

In this study, Inoue et al. investigated the regulatory mechanisms and functions of the maternal H3K27me3 mechanism. They found that maternal Eed, an essential component of the Polycomb group complex 2 (PRC2), is required for establishing H3K27me3 imprinting, and their results also reveal unique XCI dynamics in the absence of Xist imprinting.
Keywords: X chromosome inactivation, genomic imprinting, Polycomb, H3K27me3, mouse early development
Abstract
Genomic imprinting is essential for mammalian development. Recent studies have revealed that maternal histone H3 Lys27 trimethylation (H3K27me3) can mediate DNA methylation-independent genomic imprinting. However, the regulatory mechanisms and functions of this new imprinting mechanism are largely unknown. Here we demonstrate that maternal Eed, an essential component of the Polycomb group complex 2 (PRC2), is required for establishing H3K27me3 imprinting. We found that all H3K27me3-imprinted genes, including Xist, lose their imprinted expression in Eed maternal knockout (matKO) embryos, resulting in male-biased lethality. Surprisingly, although maternal X-chromosome inactivation (XmCI) occurs in Eed matKO embryos at preimplantation due to loss of Xist imprinting, it is resolved at peri-implantation. Ultimately, both X chromosomes are reactivated in the embryonic cell lineage prior to random XCI, and only a single X chromosome undergoes random XCI in the extraembryonic cell lineage. Thus, our study not only demonstrates an essential role of Eed in H3K27me3 imprinting establishment but also reveals a unique XCI dynamic in the absence of Xist imprinting.
Since the discovery of genomic imprinting in mammals, differential DNA methylation had been the only reported mechanism underlying imprinted gene expression (Ferguson-Smith 2011). DNA methylation-dependent imprinting is established during gametogenesis by DNA methyltransferase 3a (Dnmt3a) and Dnmt3L (Bourc'his et al. 2001; Kaneda et al. 2004); maintained by Dnmt1 and its associated protein, Uhrf1 (Howell et al. 2001; Sharif et al. 2007; Hirasawa et al. 2008); and erased in primordial germ cells by Tet1 coupled with DNA replication (Kagiwada et al. 2013; Yamaguchi et al. 2013). Loss of imprinting causes embryonic lethality and imprinting diseases, such as Angelman syndrome and Beckwith-Wiedemann syndrome (Lim and Maher 2010). To date, the regulatory mechanisms and functions of DNA methylation-dependent genomic imprinting have been well studied.
We discovered recently that oocyte-specific histone H3 Lys27 trimethylation (H3K27me3) can mediate genomic imprinting in a DNA methylation-independent manner (Inoue et al. 2017a). A few studies had reported previously that several paternally expressed imprinted genes (PEGs) located outside the known imprinting clusters maintain their imprinted expression in maternal Dnmt mutants (Chiba et al. 2008; Okae et al. 2012, 2014), suggesting the existence of an oocyte DNA methylation-independent imprinting mechanism. Through an integrative analysis of DNaseI-seq (DNaseI sequencing), DNA methylome, and H3K27me3 ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data sets in gametes and preimplantation embryos followed by transient overexpression of Kdm6b, an H3K27me3-specific demethylase, we revealed that maternal H3K27me3 is required for maternal allele-specific repression of dozens of PEGs that include all imprinted genes previously known to be independent of oocyte DNA methylation (Inoue et al. 2017a). We identified ∼28 PEGs whose maternal alleles are repressed by H3K27me3 in morula embryos. At least five of these genes (Gab1, Phf17/Jade1, Sfmbt2, Smoc1, and Slc38a4) maintain their imprinted expression in the extraembryonic ectoderm (ExE) at embryonic day 6.5 (E6.5) and placentae at E9.5 (Inoue et al. 2017a). Despite the discovery that maternal H3K27me3 can mediate genomic imprinting, it is largely unknown how this imprinting is established and maintained and what the functions of this imprinting are.
In preimplantation embryos of certain female mammals, such as mice and marsupials, the paternal X chromosome (Xp) is selectively inactivated, which is named imprinted X-chromosome inactivation (iXCI) (Okamoto et al. 2005). iXCI is maintained in the extraembryonic cell lineage throughout placental development (Takagi and Sasaki 1975). To achieve iXCI, Xist, whose transcripts are critical for XCI, is exclusively expressed from the Xp (Marahrens et al. 1997; Okamoto et al. 2005). We showed previously that such a paternal allele-specific Xist expression is due to the maternal H3K27me3-mediated Xist silencing (Inoue et al. 2017b). Kdm6b-assisted removal of maternal H3K27me3 induced derepression of Xist from the maternal X chromosome (Xm), leading to Xm-chromosome inactivation (XmCI) in preimplantation embryos (Inoue et al. 2017b). However, it is unknown how loss of Xist imprinting affects subsequent development and how XCI is regulated in the absence of Xist imprinting.
To address these questions, we depleted maternal H3K27me3 by generating an oocyte-specific Eed knockout mouse model and created Eed maternal knockout (matKO) embryos by fertilizing Eed knockout oocytes with wild-type sperms. We found that loss of maternally inherited H3K27me3 results in biallelic expression of all H3K27me3-mediated imprinted genes, but not canonical DNA methylation-mediated imprinted genes, in preimplantation embryos and ExEs. Although XmCI occurs in Eed matKO preimplantation embryos due to Xist derepression from the Xm, it is largely restored at the peri-implantation stage, resulting in survival of half of the embryos.
Results
Eed matKO causes loss of H3K27me3 imprinting
Analyses of public H3K27me3 ChIP-seq data sets (Liu et al. 2016; Zheng et al. 2016) revealed that all maternal H3K27me3-dependent imprinted genes are coated with H3K27me3 domains in fully grown GV stage oocytes (FGOs) and metaphase II stage (MII) oocytes (Supplemental Fig. S1). These H3K27me3 domains exhibit extremely low DNaseI-seq signals (Supplemental Fig. S1), implicating heterochromatin formation in oocytes. Importantly, H3K27me3 domains are not evident in 7-d-old growing oocytes but appear in 14-d-old growing oocytes (Supplemental Fig. S1), suggesting that H3K27me3 imprinting is established during oocyte growth.
To prevent de novo establishment of H3K27me3 during oocyte growth, we generated an oocyte-specific conditional knockout (CKO) of Eed, an essential component of Polycomb group complex 2 (PRC2) (Cao et al. 2002), by crossing an Eed flox (fl) mouse line (Xie et al. 2014) with an Gdf9Cre transgenic mouse line (Lan et al. 2004). The Gdf9Cre is an oocyte-specific Cre line expressed in early growing oocytes of mice around postnatal day 3 (Lan et al. 2004). In this study, Eedfl/fl and Gdf9Cre/Eedfl/fl females serve as wild type and CKO, respectively. Immunostaining analysis revealed that H3K27me3 is markedly decreased in both surrounded nucleolus (SN) and nonsurrounded nucleolus (NSN) types of Eed knockout FGOs (Supplemental Fig. S2A,B). The numbers of MII oocytes obtained from Eed CKO and wild-type females following superovulation were comparable (Supplemental Fig. S2C), indicating that loss of H3K27me3 does not affect oocyte growth or maturation.
Next, we analyzed the global H3K27me3 level in Eed matKO preimplantation embryos that were derived from Eed knockout oocytes fertilized with wild-type sperm. Immunostaining revealed that the H3K27me3 signal is significantly lower than that in control (CTR) embryos through the eight-cell stage but becomes comparable at the morula stage (Fig. 1A,B). This suggests that Eed may be zygotically expressed from the wild-type paternal allele, contributing to de novo deposition of H3K27me3 in Eed matKO embryos after the eight-cell stage. Thus, Eed matKO causes global loss of H3K27me3 during oogenesis and before the eight-cell stage. Nevertheless, Eed matKO embryos could develop to the blastocyst stage at a rate comparable with CTR embryos (Supplemental Fig. S2D,E), indicating that loss of H3K27me3 during this time window does not compromise preimplantation development.
Figure 1.

Maternal Eed knockout induces loss of H3K27me3 imprinting. (A) Representative images of zygotes and preimplantation embryos immunostained with an anti-H3K27me3 antibody. (M) Maternal pronucleus; (P) paternal pronucleus. Bar, 20 µm. (B) Quantification of the H3K27me3 signal intensity. The average signal intensity of CTR embryos was set as 1.0. The total numbers of embryos examined were eight CTR and 12 matKO for one-cell embryos, six CTR and five matKO for two-cell embryos, seven CTR and six matKO for four-cell embryos, nine CTR and nine matKO for eight-cell embryos, and 10 CTR and eight matKO for morula embryos, respectively. Error bars indicate SD. (***) P < 0.001, two-tailed Student t-test. (C) Scheme for assessing allele-specific gene expression and H3K27me3 enrichment in morula embryos. (+) Eed wild-type allele; (–) Eed knockout allele. (D) Heat map showing the allelic expression bias of H3K27me3-dependent and known DNA methylation-dependent PEGs in morula embryos with biological duplicates. Genes with >20 single-nucleotide polymorphism (SNP) reads in both replicates are shown. (E) Genome browser views showing loss of maternal H3K27me3 domains at the representative H3K27me3-dependent imprinted loci in CTR and Eed matKO morula embryos. (P) Paternal allele; (M) maternal allele.
To investigate whether Eed matKO causes loss of H3K27me3 imprinting, we generated hybrid zygotes using PWK strain sperm and performed RNA sequencing (RNA-seq) analysis on the embryos at the morula stage (Fig. 1C). The biological replicates of RNA-seq data sets were highly reproducible in both CTR and Eed matKO morula embryos (Supplemental Fig. S3A; Supplemental Table S1). A comparative analysis revealed that only 19 (0.18%) and 21 (0.20%) genes were up-regulated or down-regulated (fold change > 2; P < 0.05) (Supplemental Fig. S3B; Supplemental Table S1), suggesting that loss of maternal H3K27me3 had a modest effect on the whole transcriptome of morula embryos, consistent with a similar developmental rate (Supplemental Fig. S2D). Single-nucleotide polymorphism (SNP)-based analysis confirmed that Eed transcripts were all derived from the paternal allele in Eed matKO embryos (Supplemental Fig. S3C), validating efficient deletion of the maternal Eed allele. Among the 28 putative H3K27me3-dependent PEGs in morula embryos (Inoue et al. 2017a), 14 had enough SNP-containing reads in both replicates (>20 SNP reads) to allow us to reliably evaluate their allele specificity. Notably, the paternal allele expression bias of all of the 14 PEGs either was lost or became milder in Eed matKO embryos (Fig. 1D; Supplemental Fig. S3D; Supplemental Table S2). In contrast, all of the DNA methylation-dependent PEGs containing >20 SNP reads maintained their paternal allele-specific expression (Fig. 1D; Supplemental Table S2). These results demonstrate that Eed matKO causes loss of maternal H3K27me3-dependent imprinting, while DNA methylation-dependent imprinting is not affected.
To confirm that maternal H3K27me3 imprinting domains are lost in Eed matKO morula embryos, we performed a H3K27me3 Cleavage Under Targets and Release Using Nuclease (CUT&RUN) assay (Skene and Henikoff 2017) in CTR and Eed matKO morula embryos. The H3K27me3 distribution in CTR morula embryos was highly correlated with that of a public H3K27me3 ChIP-seq data set (Supplemental Fig. S4A,B; Matoba et al. 2018), validating that our CUT&RUN assay worked efficiently. As expected, the number of H3K27me3 peaks in Eed matKO embryos was fewer than that of CTR embryos (Supplemental Fig. S4C). Importantly, all of the maternal H3K27me3 domains present at the 14 H3K27me3-dependent PEGs as well as the Xist locus in CTR embryos were completely lost in the Eed matKO embryos (Fig. 1E; Supplemental Fig. S4D).
Biallelic XCI in Eed matKO morula embryos
To examine whether Eed matKO leads to Xist derepression from the Xm, we performed RNA fluorescent in situ hybridization (FISH) analysis in morula embryos. To distinguish between male and female embryos, we simultaneously labeled X chromosomes by DNA FISH using a BAC probe containing the X-linked Rnf12 locus. In this manner, males and females had one or two DNA FISH signals, respectively (Fig. 2A). As expected, this analysis revealed that female and male CTR embryos show one and zero Xist RNA clouds, respectively (Fig. 2A,B). In contrast, female and male Eed matKO embryos showed two and one RNA clouds, respectively (Fig. 2A,B), indicating that Xist is derepressed from the Xm in both male and female Eed matKO embryos.
Figure 2.

Loss of maternal H3K27me3 induces XmCI. (A) Representative images of Xist RNA FISH in female (XX) and male (XY) morula embryos. The gender of each embryo was assessed by simultaneous DNA FISH against the Rnf12 locus (green spots). (Blue) DAPI. Bars: merged images, 20 µm; enlarged images, 5 µm. (B) The ratio of blastomeres showing the indicated number of Xist RNA clouds. Each bar represents an individual embryo. The numbers of embryos examined were 25 CTR and 12 matKO for females and 16 CTR and 18 matKO for males. (C) Box plot showing the relative expression of genes on individual maternal chromosomes between CTR and Eed matKO morula embryos. Genes with enough SNP reads (SNP reads per kilobase per million mapped reads [RPKM] >0.5) were analyzed. The middle lines in the boxes represent the medians. Box edges and whiskers indicate the 25th/75th and 2.5th/97.5th percentiles, respectively. (***) P < 1.2 × 10−23, Mann-Whitney-Wilcoxon test. (D) The relative expression levels of X-linked genes between CTR and Eed matKO morula embryos. The expression levels of the maternal allele were analyzed. Each dot represents an individual gene with enough SNP reads (SNP RPKM >0.5). Known escapees were excluded. Dashed lines indicate a differential expression change of twofold.
To determine whether Xist derepression from the Xm leads to XmCI, we reanalyzed the RNA-seq data sets of Eed matKO morula embryos (Supplemental Table S1,S2). Since these RNA-seq libraries were mixtures of male and female embryos, only reads containing Xm-associated SNPs were analyzed to determine the XmCI status. We found that the overall expression level of Xm-linked genes, but not that of autosomes, is greatly down-regulated in Eed matKO embryos (Fig. 2C). The maternal expression bias [Mat/(Mat + Pat)] of the X-linked genes was significantly decreased in Eed matKO embryos (Supplemental Fig. S5A), indicating that XmCI occurred in Eed matKO embryos. Among 372 SNP-trackable Xm-linked genes, 132 (35%) were down-regulated in Eed matKO embryos, while only 24 (6.4%) were up-regulated (fold change > 2) (Fig. 2D). Interestingly, X-linked genes repressed early in iXCI tend to be more down-regulated compared with those silenced later (Supplemental Fig. S5B; Borensztein et al. 2017b), and none of the known escapees were down-regulated (Supplemental Fig. S5B,C), suggesting that XmCI somewhat recapitulates the physiological XCI. To examine the putative role of H3K27me3 in XCI at the morula stage, we closely examined nonescapee genes by analyzing our CUT&RUN and previous H3K27me3 ChIP-seq data sets (Inoue et al. 2017b; Matoba et al. 2018). Surprisingly, little H3K27me3 was enriched at the early silenced genes (e.g., Chic1 and Rlim) on the paternal allele in CTR morula of both parental alleles in Eed matKO embryos (Supplemental Fig. S5D), suggesting that initial repression of X-linked genes might be independent of H3K27me3. Taken together, these data demonstrate that Eed matKO causes XmCI in preimplantation embryos.
Male-biased lethality in Eed matKO embryos
Having demonstrated that Eed matKO causes loss of maternal H3K27me3-dependent imprinting in preimplantation embryos, we next assessed their effect on fertility. Litter size analysis of wild-type and CKO females mated with B6 males revealed that CKO females delivered some pups that were able to grow up to the weaning age and looked grossly normal (Table 1). However, the average litter size from CKO females (4.6 ± 0.7) was significantly smaller than that of wild-type females (8.7 ± 2.3; P < 0.0001, Student's t-test), and 71% of the survivors were females (Table 1), indicating that Eed matKO embryos show male-biased lethality.
Table 1.
Mating score of Eed CTR and matKO embryos

To determine when some of the Eed matKO embryos start to die during post-implantation development, we dissected E6.5 embryos from wild-type and CKO females mated with B6 males. The average number of embryos obtained from CKO females (6.1 ± 2.0) was 27% lower than wild-type females (8.4 ± 1.5), and all Eed matKO embryos showed growth retardation (Supplemental Fig. S6A). Consistent with the sex ratio at term, 65% of Eed matKO E6.5 embryos were females (Supplemental Fig. S6B). The male-biased lethality was reproducible when PWK males were used as the sires (Supplemental Fig. S6B).
We next dissected E10.5 embryos from wild-type and CKO females. The average number of embryos obtained from CKO females (5.2 ± 2.2) was 33% lower than that from wild-type females (7.8 ± 1.8). The Eed matKO survivors were again biased to females (73%) (Supplemental Fig. S7A). The gross morphologies of the survivors were normal, but the average embryonic and placental sizes were slightly smaller than that of CTR embryos (Supplemental Fig. S7B–D). Thus, analyses of Eed matKO post-implantation embryos revealed that male-biased lethality is already evident by E6.5, some embryos die even after E6.5, and, eventually, about half embryos survive to term.
Eed matKO causes loss of H3K27me3 imprinting in E6.5 embryos
To gain insights into potential causes of the lethality and assess the imprinting status of maternal H3K27me3-dependent imprinted genes in post-implantation Eed matKO embryos, we performed RNA-seq analysis in hybrid E6.5 ExEs, a cell lineage known to maintain maternal H3K27me3-dependent imprinting (Inoue et al. 2017a). The biological replicates of the ExE samples showed a high correlation (Supplemental Fig. S8A; Supplemental Table S3), and their ExE cell identity was validated by assessing cell lineage-specific marker gene expression (Supplemental Fig. S8B; Blakeley et al. 2015). Comparative transcriptome analyses revealed that, among the 10,120 reliably detectable genes (mean reads per kilobase per million mapped reads [RPKM] > 1), 33 (0.33%) and 61 (0.60%) genes are up-regulated and down-regulated (fold change > 2; P < 0.05), respectively, in Eed matKO females (Supplemental Fig. S8C). The number of differentially expressed genes (DEGs) in females was smaller than that of males in which 34 (0.34%) and 144 (1.4%) genes were up-regulated or down-regulated, respectively (fold change > 2; P < 0.05) (Supplemental Fig. S8C). Among them, four and 20 genes were commonly up-regulated and down-regulated, respectively, in both sexes (Supplemental Fig. S8D,E). Although gene ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis did not identify any significant enrichment in DEGs, the down-regulated DEGs include Cited1 (Cbp/p300-interacting transactivator with Glu/Asp-rich C-terminal domain 1) (Rodriguez et al. 2004) in both sexes and Acvr1 (Activin A receptor type 1) (Gu et al. 1999), Slc40a1 (solute carrier family 40 member 1) (Donovan et al. 2005), and Tbx20 (T-box 20) (Singh et al. 2005) in males, whose knockout results in early embryonic lethality, suggesting that down-regulation of these genes might contribute to the growth retardation and/or lethality of Eed matKO embryos.
We next examined the imprinting status in Eed matKO ExEs. Since five maternal H3K27me3-dependent autosomal PEGs maintain their imprinted expression state in E6.5 ExEs (Inoue et al. 2017a), we analyzed allelic expression of these five genes as well as the known DNA methylation-dependent imprinted genes. Consistent with our previous finding, all of the five genes—except Phf17, whose paternal expression bias is weak in the B6/PWK hybrid background (Inoue et al. 2017a)—exhibit paternal-biased expression in almost all CTR embryos examined (Fig. 3A). Importantly, Eed matKO resulted in complete loss or weaker paternal-biased expression (Fig. 3A). In contrast, all of the known DNA methylation-dependent imprinted genes largely maintained their imprinted expression state in Eed matKO ExEs (Fig. 3B,C). It is also notable that all maternally expressed imprinted genes (MEGs), including the ones in the Kcnq1ot1 cluster, such as Ascl2, Cdkn1, Osbpl5, Phlda2, Slc22a18, and Tssc4, maintained their imprinting in Eed matKO ExEs (Fig. 3C). Given that imprinting of these genes is maintained by H3K27me3 established de novo during early development (Lewis et al. 2004; Umlauf et al. 2004; Terranova et al. 2008), the above results support the notion that zygotically expressed Eed is fully functional in Eed matKO embryos. Taken together, these data demonstrate that Eed matKO causes specific loss of maternal H3K27me3-dependent imprinting, but not DNA methylation-dependent canonical imprinting, in E6.5 ExEs.
Figure 3.
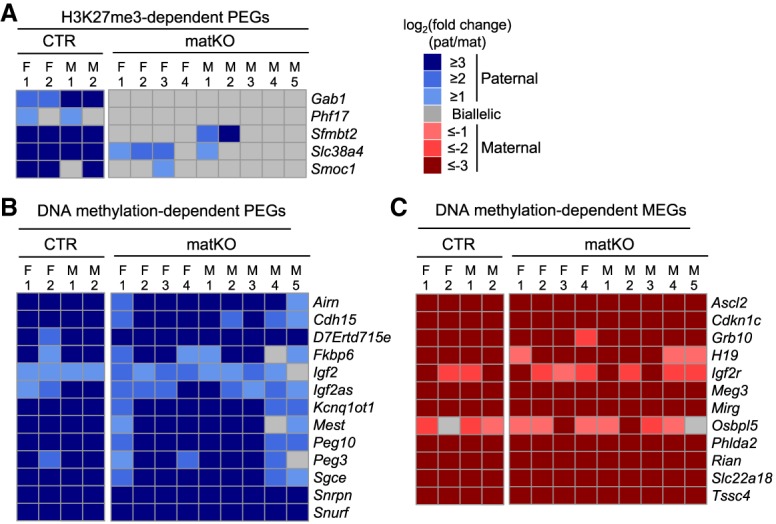
Loss of H3K27me3 imprinting in E6.5 Eed matKO embryos. (A) Heat map showing the allelic expression bias of H3K27me3-dependent PEGs in ExE at E6.5 embryos. The CTR samples include two females (F) and two males (M). The Eed matKO samples include four females and five males. (B,C) Heat maps showing the allelic expression bias of known DNA methylation-dependent PEGs (B) and MEGs (C) in ExE of E6.5 embryos. Genes with >20 SNP reads are shown.
XCI is restored in Eed matKO post-implantation embryos
Given that aberrant XmCI is incompatible with post-implantation development (Lee 2000; Sado et al. 2001), it is surprising to see that some Eed matKO embryos survive to term. Thus, it is possible that the aberrant XmCI at preimplantation is restored at a later developmental stage. Because gene expression between CTR and matKO E6.5 ExEs showed only a modest difference (Supplemental Fig. S8C), we hypothesized that aberrant XmCI has already been restored by E6.5. In support of this notion, analysis of the RNA-seq data sets revealed that down-regulation of X-linked genes in matKO embryos at E6.5 ExEs is much less pronounced than in the morula stage (Fig. 4A). Consistently, among the 370 expressed X-linked genes (RPKM > 1), only 29 (7.8%) and 41 (11%) were down-regulated (fold change > 2) in females and males, respectively (Supplemental Fig. S9A). These percentages were greatly smaller than that in morula embryos (35%) (Fig. 2D). Consistently, RNA FISH analysis revealed a single and no Xist RNA cloud in females and males, respectively, in both CTR and matKO ExEs (Fig. 4B,C). In addition, immuno-FISH analysis using an anti-H3K27me3 antibody confirmed that the H3K27me3 spot, a marker of inactive X chromosome (Xi), colocalized with the Xist RNA cloud in Eed matKO ExEs (Supplemental Fig. S9B). These results indicate that aberrant XmCI observed in matKO morula embryos is largely restored by E6.5.
Figure 4.

Aberrant XCI is restored in E6.5 Eed matKO embryos. (A) Box plot showing the relative expression of autosomes and X-linked genes between CTR and Eed matKO of morula embryos and E6.5 ExEs. The middle lines in the boxes represent the medians. Box edges and whiskers indicate the 25th/75th and 2.5th/97.5th percentiles, respectively. (***) P < 1.2 × 10−44; (**) P < 5.5 × 10−18; (*) P < 4.1 × 10−7, Mann-Whitney-Wilcoxon test. (B) Representative images of Xist RNA FISH in ExEs of female (XX) and male (XY) E6.5 embryos. (Blue) DAPI. Bars: merged images, 20 µm; enlarged images, 5 µm. (Xa) Active X chromosome. (C) The ratio of blastomeres showing the indicated number of Xist RNA clouds. Each bar represents an individual embryo. The numbers of embryos examined were nine CTR and 11 matKO for females and 16 CTR and seven matKO for males. (D) Heat map showing the allelic expression bias of X-linked genes in ExEs of female E6.5 embryos. CTR and Eed matKO groups contained two and four ExE samples, respectively. Genes with >20 SNP reads are shown. The right panel indicates a zoomed-in sector of Xist to demonstrate the anti-correlation of allelic expression of Xist and the other X-linked genes. The numbers inside the Xist row represent the actual values of log2 fold change (paternal/maternal).
To determine whether XCI is restored in an imprinted or a random manner, we analyzed allelic bias of X-linked gene expression in the female E6.5 ExE samples (Supplemental Table S4). In CTR, most of the 241 X-linked genes with enough SNP reads (>20) exhibited heavy maternal bias due to imprinted XpCI (Fig. 4D). In contrast, each ExE sample of matKO embryos showed variable parental biases of X-linked gene expression. Out of the four female embryos analyzed, one showed biallelic expression (#1), two showed maternally biased expression at a level lower than CTR (#2 and #3), and one showed paternally biased expression (#4) (Fig. 4D). As expected, the parental bias of X-linked gene expression was anti-correlated with that of Xist expression (Fig. 4D). These data suggest that XCI in ExEs of Eed matKO embryos likely occurs in a random manner.
Dynamics of XCI in Eed matKO peri-implantation embryos
To determine when and how XCI is restored in Eed matKO embryos, we performed Xist RNA FISH analysis in peri-implantation stage blastocyst embryos at E3.5 and E4.0. Simultaneous immunostaining using an anti-Nanog antibody can distinguish between Nanog-positive cells that mostly give rise to epiblast precursor (pre-Epi) cells and Nanog-negative cells that mostly give rise to trophectoderm (TE) and primitive endoderm (PrE) cells (Borensztein et al. 2017a). In CTR female embryos, the Xist RNA cloud was lost in Nanog-positive cells by E4.0, reflecting X reactivation in pre-Epi cells, while it was maintained in Nanog-negative cells (Fig. 5A,B). In Eed matKO female embryos that originally showed two Xist RNA clouds at the morula stage (Fig. 2A,B), the population of Nanog-positive cells showing two clouds was gradually decreased, and the majority of them showed no cloud at E4.0 (Fig. 5A,B). This suggests that both X chromosomes are reactivated in Nanog-positive cells of Eed matKO embryos. Interestingly, while the population of Nanog-negative cells showing two clouds was gradually decreased (as seen in Nanog-positive cells), the majority showed a single RNA cloud at E4.0 (Fig. 5A,B). Thus, while Nanog-positive cells reactivate both X chromosomes, only a single X chromosome is reactivated in the extraembryonic lineage cells of Eed matKO embryos.
Figure 5.

Aberrant XCI is restored in Eed matKO peri-implantation embryos. (A) Representative images of Xist RNA FISH (magenta) in female (XX) E3.5 and E4.0 embryos. (Green) Immunostaining signal of Nanog, a marker of pre-Epi cells; (blue) DAPI. Bars: merged images, 20 µm; enlarged images, 5 µm. (B) The ratio of blastomeres showing the indicated number of Xist RNA clouds. Each bar represents an individual embryo. The numbers of embryos examined were 13 CTR and eight matKO for E3.5 and nine CTR and nine matKO for E4.0. (C) Representative images of Xist RNA FISH in male (XY) E3.5 and E4.0 embryos. (D) The ratio of blastomeres showing the indicated number of Xist RNA clouds. Each bar represents an individual embryo. The numbers of embryos examined were eight CTR and eight matKO for E3.5 and 15 CTR and 11 matKO for E4.0.
In males, no CTR embryo showed an Xist RNA cloud at E3.5 or E4.0, as expected (Fig. 5C,D). In Eed matKO male embryos that had an Xist RNA cloud at the morula stage (Fig. 2A,B), about half of the blastomeres showed an RNA cloud in both Nanog-positive and Nanog-negative cells at E3.5, and the great majority of cells lost the clouds by E4.0 (Fig. 5C,D). This result suggests that X reactivation can also occur in males. Consistent with the Xist RNA FISH data, immunostaining of H3K27me3 revealed that 0% (CTR) and 5%–13% (Eed matKO) of blastomeres in female E3.5 embryos show two H3K27me3 foci, while 0%–5% (CTR) and 13%–38% (Eed matKO) of male embryos had one H3K27me3 foci (Supplemental Fig. S10A,B). Despite the aberrant XmCI, TUNEL assay in E3.5 Eed matKO embryos revealed that no blastomeres undergo apoptosis (Supplemental Fig. S10C), which argues against a possibility that blastomeres with aberrant XmCI die out and only the other blastomeres survive and dominate the Eed matKO embryos. Taken together, these results suggest that aberrant XmCI observed in Eed matKO morula embryos is largely restored at the peri-implantation stage in both females and males.
Discussion
H3K27me3 imprinting establishment
In this study, we revealed that Eed is essential for establishment of H3K27me3 imprinting domains during oocyte growth. After fertilization with wild-type sperm, Eed matKO embryos exhibit biallelic expression of maternal H3K27me3-dependent imprinted genes, while the monoallelic expression of canonical DNA methylation-dependent imprinted genes was not affected. This clearly demonstrates that these two types of imprinting are established independently during oocyte growth. Future studies should address how DNMTs and PRC2 select their targets and why two independent imprinting mechanisms are needed.
XCI dynamics in the absence of Xist imprinting
In the absence of maternal Eed, Xist is derepressed from the Xm, resulting in biallelic XCI in females and XCI in males at the morula embryos (Fig. 6). However, the aberrant XmCI is largely restored by E4.0. The XCI restoration is unlikely to be explained by the dying out of blastomeres that undergo aberrant XmCI, as almost all blastomeres of Eed matKO embryos undergo XmCI at the morula stage (Fig. 2B), but the embryos did not show lethality or apoptosis at the blastocyst stage (Supplemental Figs. S2, S10B). Our RNA-seq analysis suggests that XCI in the ExEs of Eed matKO female embryos likely occurs in a random manner (Fig. 4D). This is supported by a previous study analyzing XCI status in XpXp and XpY androgenetic embryos (Okamoto et al. 2000). It was demonstrated that XpXp embryos undergo biallelic XCI at the morula stage and random XCI in the extraembryonic cells at E7.5 and that XpY embryos also undergo XCI at the morula stage but not in E6.5 embryos (Okamoto et al. 2000). In our study, we used the Eed matKO model to further show that loss of Xist imprinting results in male-biased lethality (discussed below) and that XCI is restored in a cell lineage-specific manner in female embryos (Fig. 6): While both X chromosomes are reactivated in the embryonic cell lineage, only a single X chromosome appears to be reactivated in the extraembryonic cell lineage. In males, the transiently inactivated X chromosome is largely reactivated by E4.0 in both embryonic and extraembryonic cell lineages (Fig. 6).
Figure 6.
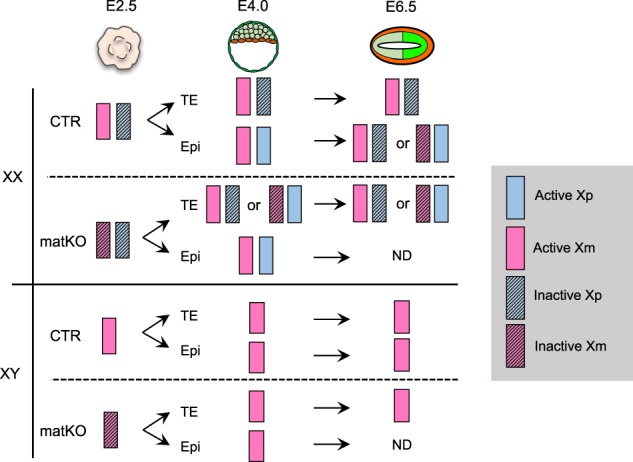
Model illustrating XCI dynamics in the absence of Xist imprinting. (Epi) Epiblast; (N.D.) “not determined” in this study.
Another interesting aspect of our study is that initial repression of X-linked genes does not seem to be associated with H3K27me3 (Supplemental Fig. S5D). It is possible that noncanonical PRC1-mediated H2A ubiquitylation may play an important role in the initiation of Xist-mediated iXCI, similar to the Pcgf3/5-PRC1-mediated XCI mechanism reported recently (Almeida et al. 2017). However, we note that this interpretation has a caveat, as our CUT&RUN and ChIP-seq samples were mixtures of male and female embryos, which caused an underestimation of H3K27me3 signals on the Xp. Thus, further analyses using only female embryos is needed.
X reactivation in the TE of Eed matKO embryos
In Eed matKO embryos, X reactivation occurred in not only Nanog-positive cells but also Nanog-negative cells (Figs. 5, 6). Since X reactivation does not take place in the TE lineage in CTR embryos, it occurs specifically in the Eed matKO context. How can this happen? We speculate that the feedback loop of Rnf12 dosage against XCI may contribute to the X reactivation. Rnf12 is a nonescapee X-linked gene and a dose-dependent trans-acting factor required for Xist expression, and its heterozygosity is known to impair initiation of XCI (Jonkers et al. 2009; Shin et al. 2010). Our RNA-seq revealed that Rnf12 is down-regulated by 5.3-fold in Eed matKO morula embryos due to XmCI (Supplemental Table S1), which is in line with our previous study demonstrating that Rnf12 expression is down-regulated by ∼10-fold in Kdm6b-injected morula embryos (Inoue et al. 2017b). Due to Rnf12 down-regulation, the Xi might be more easily reactivated in the Eed matKO embryos. As a result, X reactivation might be able to occur in TE cells where pluripotent factors implicated in Xist repression are not even expressed or are expressed at a low level (Navarro et al. 2008; Donohoe et al. 2009). The same mechanism could account for X reactivation in males.
Random X inactivation in extraembryonic cells of Eed matKO embryos
The Xi/Xi state appears to be turned into active X chromosome (Xa)/Xi without an obvious Xa/Xa intermediate in Nanog-negative cells of Eed matKO female embryos (Figs. 5, 6). The conversion from Xi/Xi to Xa/Xi could either directly take place (random X reactivation) or occur via rapid reactivation of both Xs followed by random XCI. Although further studies using live-cell imaging will be necessary to distinguish these possibilities, a similar XCI dynamic is also observed in other contexts. For example, a significant proportion of blastomeres of rabbit embryos at the early blastocyst stage exhibits an Xi/Xi state at the early blastocyst stage due to biallelic Xist expression (Okamoto et al. 2011). This is somehow resolved to become Xa/Xi at the late blastocyst stage without an obvious Xa/Xa intermediate (Okamoto et al. 2011). A recent study also demonstrated that both X chromosomes transiently adopt a partial Xi/Xi status and are resolved to an Xa/Xi state at the onset of differentiation when embryonic stem cells exit from a naive state (Sousa et al. 2018). Moreover, an Xi/Xi state artificially created by doxycycline (dox)-inducible Xist expression can also be converted to an Xa/Xi state upon dox removal (Mutzel et al. 2017). All of these observations suggest the presence of a random X-reactivation mechanism that directly converts Xi/Xi to Xa/Xi. Future studies are needed to understand this mechanism.
Male-biased lethality of Eed matKO embryos
Loss of maternal H3K27me3 causes developmental defects in males more frequently than in females. This is consistent with our RNA-seq data sets of E6.5 ExEs, showing that the number of DEGs is larger in males than in females (Supplemental Fig. S8C), and down-regulation of X-linked genes is more evident in males than in females (Supplemental Fig. S9A). Although Xist RNA FISH experiments did not detect an Xist RNA cloud in males at E6.5 (Fig. 4B,C), it is possible that aberrant XmCI during preimplantation development affects later development. One reason that females exhibit milder phenotypes than males could be that dysregulation of X-linked genes might be buffered in females due to the presence of two X chromosomes. Nevertheless, it remains possible that loss of imprinting of autosomal H3K27me3-dependent imprinted genes, rather than Xist, might contribute to the lethality of Eed matKO embryos. It will be important to distinguish between Xist-dependent and Xist-independent phenotypes of the Eed matKO embryos, which could reveal Xist-independent functions of H3K27me3 imprinting.
Evolution of Xist imprinting
It has been unclear whether Xist imprinting is required for mammalian development, since it does not seem to be conserved in most mammalian species other than rodents and marsupials (Mahadevaiah et al. 2009). In humans, for example, XIST is expressed from both the Xm and the Xp in females and from the Xm in males at the blastocyst stage. Nevertheless, XIST expression does not cause XCI (Okamoto et al. 2011), suggesting that XIST RNA function is repressed in human preimplantation embryos. The difference in Xist RNA function in mice and humans may explain the necessity of Xist imprinting that safeguards the Xm from the wave of XCI in mouse preimplantation embryos. Future studies are needed to reveal why XIST RNA expression does not induce XCI in human preimplantation embryos while it does in mouse embryos.
Materials and methods
Generation of oocyte-specific Eed knockout mice
All animal studies were performed in accordance with guidelines of the Institutional Animal Care and Use Committee at Harvard Medical School. The Eedflox (B6;129S1-Eedtm1Sho/J) and Gdf9Cre [Tg(Gdf9-icre)5092Coo/J] mouse lines were purchased from the Jackson Laboratory (022727 and 011062, respectively). The Eedflox females were crossed with the Gdf9Cre males to obtain Gdf9Cre, Eed+/flox males. The Gdf9Cre, Eed+/flox males were then crossed with Eed+/flox females to obtain Gdf9Cre, Eedflox/flox males and Eedflox/flox females. They were intercrossed to obtain Eedflox/flox (wild type) and Gdf9Cre, Eedflox/flox (CKO) females for experiments. The tail tips were used for genotyping. The primers detecting the flox allele were Eed-2f-F2 (5′-CTACCCTAGCTAAACACTGC-3′) and Eed-2f-20308 (5′-CTTGGGTGGTTTGGCTAAGA-3′; intact allele ∼160 base pairs [bp], flox allele ∼210 bp). The primers detecting the Gdf9Cre were Gdf9Cre-21218 (5′-CAGGTTTTGGTGCACAGTCA-3′) and Gdf9Cre-25494 (5′-GGCATGCTTGAGGTCTGATTAC-3′). For mating score tests, C57/BL6J (Jackson Laboratory, 000664) or PWK (Jackson Laboratory, 003715) strain males were used as the sires. The day of plug was counted as E0.5.
Collection of mouse oocytes and preimplantation embryos
The procedures of GV and MII oocyte collection and in vitro fertilization (IVF) were described previously (Inoue et al. 2017a). GV oocytes were obtained from PMSG-injected 8- to 12-wk-old females. MII oocytes were obtained from PMSG and hCG injected into 8- to 12-wk-old females. Sperm used for IVF were harvested from B6D2F1 males (BDF1) unless stated otherwise. To generate hybrid strain embryos, PWK strain males were used.
Whole-mount immunostaining
GV oocytes, one-cell zygotes, and two-cell, four-cell, eight-cell, and morula embryos were fixed with 3.7% paraformaldehyde containing 0.2% Triton X-100 for 20 min at room temperature at 10, 28, 46, 60, and 78 h post-fertilization (hpf) after removing the zona pellucida by a brief incubation in the acid Tyrode's solution (Sigma-Aldrich). After four washes with PBS containing 1% BSA (PBS/BSA), samples were treated with mouse anti-H3K27me3 antibody (1/500; Active Motif, 61017) overnight at 4°C. After washing with PBS/BSA, they were incubated with a 1:250 dilution of Alexa flour 488 donkey anti-mouse IgG (Life Technologies) for 1 h at room temperature. After washing with PBS/BSA, they were then mounted on a glass slide in VectaShield anti-bleaching solution with DAPI (Vector Laboratories). Fluorescence was detected under a laser-scanning confocal microscope (Zeiss, LSM800). The fluorescent signal intensity was quantified with Zen software (Zeiss). Briefly, the signal intensities within nuclei were determined, and the cytoplasmic signals were subtracted as the background. The average signal intensity of CTR embryos was set as 1.0.
Whole-mount RNA/DNA FISH
The procedures of probe making and the FISH experiment were described previously (Inoue et al. 2017b). Morula embryos were obtained by IVF with BDF1 sperm and fixed at 78 hpf. E3.5, E4.0, and E6.5 embryos were obtained by natural mating with BDF1 males. The day of plug was counted as E0.5. E3.5 and E4.0 embryos were flushed out from the uterus at noon of the third day, cultured in KSOM (Millipore), and fixed at noon (E3.5) or midnight (E4.0). For E6.5 embryos, ExE was dissected as described previously (Sugimoto et al. 2015). For some experiments, embryos were immunostained with rabbit anti-Nanog antibody (1/100; Cosmobio, RCAB002pF) or rabbit anti-H3K27me3 antibody (1/500; Millipore, 07449) after completion of the Xist RNA FISH procedure. The secondary antibodies were Alexa flour 488 donkey anti-rabbit IgG (Life Technologies) and Alexa flour 647 donkey anti-rabbit IgG (Life Technologies).
TUNEL assay
Embryos were first fixed in 3.7% paraformaldehyde and 0.2% Triton for 20 min at room temperature. After washing in 0.1% BSA/PBS, embryos were stained with TUNEL staining mix (9:1 label solution:enzyme solution) (Sigma-Aldrich) for 1 h at 37°C. For a positive CTR, embryos were first incubated in DNase I solution (3 U/mL DNaseI in 1× DNase I buffer) (Life Technologies) for 15 min at 37°C after the fixation. After incubating in STOP solution (0.1% BSA/PBS containing 15 mM EDTA) for 10 min at room temperature, the embryos were stained with the TUNEL mix. Embryo mounting and florescence detection were performed as described above.
RNA-seq
The hybrid morula embryos (78 hpf) for RNA-seq were obtained by IVF of MII oocytes with PWK sperm. Thirteen morula embryos per group per experiment were pooled for RNA-seq. Therefore, the morula embryo RNA-seq samples were supposed to be a mixture of male and female embryos. The hybrid E6.5 embryos were obtained by natural mating with PWK males. The day of plug was counted as E0.5. Dissection of ExE was performed as described previously (Sugimoto et al. 2015). RNA-seq libraries were prepared as described previously (Inoue et al. 2017b). Single-end 100-bp sequencing was performed on a HiSeq 2500 sequencer (Illumina). The numbers of RNA-seq reads generated are summarized in Supplemental Table S5.
RNA-seq analyses
To avoid alignment bias when mapping the F1 hybrid (B6/129 × PWK) RNA-seq reads to the mouse reference genome (mm9, B6), a custom reference sequence was constructed with all SNPs between B6/129 and PWK strains masked as “N.” The RNA-seq reads were aligned to the custom reference genome using Hisat2 version 2.1.0 (Kim et al. 2015) with default parameters, and only uniquely mapped reads were used for downstream analyses. The transcript abundance of each gene was quantified as RPKM using Cufflinks version 2.2.1 (Trapnell et al. 2010). To compute P-values for DEGs, raw RNA-seq read counts were generated using HTSeq version 0.9.1 (Anders et al. 2013) and normalized using “estimateSizeFactors” from R package DESeq (Anders and Huber 2010). The P-values were then calculated with “nbinomTest” from DESeq using a negative binomial model, and genes with P-value of <0.05 and fold change of >2 were considered as DEGs. For allele-specific gene expression analyses, the parental origin of each uniquely aligned read was determined using SNPsplit version 0.3.2 (Krueger and Andrews 2016). The allelic read counts were then summed for each gene, and only genes with >20 SNP reads were included for downstream analyses.
CUT&RUN
Eed CTR and matKO morula H3K27me3 CUT&RUN libraries were constructed as described previously (Skene et al. 2018) with a few modifications. Hybrid morula embryos were obtained by IVF of MII oocytes with PWK sperm. At 78 hpf, 30 morula embryos were treated with acid Tyrode's solution (Sigma-Aldrich) to remove the zona pellucida, washed three times with CUT&RUN wash buffer (20 mM HEPES at pH 7.5, 150 mM NaCl, 0.5 mM spermidine [Sigma-Aldrich], 1× protease inhibitor cocktails [Sigma-Aldrich]), and transferred to a 1.5-mL tube containing 50 µL of the wash buffer. The embryos were then captured with BioMagPlus Concanavalin A (Polysciences) and incubated with a rabbit anti-H3K27me3 antibody (1/100; Diagenode, C15410069) for 16 h at 4°C in antibody incubation buffer (20 mM HEPES at pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1× protease inhibitor cocktails [EDTA-free tablet; Roche], 2 mM EDTA, 0.02% digitonin [Thermo Fisher Scientific]). After unbound antibodies were washed away, 500 ng/mL protein A-MNase (pA-MN; a gift from Dr. Steven Henikoff) was added at a 1:280 ratio and incubated for 3 h at 4°C. After washing, CaCl2 was added to a final concentration of 2 mM to activate pA-MN, and the reaction was carried out for 20 min at 4°C and then stopped by adding 1/10 vol of 10× STOP buffer (1700 mM NaCl, 100 mM EDTA, 20 mM EGTA, 250 µg/mL RNase A [Thermo Fisher Scientific], 250 µg/mL glycogen [Thermo Fisher Scientific]). The protein–DNA complexes were then released by 10-min incubation at 37°C followed by centrifugation at 20,000g for 5 min at 4°C. After transferring the supernatant to a new Lo-bound tube, 1/100 vol of 10% SDS and 1/80 vol of 2.5 mg/mL Proteinase K (Thermo Fisher Scientific) were added and incubated for at least 1 h at 55°C. DNA was then precipitated by phenol/chloroform/isoamylalcohol followed by ethanol precipitation with glycogen and then dissolved in water.
Sequencing libraries were prepared using a NEBNext Ultra II DNA library preparation kit for Illumina (New England Biolabs) according to the manufacturer's instructions with a few modifications. Briefly, end repair was conducted for 30 min at 20°C followed by dA-tailing for 30 min at 65°C. After adaptor ligation for 30 min at 20°C, the DNA fragments were purified by 1.8× vol of SPRIselect beads (Beckman Coulter) followed by 16 cycles of PCR amplification with NEBNext Ultra II Q5 master mix (New England Biolabs). The PCR products were cleaned up with 0.9× vol of SPRIselect beads and quantified using Qubit dsDNA HS assay kit (Agilent Technologies). The libraries were sequenced on a HiSeq2500 with paired-end 100-bp reads (Illumina).
H3K27me3 CUT&RUN data analyses
The H3K27me3 CUT&RUN read pairs were aligned to the custom mm9 reference genome (B6/129 and PWK SNPs masked as “N”) using Bowtie2 version 2.2.9 (Langmead et al. 2009) with the following parameters: “-t -q -N 1 -L 25 --no-unal -I 150 -X 800 --no-mixed --no-discordant.” After removing nonuniquely mapped reads and PCR duplicates, MACS2 version 2.2.1 (Zhang et al. 2008) was used to identify H3K27me3 peaks with the following parameters: “-f BAMPE -g mm -q 0.01 --broad --broad-cutoff 0.1 --nolambda --SPMR --nomodel.” H3K27me3 CUT&RUN bigwig tracks were generated using BamCoverage in deepTools version 3.0.2 (Ramirez et al. 2014), and the signal intensities of Eed CTR and matKO samples were normalized as described previously (Inoue et al. 2017b). For allelic H3K27me3 enrichment analyses, the parental origins of the reads were determined using SNPsplit version 0.3.2 (Krueger and Andrews 2016). For comparison with previously published morula H3K27me3 ChIP-seq data (Matoba et al. 2018), RPKM values were calculated at 2 kb per bin resolution, and Pearson correlation coefficient was calculated in R.
Statistical analyses and data visualization
Statistical analyses were performed with R (http://www.r-project.org). Pearson's r coefficient was calculated using the “cor” function (Supplemental Figs. S3A, S4A, S8A). Mann-Whitney-Wilcoxon test was performed with the R function “wilcox.text” (Figs. 2C, 4A; Supplemental Fig. S5A,B). RNA-seq, H3K27me3 ChIP-seq, and DNaseI-seq bigwig tracks (Fig. 1; Supplemental Figs. S1, S3, S4) were generated using BamCoverage in deepTools version 3.0.2 (Ramirez et al. 2014) and visualized in the Integrative Genomics Viewer genome browser (Robinson et al. 2011). Allelic gene expression heat maps (Figs. 1D, 3, 4D) were generated using the R function “pheatmap.”
Data availability
RNA-seq and CUT&RUN data sets generated in this study have been deposited at the Gene Expression Omnibus database under accession number GSE116713. H3K27me3 ChIP-seq data sets were downloaded from GSE76687 (Zheng et al. 2016), GSE73952 (Liu et al. 2016), GSE103714 (Inoue et al. 2017b), and GSE112546 (Matoba et al. 2018). The oocyte DNaseI-seq data set was from a previous study (GSE92605) (Inoue et al. 2017a).
Supplementary Material
Acknowledgments
We thank Dr. Luis Tuesta for critical reading of the manuscript, and Dr. Osamu Masui for helpful discussion. The protein A-MNase for CUT&RUN was a kind gift from Dr. Steven Henikoff. This project was supported by the National Institutes of Health (R01HD092465) and Howard Hughes Medical Institute. Y.Z. is an Investigator of the Howard Hughes Medical Institute.
Author contributions: A.I. and Y.Z. conceived the project. A.I. and Z.C. designed and performed the experiments. Z.C. analyzed sequencing data sets. Q.Y. performed CUT&RUN. A.I., Z.C., and Y.Z. interpreted the data and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.318675.118.
References
- Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, et al. 2017. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356: 1081–1084. 10.1126/science.aal2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786. 10.1038/nprot.2013.099 [DOI] [PubMed] [Google Scholar]
- Blakeley P, Fogarty NME, del Valle I, Wamaitha SE, Hu TX, Elder K, Snell P, Christie L, Robson P, Niakan KK. 2015. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development 142: 3151–3165. 10.1242/dev.123547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borensztein M, Okamoto I, Syx L, Guilbaud G, Picard C, Ancelin K, Galupa R, Diabangouaya P, Servant N, Barillot E, et al. 2017a. Contribution of epigenetic landscapes and transcription factors to X-chromosome reactivation in the inner cell mass. Nat Commun 8: 1297 10.1038/s41467-017-01415-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borensztein M, Syx L, Ancelin K, Diabangouaya P, Picard C, Liu T, Liang JB, Vassilev I, Galupa R, Servant N, et al. 2017b. Xist-dependent imprinted X inactivation and the early developmental consequences of its failure. Nat Struct Mol Biol 24: 226–233. 10.1038/nsmb.3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. 2001. Dnmt3L and the establishment of maternal genomic imprints. Science 294: 2536–2539. 10.1126/science.1065848 [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Chiba H, Hirasawa R, Kaneda M, Amakawa Y, Li E, Sado T, Sasaki H. 2008. De novo DNA methylation independent establishment of maternal imprint on X chromosome in mouse oocytes. Genesis 46: 768–774. 10.1002/dvg.20438 [DOI] [PubMed] [Google Scholar]
- Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT. 2009. The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature 460: 128–132. 10.1038/nature08098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. 2005. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 1: 191–200. 10.1016/j.cmet.2005.01.003 [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC. 2011. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 12: 565–575. 10.1038/nrg3032 [DOI] [PubMed] [Google Scholar]
- Gu Z, Reynolds EM, Song J, Lei H, Feijen A, Yu L, He W, MacLaughlin DT, van den Eijnden-van Raaij J, Donahoe PK, et al. 1999. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development 126: 2551–2561. [DOI] [PubMed] [Google Scholar]
- Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H. 2008. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 22: 1607–1616. 10.1101/gad.1667008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. 2001. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104: 829–838. 10.1016/S0092-8674(01)00280-X [DOI] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y. 2017a. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547: 419–424. 10.1038/nature23262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Zhang Y. 2017b. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31: 1927–1932. 10.1101/gad.304113.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Barakat TS, Achame EM, Monkhorst K, Kenter A, Rentmeester E, Grosveld F, Grootegoed JA, Gribnau J. 2009. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell 139: 999–1011. 10.1016/j.cell.2009.10.034 [DOI] [PubMed] [Google Scholar]
- Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M. 2013. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J 32: 340–353. 10.1038/emboj.2012.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903. 10.1038/nature02633 [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2016. SNPsplit: allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Res 5: 1479 10.12688/f1000research.9037.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan ZJ, Xu X, Cooney AJ. 2004. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod 71: 1469–1474. 10.1095/biolreprod.104.031757 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. 2000. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell 103: 17–27. 10.1016/S0092-8674(00)00101-X [DOI] [PubMed] [Google Scholar]
- Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W. 2004. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36: 1291–1295. 10.1038/ng1468 [DOI] [PubMed] [Google Scholar]
- Lim DH, Maher ER. 2010. Genomic imprinting syndromes and cancer. Adv Genet 70: 145–175. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. 2016. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537: 558–562. 10.1038/nature19362 [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Royo H, VandeBerg JL, McCarrey JR, Mackay S, Turner JM. 2009. Key features of the X inactivation process are conserved between marsupials and eutherians. Curr Biol 19: 1478–1484. 10.1016/j.cub.2009.07.041 [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. 1997. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 11: 156–166. 10.1101/gad.11.2.156 [DOI] [PubMed] [Google Scholar]
- Matoba S, Wang H, Jiang L, Lu F, Iwabuchi KA, Wu X, Inoue K, Yang L, Press W, Lee JT, et al. 2018. Loss of H3K27me3 imprinting in somatic cell nuclear transfer embryos disrupts post-implantation development. Cell Stem Cell 23: 343–354.e5. 10.1016/j.stem.2018.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutzel V, Okamoto I, Dunkel I, Saitou M, Giorgetti M, Heard E, Schulz E. 2017. Two coupled feedback loops explain random mono-allelic Xist upregulation at the onset of X-chromosome inactivation. bioRxiv 10.1101/204909 [DOI] [Google Scholar]
- Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P. 2008. Molecular coupling of Xist regulation and pluripotency. Science 321: 1693–1695. 10.1126/science.1160952 [DOI] [PubMed] [Google Scholar]
- Okae H, Hiura H, Nishida Y, Funayama R, Tanaka S, Chiba H, Yaegashi N, Nakayama K, Sasaki H, Arima T. 2012. Re-investigation and RNA sequencing-based identification of genes with placenta-specific imprinted expression. Hum Mol Genet 21: 548–558. 10.1093/hmg/ddr488 [DOI] [PubMed] [Google Scholar]
- Okae H, Matoba S, Nagashima T, Mizutani E, Inoue K, Ogonuki N, Chiba H, Funayama R, Tanaka S, Yaegashi N, et al. 2014. RNA sequencing-based identification of aberrant imprinting in cloned mice. Hum Mol Genet 23: 992–1001. 10.1093/hmg/ddt495 [DOI] [PubMed] [Google Scholar]
- Okamoto I, Tan S, Takagi N. 2000. X-chromosome inactivation in XX androgenetic mouse embryos surviving implantation. Development 127: 4137–4145. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Arnaud D, Le Baccon P, Otte AP, Disteche CM, Avner P, Heard E. 2005. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature 438: 369–373. 10.1038/nature04155 [DOI] [PubMed] [Google Scholar]
- Okamoto I, Patrat C, Thépot D, Peynot N, Fauque P, Daniel N, Diabangouaya P, Wolf J-P, Renard J-P, Duranthon V, et al. 2011. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472: 370–374. 10.1038/nature09872 [DOI] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–W191. 10.1093/nar/gku365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez TA, Sparrow DB, Scott AN, Withington SL, Preis JI, Michalicek J, Clements M, Tsang TE, Shioda T, Beddington RSP, et al. 2004. Cited1 is required in trophoblasts for placental development and for embryo growth and survival. Mol Cell Biol 24: 228–244. 10.1128/MCB.24.1.228-244.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado T, Wang Z, Sasaki H, Li E. 2001. Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 128: 1275–1286. [DOI] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, et al. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450: 908–912. 10.1038/nature06397 [DOI] [PubMed] [Google Scholar]
- Shin J, Bossenz M, Chung Y, Ma H, Byron M, Taniguchi-Ishigaki N, Zhu X, Jiao B, Hall LL, Green MR, et al. 2010. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 467: 977–981. 10.1038/nature09457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MK, Christoffels VM, Dias JM, Trowe MO, Petry M, Schuster-Gossler K, Burger A, Ericson J, Kispert A. 2005. Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development 132: 2697–2707. 10.1242/dev.01854 [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6: e21856 10.7554/eLife.21856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, Henikoff S. 2018. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13: 1006–1019. 10.1038/nprot.2018.015 [DOI] [PubMed] [Google Scholar]
- Sousa EJ, Stuart HT, Bates LE, Ghorbani M, Nichols J, Dietmann S, Silva JCR. 2018. Exit from naive pluripotency induces a transient X chromosome inactivation-like state in males. Cell Stem Cell 22: 919–928.e6. 10.1016/j.stem.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Kondo M, Koga Y, Shiura H, Ikeda R, Hirose M, Ogura A, Murakami A, Yoshiki A, Chuva de Sousa Lopes SM, et al. 2015. A simple and robust method for establishing homogeneous mouse epiblast stem cell lines by wnt inhibition. Stem Cell Rep 4: 744–757. 10.1016/j.stemcr.2015.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi N, Sasaki M. 1975. Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 256: 640–642. 10.1038/256640a0 [DOI] [PubMed] [Google Scholar]
- Terranova R, Yokobayashi S, Stadler M, Otte A, van Lohuizen M, Orkin S, Peters A. 2008. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 15: 668–679. 10.1016/j.devcel.2008.08.015 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. 2004. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36: 1296–1300. 10.1038/ng1467 [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Stem Cell 14: 68–80. 10.1016/j.stem.2013.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Shen L, Liu Y, Sendler D, Zhang Y. 2013. Role of Tet1 in erasure of genomic imprinting. Nature 504: 460–464. 10.1038/nature12805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, et al. 2016. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63: 1066–1079. 10.1016/j.molcel.2016.08.032 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and CUT&RUN data sets generated in this study have been deposited at the Gene Expression Omnibus database under accession number GSE116713. H3K27me3 ChIP-seq data sets were downloaded from GSE76687 (Zheng et al. 2016), GSE73952 (Liu et al. 2016), GSE103714 (Inoue et al. 2017b), and GSE112546 (Matoba et al. 2018). The oocyte DNaseI-seq data set was from a previous study (GSE92605) (Inoue et al. 2017a).