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2+2] Cycloadditions. Angew. Chem., Int. Ed 2015, 54, 11918.Poplata S; Tröster A; Zou Y-Q; Bach T Recent Advances in the Synthesis of Cyclobutanes by Olefin [2+2] Photocycloaddition Reactions. Chem. Rev 2016, 116, 9748.Fructos MR; Prieto A [2+2] Cycloaddition reactions promoted by group 11 metal-based catalysts. Tetrahedron 2016, 72, 355.Wang M; Lu P Catalytic approaches to assemble cyclobutane motifs in natural product synthesis. Org. Chem. Front 2018, 5, 254.Brenninger C; Jolliffe JD; Bach T Chromophore Activation of α,β-Unsaturated Carbonyl Compounds and Its Application to Enantioselective Photochemical Reactions. Angew. Chem., Int. Ed 2018, 57, 14338.
- (5).For recent examples utilizing transition metal catalysis, see:Kumar R; Tamai E; Ohnishi A; Nishimura A; Hoshimoto Y; Ohashi M; Ogoshi S Nickel-Catalyzed Enantioselective Synthesis of Cyclobutenes via [2+2] Cycloaddition of α,β-Unsaturated Carbonyls with 1,3-Enynes. Synthesis 2016, 48, 2789.Kossler D; Cramer N Neutral chiral cyclopentadienyl Ru(II)Cl catalysts enable enantioselective [2+2]-cycloadditions. Chem. Sci 2017, 8, 1862.
- (6).García-Morales C; Ranieri B; Escofet I; López-Suarez L; Obradors C; Konovalov AI; Echavarren AM Enantioselective Synthesis of Cyclobutenes by Intermolecular [2+2] Cycloaddition with Non-C2 Symmetric Digold Catalysts. J. Am. Chem. Soc 2017, 139, 13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Teller H; Flügge S; Goddard R; Fürstner A Enantioselective Gold Catalysis: Opportunities Provided by Monodentate Phosphoramidite Ligands with an Acyclic TADDOL Backbone. Angew. Chem., Int. Ed 2010, 49, 1949. [DOI] [PubMed] [Google Scholar]; (b) González AZ; Benitez D; Tkatchouk E; Goddard WA; Toste FD Phosphoramidite Gold(I)-Catalyzed Diastereo- and Enantioselective Synthesis of 3,4-Substituted Pyrrolidines. J. Am. Chem. Soc 2011, 133, 5500. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Teller H; Corbet M; Mantilli L; Gopakumar G; Goddard R; Thiel W; Fürstner A One-Point Binding Ligands for Asymmetric Gold Catalysis: Phosphoramidites with a TADDOL-Related but Acyclic Backbone. J. Am. Chem. Soc 2012, 134, 15331. [DOI] [PubMed] [Google Scholar]; (d) Suárez-Pantiga S; Hernández-Díaz C; Rubio E; González JM Intermolecular [2+2] Reaction of N-Allenylsulfonamides with Vinylarenes: Enantioselective Gold(I)-Catalyzed Synthesis of Cyclobutane Derivatives. Angew. Chem., Int. Ed 2012, 51, 11552. [DOI] [PubMed] [Google Scholar]; (e) Mauleón P Gold-Catalyzed Intermolecular Enantioselective [2+2] Cycloadditions of Sulfonylallenamides and Styrenes. ChemCatChem 2013, 5, 2149. [Google Scholar]; (f) Wang Y; Zhang P; Liu Y; Xia F; Zhang J Enantioselective gold-catalyzed intermolecular [2+2] versus [4+2]-cycloadditions of 3-styrylindoles with N-allenamides: observation of interesting substituent effects. Chem. Sci 2015, 6, 5564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Jia M; Monari M; Yang Q-Q; Bandini M Enantioselective gold catalyzed dearomative [2+2]-cycloaddition between indoles and allenamides. Chem. Commun 2015, 51, 2320. [DOI] [PubMed] [Google Scholar]
- (8).(a) Hu J-L; Feng L-W; Wang L; Xie Z; Tang Y; Li X Enantioselective Construction of Cyclobutanes: A New and Concise Approach to the Total Synthesis of (+)-Piperarborenine B. J. Am. Chem. Soc 2016, 138, 13151. [DOI] [PubMed] [Google Scholar]; (b) Kang T; Ge S; Lin L; Lu Y; Liu X; Feng X A Chiral N,N′-Dioxide–ZnII Complex Catalyzes the Enantioselective [2+2] Cycloaddition of Alkynones with Cyclic Enol Silyl Ethers. Angew. Chem., Int. Ed 2016, 55, 5541. [DOI] [PubMed] [Google Scholar]
- (9).(a) Ishihara K; Nakano K Enantioselective [2+2] Cyclo-addition of Unactivated Alkenes with α-Acyloxyacroleins Catalyzed by Chiral Organoammonium Salts. J. Am. Chem. Soc 2007, 129, 8930. [DOI] [PubMed] [Google Scholar]; (b) Talavera G; Reyes E; Vicario JL; Carrillo L Cooperative Dienamine/Hydrogen-Bonding Catalysis: Enantioselective Formal [2+2] Cycloaddition of Enals with Nitroalkenes. Angew. Chem., Int. Ed 2012, 51, 4104. [DOI] [PubMed] [Google Scholar]; (c) Albrecht Ł; Dickmeiss G; Acosta FC; Rodríguez-Escrich C; Davis RL; Jørgensen KA Asymmetric Organocatalytic Formal [2+2]-Cycloadditions via Bifunctional H-Bond Directing Dienamine Catalysis. J. Am. Chem. Soc 2012, 134, 2543. [DOI] [PubMed] [Google Scholar]; (d) Duan G-J; Ling J-B; Wang W-P; Luo Y-C; Xu P-F Organocatalytic formal [2+2] cycloaddition initiated by vinylogous Friedel–Crafts alkylation: enantioselective synthesis of substituted cyclobutane derivatives. Chem. Commun 2013, 49, 4625. [DOI] [PubMed] [Google Scholar]
- (10).(a) Guo H; Herdtweck E; Bach T Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions of Coumarins. Angew. Chem., Int. Ed 2010, 49, 7782. [DOI] [PubMed] [Google Scholar]; (b) Brimioulle R; Guo H; Bach T Enantioselective Intramolecular [2+2] Photocycloaddition Reactions of 4-Substituted Coumarins Catalyzed by a Chiral Lewis Acid. Chem. - Eur. J 2012, 18, 7552. [DOI] [PubMed] [Google Scholar]; (c) Brimioulle R; Bauer A; Bach T Enantioselective Lewis Acid Catalysis in Intramolecular [2+2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J. Am. Chem. Soc 2015, 137, 5170. [DOI] [PubMed] [Google Scholar]; (d) Tröster A; Alonso R; Bauer A; Bach T Enantioselective Intermolecular [2+2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation. J. Am. Chem. Soc 2016, 138, 7808. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Huang X; Quinn TR; Harms K; Webster RD; Zhang L; Wiest O; Meggers E Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2 + 2] Photocycloadditions. J. Am. Chem. Soc 2017, 139, 9120. [DOI] [PubMed] [Google Scholar]; (g) Miller ZD; Lee BJ; Yoon TP Enantioselective Crossed Photocycloadditions of Styrenic Olefins by Lewis Acid Catalyzed Triplet Sensitization. Angew. Chem., Int. Ed 2017, 56, 11891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hu N; Jung H; Zheng Y; Lee J; Zhang L; Ullah Z; Xie X; Harms K; Baik M-H; Meggers E Catalytic Asymmetric Dearomatization by Visible-Light-Activated [2+2] Photocycloaddition. Angew. Chem., Int. Ed 2018, 57, 6242. [DOI] [PubMed] [Google Scholar]
- (11).(a) Conner ML; Xu Y; Brown MK Catalytic Enantioselective Allenoate–Alkene [2+2] Cycloadditions. J. Am. Chem. Soc 2015, 137, 3482. [DOI] [PubMed] [Google Scholar]; (b) Xu Y; Hong YJ; Tantillo DJ; Brown MK Intramolecular Chirality Transfer [2+2] Cycloadditions of Allenoates and Alkenes. Org. Lett 2017, 19, 3703. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wiest JM; Conner ML; Brown MK Synthesis of (−)-Hebelophyllene E: An Entry to Geminal Dimethyl-Cyclobutanes by [2+2] Cycloaddition of Alkenes and Allenoates. Angew. Chem., Int. Ed 2018, 57, 4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Snider BB; Spindell DK Lewis acid catalyzed [2+2] cycloaddition of methyl 2,3-butadienoate to alkenes. J. Org. Chem 1980, 45, 5017. [Google Scholar]; (b) Hoffmann HMR; Ismail ZM; Weber A Synthesis of cyclobutylideneacetic esters via aluminum chloride promoted [2+2] cycloadditions of ethyl 2,3-butadienoate to olefins. Tetrahedron Lett. 1981, 22, 1953. [Google Scholar]; (c) Snider BB; Ron E Lewis acid catalyzed inter- and intramolecular [2+2] cycloadditions of conjugated allenic esters to alkenes. J. Org. Chem 1986, 51, 3643. [Google Scholar]; (d) Conner ML; Brown MK Synthesis of 1,3-Substituted Cyclobutanes by Allenoate-Alkene [2+2] Cycloaddition. J. Org. Chem 2016, 81, 8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For recent reviews on the enantioselective generation of quaternary carbon centers, see:Quasdorf KW; Overman LE Catalytic enantioselective synthesis of quaternary carbon stereo-centres. Nature 2014, 516, 181.Minko Y; Marek I Stereodefined acyclic trisubstituted metal enolates towards the asymmetric formation of quaternary carbon stereocentres. Chem. Commun 2014, 50, 12597.Feng J; Holmes M; Krische MJ Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev 2017, 117, 12564.
- (14).(a) Canales E; Corey EJ Highly Enantioselective [2+2]-Cycloaddition Reactions Catalyzed by a Chiral Aluminum Bromide Complex. J. Am. Chem. Soc 2007, 129, 12686. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S; Scopton AP; Corey EJ Enantioselective Pathway for the Synthesis of Laurenditerpenol. Org. Lett 2010, 12, 1836. [DOI] [PubMed] [Google Scholar]
- (15).(a) Corey EJ; Lee TW The formyl C-H⋯O hydrogen bond as a critical factor in enantioselective Lewis-acid catalyzed reactions of aldehydes. Chem. Commun 2001, 1321. [Google Scholar]; (b) Paddon-Row MN; Anderson CD; Houk KN Computational Evaluation of Enantioselective Diels–Alder Reactions Mediated by Corey’s Cationic Oxazaborolidine Catalysts. J. Org. Chem 2009, 74, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For seminal work, see:Gompper R Cycloadditions with Polar Intermediates. Angew. Chem., Int. Ed. Engl 1969, 8, 312.Dewar MJS Aromaticity and Pericyclic Reactions. Angew. Chem., Int. Ed. Engl 1971, 10, 761.Zimmerman HE Moebius-Hueckel concept in organic chemistry. Application of organic molecules and reactions. Acc. Chem. Res 1971, 4, 272.Wang X; Houk KN Carbenoid character in transition structures for reactions of ketenes with alkenes. J. Am. Chem. Soc 1990, 112, 1754.
- (17).Enantiopurity of the carboxylic acid was determined through derivatization to the corresponding methyl ester and subsequent HPLC analysis.
- (18).Hansch C; Leo A; Taft RW A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev 1991, 91, 165. [Google Scholar]
- (19).For inspiring studies on related systems, see:Santiago CB; Guo J-Y; Sigman MS Predictive and mechanistic multivariate linear regression models for reaction development. Chem. Sci 2018, 9, 2398.
- (20).For an excellent review on Lewis acid-controlled selectivity, see:Yamamoto H From designer Lewis acid to designer Brønsted acid towards more reactive and selective acid catalysis. Proc. Jpn. Acad., Ser. B 2008, 84, 134.
- (21).For recent examples, see:Jung ME; Murakami M Total Synthesis of (±)-Hedychenone: Trimethyldecalin Terpene Systems via Stepwise Allenoate Diene Cycloaddition. Org. Lett 2006, 8, 5857.Jung ME; Murakami M Total Synthesis of (±)-Hedychilactone B: Stepwise Allenoate Diene Cycloaddition To Prepare Trimethyldecalin Systems. Org. Lett 2007, 9, 461.Jung ME; Cordova J; Murakami M Total Synthesis of (±)-Kellermanoldione: Stepwise Cycloaddition of a Functionalized Diene and Allenoate. Org. Lett 2009, 11, 3882.Henderson JR; Chesterman JP; Parvez M; Keay BA Diastereoselective Diels–Alder Reactions with an Allenyl-Containing Spiro-Amido Chiral Auxiliary. J. Org. Chem 2010, 75, 988.
- (22).In thermal cycloadditions, an initial [2+2] cycloaddition followed by a 3,3-sigmatropic rearrangement to form the [4+2] products has been suggested (ref 21a–c). However, [2+2] cyclo-adducts did in our case show no rearrangement to the respective Diels–Alder products when treated with EtAlCl2.
- (23).(a) Pappo R; Allen DS Jr.; Lemieux RU; Johnson WS Notes - Osmium Tetroxide-Catalyzed Periodate Oxidation of Olefinic Bonds. J. Org. Chem 1956, 21, 478. [Google Scholar]; (b) Yu W; Mei Y; Kang Y; Hua Z; Jin Z Improved Procedure for the Oxidative Cleavage of Olefins by OsO4–NaIO4. Org. Lett 2004, 6, 3217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.