

Summary
During sepsis, small blood vessels can become occluded by large platelet aggregates of poorly understood etiology. During Staphylococcal aureus infection, sepsis severity is linked to the bacterial alpha toxin (α-hemolysin, AT) through unclear mechanisms. In this study, we visualized intravascular events in the microcirculation and found that intravenous AT injection induces rapid platelet aggregation, forming dynamic micro-thrombi in the microcirculation. These aggregates are retained in the liver sinusoids and kidney glomeruli, causing multi-organ dysfunction. Acute staphylococcal infection results in sequestration of most bacteria by liver macrophages. Platelets are initially recruited to these macrophages and help eradicate S. aureus. However, at later time points, AT causes aberrant and damaging thrombosis throughout the liver. Treatment with an AT neutralizing antibody (MEDI4893*) prevents platelet aggregation and subsequent liver damage, without affecting the initial and beneficial platelet recruitment. Thus, AT neutralization may represent a promising approach to combat staphylococcal-induced intravascular coagulation and organ dysfunction.
Graphical Abstract
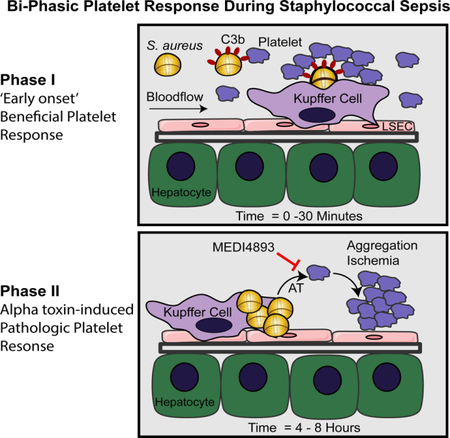
eTOC paragraph
Staphylococcal sepsis often results in thrombocytopenia and multi-organ dysfunction. Using intravital imaging, Surewaard et al. discovered that alpha-toxin (AT) directly targets platelets resulting in detrimental aggregation in the circulation. Neutralizing AT during staphylococcal sepsis does not interfere with beneficial platelet responses, while preventing microvascular dysfunction and thrombosis.
INTRODUCTION
Severe sepsis is a clinical manifestation of the host’s inflammatory reaction to bloodstream infections causing a collapse of cardiovascular function, leading to multiple organ dysfunction or failure (Rittirsch et al., 2008). One of the most significant problems in sepsis is the occlusion of small blood vessels throughout the body, resulting from dysregulated systemic activation of thrombosis, often coupled with accelerated fibrinolysis and bleeding tendency, referred to as disseminated intravascular coagulation (DIC) (Levi and ten Cate 1999). Formation of large platelet aggregates of poorly understood etiology leads to thrombotic microangiopathy, vascular occlusion, multi-organ failure, and astonishingly high mortality rates of up to 50% of patients with bacteremias such as Staphylococcus aureus (van der Poll and Opal, 2008; van der Poll et al., 2017). In fact, S. aureus is one of the most lethal, as well as one of the most common, causes of human sepsis (Lowy, 1998). The ability of S. aureus to acquire resistance to most antibiotics, along with its ability to subvert innate immune responses, makes it imperative that new antibacterial strategies are implemented to combat S. aureus pathogenesis (Chambers and DeLeo, 2009; Spaan et al., 2013). Intriguingly, S. aureus is particularly adept at causing thrombocytopenia, presumably a result of systemic platelet activation. While some antibiotics are still effective at killing S. aureus, microvascular thrombosis can persist or even progress following antimicrobial therapy, in part, due to bacterial proteins or toxins released into the blood stream from dying or lysed bacteria (Lewis and Surewaard, 2017; Powers and Bubeck Wardenburg, 2014). Several staphylococcal virulence factors, including the cytolytic α-toxin (AT), clumping factor A, fibronectin-binding protein, and cell-wall peptidoglycan have been shown to activate platelets and affect coagulation (Kessler et al., 1991; Thomer et al., 2016). Despite this clear association between S. aureus infection and coagulation, no specific targeted treatment is presently available.
Staphylococcal AT is essential for virulence in various animal infection models including, skin and soft tissue infections (SSTI), pneumonia and bacteremia (Bubeck Wardenburg et al., 2007; Sampedro et al., 2014; Wardenburg and Schneewind, 2008). In patients, AT is expressed by most clinical isolates and AT expression level correlates with disease severity (Jenkins et al., 2015). AT was originally noted for its ability to lyse rabbit red blood cells (Berube and Bubeck Wardenburg, 2013). In vitro experiments have also shown that AT can target epithelium, monocytes, neutrophils endothelium and macrophages, however which of these cells is key target for AT during sepsis is not known (Berube and Bubeck Wardenburg, 2013). Research dating back 50 years has shown that AT caused platelet swelling, dissolution of granules, as well as clotting at sub-lytic concentrations for red blood cells (Bhakdi et al., 1988; Siegel and Cohen, 1964), AT also induces platelet-neutrophil interactions in a platelet P-selectin dependent manner (Parimon et al., 2013). In addition, recent work has shown that AT can affect normal platelet function. Platelets incubated with subcytolytic concentrations of AT in vitro are impaired in function through glycoprotein VI modification, resulting in reduced platelet adhesion and aggregation to fibrinogen and von Willebrand factor (vWF) (Powers et al., 2015). Interestingly, this work also identified the cellular receptor a disintegrin and metalloproteinase 10 (ADAM10) being involved in AT mediated platelet dysfunction. ADAM10 a cell membrane-associated protease was shown to facilitate AT binding and cleavage of GPVI. Compound knockout of ADAM10 on myeloid cells and platelets blocked mortality from a lethal staphylococcal sepsis indicating a crucial role for this toxin and its interplay with ADAM10 on several cell types (Powers et al., 2015). In addition to AT, other staphylococcal toxins such as the bi-component pore forming toxins; e.g. leukotoxin ED(Alonzo et al., 2013; Spaan et al., 2017), superantigens (Xu et al., 2014) and α-type phenol-soluble modulins (PSMs)(Wang et al., 2007) have all been attributed to play a role during bloodstream infections and facilitating organ damage, however none of these toxins appear to target platelets directly.
Although depleting or impairing platelet function could be a strategy to prevent S. aureus-induced thrombosis, we previously reported that platelets cooperate with macrophages of the liver (Kupffer cells) to help eradicate S. aureus (Wong et al., 2013). Kupffer cells catch S. aureus then platelets rapidly bind to the macrophage, preventing escape of the pathogen, thereby making platelet-depletion a deadly proposition (Wong et al., 2013). By contrast, neutralizing AT with monoclonal antibodies (mAbs) increased epithelial integrity, survival and bacterial clearance in a murine pneumonia model leading to current clinical trials for treatment of S. aureus pneumonia (Cohen et al., 2016; Hua et al., 2014; Ragle and Bubeck Wardenburg, 2009). While the latter work did not examine vascular pathology, understanding how AT works and how it contributes to the vascular dysfunction associated with severe sepsis would be extremely useful. To date, much of the work on AT has been performed in vitro in isolated systems lacking endothelium which has both pro- and anti-coagulation capacity (Nachman and Rafii 2008). Also, the heterogeneity of different organs (e.g. intravascular macrophages adherent in the liver vasculature), is impossible to recapitulate in vitro. In fact, AT has been shown to exert disparate effects in different tissue compartments (Powers et al., 2015), making it imperative to study these dynamic events in vivo in different organ systems. We systematically evaluated the role of AT using spinning-disk intravital microscopy (SD-IVM) to gain insight into the key intravascular and cellular interactions that occur following AT intoxication within the bloodstream of various organs. We also visualized the intravascular responses to S. aureus infection and determined the importance of inhibiting AT on platelet function, thrombosis and propensity to survive sepsis.
RESULTS
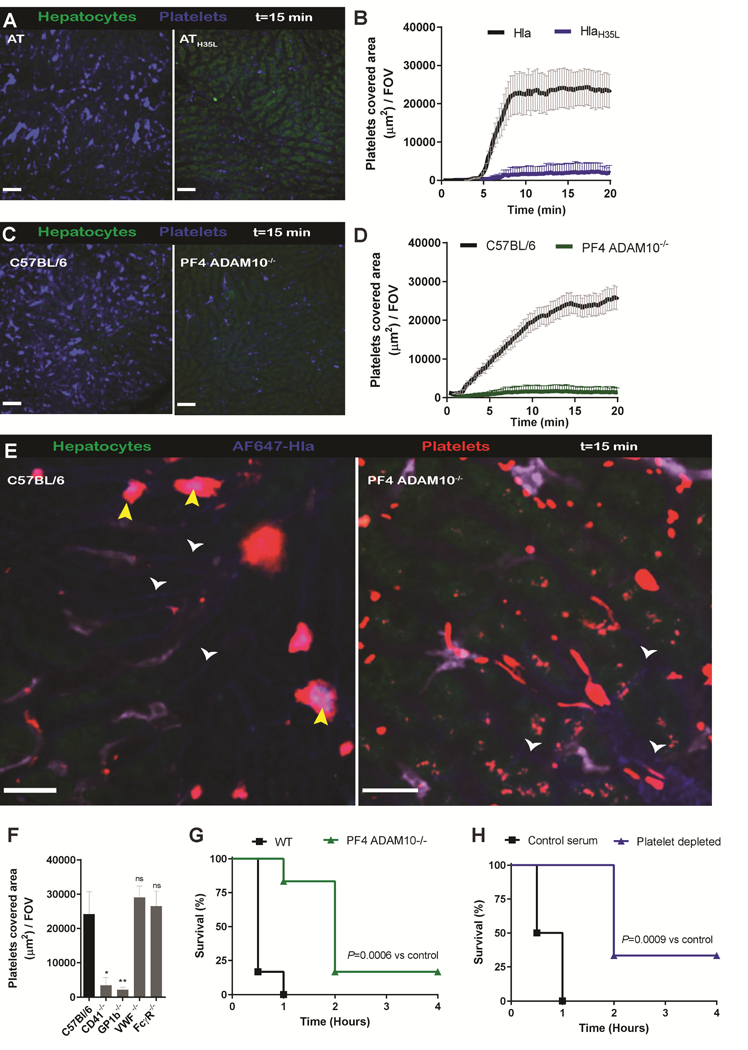
AT intoxication leads to systemic platelet aggregation and subsequent deposition in the liver.
With SD-IVM of the liver microvasculature, we recently showed the continuous ‘touch-and-go’ behavior of circulating platelets binding to and letting go of intravascular Kupffer cells of the liver (Jenne et al., 2011; Wong et al., 2013). After intravenous saline administration, very few platelets adhered to the intravascular macrophages and only a few aggregates were formed suggesting little or no activation. In stark contrast, mice intravenously injected with AT (1 μg/mouse) had massive platelet aggregation in the bloodstream with subsequent deposition of platelet aggregates in the liver (Figure 1A, B and Video 1). Approximately 50% of these platelet aggregates were present on the surface of Kupffer cells, with the remaining aggregates adhering to the sinusoidal endothelium (Figure 1C). Indeed, depletion of Kupffer cells with clodronate liposomes reduced sequestration of platelet aggregates in the liver by about half (Figure 1D) in line with 50% of aggregates adhering to endothelium. High-magnification images of the aggregates showed a core of stably adherent platelets that were P-selectin-positive, consistent with α- granule secretion and irreversible activation of the platelets (Figure 1E). We observed minimal expression of P-selectin on the sinusoidal endothelium consistent with this molecule being primarily upregulated on platelets. However, expression of P-selectin following AT administration lagged platelet accumulation by 5–10 min, suggesting that P-selectin was not required for the initial adhesion of platelets. The thrombotic core was surrounded by a loose shell of P-selectin negative platelets not dissimilar to hemostatic plugs observed in the cremaster muscle after laser-induced endothelial damage (Stalker et al., 2013).
Figure 1.
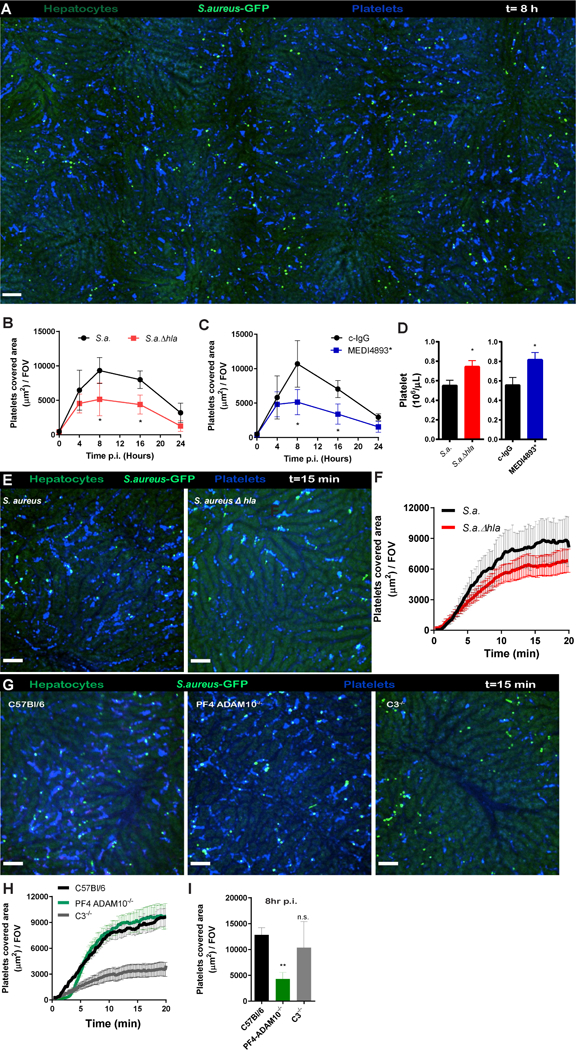
(A) SD-IVM image of the liver before and 15 min after AT or saline infusion in C57Bl/6 mice. Images were taken from (Video 1). Autofluorescent hepatocytes (green), Kupffer cells (F4/80; purple), platelets (CD49b; blue). Bars, 50 μm. (B) Enumeration of SD-IVM videos form (A) n = 4 mice; Line represents mean ± SEM. (C) High-magnification SD-IVM image of platelet (CD49b; blue) aggregates adhering to liver Kupffer cells; (F4/80; purple) white arrowhead or to the sinusoidal endothelium; yellow arrowhead. (D) Quantification of SD-IVM images of mice treated with control or clodronate liposomes (CCL) following AT intoxication. n = 4 mice; line represents mean ± SEM (E) SD-IVM image of image of platelet (CD49b; blue) aggregate co-localizing with P-selectin (CD62p; green), P-selectin expression was used as marker for α-granule degranulation and irreversible platelet aggregation, degranulation followed platelet aggregation with a 5 min delay. Representative SD-IVM images were taken 15min (D) and 25 min (E) post-AT administration from three independent experiments. Bars, 25 μm. (F) Enumeration of platelet aggregates by SD-IVM in different organs 15 min after AT intoxication of C57BL/6 mice. (Video 2) n = 3 mice with 5 FOV analyzed per organ. Data are represented as mean ± SEM. (G) Multi-photon image of kidney 15 min post-AT intoxication. Vasculature (cyan, Q-tracker 596), platelets (cd49b; red) Representative images of two independent experiments.
We performed SD-IVM on three additional vascular beds. Interestingly, AT administration induced intravascular platelet aggregate formation in the lung and skin microvasculature, but the aggregates adhered only transiently and were rapidly washed away by the blood flow. Additionally, we observed no aggregates forming in blood vessels of the tubules of the kidney (Figure. 1F and Video 2), but using multi-photon microscopy, we observed significant platelet aggregation in glomeruli following AT administration (Figure 1G). Importantly, at the injected dosage of AT (1μg/mouse) no signs of hemolysis was observed visually (Figure S1A) or using the hemolysis biomarker haptoglobulin (Figure S1B), in line with the relative insensitivity of murine erythrocytes to AT (Cooper et al., 1966). In addition, at the concentration of AT used, there was very little evidence of cell death determined by the lack of staining of endothelium and liver tissue with propidium iodide (Figure S1C). Titrating down the AT-dosage to 250ng per mouse still showed substantial platelet aggregation in the liver (Figure S1D) suggesting exquisite platelet sensitivity to AT. Both AT preparations tested from a commercial vendor (data not shown) and ultrapure recombinant AT (with no detectable levels of LPS or other contaminants), induced platelet aggregation suggesting a specific effect of AT and not a contaminant. These results show that sub-lytic AT concentrations mediated rapid platelet aggregation in the circulation which accumulated primarily in the liver and to more limited extent in kidney glomeruli. The lack of aggregates adhering in skin and lack of retention of aggregates in lung suggests significant organ heterogeneity.
AT binds and activates platelets in an ADAM10 dependent fashion
Previous work reported that ADAM10 functions as the AT receptor, promoting oligomerization of seven toxin monomers into a transmembrane pore to facilitate cell lysis (Inoshima et al., 2011). To investigate whether AT oligomerization was also required for AT-induced platelet aggregation, we infused non-toxigenic AT (ATH35L) which cannot oligomerize or activate ADAM10, yet retains binding to ADAM10 (Powers et al., 2015). Despite still binding platelets and purified in similar manner as WT AT, no platelet aggregation was noted with ATH35L (Figure 2A,B) again suggesting that the activation was not due to a contamination. Because many cell types, including platelets and endothelial cells, are reported to express ADAM10 we utilized PF4 ADAM10−/− mice which lack ADAM10 exclusively on platelets to examine whether AT exerts platelet aggregation directly, or by eliciting endothelial damage. Administration of AT to these animals failed to induce any platelet aggregation (Figure 2C,D), despite knowledge that these platelets responded normally to other physiologic stimuli (Powers et al., 2015). Use of fluorescent labeled AT, revealed intense staining at the core of the platelet aggregates, showing AT binding directly to platelets to form the large aggregates. Administration of the AF647 labelled-AT in PF4 ADAM10−/− mice resulted in no toxin binding to platelets, and little aggregate formation (Figure 2E). It is worth noting that in both wild-type and PF4 ADAM10−/− mice some of the AF647 AT did bind to the endothelium but caused minimal platelet aggregate formation in those localized areas.
Figure 2.

(A) SD-IVM images of the liver 15 min after AT or ATH35L infusion in C57Bl/6 mice. Autofluorescent hepatocytes (green), Kupffer cells (F4/80; purple), platelets (CD49b; blue). (B) Quantification of SD-IVM images form (A) n = 4 mice; Line represents mean ± SEM. (C) SD-IVM images of the liver 15 min after AT infusion in C57Bl/6 or PF4-ADAM10−/− mice, (D) Quantification of SD-IVM videos form (C) n = 5 mice; Line represents mean ± SEM. (E) High magnification SD-IVM image of the liver after infusion of N-terminally labelled AT (AT-AF647; blue) in WT C57Bl/6 (left) or PF4-ADAM10−/− mice (right). Platelet aggregates (CD49b; red), Kupffer cells (F4/80; purple). Yellow arrowheads; blue AT staining on platelet aggregates. White arrowheads; AT binding to the sinusoidal endothelium. (F) Quantification of platelet aggregation after AT intoxication in C57Bl/6, CD41−/−, GP1b−/−, VWF−/− or FcyR−/− mice. * P < 0.05 or** P < 0.01, NS; not significant, versus C57Bl/6. One-way ANOVA with Bonferroni’s posttest. (G) Survival of AT intoxicated C57Bl/6 or PF4-ADAM10−/− mice. n = 5 mice per treatment group; log-rank test. (H) Survival of AT intoxicated control or platelet depleted mice. n = 5 mice per treatment group; log-rank test
Recently, we showed that during S. aureus infection Glycoprotein 1b (GPIb) bound to vWF to allow platelet tethering followed by sustained GPIIb-GPIIIa (CD41)-dependent platelet aggregation on Kupffer cells (Wong et al., 2013). In this study, we exposed GP1ba−/− and CD41 deficient-mice to intravenous AT intoxication and both strains of mice were completely unable to support AT-induced platelet aggregation (Figure 2F). It is important to note that GP1b−/− mice have enlarged platelets and are partially thrombocytopenic making definitive conclusions difficult. Indeed, mice deficient in vWF, the ligand for GPIb, retained WT levels of AT-induced platelet aggregation (Figure 2F) suggesting a different platelet aggregation mechanism than that involved in platelet binding to vWF immobilized on Kupffer cells. In addition, FcRy−/− mice, lacking the Fc receptor common y-chain involved in immunoreceptor tyrosine-based activation motif signaling downstream of GPVI and also mediating a role in GPIb signaling (Wu et al., 2001) showed unaltered platelet accumulation after AT challenge (Figure 2F). These data suggest a predominant role for GPIIb-GPIIIa but most likely not other platelet adhesion molecules.
Platelet aggregation could be either an essential scavenging mechanism providing benefit or a subversive mechanism utilized by S. aureus to cause host injury. Despite no overt signs of cell lysis with the concentration of AT used in our studies, platelet aggregation was associated with rapid mortality in WT mice and more delayed mortality in PF4 ADAM10−/− mice (Figure 2G). Mice in which platelets were depleted survived longer after AT administration indicating a clear contribution of platelet aggregation to the associated mortality (Figure 2H). This phenomenon was opposite to what was observed with intravenous S. aureus or Bacillus cereus injection, which caused localized platelet accumulation on Kupffer cells and this innate immune mechanism helped with eradication of the bacteria and prevented mortality (Wong et al., 2013). Therefore, it would seem likely that there were two phases of platelet aggregation; an early AT independent beneficial phase and a later AT-dependent detrimental phase. Given AT direct intravenously bypassed the beneficial phase.
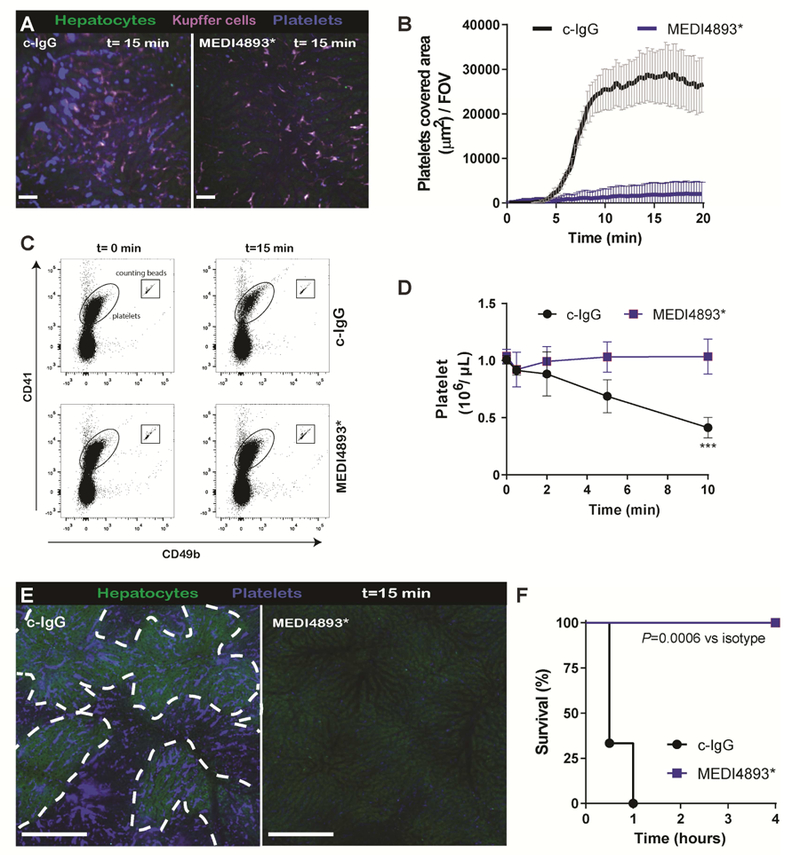
Anti-AT MEDI4893* monoclonal antibody prevents AT-induced platelet aggregation.
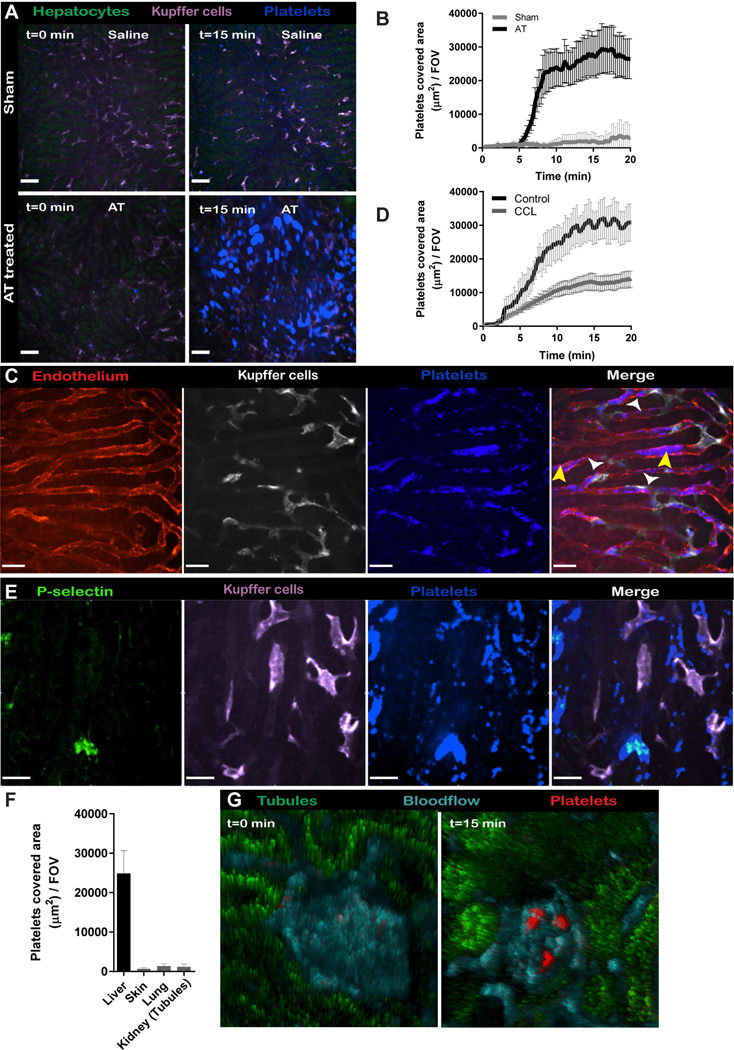
Since platelets contributed to protection against S. aureus infections, targeting platelets directly would appear likely to increase susceptibility to lethal disease. To determine whether we could prophylactically overcome AT-induced platelet aggregation and associated lethality in mice, we passively immunized mice with an AT neutralizing monoclonal antibody (mAb), MEDI4893* which essentially abolished AT-induced platelet aggregation (Figure 3A,B and Video 3). Furthermore, administration of AT compared to isotype control IgG (c-IgG) treated mice showed a clear 50% drop in circulating blood platelets within 10 min, whereas the platelet levels remained unchanged in MEDI4893* treated mice (Figure 3C,D). Twenty min post-AT injection, in MEDI4893* treated mice the mAb prevented the platelet aggregates, and less non-perfused ischemic liver was observed (Figure 3E). Importantly, non-perfused liver areas will ultimately lead to tissue damage, focal necrotic lesions and organ failure (Kolaczkowska et al., 2015). Mice treated with MEDI4893* antibodies were completely protected against AT-induced mortality (Figure 3F). Having a tool to effectively inhibit AT, we turned our attention to studying S. aureus infections.
Figure 3.
(A) SD-IVM images of the liver 15 min after AT intoxication in prophylactically treated mice with c-IgG or MEDI4893*. Autofluorescent hepatocytes (green), Kupffer cells (F4/80; purple), platelets (CD49b; blue). c-IgG or MEDI4893* (Video 3). (B) Quantification of SD-IVM videos form (A) n = 4 mice; Line represents mean ± SEM. (C) Representative flow cytometry plots of blood platelets before and 10 min after AT intoxication in c-IgG or MEDI4893* treated C57Bl/6 mice. Platelets and quantification beads were gated on side scatter, CD41 and CD49b expression levels. (D) Quantification of blood platelet levels as shown in (C) at different time points post-AT intoxication. n=3 mice. Two way ANOVA with multiple comparison. *** P<0.001. (E) Representative stitched SD-IVM image of liver prophylactically treated mice with c-IgG or MEDI4893*, 20 min after AT intoxication. Encircled dark spots represent non-perfused liver areas which lack the green autofluorescence of hepatocytes. Platelets (CD49b; blue). Bar; 200μm (F) Survival of prophylactically treated mice with c-IgG or MEDI4893* following AT intoxication n = 5 mice per treatment group; log-rank test. In all experiments mAbs were i.p. administered at 15MPk 24h before intoxication.
S. aureus-induced platelet aggregation via AT-dependent and complement-dependent mechanisms.
To investigate whether AT is the major secreted factor from S. aureus that induces platelet aggregation, we injected culture supernatants from WT and AT-deficient staphylococci into mice. Very little platelet aggregation was observed when mice were infused with sterile culture media. Profound platelet aggregation, comparable to that observed after AT injection, was noted in the livers of mice that received WT staphylococcal supernatant. By contrast, platelet aggregation was greatly reduced in mice injected with supernatant from a S. aureus AT knockout strain (S. aureusΔhla) Figure S2A,B and Video 4).
Upon infection, it is likely that S. aureus needs to adjust to its new environment and does not secrete critical concentrations of AT instantaneously. Therefore its effects may be delayed compared to intravenous injection of purified AT or culture supernatant. As there was greater variability and patchiness in platelet aggregation with the bacterium, the data were analyzed across large liver areas (Figure 4A). Intravenous injection of viable staphylococci caused significant aggregate formation at 4 h (Figure 4A), and by 8 and 16 h post-infection there was an additional two-fold increase in platelet aggregation (Figure 4B). By contrast, infection with an AT-deficient mutant resulted in an initial increase in platelet aggregation at 4 h which did not increase further at 8 or 16 h post-infection (Figure 4B), suggesting that S. aureus needs approximately 4–8 h to ramp up AT production and achieve maximal platelet aggregation. As with AT-mutant, iv S. aureus induced a similar platelet aggregation profile was observed with MEDI4893* treated animals (Figure 4C). Again, significant reduction of platelet aggregates was observed at the 8 and 16 h time points but not at 4 h post-infection with the anti-AT antibody treatment. No sex differences were observed in platelet aggregation post-staphylococcal infection (Figure S4). Additionally, flow cytometric analysis of the blood at 8 h post-infection revealed increased thrombocytopenia in WT infected or c-IgG treated animals when compared to the AT mutant or MEDI4893*treated animals (Figure 4D).
Figure 4.
(A) Stitched SD-IVM image of mouse liver at 8 h after i.v. infection with S. aureus GFP (5×107 CFU, USA300 LAC-GFP; bright green), autofluorescent hepatocytes (dull green) and platelets (CD49b; blue). Bar, 250 μm. (B) Quantification of platelet accumulation in the mouse livers at different time points after infection with wild-type S. aureus (USA300 LAC) or S. aureusΔhla. n = 4 per group. Data represents mean ± SEM. (C) Quantification of platelet accumulation in the mouse livers at different time points after infection with S. aureus in prophylactically treated mice with c-IgG or MEDI4893* mAb. c-IgG or MEDI4893* mAbs were i.v. administered at 30MPk 3h prior to AT intoxication. n = 4 per group. (B-C) Two- way ANOVA with multiple comparison. * P<0.05. Data represents mean ± SEM. (D) Quantification of blood platelet levels measured by flowcytometry at 8h post-infection with wild-type S. aureus or S. aureusΔhla (left) or prophylactically treated mice with c-IgG or MEDI4893* infected with wild-type S. aureus (right).Mice were injected with 30MPK mAbs at 2hrs prior to infection. n = 5 mice. *P<0.05. Data represents mean ± SEM.t-test.* p<0.05. (E) SD-IVM images of the liver 15 min after infection with wild-type S. aureus (USA300 LAC) or S. aureusΔhla in C57Bl/6 mice. S. aureus-GFP (bright-green), platelets (CD49b; blue). (F) Quantification of SD-IVM videos of mice infected with wild-type S. aureus (USA300 LAC) or S. aureusΔhla in C57Bl/6 mice. n = 4 mice Data represents mean ± SEM. Video 5 (G) SD-IVM images of the liver 15 min after infection with wild-type S. aureus-GFP in C57Bl/6, PF4ADAM10−/−or C3−/−-mice. (H) Quantification of platelet aggregation in mouse livers of SD-IVM images form (G) n = 4 mice; Line represents mean ± SEM. (I) Quantification of platelet accumulation in the mouse livers at 8h after infection with wild-type S. aureus-GFP in C57Bl/6, PF4ADAM10−/−or C3−/−-mice. n = 4 mice; Data represents mean ± SEM. One-way ANOVA with multiple comparison. ** P<0.01.
Clearly, platelet aggregation at later time points (8 and 16 h) was AT-dependent. To demonstrate the early recruitment of platelets on Kupffer cells was independent of AT, WT S. aureus or the isogenic S. aureusΔhla mutant were injected and platelet aggregation was followed using SD-IVM. Intravenous staphylococcal injection showed no significant difference in platelet aggregation on the surface of Kupffer cells between the two staphylococcal strains, indicating that AT does not influence the previously described early protective platelet response (Figure 4E,F and Video 5). In support of this, we found that the protective platelet response was still present in PF4-ADAM10−/− mice. (Figure 4G,H). Surprisingly, heat-inactivated staphylococci were still able to induce the early platelet accumulation on Kupffer cells, indicating that this aggregation is not an active bacterial virulence mechanism, but rather an innate immune-mediated defense (data not shown). Recently, we showed that complement component 3 (C3) is not necessary for staphylococcal capture by Kupffer cells (Zeng et al., 2016). Interestingly, early onset platelet accumulation on Kupffer cells was diminished in C3−/− mice following S. aureus infection, despite normal catching of bacteria (Figure 4G,H). Importantly, platelets from C3−/− mice did not exhibit reduced aggregation in response to recombinant AT (data not shown). Eight hours post-staphylococcal infection there was similar numbers of platelet aggregates were present in the liver of the C3−/− mice and WT mice (Figure 4I). Suggesting complement played no role in AT-dependent platelet aggregation. By contrast PF4-ADAM10−/− had greatly reduced platelet aggregates at 8 h post infection. −Collectively, these data suggest that staphylococcal infection induces an early C3-dependent protective platelet response, followed by pathological AT-dependent platelet aggregation in the liver.
S. aureus-induced platelet aggregation causes AT-dependent pathology
To initially visualize the platelet responses by SD-IVM following staphylococcal infection, a non-lethal dose of 5×107CFU S. aureus was first used. As such, no mice died post-infection, however pathologically, S. aureus caused large focal necrotic lesions in the liver and increased ALT levels 24 hours after infection (Figure 5A,B) along with an increase in creatinine levels (data not shown). The AT mutant caused notably fewer lesions and resulted in a 50% reduction in ALT levels. Similarly, prophylactic treatment of infected mice with MEDI4893* reduced focal necrotic lesions and resulted in, near significant (p=0.06), reduction in liver damage measured by ALT in the blood (Figure S2C).
Figure 5.
(A) Representative images of the liver at 24 hours post infection with wild-type S. aureus, S. aureus agr, S. aureusΔhla or S. aureusΔpsmαβγ. Corresponding serum ALT levels (B) or CFU levels of liver (C) and kidney (D) at 24h in mice n = 7 per treatment group. *, P < 0.05; one-way ANOVA with Bonferroni’s posttest. Data shown are compiled from 2 independent experiments (E) Representative SD-IVM image of mouse liver at 8 h after i.v. infection with S. aureus GFP (USA300-GFP; bright green) and treated with c-IgG or MEDI4893* (30MPK) mAb 4 hours post-infection. Autofluorescent hepatocytes (dull green), platelets (CD49b; blue). Bar, 50 μm (F) Quantification of platelet accumulation in the mouse livers treated as in panel E. Data represents mean of >20 FOV in 5 mice ± SEM. (G) Representative images of the liver at 24 hours post-infection with S. aureus (USA300 LAC) or (H) serum ALT levels in mice treated 4 hours after infection with c-IgG or MEDI4893* (30MPK) mAb. n = 8, t-test, P < 0.05). (I) Survival of mice receiving a lethal dose of S. aureus (8×107 CFU USA300 LAC) treated with c-IgG of MEDI4893*(30MPK) 4 h post infection. n = 10 per treatment group, Log-rank test versus c-IgG. (J) Serum ALT levels 24 hours post-infection with S. aureus USA100 (NRS382), USA200 (NRS261) and USA400 (MW2) and treated 4 hours after infection with c-IgG or MEDI4893* (30MPK) mAbs. n = 6, t-test, *P < 0.05 **P<0.01, n.s.; not significant.
We previously reported that some S. aureus survive and even replicate inside Kupffer cells and subsequently disseminate to kidneys (Surewaard et al., 2016). S. aureusΔhla did not survive as effectively inside the liver as the WT strain (Figure 5C) and dissemination to kidneys trended towards a lower bacterial burden (Figure 5D). Blocking AT with MEDI4893* decreased staphylococcal survival in both liver and kidneys (Figure S2 D, E). Additionally, a mutation in the main regulator of staphylococcus virulence, Agr, showed a similar reduction in liver damage (Figure 5A,B), whereas an isogenic triple mutant in the operons encoding all the phenol soluble modulins (PSMs), had no impact on liver damage (Figure 5A,B). Agr and AT mutants also showed a similar reduction in the liver bacterial burden, again confirming that AT is an important Agr driven virulence factor aiding bacterial survival (Figure 5C,D).
Interestingly, our data showed a clear delay in AT induced platelet aggregation following staphylococcal infection (Figure 4 B-C). This potentially creates a window of opportunity to treat this deleterious platelet response to AT after initiation of infection. Indeed, mice treated with MEDI4893 at 4 h post-infection developed significantly less platelet aggregates in the livers (Figure 5E,F) and had a reduction in liver damage (Figure 5 G,H). Most important, mice treated 4h post infection with MEDI4893 had survival benefit compared to control mice (Figure 5I).
To test the impact of AT neutralization on a broader selection of clinically important strains we analyzed three additional strains from various staphylococcal lineages that caused a significant burden of disease in the last decades (Figure 5J). Interestingly, the in vitro production for strains NRS382 (USA100) and NRS261 (USA200) were among the lowest AT producers investigated in vitro; 0.34 and 1.1 μg/mL respectively. Nevertheless, both these strains showed significant liver damage that was reduced by MEDI4893* treatment after infection. Mice infected with MW2 (USA400), a moderate in vitro AT producer (2.2 ug/mL) also caused liver damage but MEDI14893* did not statistically significant (P=0.09) reduce this damage. Interestingly, in vivo AT treatments did not correspond to in vitro AT levels suggesting differential upregulation of AT inside the host. Collectively, neutralization of AT provides therapeutic benefit for the majority of staphylococcal strains investigated.
AT is a major staphylococcal platelet activator in humans
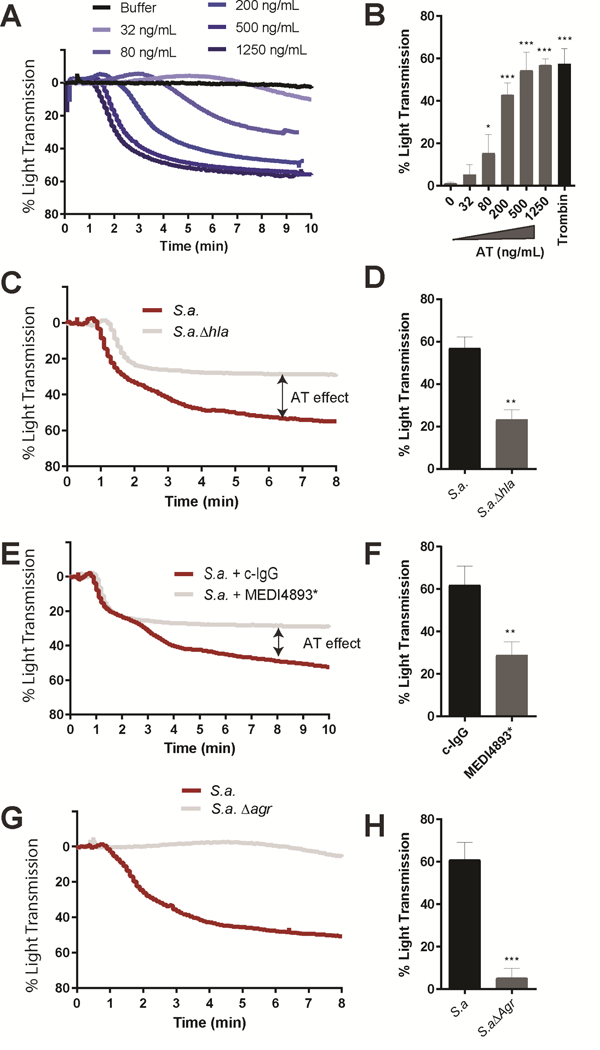
Consistent with our mouse work, recombinant AT potently aggregated human platelets (Figure 6A). By dose limiting titration of AT we calculated an EC50 of 85 ng/ml for human platelets which was consistently observed across 5 independent healthy donors (Figure 6B). When human platelets were treated with S. aureus culture supernatant, two phases in the aggregation graphs were observed, as light transmission increases proportional to platelet aggregation (Figure 6C). We first observed a rapid AT-independent aggregation up to 25% aggregation, followed by a second AT-dependent aggregation accounting for an addition 35% aggregation (Figure 6D). Supernatant from the S. aureusΔhla or MEDI4893* incubated WT supernatant lacked the second AT-dependent phase of platelet aggregation (Figure 6C–F). The first AT-independent phase and second AT-dependent phase of platelet aggregation were both dependent on an Agr-regulated factor as culture supernatant from a S. aureusΔagr did not aggregate human platelets suggesting a second, yet to be identified, Agr-regulated pro-aggregator molecule.
Figure 6. Aggregation of human platelets after AT intoxication.
(A) Aggregation traces of human platelets after exposure with for various concentrations of recombinant AT. (B) AT (0 – 1250 ng/mL) or thrombin (1 IU/mL) was added to platelets, and percentage of light transmission was monitored for 8 minutes. Data are reported means ± SD of 5 independent experiments from different donors. One-way ANOVA with Dunnet’s posttest. * P<0.05, *** P<0.001 (C) Aggregation traces of human platelet following exposure to culture supernatants of wild type S. aureus (USA300 LAC) or S. aureus hla. (D) Percentage of light transmission at 8 minutes with culture supernatants as described in panel C. Data represents mean ± SD of 3 independent experiments from different donors. (E) Aggregation traces of human platelet following injection of culture supernatants of wild type S. aureus (USA300 LAC) pre-incubated with c-IgG or MEDI4893*mAb (10 µg/mL). (F) Percentage of light transmission at 8 minutes with treated culture supernatants as described in panel E. Data represents mean ± SD of 3 independent experiments from different donors. (G) Aggregation traces of human platelet following injection of culture supernatants of wild type S. aureus (USA300 LAC) or S. aureusΔagr. (H) Percentage of light transmission at 8 minutes with treated culture supernatants as described in panel G. Data represents mean ± SD of 3 independent experiments from different donors. D,F,H Data represents mean ± SD; t-test, **P = 0.01 and*** P<0.001
DISCUSSION
During staphylococcal sepsis, patients present with microvascular clotting and a marked disposition towards systemic bleeding, consistent with the notion that platelets and coagulation are potentially dysregulated. Innovative, SD-IVM enables us to visualize rapid and dynamic responses to S. aureus in the microvasculature and unveiled molecular mechanisms in the field of staphylococcal pathophysiology and intravascular coagulopathy. We demonstrated a direct causal link between the administration of purified AT or WT S. aureus bacteremia and thrombotic events in sinusoidal vessels of the liver. There was also a clear link to organ injury in both liver and kidney (data not shown) and disease severity. It is also worth mentioning that, while AT can lyse platelets at high concentration, there is significant thrombosis at sub-lytic concentrations, suggesting that thrombosis may be the primary AT mechanism of action on platelets. Importantly, we also described an approach to mitigate S. aureus coagulopathy and pathology with an AT neutralizing antibody.
Although targeting platelets and their adhesion receptors could disrupt this inappropriate and damaging thrombosis, there is increasing evidence that suggests platelets help fight infections. For example, multiple in vitro reports have shown that platelets can attach to and kill microbes (McMorran et al., 2009; Tohidnezhad et al., 2012). In vivo, intravascular Kupffer cells in the liver capture S. aureus as it flows through the sinusoids and rapidly recruit platelets to encase and limit the infection (Wong et al., 2013). This evolutionarily-driven mechanism is facilitated by Kupffer cells, which utilize CRIg as a Pathogen Recognition Receptor for binding lipotechoic acid, found on Gram-positive bacteria, and quickly sequester bacteria from the blood (Helmy et al.; Zeng et al., 2016). While platelets have limited time to bind bacteria in the bloodstream, we saw them avidly bind Kupffer cells that had captured bacteria to prevent the escape of the pathogen from the macrophage. Interrupting this process by targeting platelets or their adhesion mechanisms led to a detrimental outcome for the host (Wong et al., 2013) this was recently corroborated by another group (Powers et al., 2015). Therefore, we chose to target AT as a key inducer of platelet aggregation in both mice and humans to prevent the damaging thrombosis but leave the protective effects of platelets intact. Targeting AT genetically or therapeutically with MEDI4893* did not affect early platelet-Kupffer cell protective mechanisms but disrupted the later bacterial-induced pathogenic thrombotic response. It is conceivable that AT secretion causes thrombosis at a site distal from the bacteria as an important virulence strategy, to consume or distract platelets.
Our previous work suggested that 90% of bacteria were killed in Kupffer cells, however 10% of the S. aureus inside the macrophage overcame the phagocytic machinery, and lysed the macrophage leading to bacterial dissemination (Surewaard et al., 2016). MEDI4893*prophylaxis, did not affect uptake of the S. aureus consistent with the view that AT is secreted and not displayed on the staphylococcal surface AND in line with previous reports that MEDI4893* has no opsonophagocytic properties (Tkaczyk et al., 2016; Tkaczyk et al., 2017). S. aureus required 4–8 h to secrete sufficient AT, as AT-dependent platelet aggregation was only observed at 8 h while direct administration of AT caused instantaneous aggregation. Remarkably, this timeframe coincided directly with the time it takes for S. aureus to escape from Kupffer cells (Surewaard et al., 2016). Blocking AT reduced the staphylococcal burden inside the liver as well as the kidney. Although it is unclear how the anti-AT mAb indirectly reduced growth of the bacterium in liver and kidney, we do not think it is due to intracellular activity. Rather, we speculate that the reduced thrombosis, improved blood flow, and platelet access to infected areas, increased neutrophil recruitment and macrophage viability resulting in reduced local infection as well as less bacterial dissemination.
There is growing evidence that AT may be important for human disease. Indeed, there is literature regarding the putative AT receptor ADAM10. Analysis of ADAM10 polymorphisms in human sepsis revealed that the C allele of rs653765 ADAM10 promoter conferred susceptibility to the most severe septic episodes due to increased ADAM10 levels (Cui et al., 2015). In addition, we observed that AT and S. aureus culture supernatant induced rapid human platelet aggregation that was inhibited by MEDI4893*. While it is not possible to visualize the role of AT in humans, previous mouse sepsis models also support a role for AT and ADAM10 (Powers et al., 2015). Indeed, in a lethal sepsis model a lack of ADAM10 expression on platelets was sufficient to reduce the lung injury but not mortality (Powers et al., 2015). Interestingly, it was only when both platelets and myeloid cells lacked ADAM10 that an improvement in mortality was observed, suggesting the involvement of myeloid cells in addition to platelets at lethal doses of bacteria. Using a non-lethal dose of S. aureus, revealed that AT caused thrombosis and DIC rather than overt hemorrhage, but liver injury was still observed (Yu et al., 2016). Collectively, these results confirm that AT is a key virulence determinant in S. aureus sepsis and demonstrates the role of AT in liver injury associated with sepsis. Interestingly, S. aureus infection required at least 8 h to result in AT levels that can induce platelet aggregation making it an attractive target for therapeutic administration. Therefore, AT is a valid target for immunotherapy and molecules such as MEDI4893*, could prevent microvascular dysfunction, thrombosis and DIC in sepsis. Such a therapeutic could also be used in combination with bactericidal antibiotics. The latter would kill the bacteria but fail to neutralize the effects of the secreted AT that could be blocked by a toxin neutralizing antibody. Multiple studies from independent laboratories with different anti-AT mAbs have demonstrated additive or synergistic protective activity with multiple antibiotics in different S. aureus animal infection models (Foletti et al., 2013; Hua et al., 2015; Hua et al., 2014; Le et al., 2016). The results herein offer one possible explanation as to how targeting AT may contribute to enhanced protection from acute lethality on top of traditional antibiotic antibacterial activity.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Paul Kubes,pkubes@ucalgary.ca
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animal experiments were carried out with male adult 8–12-wk-old mice (unless otherwise indicated) and all experimental animal protocols were approved by the University of Calgary Animal Care Committee and were in compliance with the Canadian Council for Animal Care Guidelines (protocol nr.AC16–0148). All mice were co-house bread in a specific pathogen-free, double-barrier unit at the University of Calgary. F4cre transgenic C57Bl/6 mice were bred to Adam10loxP/loxP transgenic C57Bl/6 mice to generate PF4cre Adam10loxP/loxPmice (PF4 ADAM10−/−)(Powers et al., 2015).
Staphylococcal strains and culture conditions
Staphylococcus aureus strains USA300 LAC, its isogenic AT mutant was used for all the experiments unless described otherwise. (Inoshima et al., 2011). Bacteria were grown in Brain Heart Infusion (BHI) media at 37°C while shaking. When appropriate, chloramphenicol (10 μg/ml) was added for overnight maintenance of the plasmids. For infection experiments, S. aureus strains/mutants were sub-cultured without antibiotics until exponential phase (OD660nm 1.0) washed with saline once, resuspended in saline and injected intravenously in the tail vein at 5×107 CFU in 200µL unless otherwise indicated. Alternatively, S. aureus-GFP were washed and resuspended as above, but heat-inactivated at 65°C or 95°C for 20 min. Bacterial culture supernatants were clarified by centrifugation, filtered through 0.22 μm pore size filter, concentrated 10× with Amicon-ultra 10kD cutoff filter (Millipore) and stored at −20°C in aliquots until use as described1.
Strains USA300 LAC, MW2, NRS382 and NRS261 produced AT, as measured in overnight culture supernatants (at 5.8, 2.2, 0.34 and 1.1 μg/ml, respectively). All strains, were cultured and transformed to constitutively express GFP as was described (Surewaard et al., 2016; Surewaard et al., 2013).
METHOD DETAILS
Antibodies and Reagents
AT and MEDI4893* were recombinant expressed and purified as described (Cohen et al., 2016), Lipopolysaccharide levels were below 1pg/mL. AT was labelled on the C-terminus with AF647 as recently described (Virreira Winter et al., 2016).
Mouse infections and in vivo treatments
Kupffer cell depletion was performed by intravenous injection of 200 μL of Clodronate Liposomes (CLL) per mouse (0.69 mol/L) 48 hours prior to the experiment. Mice were passively immunized by either intraperitoneal or intravenous injection of c-IgG, the anti-AT mAB MEDI4893* and then challenged 24 h or 2h later by intravenous injection with USA300 LAC. Alternatively mice were treated 4hrs after infection with c-IgG, the anti-AT mAB MEDI4893*. For survival studies mice were injected with a lethal dose of S.aureus (8×107 CFU) with treatment 4hrs post infection with c-IgG, or the anti-AT mAB MEDI4893*. A murine sepsis score system was introduced to monitor the mice based on their appearance, level of consciousness, activity, response to stimuli, eyes, respiration rate and quality (from 0 to 4 points for each criteria). An overall score of 18 or a score of 4 in any of the following criteria was considered as a humane endpoint: level of consciousness, activity, response to stimulus, respiration rate and respiration quality. For survival AT intoxication studies, mice were anesthetized as described above and i.v. injected with 1ug of AT, with or without prophylactic treatment of control IgG or MEDI1493*. S
Bacteriological analysis.
The needle insertion site on anesthetized mice was washed with 70% ethanol. Blood was collected in a heparinized syringe by cardiac puncture. Samples were then centrifuged at 400g for 10 min for the retrieval of plasma. Alanine Transaminase (ALT) in the plasma was analyzed by Calgary Laboratory Services. Haptoglobulin was analized using commercial ELISA kits according to the manufactures protocols. The liver and kidneys were removed after thoracotomy, weighed and homogenized. For determination of colony forming units (CFU), 30 μl of tissue homogenate was serially diluted, plated onto BHI agar plates, incubated at 37 °C for 18 h, and bacterial colonies were counted.
Intravital microscopy
Multichannel spinning-disk confocal microscopy was used to image the liver, lungs, kidney (tubules) and skin, two-photon confocal microscopy was used to image the glomeruli of kidneys. Mice were anesthetized and livers and lungs were prepared for intravital microscopy as previously described (Surewaard and Kubes, 2017; Thanabalasuriar et al., 2017). Skin was prepared as described recently (Jenne et al., 2011; Yipp et al., 2012). Kidneys were prepared as described (Devi et al., 2013). Mice were anaesthetized (10 mg/kg xylazine hydrochloride and 200 mg/kg ketamine hydrochloride). A tail vein catheter was inserted to inject fluorescent antibodies and to maintain anesthesia. Image acquisition of the liver, skin, and kidney (tubules) was performed using an inverted microscope (IX81; Olympus), equipped with a focus drive (Olympus) and a motorized stage (Applied Scientific Instrumentation) and fitted with a motorized objective turret equipped with 4×/0.16 UPLANSAPO, 10×/0.40 UPLANSAPO, and 20×/0.70 UPLANSAPO objective lenses coupled to a confocal light path (WaveFx; Quorum Technologies) based on a modified CSU-10 head (Yokogawa Electric Corporation). Target cells were visualized using fluorescently stained antibodies or fluorescent reporter bacteria. Typically, Kupffer cells, platelets and neutrophils were stained by iv injection of 2.5 µg anti-F4/80 or 3.0 µg anti-ly6G or anti-CD49b fluorescent conjugated mAbs. For each animal, platelet aggregates or bacteria were analyzed in 5 Fields Of View (FOV) selected at random prior to image acquisition. Acquisition of images was initiated and after 1 min of background image recording, 1ug AT, culture supernatants or live bacteria (5*107 CFU) were injected i.v. and platelet aggregation was observed. Find Objects function in the Volocity software was used to identify and quantify the amount of aggregated platelets and or individual captured bacteria.
Imaging of the glomeruli was performed in 4-week old mice as described previously 2.It was impossible to visualize glomeruli of older mice. In brief, mice were anesthetized as described above. The right jugular vein of anesthetized mice was cannulated to enable application of antibodies and additional anesthetics. Mice were shaved to remove the fur from the upper left flank region, an incision through the skin and body cavity wall was performed and the left kidney was exposed without removing the renal capsule or interrupting the blood flow. Mice were placed on a heating pad and the kidney was immobilized in a heated well built into a custom microscope stage and stabilized with vacuum grease. Tissue was bathed in warmed normal saline and covered with a coverslip.
The renal microvasculature was observed with a Leica SP8 2-photon microscope (Leica Microsystems), equipped with 25×0.95 NA water objective lens, 2 InSight DeepSee pulsed infrared lasers (fixed 1040 nm and tunable 680–1300 nm; Spectra-Physics) and a high speed 8 kHz resonant scanner. Emitted fluorescence was detected by non-descanted HyD-detectors with 650–700 nm, 565–620 nm and 500–550 nm. Qdots655 (Thermo Fisher) was used to label the vasculature and plates were labeled with PE-conjugated anti-CD49b.
Donor consent and human platelets isolation and aggregometry
Ethical approval for obtaining healthy human volunteer blood was provided by The University of Calgary research ethics committee, in accordance with the Declaration of Helsinki and each human subject provided informed consent. Whole blood was drawn from healthy donors who had not taken aspirin products for at least 48 hours. Blood was collected via a 21G butterfly needle into a 30 ml propylene syringe. The first 1ml of blood was discarded to avoid tissue contaminants. Blood was transferred into 50 ml polypropylene tubes containing 1 ml of acid-citrate dextrose (ACD) for every 5 ml of blood, and gently inverted to mix. Samples were spun at 140 × g for 10 minutes at room temperature (21°C) and supernatants (containing plasma and platelets) were transferred to new 50 ml polypropylene tubes containing 2 ml of ACD with a wide mouth transfer pipet, taking caution not to disrupt the red blood cell pellet. Supernatants with ACD were further diluted to 50 ml total volume with room temperature phosphate buffered saline (PBS), and spun at 700 ×g for 10 minutes at room temperature. Following centrifugation, the supernatant (plasma, ACD, PBS) was discarded and pellet (platelets) was resuspended in 10 ml of room temperature Hepes containing calcium chloride. Sample was allowed to sit for 10 minutes at 37°C prior to challenge. Platelets were challenged with thrombin (1.0 U/ml) as positive control or AT (0.01–1 μg/ml), or with cleared culture supernatants, while being stirred at 1200 rpm at 37°C in a Chronolog model 700 aggregometer. All samples contained a stir bar to keep platelets evenly suspended throughout the sample, and all samples were warmed to 37°C prior to challenge.
QUANTIFICATION AND STATISTICAL ANALYSIS
In vivo Image Analysis
All videos and images were acquired and processed using Volocity software (PerkinElmer) Leica software (Leica Microsystems). Volocity software (PerkinElmer) was used to quantify the accumulation of platelet aggregation and to quantify the size of the objects using the find objects tool.
Statistics
All experiments were performed a minimal of three independent replications. For in vivo results, each n value represents an individual experiment and mouse. For the human in vitro experiments, each n refers to a separate independent experiment with platelets isolated from independent healthy donors. The number of experiments for each figure can be found in the figure legends. Data are presented as mean ± SEM. Data were tested for Gaussian distribution using a Shapiro-Wilk normality analysis. For normally distributed data unpaired Student’s t test was used and for non-Gaussian distributions unpaired Mann-Whitney tests were used to compute statistical significance between two groups. One-way ANOVA with post-hoc testing was used to compute statistical significance between multiple groups. All statistical analysis was performed using GraphPad PRISM 7.2 software
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti - F4/80 (clone BM8) | Ablab | AbLab Cat. No.75– 0035–05 |
| Anti - CD49b (clone HMα2) | Biolegend | Biolegend Cat. No. 83598 |
| Anti - CD31 (clone MEC13.3) | Biolegend | RRID: AB_2161030 BioLegend Cat. No. 102515 |
| Anti - P-selectin (clone RP40.3) | BD Biosciences | RRID:AB_395026 BD Biosciences Cat.no. 553744, |
| Bacterial and Virus Strains | ||
| Staphylococcus aureus USA300 LAC | (Inoshima et al., 2011) | N/A |
| Staphylococcus aureus USA300 LACΔhla | (Inoshima et al., 2011) | N/A |
| Staphylococcus aureus USA300 LACΔagr | Periasamy et al., 2012) | N/A |
| Staphylococcus aureus USA300 LACΔαβhld | Periasamy et al., 2012) | N/A |
| Staphylococcus aureus USA400 MW2 | The Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) |
NARSA Cat.nr. NRS123 |
| Staphylococcus aureus USA100 | NARSA | NARSA Cat.nr. NRS382 |
| Staphylococcus aureus USA200 | NARSA | NARSA Cat.nr. NRS261 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Qdots 655 | ThermoFisher | Cat. Nr. Q21021MP |
| Recombinant alpha toxin (AT) | Medimmune (Cohen et al., 2016) | N/A |
| Recombinant non toxigenic ATH35L | Medimmune (Cohen et al., 2016) | N/A |
| MEDI4893* (Suvratoxumab) | Medimmune (Cohen et al., 2016) | N/A |
| Clodronate liposomes | Liposoma | Cat.Nr. CP-005–005 |
| Critical Commercial Assays | ||
| Mouse Haptoglobin ELISA Kit | Abcam | Cat.Nr. ab157714 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory |
Stock No: 000664 |
| C3−/−; (B6;129S4-C3tm1Crr/J) | The Jackson Laboratory |
Stock No: 003641 |
| FcyR−/− B6;129P2-Fcer1gtm1Rav/J | The Jackson Laboratory |
Stock No: 002847 |
| VWF −/− B6.129S2-Vwftm1Wgr/J | Gift from D Lillicrap (Queens University) |
TheJackson Laboratory, Stock No: 003795 |
| GpIbα−/− | Gift from T. Chavakis (Dresden University of Technology) |
N/A |
| PF4 ADAM10−/− (PF4cre Adam10loxP/loxPmice) | (Powers et al., 2015). | N/A |
| Cd41-yfpki/ki | Gift from Kelly McNagny, University of British Columbia) |
N/A |
| Software and Algorithms | ||
| Volocity | PerkinElmer | N/A |
| Image J | National Institute of Health |
https://imagej.nih.gov/ij/ |
| Prism 7 | GraphPad | V7.02 |
| FlowJo | FlowJo | V10.1 |
Highlights.
S. aureus AT intoxication leads to platelet aggregates that are deposited in the liver
Infection induces beneficial platelet responses followed by damaging AT-induced thrombosis
Antibody-mediated neutralization of AT prevents microvascular dysfunction and thrombosis
ACKNOWLEDGEMENTS
We thank T. Nussbaumer for mice husbandry. We also thank the staff at the University of Calgary Live Cell Imaging Facility for their assistance and K. Poon at the Nicole Perkins Microbial Communities Core Labs for assistance with flow cytometry. This work was supported by Canadian Institutes of Health Research (CIHR), CIHR fellowship (BGJS), Marie Currie actions FP7-PEOPLE-2013-IOF (grant no. 627575) (BGJS) and a grant from the Heart and Stroke Foundation of Canada (PK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration interest
Conflict of interest statement: PK received grant support and consulting fees from MedImmune, LLC for the work reported in this paper. CT, TSC, CKS and BRS are employed by MedImmune, LLC, a subsidiary of AstraZeneca, and may hold AstraZeneca stock.
References
- Alonzo F 3rd, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, and Torres VJ (2013). CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube BJ, and Bubeck Wardenburg J (2013). Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins 5, 1140–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Muhly M, Mannhardt U, Hugo F, Klapettek K, Mueller-Eckhardt C, and Roka L (1988). Staphylococcal α toxin promotes blood coagulation via attack on human platelets. The Journal of Experimental Medicine 168, 527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, and Schneewind O (2007). Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med 13, 1405–1406. [DOI] [PubMed] [Google Scholar]
- Chambers HF, and DeLeo FR (2009). Waves of resistance: Staphylococcus aureus in the antibioticera. Nature Reviews Microbiology 7, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TS, Hilliard JJ, Jones-Nelson O, Keller AE, O’Day T, Tkaczyk C, DiGiandomenico A, Hamilton M, Pelletier M, Wang Q, et al. (2016). Staphylococcus aureus alpha toxin potentiates opportunistic bacterial lung infections. Sci Transl Med 8, 329ra331. [DOI] [PubMed] [Google Scholar]
- Cooper LZ, Madoff MA, and Weinstein L (1966). Heat Stability and Species Range of Purified Staphylococcal α-Toxin. Journal of Bacteriology 91, 1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Gao Y, Xie Y, Wang Y, Cai Y, Shao X, Ma X, LI Y, Ma G, Liu G, et al. (2015). An ADAM10 promoter polymorphism is a functional variant in severe sepsis patients and confers susceptibility to the development of sepsis. Critical Care 19, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi S, Li A, Westhorpe CLV, Lo CY, Abeynaike LD, Snelgrove SL, Hall P, Ooi JD, Sobey CG, Kitching AR, et al. (2013). Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med 19, 107–112. [DOI] [PubMed] [Google Scholar]
- Foletti D, Strop P, Shaughnessy L, Hasa-Moreno A, Casas MG, Russell M, Bee C, Wu S, Pham A, Zeng Z, et al. (2013). Mechanism of Action and In Vivo Efficacy of a Human-Derived Antibody against Staphylococcus aureus α-Hemolysin. Journal of Molecular Biology 425, 1641–1654. [DOI] [PubMed] [Google Scholar]
- Helmy KY, Katschke KJ Jr., Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, and van Lookeren Campagne M CRIg: A Macrophage Complement Receptor Required for Phagocytosis of Circulating Pathogens. Cell 124, 915–927. [DOI] [PubMed] [Google Scholar]
- Hua L, Cohen TS, Shi Y, Datta V, Hilliard JJ, Tkaczyk C, Suzich J, Stover CK, and Sellman BR (2015). MEDI4893* Promotes Survival and Extends the Antibiotic Treatment Window in a Staphylococcus aureus Immunocompromised Pneumonia Model. Antimicrobial Agents and Chemotherapy 59, 4526–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua L, Hilliard JJ, Shi Y, Tkaczyk C, Cheng LI, Yu X, Datta V, Ren S, Feng H, Zinsou R, et al. (2014). Assessment of an Anti-Alpha-Toxin Monoclonal Antibody for Prevention and Treatment of Staphylococcus aureus-Induced Pneumonia. Antimicrobial Agents and Chemotherapy 58, 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, and Bubeck Wardenburg J (2011). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med 17, 1310–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins A, Diep BA, Mai TT, Vo NH, Warrener P, Suzich J, Stover CK, and Sellman BR (2015). Differential Expression and Roles of Staphylococcus aureus Virulence Determinants during Colonization and Disease. mBio 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenne CN, Wong CH, Petri B, and Kubes P (2011). The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PLoS One 6, e25109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler CM, Nussbaum E, and Tuazon CU (1991). Disseminated intravascular coagulation associated with Staphylococcus aureus septicemia is mediated by peptidoglycan-induced platelet aggregation. J Infect Dis 164, 101–107. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G, and Kubes P (2015). Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 6, 6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le VTM, Tkaczyk C, Chau S, Rao RL, Dip EC, Pereira-Franchi EP, Cheng L, Lee S, Koelkebeck H, Hilliard JJ, et al. (2016). Critical Role of Alpha-Toxin and Protective Effects of Its Neutralization by a Human Antibody in Acute Bacterial Skin and Skin Structure Infections. Antimicrobial Agents and Chemotherapy 60, 5640–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M, and ten Cate H (1999). Disseminated Intravascular Coagulation. New England Journal of Medicine 341, 586–592. [DOI] [PubMed] [Google Scholar]
- Lewis ML, and Surewaard BGJ (2017). Neutrophil evasion strategies by Streptococcus pneumoniae and Staphylococcus aureus. Cell Tissue Res [DOI] [PubMed]
- Lowy FD (1998). Staphylococcus aureus infections. N Engl J Med 339, 520–532. [DOI] [PubMed] [Google Scholar]
- McMorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, Corbin JE, Alexander WS, and Foote SJ (2009). Platelets Kill Intraerythrocytic Malarial Parasites and Mediate Survival to Infection. Science 323, 797–800. [DOI] [PubMed] [Google Scholar]
- Nachman RL, and Rafii S (2008). Platelets, Petechiae, and Preservation of the Vascular Wall. New England Journal of Medicine 359, 1261–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parimon T, Li Z, Bolz DD, McIndoo ER, Bayer CR, Stevens DL, and Bryant AE (2013). Staphylococcusaureus α-Hemolysin Promotes Platelet-Neutrophil Aggregate Formation. The Journal of Infectious Diseases 208, 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, Cheung GY, and Otto M (2012). How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A 109, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ME, Becker RE, Sailer A, Turner JR, and Bubeck Wardenburg J (2015). Synergistic Action of Staphylococcus aureus alpha-Toxin on Platelets and Myeloid Lineage Cells Contributes to Lethal Sepsis. Cell Host Microbe 17, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ME, and Bubeck Wardenburg J (2014). Igniting the fire: Staphylococcus aureus virulence factors in the pathogenesis of sepsis. PLoS Pathog 10, e1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragle BE, and Bubeck Wardenburg J (2009). Anti-Alpha-Hemolysin Monoclonal Antibodies Mediate Protection against Staphylococcus aureus Pneumonia. Infection and Immunity 77, 2712–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, and Ward PA (2008). Harmful molecular mechanisms in sepsis 8, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, and Bubeck Wardenburg J (2014). Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J Infect Dis 210, 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel I, and Cohen S (1964). Action of Staphylococcal Toxin on Human Platelets. J Infect Dis 114, 488–502. [DOI] [PubMed] [Google Scholar]
- Spaan AN, Surewaard BGJ, Nijland R, and van Strijp JAG (2013). Neutrophils versus Staphylococcus aureus: A Biological Tug of War. Annual Review of Microbiology 67, null. [DOI] [PubMed] [Google Scholar]
- Spaan AN, van Strijp JAG, and Torres VJ (2017). Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, and Brass LF (2013). Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 121, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewaard BG, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, Omri A, Yates RM, and Kubes P (2016). Identification and treatment of the Staphylococcus aureus reservoir in vivo. J Exp Med 213, 1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewaard BGJ, de Haas CJC, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JAG, and Nijland R (2013). Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cellular Microbiology, n/a-n/a. [DOI] [PMC free article] [PubMed]
- Surewaard BGJ, and Kubes P (2017). Measurement of bacterial capture and phagosome maturation of Kupffer cells by intravital microscopy. Methods [DOI] [PubMed]
- Thanabalasuriar A, Surewaard BG, Willson ME, Neupane AS, Stover CK, Warrener P, Wilson G, Keller AE, Sellman BR, DiGiandomenico A, et al. (2017). Bispecific antibody targets multiple Pseudomonas aeruginosa evasion mechanisms in the lung vasculature. J Clin Invest 127, 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomer L, Schneewind O, and Missiakas D (2016). Pathogenesis of Staphylococcus aureus Bloodstream Infections. Annual Review of Pathology: Mechanisms of Disease 11, 343–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkaczyk C, Hamilton MM, Sadowska A, Shi Y, Chang CS, Chowdhury P, Buonapane R, Xiao X, Warrener P, Mediavilla J, et al. (2016). Targeting Alpha Toxin and ClfA with a Multimechanistic Monoclonal-Antibody-Based Approach for Prophylaxis of Serious Staphylococcus aureus Disease. mBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkaczyk C, Kasturirangan S, Minola A, Jones-Nelson O, Gunter V, Shi YY, Rosenthal K, Aleti V, Semenova E, Warrener P, et al. (2017). Multimechanistic Monoclonal Antibodies (MAbs) Targeting Staphylococcus aureus Alpha-Toxin and Clumping Factor A: Activity and Efficacy Comparisons of a Mab Combination and an Engineered Bispecific Antibody Approach. Antimicrobial Agents and Chemotherapy 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohidnezhad M, Varoga D, Wruck CJ, Podschun R, Sachweh BH, Bornemann J, Bovi M, Sönmez TT, Slowik A, Houben A, et al. (2012). Platelets display potent antimicrobial activity and release human beta-defensin 2. Platelets 23, 217–223. [DOI] [PubMed] [Google Scholar]
- van der Poll T, and Opal SM (2008). Host–pathogen interactions in sepsis. The Lancet Infectious Diseases 8, 32–43. [DOI] [PubMed] [Google Scholar]
- van der Poll T, van de Veerdonk FL, Scicluna BP, and Netea MG (2017). The immunopathology of sepsis and potential therapeutic targets 17, 407. [DOI] [PubMed] [Google Scholar]
- Virreira Winter S, Zychlinsky A, and Bardoel BW (2016). Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus alpha-hemolysin-mediated toxicity. Sci Rep 6, 24242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, et al. (2007). Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13, 1510–1514. [DOI] [PubMed] [Google Scholar]
- Wardenburg JB, and Schneewind O (2008). Vaccine protection against Staphylococcus aureus pneumonia. The Journal of Experimental Medicine 205, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CHY, Jenne CN, Petri B, Chrobok NL, and Kubes P (2013). Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance 14, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Suzuki-Inoue K, Satoh K, Asazuma N, Yatomi Y, Berndt MC, and Ozaki Y (2001). Role of Fc receptor gamma-chain in platelet glycoprotein Ib-mediated signaling. Blood 97, 3836–3845. [DOI] [PubMed] [Google Scholar]
- Xu SX, Gilmore KJ, Szabo PA, Zeppa JJ, Baroja ML, Haeryfar SMM, and McCormick JK (2014). Superantigens Subvert the Neutrophil Response To Promote Abscess Formation and Enhance Staphylococcus aureus Survival In Vivo. Infection and Immunity 82, 3588–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, et al. (2012). Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18, 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KO, Randolph AG, Agan AA, Yip WK, Truemper EJ, Weiss SL, Ackerman KG, Schwarz AJ, Giuliano JS Jr., Hall MW, et al. (2016). Staphylococcus aureus alpha-Toxin Response Distinguishes Respiratory Virus-Methicillin-Resistant S. aureus Coinfection in Children. J Infect Dis 214, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Surewaard BG, Wong CH, Geoghegan JA, Jenne CN, and Kubes P (2016). CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 20, 99–106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.