

Here, Coutelier et al. used a microfluidic-based approach and live-cell imaging in yeast to capture early mutation events during replicative senescence and observed that prolonged checkpoint arrests occurred frequently in telomerase-negative lineages. Their results demonstrate that the adaptation pathway is a major contributor to the genome instability induced during replicative senescence.
Keywords: telomere, senescence, DNA damage checkpoint, adaptation, genomic instability, Cdc5
Abstract
In cells lacking telomerase, telomeres gradually shorten during each cell division to reach a critically short length, permanently activate the DNA damage checkpoint, and trigger replicative senescence. The increase in genome instability that occurs as a consequence may contribute to the early steps of tumorigenesis. However, because of the low frequency of mutations and the heterogeneity of telomere-induced senescence, the timing and mechanisms of genome instability increase remain elusive. Here, to capture early mutation events during replicative senescence, we used a combined microfluidic-based approach and live-cell imaging in yeast. We analyzed DNA damage checkpoint activation in consecutive cell divisions of individual cell lineages in telomerase-negative yeast cells and observed that prolonged checkpoint arrests occurred frequently in telomerase-negative lineages. Cells relied on the adaptation to the DNA damage pathway to bypass the prolonged checkpoint arrests, allowing further cell divisions despite the presence of unrepaired DNA damage. We demonstrate that the adaptation pathway is a major contributor to the genome instability induced during replicative senescence. Therefore, adaptation plays a critical role in shaping the dynamics of genome instability during replicative senescence.
Eukaryotic cells respond to DNA damage by activating a checkpoint that arrests the cell cycle and permits the cell to attempt DNA damage repair. This pathway is activated at specific phases of the cell cycle and limits genome instability (Harrison and Haber 2006). However, if DNA repair pathways fail or are impeded, cells can avoid death by bypassing the checkpoint arrest and undergoing cell division despite persistent DNA damage, a process known as adaptation (Sandell and Zakian 1993; Toczyski et al. 1997; Lee et al. 1998b). Adaptation is thought to provide the cell with an opportunity to survive and produce progeny by allowing DNA repair to occur in other cell cycle phases and/or by tolerating nonlethal loss of genetic material (Galgoczy and Toczyski 2001; Kaye et al. 2004). For unicellular organisms, adaptation could generate genetic and phenotypic diversity and allow survival of the population under stress conditions. In human cells, adaptation is also a proposed source of genome instability and can contribute to the development of therapy resistance in cancer cells (Syljuasen et al. 2006; Kalsbeek and Golsteyn 2017).
Double-strand breaks (DSBs) and other DNA lesions lead to cell cycle arrest by activating the DNA damage checkpoint signaling cascade. The Mec1/ATR kinase is recruited along with its interacting partner, Ddc2/ATRIP, to the DSB, where they bind to newly formed ssDNA. This complex phosphorylates Rad9 (functionally related to 53BP1 in mammals), which then binds the forkhead-associated (FHA) domains of the Rad53 and Chk1 kinases (Durocher et al. 1999). Once bound to Rad9, Rad53 is phosphorylated by Mec1, which initiates autohyperphosphorylation of Rad53, which is required to fully activate and amplify the checkpoint signal, resulting in cell cycle arrest in G2/M (Harrison and Haber 2006).
If the cell can repair DNA damage during the checkpoint arrest, it can resume the cell cycle and proceed with mitosis. This process, known as recovery, requires inactivation of the checkpoint factors, including reversal of Rad53 phosphorylation (Pellicioli et al. 2001; Leroy et al. 2003). If DNA damage persists, the cell either is permanently arrested and eventually dies or initiates adaptation. In the latter case, only Rad53 is dephosphorylated and inactivated, whereas the upstream checkpoint factors remain active, with factors such as Ddc2/ATRIP remaining at the DNA damage site (Melo et al. 2001) and phosphorylated Rad9 persisting in the nucleus (Donnianni et al. 2010; Vidanes et al. 2010).
Although telomeres, which cap the ends of linear eukaryotic chromosomes, are structurally similar to the ends of DSBs, they do not activate the DNA damage response under normal conditions and are intrinsically refractory to repair. This prevents fusion of chromosomes and genome instability (Pardo and Marcand 2005; Jain and Cooper 2010). Cellular DNA polymerases cannot fully replicate telomeric DNA, which shortens with each cell division, compromising telomere protective functions (Lingner et al. 1995; Soudet et al. 2014). Telomere shortening is counteracted by telomerase, a reverse transcriptase that iteratively synthesizes telomeric repeats (Wu et al. 2017). However, in some vertebrates, including humans, telomerase is absent from many somatic tissues, and the telomeres eventually shorten sufficiently to induce permanent activation of the DNA damage checkpoint and trigger replicative senescence (d'Adda di Fagagna et al. 2003; Herbig et al. 2006; Campisi and d'Adda di Fagagna 2007; Carneiro et al. 2016). In other multicellular organisms that have constitutively active telomerase, such as mice, and most unicellular experimental model organisms, such as budding yeast, this senescence phenotype can be recapitulated by deleting or inhibiting telomerase activity (Lundblad and Szostak 1989; Blasco et al. 1997; Enomoto et al. 2002; Ijpma and Greider 2003; Teixeira 2013; Gonzalez-Garcia et al. 2015). Thus, telomere shortening limits the life span of eukaryotic cells (Suram and Herbig 2014). Cancer cells can avoid senescence by altering the DNA damage checkpoint pathway response; for example, through p53 inactivation and abnormal elongation of telomeres, by re-expression of telomerase, or by up-regulation of homology-dependent repair between telomeres (Lundblad and Blackburn 1993; Greider 1998; Artandi and DePinho 2000; Shay et al. 2012).
Although senescence was first described as a tumor suppressor mechanism, there is also evidence that telomerase deficiency/inactivation is linked to increased genome instability, a hallmark of cancer (Blasco et al. 1997; Lee et al. 1998a; Chin et al. 1999; Hanahan and Weinberg 2000; Hackett et al. 2001; Hackett and Greider 2003; Maciejowski et al. 2015; Aunan et al. 2017; Maciejowski and de Lange 2017). However, how genome instability arises in a cell with eroding telomeres but functional DNA damage checkpoints, as it occurs physiologically in telomerase-negative somatic cells, has remained elusive. This is a crucial question for understanding how mutations and chromosomal aberrations are introduced during the early stages of tumorigenesis.
Here, we used telomerase-negative Saccharomyces cerevisiae cells to investigate sources of genome instability occurring before the onset of replicative senescence. We tracked individual cell lineages over time using a microfluidic/single-cell imaging approach and found that the process of adaptation occurs frequently in response to DNA damage in checkpoint-proficient cells during senescence. Moreover, we show that frequent prolonged arrests and adaptation shape senescence dynamics and are a major contributor to the increase in genome instability associated with replicative senescence.
Results
Prolonged nonterminal cell cycle arrests in cells lacking telomerase activity
To understand the origin of genome instability during replicative senescence in DNA damage checkpoint-proficient cells, we used microfluidics coupled to live-cell imaging, allowing us to monitor successive divisions of single yeast cells (Fig. 1A; Supplemental Fig. S1; Supplemental Movie S1; Fehrmann et al. 2013; Xu et al. 2015). In our previous study (Xu et al. 2015), we examined individual senescent yeast lineages using a TetO2-TLC1 strain in which expression of TLC1 telomerase RNA is conditionally repressed by addition of doxycycline (dox) to the medium. We showed that terminal senescence and cell death are often preceded by intermittent and stochastic long cell cycles followed by resumption of cell cycling, suggesting that the onset of replicative senescence is a complex multistep pathway.
Figure 1.

Analysis of individual telomerase-deficient lineages reveals frequent prolonged nonterminal arrests. (A) Sequential images of telomerase-negative (TetO2-TLC1) cells trapped in a microcavity of the microfluidic device. Hours spent after addition of 30 µg/mL dox to repress telomerase activity are shown in yellow. (B) Display of consecutive cell cycle durations of TetO2-TLC1 lineages grown in the microfluidic device as in A (n = 187, 40 of which were already published in our previous work) (Xu et al. 2015). Cells were monitored overnight before (−dox) and then for successive generations after (+dox) addition of 30 µg/mL dox to inactivate telomerase (designated generation 0). Each horizontal line is an individual cell lineage, and each segment is a cell cycle. Cell cycle duration (in minutes) is indicated by the color bar. X at the end of the lineage indicates cell death, whereas an ellipsis (…) indicates that the cell was alive at the end of the experiment. (Inset) Magnified view of several lineages showing nonterminal arrests (black). (C) Distribution of all cell cycle durations taken from telomerase-negative (red; n = 5962) and telomerase-positive (black; n = 1895) lineages shown in B and Supplemental Figure S1. Percentages indicate the fraction of cell cycles >150 min (first vertical black line) or >360 min (second vertical black line) for each lineage. (D) Distribution of nonterminal cell cycle durations as a function of generation for telomerase-negative (left; n = 5775) and telomerase-positive (right, n = 1887) cells extracted from B and Supplemental Figure S1. The color bar indicates the frequency. (E) Frequency of all nonterminal arrests (cell cycle >150 min; blue circles) and prolonged nonterminal arrests (cell cycle >360 min; red triangles) extracted from B and Supplemental Figure S1 as a function of generation for telomerase-negative (left) and telomerase-positive (right) lineages. The gray-shaded area encompasses points based on less than five lineages, for which the data may be less reliable. See also Supplemental Figure S1.
To investigate the relationship between long cell cycles and genome instability, we compared cell division cycles in telomerase-positive (−dox) (Supplemental Fig. S1) and telomerase-negative (+dox) (Fig. 1B) TetO2-TLC1 lineages. We detected a significant difference between the distribution of cell cycle durations of telomerase-positive and telomerase-negative cells (n = 1895 and n = 5962, respectively; P = 3.10−61 by two-sample Kolmogorov-Smirnov test) (Fig. 1B; Supplemental Fig. S1). The average cell cycle duration of telomerase-positive cells was ∼90 min, and only 1.3% of cycles were considered “long” (defined as >150 min [mean + 3 SD duration of telomerase-positive cell division]). In contrast, the mean cell cycle duration for telomerase-negative cells was ∼140 min, and “long” cycles were much more frequent (>150 min for 19% of cycles) (Fig. 1B,C). Thus, repression of telomere activity substantially increased the frequency of “long” cell cycles.
Because cell cycle arrests found at the termini of the lineages lead to cell death, these events cannot contribute to genome instability at a population level. Therefore, we focused on nonterminal arrests, which we defined as a long (>150 min) cycle followed by at least one more cell division. When the duration and frequency of nonterminal cell cycles were analyzed as a function of generation number, we observed that the frequency of nonterminal arrests increased with generations in telomerase-negative but not in telomerase-positive cells (Fig. 1D,E). We proposed previously that nonterminal arrests could be attributed at least partially to telomeric DNA damage signaling and an attempt by the cell to effect a repair (Xu et al. 2015). However, close inspection of our larger data set here revealed that a subset of the nonterminal arrests was extremely long (>6 h, which we termed “prolonged” arrests) (Fig. 1B, black segments). In telomerase-negative cells, these prolonged arrests represented ∼20% of all nonterminal arrests and also increased in frequency with successive generations. In contrast, they were present at very low frequency in telomerase-positive cells (Fig. 1E, red triangles). The duration of these prolonged arrests was inconsistent with the time scale of DNA repair, even that mediated by break-induced replication, which induces a delay of 2–4 h for repair of a single DSB (Malkova et al. 2005). Thus, our single-cell analyses establish that telomerase-deficient lineages experience many transient and prolonged cell cycle arrests, suggesting that processes distinct from typical DNA repair may occur during these arrests.
Adaptation occurs in response to telomerase inactivation
Adaptation to unrepaired DNA damage might account for the ability of cells to resume division after such prolonged arrests (Sandell and Zakian 1993; Toczyski et al. 1997; Lee et al. 1998b). To determine whether this is occurring in telomerase-negative cells, we analyzed the kinetics of senescence in two adaptation-deficient mutants: cdc5-ad, which carries a point mutation in the polo-like kinase gene CDC5 (Supplemental Movie S2; Toczyski et al. 1997), and tid1Δ, which lacks the yeast paralog of the recombination and repair enzyme Rad54 (Lee et al. 2001).
When the cdc5-ad allele was introduced into our TetO2-TLC1 strain and the cells were grown in the absence of dox (i.e., telomerase-positive), we observed no difference between cdc5-ad and wild-type lineages in terms of cell cycle duration (mean ± SD: 98 ± 26 min and 91 ± 37 min, respectively) (Supplemental Fig. S2A,E), basal mortality (rate <5.3 × 10−3 and <4.3 × 10−3 per generation, respectively), or frequency of nonterminal arrests (Supplemental Fig. S2A,C,D). However, telomerase-negative cdc5-ad lineages showed a significantly reduced frequency of prolonged nonterminal arrests (Fig. 2A,C,D,G–I; Supplemental Movie S3) compared with telomerase-negative CDC5 lineages (Fig. 1B,D [left panel], E [left panel]), suggesting that adaptation may be responsible for the high frequency of prolonged nonterminal arrests in telomere-negative cells.
Figure 2.

Adaptation-deficient mutants display a reduced frequency of prolonged nonterminal arrests. (A,B) Display of TetO2-TLC1 cdc5-ad (A; n = 116) and tid1Δ (B; n = 103) lineages before and after dox addition. See Figure 1B for a description of the plot features. (C,E) Distribution of nonterminal cell cycle durations as a function of generation for telomerase-negative cdc5-ad (C, n = 2397) and tid1Δ (E, n = 999) cells, as extracted from experiment shown in A and B, respectively. The color bar indicates the frequency. (D,F) Frequency of all nonterminal arrests (cell cycle >150 min; blue circles) and prolonged nonterminal arrests (cell cycle >360 min; red triangles) as a function of generation for telomerase-negative cdc5-ad (D) and tid1Δ (F) cells, as extracted from experiments shown in A and B, respectively. The gray-shaded area encompasses points based on less than five lineages, for which the data may be less reliable. (G–I) Percentage of nonterminal arrests (G) and prolonged nonterminal arrests (H) and the ratio of nonterminal to all prolonged (nonterminal + terminal) arrests (I) over all cell cycles for telomerase-negative lineages of the indicated genotypes (wild type: TetO2-TLC1), as extracted from experiment shown in A and B and Figure 1B. (n.s.) Not significant; (**) P < 0.01; (****) P < 0.0001 by χ2 goodness of fit test. See also Supplemental Figure S2.
To verify this, we examined the tid1Δ mutant, which likely plays a distinct role in adaptation (Lee et al. 2001; Shah et al. 2010). Like the cdc5-ad strain, telomerase-positive tid1Δ cells displayed normal cell cycle durations (mean ± SD: 85 ± 26 min) (Supplemental Fig. S2B,E), basal mortality level (rate <6 × 10−3 per generation), and frequency of nonterminal arrests (Supplemental Fig. S2B–D) compared with wild-type cells. We also observed a significant decrease in the frequency of prolonged nonterminal arrests in telomerase-negative tid1Δ lineages compared with wild-type lineages, albeit to a lesser extent than telomerase-negative cdc5-ad cells (Fig. 2B,E,F,G–I; Supplemental Movie S4). Both cdc5-ad and tid1Δ mutants also showed a reduced frequency of nonterminal arrests <6 h (Fig. 2A,B,G), suggesting that adaptation in response to telomere shortening might also occur during briefer arrests. Collectively, these data indicate that adaptation mediates the resumption of cell cycling after prolonged arrests.
Adaptation does not affect telomere homeostasis, checkpoint activation, or homology-dependent repair
The absence of prolonged arrests in adaptation mutants could result from altered telomere length or structure. To verify that telomere homeostasis was not disrupted in the adaptation-defective mutants used here, we measured telomere lengths in telomerase-positive (reflecting steady-state length) and telomerase-negative (indicating the kinetics of telomere shortening) backgrounds. A heterozygous diploid TLC1/tlc1Δ CDC5/cdc5-ad strain was allowed to sporulate, and spores of all four resulting genotypes were selected. Cultures were grown and diluted daily, and samples were taken throughout senescence (days 4–8) and post-senescence (days 9–10) (Fig. 3A). Telomere length was measured using XhoI digestion of genomic DNA followed by Southern blot analysis. We observed no significant differences between cdc5-ad and wild-type cells in steady-state telomere length distribution, telomere shortening kinetics, or the ability of post-senescent survivors to maintain typical long and heterogeneous telomeres (Fig. 3B,E; Lundblad and Blackburn 1993). Similar results were found for tid1Δ mutants (Fig. 3C–E). Thus, aberrant telomere homeostasis does not explain the lack of prolonged arrests in the adaptation mutants.
Figure 3.

Adaptation does not affect telomere length or its regulation. (A) Growth of tlc1Δ CDC5 and tlc1Δ cdc5-ad cells selected after sporulation of a heterozygous TLC1/tlc1Δ CDC5/cdc5-ad diploid strain. After dissection, spores were grown on plates for 2 d, genotyped, and transferred to liquid culture (indicated as day 3). Cells were sampled daily and normalized to the same density (OD600 nm = 0.01). (B) XhoI terminal restriction fragment Southern blot analysis of telomere lengths in the cells described in A. (PD) Population doublings. (C,D) As described for A and B except that TLC1/tlc1Δ TID1/tid1Δ heterozygotes were allowed to sporulate, and the growth rates and telomere lengths of tlc1Δ TID1 and tlc1Δ tid1Δ genotypes were analyzed. (E) Mean telomere shortening rates (base pair/population doubling [bp/PD]) of the genotypes analyzed in A–D over days 3–5. Mean ± SD of n = 3 independent experiments per genotype. See also Supplemental Figure S3.
We also asked whether the adaptation mutants might display defective checkpoint activation in response to telomeric damage. We considered this unlikely because while many proteins involved in adaptation also play roles in DNA damage repair and checkpoint activation, this is not the case for Cdc5 and Tid1 (Toczyski et al. 1997; Lee et al. 2001; Melo et al. 2001; Pellicioli et al. 2001; Harrison and Haber 2006; Clerici et al. 2014). Indeed, we specifically chose them to avoid confounding effects on checkpoint activation. Consistent with this, we found that cdc5-ad mutants and wild-type cells showed indistinguishable abilities to (1) form Ddc2-eGFP foci in the nucleus, (2) arrest in G2/M phase of the cell cycle, (3) undergo Rad53 hyperphosphorylation in response to DSBs induced by zeocin, and (4) undergo G2/M arrest at the restrictive temperature in a cdc13-1 background (in which telomeres become uncapped at high temperature, inducing checkpoint activation) (Supplemental Fig. S3A–D; Supplemental Movie S2). Therefore, we conclude that the reduction in prolonged nonterminal arrests during replicative senescence in the cdc5-ad mutant is not caused by disruption of DNA damage checkpoint activation.
Finally, we wondered whether adaptation mutants might alter homology-dependent repair efficiency. To test this, we performed a recombination assay based on cell survival following repair of a DSB by homologous recombination (Batte et al. 2017). A single DSB was induced by expression of the I-SceI endonuclease, which is driven by the inducible GAL promoter, in a ura3 allele bearing the I-SceI recognition site. Cells survived by homology-dependent repair with a donor ura3-1 allele. Survival frequency normalized to the total number of plated cells provides a measure of repair efficiency. We detected no significant difference in survival frequency after DSB induction between the three genotypes (wild type, cdc5-ad, and tid1Δ) regardless of where the cutting site was placed; i.e., at an intrachromosomal locus (LYS2 gene) or the 6R subtelomere (Supplemental Fig. S3E). Thus, the observed phenotypes of cdc5-ad and tid1Δ mutants in replicative senescence are probably not due to a change of homology-dependent repair efficiency.
A checkpoint activation reporter for monitoring adaptation in single cells
To detect adaptation events in senescing cells in real-time, we developed a DNA damage checkpoint activation reporter. We reasoned that the DNA damage checkpoint machinery would remain active during the cell cycle immediately following prolonged arrest if the cell underwent adaptation, whereas the checkpoint would be inactivated if the cell successfully repaired the damage and recovered. We monitored Rad9 phosphorylation, which is maintained at high levels in adapted cells despite Rad53 dephosphorylation (Donnianni et al. 2010; Vidanes et al. 2010). Rad9 is recruited to the DNA damage site and phosphorylated in a Mec1-dependent manner (Emili 1998; Vialard et al. 1998; Schwartz et al. 2002). Phosphorylated Rad9 then binds to Rad53 via its FHA domains (Durocher et al. 1999; Schwartz et al. 2002; Sweeney et al. 2005). We generated strains harboring an mCherry-tagged Rad53 FHA1 domain, which should remain cytoplasmic until checkpoint activation, when it should interact with phosphorylated Rad9 in the nucleus (Fig. 4A). The cells also expressed histone Hta2 fused to yECFP as a nuclear marker.
Figure 4.

Detection of DNA damage checkpoint activation with a fluorescent biosensor. (A) Schematic showing the mCherry-coupled Rad53-FHA1 fluorescent protein interacting with phosphorylated Rad9 in the nucleus in response to a DSB. (B) Representative phase contrast and fluorescence images of cells carrying the nuclear marker Hta2-yECFP and either the wild-type FHA1-mCherry (wt) or a mutant version that cannot bind to phosphorylated Rad9 (FHA1-H75A-mCherry). Cells were imaged 3.5 h after treatment with 300 µg/mL zeocin or nontreated. (C) Quantification of the experiment in B. Data are presented as the nuclear mCherry signal normalized to the untreated cells carrying wild-type FHA1-mCherry. N ≥ 50 untreated cells; N ≥ 160 zeocin-treated cells. (****) P < 0.0001; (n.s.) not significant by the Mann-Whitney U-test. (D, bottom panel) The experimental scheme: cdc13-1 cells carrying the FHA1-mCherry reporter were grown at 23°C (“unchallenged cells”) and then placed for 3 h at the restrictive temperature of 32°C (“arrested cells”). The cells were then split into two cultures: One was incubated for 2 h at 23°C (“recovered cells”), and the other was incubated for an additional 21 h at 32°C (“adapted cells”). (Top panel) Quantification of nuclear mCherry fluorescence normalized to the unchallenged cells. N = 279 unchallenged cells; N = 191 arrested cells; N = 255 recovered cells; N = 59 adapted cells. (*) P < 0.05; (****) P < 0.0001 by the Mann-Whitney U-test. See also Supplemental Figure S4.
We treated cells harboring both FHA1-mCherry and Hta2-yECFP (the “FHA1-mCherry reporter”) with zeocin to generate DSBs. DNA damage resulted in the accumulation of FHA1-mCherry in the nucleus (Fig. 4B,C). Most cells arrested in G2/M (Supplemental Fig. S4A), indicating that expression of the reporter did not interfere with the checkpoint response. A mutant FHA1 that cannot interact with phosphorylated Rad9 (His75 to alanine [H75A], corresponding to H88A in the full-length Rad53 protein) (Durocher et al. 1999) abolished nuclear localization of mCherry upon DNA damage induction (Fig. 4B,C). Similar results were obtained upon induction of a single DSB in a strain containing a unique cleavage site for the endonuclease HO (Supplemental Fig. S4B,C; Lee et al. 1998b). Thus, the FHA1-mCherry reporter specifically detects checkpoint activation in response to DNA damage.
We asked whether the FHA1-mCherry reporter could distinguish between adaptation and recovery events. We used the cdc13-1 mutant model of adaptation, in which telomeres are uncapped at high temperatures, leading to checkpoint activation and either adaptation or death (Garvik et al. 1995; Paschini et al. 2012). However, checkpoint activation can be reversed if the cells are returned to the permissive temperature (23°C), allowing full recovery (Lydall and Weinert 1995; Toczyski et al. 1997). We incubated cdc13-1 cells expressing FHA1-mCherry reporter for 3 h at 32°C and then either incubated them for an additional 21 h at 32°C or returned them for 2 h to 23°C (Fig. 4D). As expected, the nuclear mCherry signal increased significantly after 3 h at the restrictive temperature, indicative of checkpoint activation, and then decreased to basal levels after 2 h at 23°C. However, cells remaining for 24 h at 32°C showed sustained elevation of the nuclear mCherry signal upon adaptation compared with basal levels (Fig. 4D; Supplemental Fig. S4D,E). These data demonstrate that the FHA1-mCherry reporter can distinguish between recovery and adaptation events in live single cells.
We used this reporter to assess checkpoint activation in individual telomerase-negative cell lineages. We confirmed that the senescence profile of reporter-expressing TetO2-TLC1 cells was comparable with that of control TetO2-TLC1 cells under the microfluidic imaging conditions (median ± SD: 34 ± 18 vs. 31 ± 13 generations, respectively) (Supplemental Fig. S5A; Fig. 1B). The average nuclear mCherry signal before senescence was low and displayed oscillatory behavior (Fig. 5A,B), which could reflect Rad9 phosphorylation during S and G2 phases of the cell cycle (Vialard et al. 1998). In contrast, the reporter signal increased substantially during cell cycle arrest, indicating DNA damage checkpoint activation, consistent with our finding that deletion of MEC1 suppressed these arrests (Fig. 5A,B; Xu et al. 2015).
Figure 5.

A novel checkpoint activation reporter reveals adaptation events at the single-cell level in telomerase-negative lineages. (A,B) Average nuclear FHA1-mCherry signal in representative telomerase-negative lineages monitored over the course of senescence shown in Supplemental Figure S5A. Vertical lines represent budding events. The gray-shaded area represents the basal nuclear FHA1-mCherry signal in normally dividing cells ± 3 SD. (C) Average nuclear FHA1-mCherry signal per cell cycle quantified in telomerase-negative single cells with normal cell cycle durations (<2.5 h; n = 31 cell cycles), cell cycles showing prolonged arrest (>5 h, n = 32 cell cycles) (see Supplemental Fig. S5B, indicated by white arrows in Supplemental Fig. S5A), and the first cell cycle immediately following prolonged arrest (n = 64 cell cycles) (see Supplemental Fig. S5B). Data were taken from the experiment shown in Supplemental Figure S5A. (****) P < 0.0001; (n.s.) not significant by the Mann-Whitney U test. (D,F) The analysis in C was further divided into two subsets: adaptation (D; n = 38 pairs of two consecutive cell cycles) and recovery (F; n = 10 pairs of two consecutive cell cycles); only cells arrested for >5 h and displaying increased mCherry signal were included in this analysis, thus excluding 16 out of the 64 prolonged arrests from further analysis (Supplemental Fig. S5B). Adaptation and recovery are defined as a high nuclear mCherry signal that is maintained (adaptation; D) or decreases to a basal level (recovery; F) in the cell cycle immediately following prolonged arrest. (**) P < 0.01; (n.s.) not significant by the Mann-Whitney U-test. (E,G) Representative plots of the average nuclear FHA1-mCherry signal showing an adaptation (E) and a recovery (G) event. Blue lines show the prolonged arrest (>5 h) followed by cell division, budding, and the second cell cycle of the resulting mother (red) and daughter (green) cells. The gray-shaded area represents the average basal reporter signal during normal cell cycles, and the vertical lines indicate cytokinesis and budding. (H) Schematic summarizing the behavior of the FHA1-mCherry reporter during an arrest >5 h, the immediate following cell cycle, and its interpretation into adaptation (79% of the events) or recovery (21% of the events), as plotted in D–G. These particular cell cycles are placed in the context of cell lineages from telomerase inactivation to senescence (red cells). A red nucleus indicates that the nuclear signal of FHA1-mCherry fusion protein is above background as in Figure 4A. See also Supplemental Figure S5. (I) Distribution of the number of cell divisions after prolonged arrest. n = 32.
We observed two distinct patterns of nuclear mCherry signal intensity in the cell cycle immediately following nonterminal arrests: maintenance of high levels (79%; 38 out of 48 cycles) (Fig. 5C,D,E,H; Supplemental Fig. S5B) and return to basal levels (21%; 10 out of 48 cycles) (Fig. 5C,F,G,H; Supplemental Fig. S5B), consistent with adaptation and recovery events, respectively. The fraction of cells that adapted after experiencing a prolonged arrest (79%) was in agreement with the 83% reduction in prolonged nonterminal arrests found in the cdc5-ad mutant compared with wild-type cells (Fig. 2A,H). Thus, adaptation events occur frequently during senescence, suggesting that telomeric damage is not always repaired before the affected cells resume division and reach a terminal senescent state.
Of 32 prolonged cell cycles, 23 (72%) were followed at some point in the subsequent divisions by a cell cycle of normal duration (Fig. 5I; Supplemental Fig. S5C,D). The mean ± SD number of cell divisions that followed a prolonged arrest was 9.4 ± 10.1 divisions, with a maximum of 33. Therefore, in a majority of lineages, adaptation eventually led to normal cell cycles with inactive DNA damage checkpoints, suggesting that repair pathways operated to silence the initial damage signal in subsequent cell divisions and that post-adapted cells would not die out in the population of senescent cells. Instead, post-adapted cells display substantial proliferation potential, up to billions of cells in the progeny.
Adaptation drives genome instability in senescence
Based on our observation of a high frequency of adaptation events in telomerase-negative lineages (∼2%–7% of all cell divisions) and the proliferative potential of adapted cell lineages, we hypothesized that adaptation might contribute to the increase in genome instability observed as cells advance into senescence, since adaptation can be followed by genome instability (Galgoczy and Toczyski 2001). To test this hypothesis, we measured the mutation rate of the endogenous CAN1 gene in the TetO2-TLC1 strain using fluctuation assays (Hackett et al. 2001). CAN1 encodes an arginine permease such that cells carrying mutations in the gene are insensitive to the toxic arginine analog canavanine. Growth in arginine-deficient canavanine-supplemented medium thus selects for CAN1 mutants, and the mutation rate can be estimated by enumerating CanR clones (Lea and Coulson 1949; Chen et al. 1998). We observed no difference in the CAN1 mutation rates of telomerase-positive cdc5-ad and wild-type strains (mean [95% confidence intervals]: 1.4 [1.0–1.7] × 10−7 and 1.6 [1.2–2.0] × 10−7 mutations per generation, respectively).
To evaluate CAN1 mutation rates during senescence, independent cultures of TetO2-TLC1 wild-type and cdc5-ad cells were grown in rich liquid dox-containing medium and diluted daily to 104 cells per milliliter. Each day, a sample of each culture was plated on canavanine-containing plates to score CanR colonies and, in parallel, on rich medium plates to normalize the number of cells plated (Fig. 6A). As telomerase-negative cells lose their proliferation potential over time, CAN1 mutants were selected on plates lacking dox to allow telomerase re-expression and thus ensure accurate quantification of colony number. Wild-type telomerase-negative cultures displayed an increase in CAN1 mutation rate over time, which peaked at approximately ninefold the basal rate on day 6, concomitant with the senescence crisis (Fig. 6B,D; Supplemental Fig. S6A–F; Hackett et al. 2001). In contrast, the cdc5-ad strain mutation rate was more modest and peaked at approximately fourfold the basal rate at senescence crisis, corresponding to an ∼56% decrease compared with the wild-type strain (Fig. 6D). Similar results were obtained with the tid1Δ telomerase-negative strain, which displayed a mutation rate at senescence crisis ∼40% lower than the wild-type telomerase-negative cultures (approximately sixfold vs. ∼10-fold increased rate, respectively) (Fig. 6C,E; Supplemental Fig. S6G–J). These data indicate that adaptation contributes nearly half of the increase in mutation rate observed in senescence.
Figure 6.
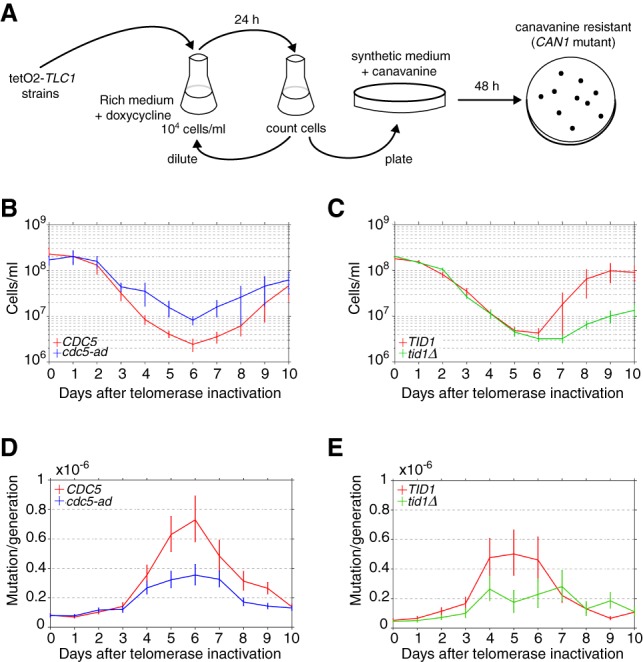
Adaptation contributes to the increased mutation rate observed in senescence. (A) Experimental protocol for the fluctuation assay of CAN1 gene mutation during senescence. Cells were grown in liquid YPD medium and 30 µg/mL dox for 10 d. Cells were counted and diluted daily to ensure that CanR mutants were not transferred from the previous culture. A sample of cells was plated on canavanine-containing medium lacking arginine or on YPD plates (for normalization). The colonies formed after 2 d at 30°C were counted, and the mutation rate was calculated. (B,C) Average growth curves of 20 independent cultures of telomerase-inactivated CDC5 and cdc5-ad strains (B) and 10 independent cultures of telomerase-inactivated TID1 and tid1Δ strains (C). Data are presented as the means ± SD. (D,E) Rates of CAN1 gene mutation in the cultures shown in B and C, respectively. Data are presented as the mutation rate ±95% confidence interval. See also Supplemental Figure S6.
Post-adapted cells display gross genome rearrangements
After adaptation to DNA damage, repair pathways available to the cell in the next G1 phase may differ from those in G2/M. We thus asked whether adaptation modified the spectrum of mutations that led to CanR cells obtained in the fluctuation assays (Fig. 6A).
The CanR colonies obtained in both wild-type and cdc5-ad backgrounds displayed size heterogeneity, possibly reflecting diverse mutation mechanisms. Most small colonies recovered normal growth when restreaked, reminiscent of the transient lower growth/high mortality seen immediately after the appearance of unstable chromosomes (Pobiega and Marcand 2010; Vasan et al. 2014; Beyer and Weinert 2016). To identify altered chromosomes, we separated the whole chromosomes of CanR clones by pulsed-field gel electrophoresis (PFGE). Most CanR colonies displayed changes in the mobility of one or several chromosomes compared with the initial wild-type or cdc5-ad CAN1 strains before telomerase inactivation (Fig. 7A; Supplemental Fig. S7A), showing that gross chromosomal rearrangements (GCRs) occur in senescent cells and not only on the chromosome carrying the reporter gene for which we selected. However, the extent of these abnormalities appeared similar between wild-type and cdc5-ad strains.
Figure 7.
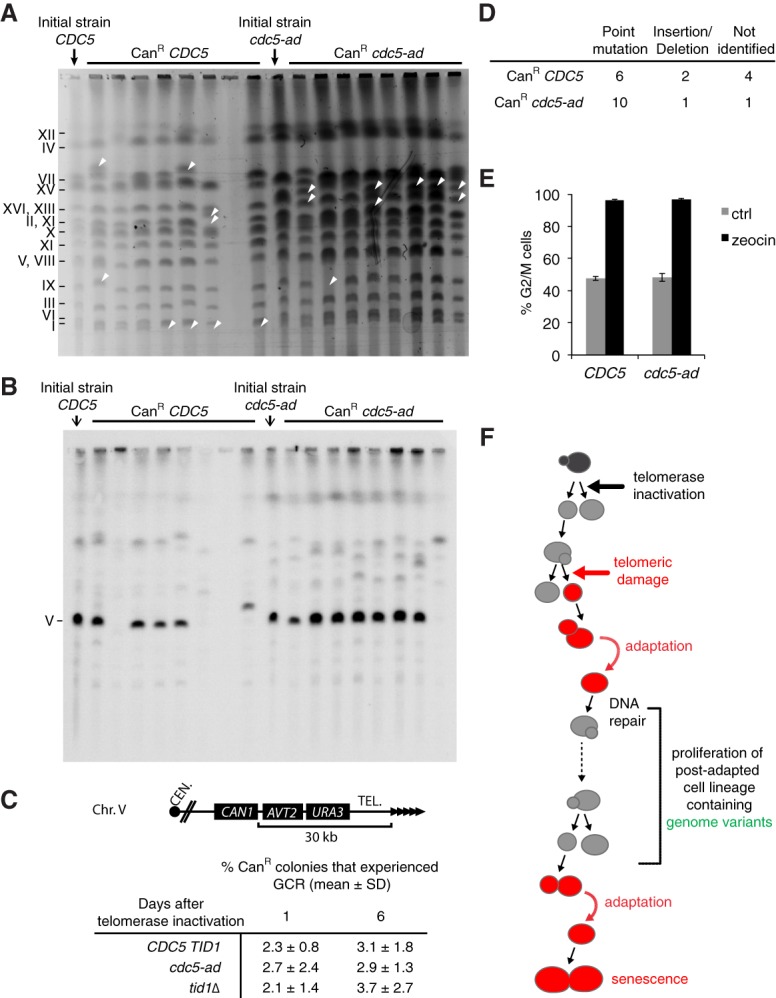
Adaptation drives genome instability in senescence. (A) Chromosomes of CanR colonies of the indicated genotypes were separated by PFGE and visualized by ethidium bromide staining. Arrows illustrate rearranged chromosomes. (B) Southern blot of the PFGE shown in A hybridized with a probe against the essential gene PCM1 on chromosome 5. (C) Fraction of the CanR colonies that grew on plates containing both canavanine and 5-fluoroorotic acid (5-FOA), selecting for loss of CAN1 and URA3 functions, events that were previously characterized as GCRs (Chen and Kolodner 1999). The percentages were derived from n > 2500 CanR colonies for each genotype and day and from N = 5–6 independent cultures per genotype. (Top panel) A scheme of the region bearing the CAN1 and URA3 reporter genes is shown. (D) Types of mutations found by sequencing the CAN1 gene of CanR colonies. (E) CanR colonies of the indicated genotypes were tested for the ability to arrest in G2/M following exposure to 300 µg/mL zeocin for 3.5 h. Data are presented as the means ± SD. (F) Model for the emergence of checkpoint activation-proficient adapted cells with persistent DNA damage. Upon telomerase inactivation, the DNA damage checkpoint is activated in response to telomere replication defects or telomere shortening (red cells), which interrupts cell proliferation and results in their progressive dilution in the population. Even if DNA repair fails, the cells may undergo adaptation by bypassing downstream checkpoint signaling. The proliferation capacity of these lineages is thus extended despite the persistence of the initial damage or repair intermediates. Some genome variants may arise that result in viable progeny. See also Supplemental Figure S7.
To better quantify the gross chromosomal rearrangements leading to canavanine resistance, we replica-plated CanR colonies on plates containing both canavanine and 5-fluoroorotic acid (5-FOA), thus additionally selecting for loss of URA3 expression (Fig. 7C). The simultaneous loss of CAN1 and URA3 was characterized previously as due to GCRs (Chen and Kolodner 1999). We observed that in all three genotypes (CDC5 TID1, cdc5-ad, and tid1Δ), 2%–4% of CanR colonies experienced GCRs. Importantly, we found no significant differences between adaptation-proficient and adaptation-deficient strains at both day 1 and day 6 after telomerase inactivation. Therefore, adaptation did not influence the mechanisms by which senescent cells generate aberrant genome rearrangements.
In small CanR colonies, PCR designed to amplify the CAN1 gene frequently failed (Supplemental Fig. S7B,C), consistent with a terminal deletion of chromosome 5 (Hackett et al. 2001), although other possibilities exist, such as mutations of one of the primer annealing sites, large insertions in the CAN1 gene, or chromosome fusions disrupting the CAN1 gene. In contrast, in normal-sized colonies, the CAN1 gene could be amplified most of the time (Supplemental Fig. S7B,D), and chromosome 5 was frequently found to be intact (Fig. 7B). We sequenced the PCR products (Fig. 7D) and identified point mutations, insertions, and deletions. Overall, they were not significantly different between wild-type cells and cdc5-ad mutants. Therefore, adaptation did not influence the nature and spectrum of CAN1 mutations formed during senescence.
Since adaptation is known to transiently alter checkpoint activation, we wondered whether CanR strains—roughly half of which have previously undergone adaptation in senescence—still had an intact checkpoint pathway. To test this, CanR strains were treated with zeocin and tested for their ability to arrest in G2/M. All strains of both wild-type and cdc5-ad genotypes were arrested in G2/M to the same extent as the parental strains (Fig. 7E). As a negative control, RAD9 deletion prevented the G2/M arrest in cells treated by zeocin (Supplemental Fig. S3B). This result suggested that although half of the CanR mutants of wild-type genotype previously experienced adaptation, none irreversibly mutated their checkpoint signaling pathway. Therefore, the increase in mutagenesis and chromosomal rearrangements in senescence is not the result of pre-existing mutations in genes encoding checkpoint factors.
Discussion
The molecular events occurring in individual cells as they divide and progress toward senescence are difficult to study in bulk cell populations. Mutations and other genome-destabilizing events are rare but can have major consequences for the whole cell population. Here, we studied cell division dynamics in individual telomerase-deficient cell lineages to understand when and how genome instability is initiated upon telomerase inactivation. We used a microfluidic-based approach coupled to live-cell imaging to monitor consecutive divisions of individual telomere-deficient cells until they entered senescence and died. We found that many lineages display frequent arrests prior to the terminal arrest at replicative senescence. These nonterminal arrests increased in frequency with time and mirrored the increase in genome instability during senescence. About 20% of the nonterminal arrests, which we called prolonged arrests, were extremely long (>5–6 h), a time scale inconsistent with known DNA repair mechanisms (Fig. 1). Instead, the cells underwent adaptation, entered mitosis, and proceeded to divide in the presence of unrepaired DNA damage (Sandell and Zakian 1993; Toczyski et al. 1997). We showed that this mechanism accounted for nearly half of the increase in genome instability of senescent cells. Our results support the following sequence of events (Fig. 7F): A critically short or damaged telomere induces a checkpoint arrest, and, because repair pathways might not be efficient, adaptation would allow cell division after an extended period of time and lead to genome instability in the following cell cycles. This opens the way for genome variants to arise. Most variants are detrimental to cell growth, but rare ones might potentially stabilize and propagate and thus serve as a precursor for senescence escape.
We used two independent approaches to demonstrate that adaptation occurred in the absence of telomerase. In the first approach, we analyzed adaptation-deficient mutants and found that they displayed significantly fewer nonterminal arrests, particularly the prolonged arrests, compared with their wild-type counterparts, suggesting that adaptation was required for telomerase-negative cells to escape from these arrests (Fig. 2). Interestingly, telomerase-negative cdc5-ad lineages display a median survival similar to telomerase-negative CDC5 lineages, which is higher than expected if adaptation mutants were unable to resume cell cycle after an early arrest. Since telomerase-negative tid1Δ lineages have a significantly reduced median survival compared with cdc5-ad (median ± SD: 22 ± 12 and 30 ± 13, respectively), we speculate that the higher survival of the cdc5-ad mutant might be due to an additional or altered function unrelated to adaptation. Furthermore, in response to DNA damage, the cdc5-ad mutant was as proficient for checkpoint activation, cell cycle arrest, recruitment of repair factors (Toczyski et al. 1997; Melo et al. 2001; Pellicioli et al. 2001; this study), and homology-dependent repair as wild type. This rules out inefficient checkpoint activation or repair as a possible explanation for the unexpectedly high survival of telomerase-negative cdc5-ad lineages. Further studies shall clarify this cdc5-ad-specific phenotype observed only in the absence of telomerase. In the second approach, we developed a fluorescent biosensor to monitor checkpoint activation in live cells (Figs. 4, 5). In the majority of prolonged arrests (∼80%), Rad9 phosphorylation was maintained at high levels through the next cell cycle, as expected after adaptation (Donnianni et al. 2010; Vidanes et al. 2010). The remaining prolonged arrests (∼20%) were followed immediately by a cell cycle in which the checkpoint reporter signal returned to basal levels, consistent with recovery after repair of the initial DNA damage. The two independent approaches led to similar estimates of the fraction of prolonged nonterminal arrests followed by adaptation (∼80%). Taken together, the results revealed that adaptation occurred in telomerase-negative cells at a frequency of ∼2%–7% of all cell divisions and therefore contributed substantially to the kinetics of senescence.
Adaptation has been observed previously in response to various mechanisms of DNA damage, including an endonuclease-induced single DSB, DSB-inducing drugs (e.g., zeocin), replication-inhibiting drugs (e.g., aphidicolin), and ionizing radiation (Serrano and D'Amours 2014). However, not all DNA damages can support adaptation; for instance, cells experiencing two induced DSBs cannot adapt, suggesting that adaptation relies on a quantitative and/or qualitative assessment of the initial damage by the cell (Lee et al. 1998b). In contrast, telomeres lacking functional Cdc13 accumulate large amounts of ssDNA, trigger a potent DNA damage checkpoint activation, and can still support adaptation (Garvik et al. 1995; Toczyski et al. 1997). Furthermore, Cdc13-independent survivor cells require factors involved in adaptation (Mersaoui et al. 2015). Our results add critically short and/or broken telomeres resulting simply from telomerase deficiency to the list of DNA damage mechanisms that can lead to adaptation.
We found that some telomerase-negative lineages outlived their first prolonged arrest by many generations (up to 33, representing ∼9 billion cells in the progeny) (Fig. 5). Several scenarios might explain such longevity in the face of persistent DNA damage. First, a different set of telomere repair mechanisms might be available in a subsequent cell cycle to repair the dysfunctional telomeres that elicited the prolonged arrest. Second, adaptation might result in at least temporary deficiency in checkpoint activation events downstream from Rad9 phosphorylation. Third, consecutive adaptation events might occur, which is plausible in the case of critically short telomeres because they are not lethal per se. If so, this would raise the question of whether the first adaptation event could favor or even accelerate subsequent ones, perhaps through a priming mechanism or simply because adaptation-promoting factors such as Cdc5 would already be present at high levels. In this context, the initial telomeric damage and subsequent genome rearrangements might be less detrimental to cell growth than other chromosomal breaks leading to loss of essential genes, for instance.
In adaptation-deficient strains, the mutation rate was approximately twofold lower than in adaptation-proficient strains at senescence crisis (Figs. 6, 7). Several non-mutually exclusive mechanisms have been suggested to explain genome instability in senescence. Dysfunctional or short telomeres can lead to extensive resection and end degradation, which would initiate genome instability close to chromosome ends (Garvik et al. 1995; Hackett and Greider 2003; Fallet et al. 2014). Exposed ssDNA or telomere-free ends can also cause global genome instability by recombination with homologous sequences or telomere fusions and subsequent breakage–fusion–bridge cycles (Murnane 2006). Alternatively, sister dysfunctional/short telomeres can fuse, perhaps through faulty replication and fork closure, and form dicentric or other types of unstable chromosomes (Beyer and Weinert 2016). In addition, repair attempts may leave toxic or unstable repair intermediates such as half crossover-initiated cascades, as observed when break-induced replication is ineffective (Vasan et al. 2014). Resolution of chromosomal instability by successive recombination and breakage events over many cell divisions may then spread genome instability across chromosomes. The possibility that the stabilization of the genome spans over several cell divisions is consistent with the nonterminal arrests that we describe, which increase the chances of subsequent arrests, as we noticed previously (Xu et al. 2015). We thus propose that, although adaptation does not change DNA repair options or mechanisms, it could be an important mechanism to force mitosis in cells that might otherwise be permanently arrested, thus promoting progress toward resolution of the instability.
In conclusion, the role of adaptation in senescence described here may have implications for understanding of the early steps of tumorigenesis. We provide a plausible scenario through which mutations and genome instability in general, key enabling characteristics of cancer, can arise in the absence of prior mutations, since telomerase is physiologically repressed in the vast majority of somatic cells in humans. This scenario is consistent with evidence of telomere exhaustion, checkpoint activation, and genome instability found in precancerous lesions (Artandi and DePinho 2010; Hanahan and Weinberg 2011). In addition, because senescent cells accumulate with age (Jeyapalan et al. 2007), adaptation to telomere signaling could be involved in the association between an increased risk of cancer and age.
Materials and methods
Yeast strains
All yeast strains used in this study have a W303 background corrected for RAD5 and ADE2 (rad5-535 and ade2-1 in the original strain) (see Supplemental Table S1).
Terminal restriction Southern blot analysis
Genomic DNA was extracted from cultures using a standard phenol:chloroform:isoamyl (25:24:1) purification procedure and isopropanol precipitation. A sample of 500 ng of genomic DNA was digested with XhoI, and the products were ethanol-precipitated, resuspended in loading buffer (gel loading dye, Purple 6X, New England Biolabs), and resolved on a 1% agarose gel for 4 h at 150 V. The gel was then soaked in a denaturation bath (0.4 M NaOH, 1 M NaCl) for 20 min and transferred by capillary action to a charged nylon membrane (Hybond XL, GE Healthcare). The telomere-specific oligonucleotide probe (5′-GGGTGTGGGTGTGTGTGGTGGG-3′) was 32P-labeled at the 5′ terminus with ATP (γ-32P) using T4 polynucleotide kinase (New England Biolabs). The membrane was then hybridized using the Rapid-hyb buffer protocol (GE Healthcare). In brief, the membrane was prehybridized for 1 h at 42°C in Rapid-hyb buffer, 20 pmol of the radioactive probe was added, and the incubation was continued for 1 h. The membrane was washed consecutively with 5× SSC and 0.5% SDS for 10 min at 42°C, 5× SSC and 0.1% SDS for 20 min at 42°C, and 1× SSC and 0.1% SDS for 30 min at 25°C. The membrane was then imaged with a Typhoon FLA 9500 scanner (GE Healthcare).
SDS-PAGE and Western blot analysis
Aliquots of 5 × 107 cells were harvested by centrifugation. The pellets were lysed in 0.2 M NaOH for 10 min on ice, and proteins were precipitated by the addition of 50 µL of 50% trichloroacetic acid. The samples were centrifuged at 16,100g for 10 min at 4°C, and the pellets were resuspended in 4× Laemmli buffer and heated for 5 min at 95°C. Samples were separated in a denaturing 7.5% 37.5:1 polyacrylamide gel, and proteins were transferred to a nitrocellulose membrane (Amersham Protran 0.45 NC, GE Healthcare). The membranes were stained with Ponceau Red and immunoblotted with anti-Rad53 primary antibody (Abcam, EL7.E1) that recognizes both the unphosphorylated and phosphorylated forms of Rad53. Blots were then incubated with a horseradish peroxidase-coupled secondary antibody, and the signal was detected using ECL reagent (Amersham, GE Healthcare).
Recombination efficiency measurement
The recombination efficiency measurement upon induction of a single DSB was performed as described (Batte et al. 2017). Briefly, the yeast strains were grown overnight in rich medium containing 2% lactate, 3% glycerol, and 0.05% glucose; counted; and then plated on 2% galactose plates to induce I-Sce I expression and on 2% glucose plates to normalize the cell count and plating efficiency.
PFGE
To prepare DNA samples for PFGE, exponentially growing cells were harvested, washed in 10 mM Tris-HCl and 50 mM EDTA (pH 8.0), and cast in plugs of 0.56% low-melting-point agarose (MP Biomedicals) in 50 mM potassium phosphate and 50 mM EDTA (pH 7.0). Plugs were incubated in the same buffer containing 10 mM dithiothreitol and 0.4 mg/mL Zymolyase 20T (Seikagaku) for 24 h at 37°C. The solution was then replaced with 10 mM Tris-HCl (pH 8.0) containing 50 mM EDTA, 1% sarkosyl, and 2 mg/mL proteinase K (New England Biolabs), and the plugs were incubated for 24 h at 50°C. After extensive washing with 10 mM Tris-HCl and 50 mM EDTA (pH 8.0), the plugs were loaded into the wells of a 0.9% agarose gel (Seakem Gold, Ozyme) and electrophoresed at 13°C in a rotating PFGE apparatus (Rotaphor 6.0, Biometra) according to the manufacturer's instructions for separation of S. cerevisiae chromosomes. DNA was visualized by staining with 0.5 µg/mL ethidium bromide under UV illumination (Gel Doc, Bio-Rad).
Fluctuation assay for mutation rate estimation
Cells bearing the TetO2-TLC1 construct and the endogenous CAN1 gene were used for this assay. Five independent clones of each genotype (wild-type, cdc5-ad, and tid1Δ) were selected, grown in 5 or 10 mL of medium containing 30 µg/mL dox for 22 h at 30°C, and counted. Samples of 200–500 cells were plated on standard (control) YPD plates, and 107 or 108 cells were plated on synthetic complete medium-containing 2% glucose (SD) plates lacking arginine and supplemented with 60 µg/mL canavanine. Each day, the liquid cultures were diluted to 1 × 104 cells per milliliter. Plates were incubated for 48 h at 30°C, and the colonies were counted. The number of CanR colonies growing on the canavanine plates gives a value r corresponding to the number of mutants. The colony count on the control YPD plates was for correction of the cell number added to the canavanine plates. From the r values of the five independent cultures, which theoretically follows the Luria and Delbrück distribution, the mutation rate was estimated using the maximum likelihood method (MSS algorithm) (Sarkar et al. 1992; Foster 2006). This algorithm is based on the Lea and Coulson function that gives the different terms (pr)r≥0 of the Luria and Delbrück distribution for a given number of mutations m:
where (pr)r≥0 is the probability of obtaining a culture containing r mutants from a number of mutations m. To estimate the value of m that best fits the probability (pr)r≥0 from the experimental measure of r, the algorithm maximizes the likelihood function f:
where f(r∣m) = pr , and C is the number of cultures. After obtaining an estimated value of m, a correction was applied to take into account that only a fraction of the culture was used to estimate the number of mutants, which would lead to a sampling bias of mact = mobs[(z − 1)/zln(z)] , where mact is the corrected number of mutants, mobs is the number of mutants before correction, and z is the fraction of plated cells. The mutation rate was then obtained by dividing mact by the total number of cells in the culture.
The 95% confidence interval (mdown,mup) of mact was estimated according to the Stewart method (Stewart 1994; Foster 2006): mup = exp[ln(mact) + 1.96σ(e1.96σ)−0.315] and mdown = exp[ln(mact) − 1.96σ(e1.96σ)+0.315], where σ is the standard deviation of ln(mact), calculated with the following approximation:
where C is the number of cultures. Finally, mup and mdown were divided by the average total number of cells in the cultures to obtain the 95% confidence interval of the estimated mutation rate.
Microfluidic experiments
The microfluidic mold was fabricated using standard soft lithography techniques as described (Fehrmann et al. 2013). Epoxy replicates of the mold were used to make the chips. To cast the chip, polydimethylsiloxane (PDMS; Sylgard 184) and curing agent were mixed in a 10:1 ratio, degassed with a vacuum pump for 30 min, and poured into the mold. The PDMS was cured by baking for 5 h at 70°C and then carefully removed from the mold. A biopsy puncher (1.5 mm; Harris Unicore) was used to create holes to connect the tubing. The PDMS and a glass coverslip (24 × 50 mm) were surface-activated by plasma (Diener Electronic) to covalently bond the two parts. SD medium was filtered using a 0.22-µm polyethersulfone filter (Millipore) and loaded into the device using a peristaltic pump (IPCN, Ismatec). Cells from a log-phase culture (OD600 = 0.5) were gently injected into the device using a 1-mL syringe. A constant medium flow (28 µL/min) was maintained throughout the experiment. For experiments with strains expressing the TetO2-TLC1 construct, cells were allowed to divide and invade the cavities for 12–24 h before the medium was switched to SD containing 30 µg/mL dox.
Time-lapse and fluorescence microscopy
Cells in the microfluidic device were imaged using a fully motorized Axio Observer Z1 inverted microscope (Zeiss) with a 100× immersion objective, a Hamamatsu Orca R2 camera, and constant focus maintained with focus stabilization hardware (Definite focus, Zeiss). To minimize phototoxicity, we used light-emitting diode light sources for both phase contrast and fluorescence images (Colibri 2, Zeiss) with the following settings: for standard microfluidic time-lapse experiments, 4.0 V for 70 msec; for time-lapse experiments using the FHA1-mCherry/Hta2-yECFP reporters, 3.0 V for 40 msec; and for phase contrast, 10% of maximum intensity for 200 msec for the CFP channel and 15% of maximum intensity for 500 msec for the mCherry channel, with 2 × 2 binning in all three. The temperature was maintained at 30°C with a controlled heating unit and an incubation chamber that held the entire microscope base, including the stage and the objectives. Images were acquired every 10 min using Zen software (Zeiss). All aspects of image acquisition were fully automated and controlled, including temperature, focus, stage position, and time-lapse imaging. Images were acquired for up to 120 h in standard experiments. Static fluorescence microscopy experiments using the FHA1-mCherry reporter were performed with the following settings: 4.0 V for 70 msec for phase contrast, 100% intensity for 300 msec for the CFP channel, and 100% intensity for 1000 msec for the mCherry channel without binning. Ddc2-eGFP was detected similarly with the GFP filter (100% intensity for 400 msec). For the experiment using the single inducible HO cut in Supplemental Figure S4, B and C, live-cell images were acquired at the indicated time points after induction using a wide-field inverted microscope (Leica, DMI-6000B) equipped with adaptive focus control to eliminate Z drift, a 100×/1.4 NA immersion objective with a Prior NanoScanZ Nanopositioning Piezo Z stage system, a CMOS camera (ORCA-Flash 4.0, Hamamatsu), and a solid-state light source (SpectraX, Lumencore). The system was piloted by MetaMorph software (Molecular Devices). Fifteen focal steps of 0.25 µm were acquired sequentially for CFP, YFP, and mCherry, with an exposure time of 100 msec using solid state 434-, 475-, and 575-nm diodes and appropriate filters (GFP-mCherry filter: excitation double BP, 450–490/550–590 nm and dichroic double BP 500–550/600–665 nm; ECFP/EYFP/mCherry filter: excitation triple BP 420–450/490–515/560–590 and dichroic triple BP 455–485/520–550/600–670; Chroma Technology Corp.).
Image analysis and single-cell lineage tracking
Custom software written in Matlab, phyloCell 2.1 was used to segment and track cells and assign mother–daughter links. Time-lapse images were exported as high-resolution TIF files and analyzed with the graphical user interface of phyloCell. For details about the routines and algorithms implemented in phyloCell, see Fehrmann et al. (2013). In contrast to Fehrmann et al. (2013), the cell at the tip of the cavity would frequently be replaced by its daughter cell, which was intended in our approach. To efficiently track such lineages, in which we frequently switched focus from a given cell to its daughter cell, the time-lapse images were retrospectively analyzed starting from the last image. This avoided tracking of lineages in which the cells were ejected from the microcavity. In experiments involving the FHA1-mCherry/Hta2-yECFP reporter, the nucleus was segmented using the CFP channel with an Otsu threshold method, and the cell was segmented with either phyloCell for microfluidic experiments or a custom watershed-based method for phase contrast images. The nuclear mCherry signal was measured in the overlap of the cellular and nuclear masks and averaged over the measured area of the nucleus. For the experiment using the single inducible HO cut, image analysis was performed on the median slice of each image. Nuclei were segmented based on the CFP signal (automatic Otsu threshold), and the mean mCherry intensity was measured for each nucleus using Fiji software (Schindelin et al. 2012).
Checkpoint activation reporter system
The checkpoint activation reporter system consisted of the Rad53 FHA1 domain coupled to mCherry and the nuclear marker Hta2-yECFP. A codon-shuffled sequence encoding the Rad53 FHA1 domain (amino acids 14–164) flanked by two 15-amino-acid linkers was cloned into a pSIV URA3 vector under the pRPS20 promoter and fused with mCherry (pNM66) (Wosika et al. 2016). For Supplemental Figure S4, B and C, the wild-type and H75A mutant versions of FHA1 were cloned into the pSIV URA3 vector under the pRPL24A promoter (pNM65 and pNM142). BstBI-digested fragments of these plasmids were then transformed into the strains of interest and inserted into the URA3 locus. The endogenous HTA2 gene was tagged with yECFP-Kan amplified from the pKT102 plasmid (Sheff and Thorn 2004).
Computational and statistical analyses
All computational and statistical analyses were performed with the statistical toolbox of Matlab R2013a. Data are represented as the median, first and third quartiles, 99th percentiles (box plots), the mean ± SD, or the mean and 95% confidence interval, as indicated in the legends. Differences were compared using Student's t-test, the Mann-Whiney U test, and two-sample Kolmogorov-Smirnov test as indicated.
Supplementary Material
Acknowledgments
We thank David Toczyski, Jim Haber, and Naïma Belgareh Touzé for yeast strains and plasmids; Pascale Jolivet, Aubin Fleiss, and Gilles Fischer for technical help; and Eric Vandjour for medium preparation. We also thank members of M.T.T.’s laboratory, Miguel Ferreira, Claus Azzalin, Ted Weinert, Titia de Lange, Joachim Lingner, Jim Haber, and Damien D'Amours for fruitful discussions. K.D. thanks the European Research Council under the European Community's Seventh Framework Program (FP7/2007 2013/European Research Council grant agreement 281287) for funding. S.P. thanks the Swiss National Science Foundation and the University of Lausanne for funding, and Nadim Mira for technical assistance. This work was supported by “Fondation de la Recherche Medicale” (“MTT Équipe Labellisée”) and the French National Research Agency (ANR) as part of the “Investissements d'Avenir” Program (LabEx Dynamo) and ANR-16-CE12-0026 to M.T.T. and G.C. H.C. has been supported by a doctoral grant from the Paris Sciences et Lettres (PSL) Idex program implemented by the ANR (ANR-10-IDEX-0001-02 PSL).
Author contributions: Z.X. and M.T.T. conceived the study. H.C., Z.X., S.P., G.C., K.D., and M.T.T. performed the methodology. Z.X. and G.C. were responsible for the software used. H.C. and Z.X. performed the formal analysis. H.C., Z.X., M.C.M., M.L.-A., S.P., and K.D. performed the investigations. H.C., Z.X., and M.T.T. wrote the original draft of the manuscript. H.C. and Z.X. visualized the study. Z.X. and M.T.T. supervised the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.318485.118.
Freely available online through the Genes & Development Open Access option.
References
- Artandi SE, DePinho RA. 2000. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr Opin Genet Dev 10: 39–46. [DOI] [PubMed] [Google Scholar]
- Artandi SE, DePinho RA. 2010. Telomeres and telomerase in cancer. Carcinogenesis 31: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunan JR, Cho WC, Soreide K. 2017. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis 8: 628–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batte A, Brocas C, Bordelet H, Hocher A, Ruault M, Adjiri A, Taddei A, Dubrana K. 2017. Recombination at subtelomeres is regulated by physical distance, double-strand break resection and chromatin status. EMBO J 36: 2609–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer T, Weinert T. 2016. Ontogeny of unstable chromosomes generated by telomere error in budding yeast. PLoS Genet 12: e1006345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34. [DOI] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740. [DOI] [PubMed] [Google Scholar]
- Carneiro MC, de Castro IP, Ferreira MG. 2016. Telomeres in aging and disease: lessons from zebrafish. Dis Model Mech 9: 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. 1999. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet 23: 81–85. [DOI] [PubMed] [Google Scholar]
- Chen C, Umezu K, Kolodner RD. 1998. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell 2: 9–22. [DOI] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. 1999. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538. [DOI] [PubMed] [Google Scholar]
- Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP. 2014. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J 33: 198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198. [DOI] [PubMed] [Google Scholar]
- Donnianni RA, Ferrari M, Lazzaro F, Clerici M, Tamilselvan Nachimuthu B, Plevani P, Muzi-Falconi M, Pellicioli A. 2010. Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet 6: e1000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher D, Henckel J, Fersht AR, Jackson SP. 1999. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell 4: 387–394. [DOI] [PubMed] [Google Scholar]
- Emili A. 1998. MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 2: 183–189. [DOI] [PubMed] [Google Scholar]
- Enomoto S, Glowczewski L, Berman J. 2002. MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Mol Biol Cell 13: 2626–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallet E, Jolivet P, Soudet J, Lisby M, Gilson E, Teixeira MT. 2014. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res 42: 3648–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann S, Paoletti C, Goulev Y, Ungureanu A, Aguilaniu H, Charvin G. 2013. Aging yeast cells undergo a sharp entry into senescence unrelated to the loss of mitochondrial membrane potential. Cell Rep 5: 1589–1599. [DOI] [PubMed] [Google Scholar]
- Foster PL. 2006. Methods for determining spontaneous mutation rates. Methods Enzymol 409: 195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgoczy DJ, Toczyski DP. 2001. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol 21: 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik B, Carson M, Hartwell L. 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 15: 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Garcia MP, Pavelescu I, Canela A, Sevillano X, Leehy KA, Nelson AD, Ibanes M, Shippen DE, Blasco MA, Cano-Delgado AI. 2015. Single-cell telomere-length quantification couples telomere length to meristem activity and stem cell development in Arabidopsis. Cell Rep 11: 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW. 1998. Telomeres and senescence: the history, the experiment, the future. Curr Biol 8: R178–R181. [DOI] [PubMed] [Google Scholar]
- Hackett JA, Greider CW. 2003. End resection initiates genomic instability in the absence of telomerase. Mol Cell Biol 23: 8450–8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Feldser DM, Greider CW. 2001. Telomere dysfunction increases mutation rate and genomic instability. Cell 106: 275–286. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100: 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Harrison JC, Haber JE. 2006. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40: 209–235. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. 2006. Cellular senescence in aging primates. Science 311: 1257. [DOI] [PubMed] [Google Scholar]
- Ijpma AS, Greider CW. 2003. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell 14: 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Cooper JP. 2010. Telomeric strategies: means to an end. Annu Rev Genet 44: 243–269. [DOI] [PubMed] [Google Scholar]
- Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. 2007. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 128: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsbeek D, Golsteyn RM. 2017. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int J Mol Sci 18: E2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP. 2004. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol 14: 2096–2106. [DOI] [PubMed] [Google Scholar]
- Lea DE, Coulson CA. 1949. The distribution of the numbers of mutants in bacterial populations. J Genet 49: 264–285. [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW II, Greider CW, DePinho RA. 1998a. Essential role of mouse telomerase in highly proliferative organs. Nature 392: 569–574. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. 1998b. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409. [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Malkova A, Foiani M, Haber JE. 2001. The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr Biol 11: 1053–1057. [DOI] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, Marsolier-Kergoat MC. 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell 11: 827–835. [DOI] [PubMed] [Google Scholar]
- Lingner J, Cooper JP, Cech TR. 1995. Telomerase and DNA end replication: no longer a lagging strand problem? Science 269: 1533–1534. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH. 1993. An alternative pathway for yeast telomere maintenance rescues est1− senescence. Cell 73: 347–360. [DOI] [PubMed] [Google Scholar]
- Lundblad V, Szostak JW. 1989. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57: 633–643. [DOI] [PubMed] [Google Scholar]
- Lydall D, Weinert T. 1995. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270: 1488–1491. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, de Lange T. 2017. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. 2015. Chromothripsis and kataegis induced by telomere crisis. Cell 163: 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE. 2005. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP. 2001. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersaoui SY, Gravel S, Karpov V, Wellinger RJ. 2015. DNA damage checkpoint adaptation genes are required for division of cells harbouring eroded telomeres. Microb Cell 2: 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murnane JP. 2006. Telomeres and chromosome instability. DNA Repair 5: 1082–1092. [DOI] [PubMed] [Google Scholar]
- Pardo B, Marcand S. 2005. Rap1 prevents telomere fusions by nonhomologous end joining. EMBO J 24: 3117–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschini M, Toro TB, Lubin JW, Braunstein-Ballew B, Morris DK, Lundblad V. 2012. A naturally thermolabile activity compromises genetic analysis of telomere function in Saccharomyces cerevisiae. Genetics 191: 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. 2001. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7: 293–300. [DOI] [PubMed] [Google Scholar]
- Pobiega S, Marcand S. 2010. Dicentric breakage at telomere fusions. Genes Dev 24: 720–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA. 1993. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 75: 729–739. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ma WT, Sandri GH. 1992. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85: 173–179. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. 2002. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9: 1055–1065. [DOI] [PubMed] [Google Scholar]
- Serrano D, D'Amours D. 2014. When genome integrity and cell cycle decisions collide: roles of polo kinases in cellular adaptation to DNA damage. Syst Synth Biol 8: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Zheng X, Epshtein A, Carey JN, Bishop DK, Klein HL. 2010. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Mol Cell 39: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Reddel RR, Wright WE. 2012. Cancer. Cancer and telomeres—an ALTernative to telomerase. Science 336: 1388–1390. [DOI] [PubMed] [Google Scholar]
- Sheff MA, Thorn KS. 2004. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21: 661–670. [DOI] [PubMed] [Google Scholar]
- Soudet J, Jolivet P, Teixeira MT. 2014. Elucidation of the DNA end-replication problem in Saccharomyces cerevisiae. Mol Cell 53: 954–964. [DOI] [PubMed] [Google Scholar]
- Stewart FM. 1994. Fluctuation tests: how reliable are the estimates of mutation rates? Genetics 137: 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suram A, Herbig U. 2014. The replicometer is broken: telomeres activate cellular senescence in response to genotoxic stresses. Aging Cell 13: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. 2005. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 15: 1364–1375. [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Jensen S, Bartek J, Lukas J. 2006. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res 66: 10253–10257. [DOI] [PubMed] [Google Scholar]
- Teixeira MT. 2013. Saccharomyces cerevisiae as a model to study replicative senescence triggered by telomere shortening. Front Oncol 3: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH. 1997. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90: 1097–1106. [DOI] [PubMed] [Google Scholar]
- Vasan S, Deem A, Ramakrishnan S, Argueso JL, Malkova A. 2014. Cascades of genetic instability resulting from compromised break-induced replication. PLoS Genet 10: e1004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF. 1998. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 17: 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidanes GM, Sweeney FD, Galicia S, Cheung S, Doyle JP, Durocher D, Toczyski DP. 2010. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol 8: e1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosika V, Durandau E, Varidel C, Aymoz D, Schmitt M, Pelet S. 2016. New families of single integration vectors and gene tagging plasmids for genetic manipulations in budding yeast. Mol Genet Genomics 291: 2231–2240. [DOI] [PubMed] [Google Scholar]
- Wu RA, Upton HE, Vogan JM, Collins K. 2017. Telomerase mechanism of telomere synthesis. Annu Rev Biochem 86: 439–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Fallet E, Paoletti C, Fehrmann S, Charvin G, Teixeira MT. 2015. Two routes to senescence revealed by real-time analysis of telomerase-negative single lineages. Nat Commun 6: 7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.