

SUMMARY
Nervous system function depends on proper myelination for insulation and critical trophic support for axons. Myelination is tightly regulated spatially and temporally, but how it is controlled molecularly remains largely unknown. Here, we identified key molecular mechanisms governing the regional and temporal specificity of central nervous system (CNS) myelination. We show that transcription factor EB (TFEB) is highly expressed by differentiating oligodendrocytes and that its loss causes precocious and ectopic myelination in many parts of the murine brain. TFEB functions cell-autonomously through PUMA induction and Bax/Bak activation to promote programmed cell death of a subset of premyelinating oligodendrocytes, allowing for selective elimination of oligodendrocytes in normally unmyelinated brain regions. This pathway is conserved across diverse brain areas and is critical for myelination timing. Our findings define an oligodendrocyte-intrinsic mechanism underlying the spatiotemporal specificity of CNS myelination, shedding light on how myelinating glia sculpt the nervous system during development.
Keywords: Oligodendrocyte, Myelination, Temporal and Regional Specificity, Central Nervous System, Transcription Factor EB, PUMA, Stress Response, Programmed Cell Death
Graphical Abstract
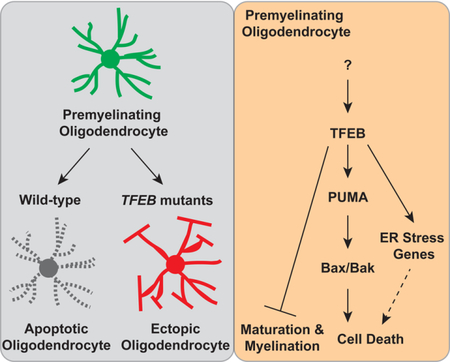
IN BRIEF
Programmed cell death actively eliminates oligodendrocytes in non-myelinated brain regions to ensure region specific myelination
eTOC Blurb
Transcription Factor EB activates the PUMA-Bax/Bak pathway to promote programmed cell death of subsets of premyelinating oligodendrocytes, thus allowing for the temporal and regional specificity of CNS myelination.
INTRODUCTION
Myelination provides insulation and trophic support for axons and allows for normal nervous system function (Saab and Nave, 2017). Disruption of myelination in the central nervous system (CNS) leads to many demyelinating diseases, including multiple sclerosis (MS) and leukodystrophies (Birey et al., 2017). Throughout development, CNS myelination is tightly controlled temporally (Bergles and Richardson, 2015). For instance, in the developing cortex the axons within deep cortical layers are myelinated first; myelination then continues from the deep cortical layers to the upper cortical layers, despite the presence of axons within each cortical layer during this developmental window (Tomassy et al., 2014; Trapp et al., 1997). Myelination also exhibits stringent regional specificity. For example, myelination in the cerebellum is largely confined beneath the molecular layer, the outermost layer that contains numerous axons including those originating from cerebellar granule cells (the parallel fibers) and from inferior olive neurons (the climbing fibers). Most of these axons in the molecular layer remain unmyelinated through development and into adulthood (Wyatt et al., 2005). A previous study showed that ectopically increasing axon diameters of cerebellar granule cells promotes de novo myelination in the molecular layer (Goebbels et al., 2017), however, the intrinsic mechanisms governing regional specificity of myelination remain largely unknown.
In the CNS, myelin is solely produced by a glial cell type called oligodendrocytes (OLs). To generate mature OLs, oligodendrocyte precursor cells (OPCs) must first stop dividing and differentiate into premyelinating OLs (pre-OLs), an intermediate stage in which these cells extend numerous radial processes but do not yet wrap nearby axons (Zuchero and Barres, 2013). During development, pre-OLs are over-generated and a significant portion of them undergo programmed cell death (Raff et al., 1993). In the developing rat optic nerve, at least 50% of newly formed OLs undergo apoptosis within 2–3 days after they are generated, likely still at the premyelinating stage (Barres et al., 1992). Additionally, in the rat cortex, approximately 20–30% of pre-OLs die by postnatal day 21 (P21)(Trapp et al., 1997). Recent in vivo imaging work suggests that OL apoptosis also occurs in the adult rodent brain, where most pre-OLs die prior to committing to myelination (Hill et al., 2018; Hughes et al., 2018). To account for the vulnerability of pre-OLs, a model was proposed hypothesizing that when OPCs differentiate to pre-OLs, they rapidly lose their PDGF receptors that typically promote cell survival, thus becoming sensitized to cell death cues (Barres and Raff, 1994). Meanwhile, pre-OLs have a narrow window to compete against each other for a limited supply of trophic factors and for contacting non-myelinated axonal regions, which provide further survival support (Klingseisen and Lyons, 2018). This model, however, leaves unanswered the identity of the critical intrinsic pathways that link extracellular trophic cues with downstream cell death events, and the other molecular mechanisms that facilitate pre-OL cell death. It also remains unclear whether programmed cell death of pre-OLs actively determines the timing and location of myelination.
Here, we discovered an intrinsic pathway that dictates programmed cell death events in developing OLs and powerfully controls CNS myelination. We identified a novel role for Transcription Factor EB (TFEB), a transcription factor previously identified as a master regulator for lysosome biogenesis, in controlling the spatial and temporal specificity of CNS myelination. We found that TFEB is highly expressed by pre-OLs early postnatally, and that genetic deletion of TFEB in OL lineage cells causes survival of ectopic pre-OLs during development. This in turn leads to aberrant myelination in brain regions that normally remain unmyelinated during development and into adulthood. Death of pre-OLs is achieved through transcriptional induction of PUMA by TFEB and subsequent activation of Bax/Bak-dependent programmed cell death. Finally, the continuous expression of TFEB in myelinating OLs acts as a “brake” on OL maturation and myelination. Our findings reveal that the TFEB-PUMA-Bax/Bak pathway is essential for selective OL elimination during development, allowing for the spatiotemporal specificity of CNS myelination.
RESULTS
Transcription Factor EB (TFEB) is Enriched in Premyelinating Oligodendrocytes
To search for new regulators of OL development, we leveraged our whole genome RNA sequencing transcriptome datasets (Zhang et al., 2014) and identified an OL-enriched transcription factor, Transcription Factor EB (TFEB), that has not been previously characterized in OL development and myelination. TFEB mRNA is highly expressed by OL lineage cells, with the peak expression specifically at the pre-OL stage (Figure S1A). To elucidate TFEB expression pattern in vivo, we employed double fluorescence in situ hybridization with probes recognizing endogenous TFEB mRNA and the mRNAs of several OL lineage-specific markers (Figure 1A). We found that TFEB mRNA is highly expressed in murine white matter including the corpus callosum, cerebellar white matter tracts, and internal capsule (Figures 1B–D). In these brain regions most OL lineage cells including Olig2+ (a pan-OL lineage marker) and ENPP6+ cells (a pre-OL and myelinating OL marker)(Xiao et al., 2016) express TFEB mRNA (Figures 1B’–B” and C’– D”, respectively; quantified in Figure 1E). To validate TFEB spatial and temporal expression, we obtained a TFEBKI mouse line from the Knockout Mouse Project (KOMP) Repository and subsequently generated a TFEBLacZ allele, in which the endogenous TFEB promoter drives the expression of cytosolic β galactosidase (β-gal; Figure S1C). We utilized the X-gal colorimetric reactions in the TFEBLacZ/+ heterozygous mice, and found that many β-gal+ cells reside in brain white matter areas where OLs are enriched (Figure 1F), however they are also present in grey matter brain regions including the cortex, hippocampus, and cerebellum (Figure 1F and Figures S1H–K). To better characterize the identities of β-gal+ cells, we employed double immunohistochemistry using antibodies directed against β-gal and CC1 (a post-mitotic OL marker) and found that β-gal immunoreactivity is localized to most CC1+ OLs in both white matter and grey matter brain regions (Figures 1G–H“; quantified in Figure 1I)
Figure 1. Transcription Factor EB (TFEB) is Expressed by Oligodendrocyte Lineage Cells during Developmental Myelination.

(A) Diagram of oligodendrocyte (OL) differentiation and the molecular markers that delineate each differentiation stage. (B-D”) Double fluorescent in situ hybridization of P12 mouse brain sections with probes against the mRNAs of TFEB (B, C, and D), Olig2 (B’), and ENPP6 (C’ and D’) in white matter brain regions including the corpus callosum (B-C”) and cerebellar white matter (D-D”). (E) Quantification of double fluorescent in situ signals in P12 mouse corpus callosum (CC), cerebellar white matter (Cb), and internal capsule (IC). (F) X-gal staining on P21 TFEBLacZ/+ brain sections (see Figure S1 for details on the TFEBLacZ allele). D, dorsal; A, anterior. (G-H”) Double immunolabeling of P14 mouse brain sections by antibodies directed against β-gal (G and H) and CC1 (G’ and H’), showing that most β-gal+ cells are CC1 immuno-positive (G” and H”). (I) Quantification of the ratio of CC1 and β-gal double immuno-positive cells to the total β-gal+ cells on P14-P21 mouse brain sections. (J) Westernblot analysis using cell lysates from acutely purified OPCs, pre-OLs, and OLs in P12 TFEBF/F (Ctl) or Olig2-Cre; TFEBF/F (cKO) mice, showing that TFEB is expressed in all OL lineage stages and is enriched in pre-OLs. Each lane was loaded with the same amount of total proteins. n=4 independent experiments. Error bars indicates SEM. Scale bars: 100 μm in (D”) for (B)-(D”); 10 μm in the inset of (D”) for insets in (B”), (C”), and (D”); 1 mm in (F); and 100 μm in (H”) for (G)-(H”).
To determine the temporal expression pattern of TFEB, we performed X-gal staining on the TFEBLacZ/+ mouse brain sections at various developmental time points (Figures S1D–G and Figure 1F), and found highest staining between P14 and P21 (Figures 1F and S1F) when the mouse brain is actively being myelinated (Yang et al., 2016). To further pinpoint the differentiation phase at which TFEB protein is enriched, we used immunopanning to acutely purify OPCs, pre-OLs, and myelinating OLs from P12 mouse brains (Emery and Dugas, 2013). Consistent with published RNAseq results (Figure S1A)(Zhang et al., 2014), we found expression of TFEB in all three stages, with highest expression levels in pre-OLs (Figure 1J). Therefore, TFEB is expressed by OL lineage cells and enriched in pre-OLs during early postnatal development.
TFEB is Required to Eliminate Transiently Differentiated Oligodendrocytes in the Cerebellar Molecular Layer
To test if TFEB regulates OL development and myelination, we generated a TFEB conditional allele (TFEBFlox or TFEBF; Figure S1C) and subsequently generated TFEB OL conditional KO (TFEB cKO) animals by crossing the TFEBF allele with a battery of mouse lines that each express Cre recombinase during specific OL differentiation stages (Figure S1C and Figure 1A). These mouse lines include Olig2-Cre and CNP-Cre that begin expressing Cre recombinase in the earliest OL lineage and throughout the duration of OL differentiation and myelination (Zuchero et al., 2015), as well as an inducible PDGFRa-CreER line that expresses Cre recombinase in OPCs (Kang et al., 2010). TFEB cKO animals are viable, fertile, and have normal lifespan (data not shown). Cell lysates from OPCs, pre-OLs, and myelinating OLs purified from Olig2-Cre; TFEBF/F mice shows that TFEB proteins are absent from all differentiation stages (Figure 1J), confirming that following Cre-mediated recombination the TFEBF allele becomes a TFEB null allele.
To determine TFEB function in OL development and myelination, we first analyzed the developing cerebellum where TFEB is highly expressed (Figures 1D–D“, Figures 1H–H“, and Figure S1K). In P14 control animals, OPCs are present in both the granule cell layer (GL) and the molecular layer (ML; white arrowheads in Figures 2A and 2B; see also (Goebbels et al., 2017; Lin et al., 2005)), however myelination, marked by myelin basic protein (MBP) and myelin proteolipid protein (PLP), is strictly constrained beneath the ML (Figures 2C and 2D, respectively). In strong contrast, TFEB cKO mice (Olig2-Cre; TFEBF/F) exhibit fully-penetrant, ectopic myelin in the ML across the entire cerebellum at P14 (Figures 2C’–D’; quantified in Figure 2G) and in adulthood (Figures S2I-J). To determine if the aberrant myelin seen in the ML is caused by the ectopic presence of OLs in the ML, we utilized in situ hybridization with probes recognizing MBP and ASPA mRNAs, two markers expressed by pre-OLs and myelinating OLs (Madhavarao et al., 2004; Zuchero et al., 2015). We found that P56 control mice do not contain MBP+ or ASPA+ OLs in the ML, whereas TFEB cKO mice exhibit significantly increased numbers of MBP+ and ASPA+ cells in the ML (Figures 2E–E’ and Figures S2G–G’, respectively; quantified in Figures 2H and S2H). To further investigate if ectopic myelin wrapping is formed in TFEB cKO ML, we performed transmission electron microscopy (TEM) analysis in both the granule cell layer (GL) and the ML. In littermate controls, myelination occurs in the GL but not in the ML (Figures 2F, S2K and S2L; n=3 animals). Conversely, TFEB cKO mice exhibit ectopic myelin wrapping in the ML (Figure 2F’, and Figures S2K’–O; n=3 animals). We found ~58% ectopic myelin wrapping occur on axons (Figure 2F and Figures S2P–P’) which likely belong to cerebellar granule cell (Figures S2C–C’), whereas ~42% ectopic myelin wraps do not contain obvious axonal structures (Figure S2Q).
Figure 2. Conditional Deletion of TFEB in Oligodendrocyte Lineage Cells Causes Ectopic Myelination in the Cerebellar Molecular Layer.

(A and B) Genetic labeling reveals OPCs in P14 mouse cerebellar molecular layer (red in A, indicated by white arrowheads). ML: molecular layer. GL: granule layer. (C-D’) Genetic deletion of TFEB in OL lineage cells elicits fully-penetrant (6/6 animals), ectopic myelin presence in the cerebellar ML. MBP, myelin basic protein; PLP, myelin proteolipid protein. (E and E’) In situ hybridization using probes recognizing MBP mRNAs shows that MBP+ cells are present in the cerebellar ML of P56 TFEB cKO mice (red arrows in E’). (F and F’) Transmission electron microscopy (TEM) analysis reveals ectopic myelin wrapping in the cerebellar ML of P56 TFEB cKO mice. See more examples in Figures S2K–Q. (G) Quantification of the averaged MBP fluorescence intensity in the cerebellar ML of P14 control (Ctl) and TFEB cKO mice. (H) Quantification of MBP+ cell number in the cerebellar ML of P56 mice. (I-M’) Characterization of OL development in the developing cerebellar ML at P6 (I and I’), P8 (J and J’), and P11 (K-M’) from control and TFEB cKO mice. L and L’ represent the inset in K, and M and M’ represent the inset in K’. (N) Quantification of the averaged MBP immunofluorescence in control and TFEB cKO ML. (O) Quantification of the ratio of cleaved Caspase-3 and MBP double immune-positive cells to the total MBP+ cells in the ML at P11. (P-Q’) Double fluorescent in situ hybridization using probes against TFEB (green in P and Q) and ENPP6 (red in P and Q’) showing a transiently differentiated OL in the ML that also expresses TFEB mRNA. (R-S) Characterization and quantification of ectopic OLs in the ML of 15-month-old control (R) and TFEB cKO mice (R’). (T and T’) Conditional and inducible deletion of TFEB in OL lineage cells (PDGFRα-CreER; TFEBF/F) from P4 leads to ectopic myelination in the cerebellar ML. (U) Quantification of the averaged MBP fluorescence intensity in the ML of PDGFRα-CreER; TFEBF/+ (Ctl) and PDGFRα-CreER; TFEBF/F(cKO) mice. Error bars represent SEM. Scale bars: 100 μm in (E’) for (A)-(E’); 0.5 μm in (F’) for (F) and (F’); 100 μm in (K’) forI(I)-(K’); 20 μm in (M’) for (L)-(M’); 10 μm in (P); 10 μm in (Q’) for (Q) and (Q’); and 100 μmin (T’) for (R), (R’), (T), and (T’).
OL differentiation and myelination proceeds in multiple stages, including OPC migration, pre-OL differentiation and elimination, OL maturation and myelination (Bergles and Richardson, 2015). We next asked in which phase TFEB acts to pattern myelination in the cerebellar ML. At P6, MBP+ OLs are only present within the inner granule cell layer (Figure 2I). By P8, we observed several MBP+ cells in the ML of control mice (white arrowheads in Figure 2J; quantified in Figure 2N), suggesting that a small number of OLs normally differentiate in the ML at this stage. By P11, very few MBP+ OLs are present in control ML and ~35.3% of them are immunopositive for cleaved Caspase-3, a marker for cells undergoing apoptosis (Figures K-L’; quantified in Figures 2N and 2O). Importantly, most of these cells co-express TFEB (Figures 2P–Q’; 14/15 ENPP6+ cells are TFEB+ in 3 TFEBF/F mice), suggesting that TFEB plays a role in transiently differentiated OLs in the ML. Indeed, we observed elevated number of MBP+ OLs in TFEB cKO ML at P11 (Figure 2K’) and very sparse expression of cleaved Caspase-3+ in this population (Figures 2M–M’; quantified in Figures 2N and 2O; 1.6%±0.1% MBP+ cells are also cleaved Caspase-3+, mean±SEM, n=4 animals). The ectopic presence of OLs persist in the ML of 15-month-old TFEB cKO mice (Figures 2R–S). Importantly, this defect is not caused by altered OPC proliferation or migration, as the number and spatial pattern of PDGFRa+ OPCs remain the same in TFEB cKO cerebellum (Figures S2E–F). Finally, to rule out the possibility that the aberrant myelination phenotype is caused by Olig2-Cre expression and TFEB function in cerebellar granule cells, we generated a PDGFRa-CreER; TFEBF/F mouse line that allows us to ablate TFEB specifically in OPCs (Kang et al., 2010). We administered 4-hydrotamoxifen (4HT) immediately prior to the onset of the phenotype and again observed ectopic myelin presence in the ML (Figures 2T–U). Taken together, TFEB promotes OL programmed cell death in the cerebellar ML.
TFEB cKO Brain is Precociously Myelinated and Exhibits Increased OL Number
Outside the cerebellum, TFEB is widely expressed by OL lineage cells in the forebrain, including the cortex and hippocampus (Figures 1F–G“, Figures S1H and S1J, and Figures S3A–B). Therefore, we next asked whether TFEB might function in these brain regions in regulating OL development and myelination. TFEB cKO mice exhibit “precociously” differentiated MBP+ OLs in the midbrain and deep cortical layers at P6, a developmental stage earlier than typical myelination (Figures 3A and 3A’). Consistent with this finding, as compared to littermate controls (TFEBF/F), P8 TFEB cKO mice express increased levels of myelin proteins, including MBP and CNPase (Figure 3B; quantified in Figure 3G). At P14, OL differentiation typically occurs in deep cortical layers including layers IV-VI (Figures 3C–E), delineated by a cortical layer maker Brn2 (red in Figure 3D, the marker for layer II, III, and IV). In strong contrast, TFEB cKO mice exhibit ectopic OLs in upper cortical layers (Figures 3C’–E’; quantified in Figure 3H) including layer I (red arrows in Figure 3C’ and white arrows in Figure 3D’). We observed a similar phenotype in CNP-Cre; TFEBF/F mice, a separate TFEB cKO line driven by CNP-Cre that expresses Cre recombinase in all OL lineage cells (Lappe-Siefke et al., 2003) (Figures S3L–L‘). Importantly, cortical lamination is not disrupted in TFEB cKO mice (Figures S3C–C’), demonstrating that the phenotype is not due to cortical layer disorganization, which can cause ectopic myelination (Tomassy et al., 2014). To determine if precocious myelination occurs in layer I, we performed TEM analysis and found that indeed a subset of axons within this layer are myelinated in TFEB cKO mice (red arrows in Figure 3F’; n=3 animals). We further found that OLs are precociously present in other brain regions, including the piriform cortex (Figures S3D–D’) and hippocampus (Figures S3E–E‘).
Figure 3. TFEB cKO Mice Exhibit Precious Myelination and Increased Oligodendrocyte Number Across the Entire Brain.

(A and A’) Sagittal brain sections immunostained with the antibody directed against MBP showing that TFEB cKO mice (Olig2-Cre; TFEBF/F in A’) harbors differentiated OLs preciously in the forebrain and midbrain (compare A’ to A). n=4 animals per genotype. (B) Western immunoblot of myelin proteins from P8 control (TFEBF/F) and TFEB cKO (Olig2-Cre; TFEBF/F) brain lysates. (C-D’) Characterization of cortical OL development with a differentiated OL marker (MBP, green in D and D’) and cortical layer marker Brn2 (labels layer II, III, and V; red in D and D’) at P14. TFEB cKO mice exhibit differentiated OLs in cortical layer I (red arrows in C’ and white arrows in D’) and layers II&III at this stage (red arrowheads in C’ and white arrowheads in D’). See quantification in (H). (E and E’) Confocal images of P14 cortical layer I stained with MBP antibody (green). Red arrows show ectopically differentiated OLs in layer I. Red arrowheads indicate OL longitudinal processes that may begin ensheathing axons. White dash lines delineate the brain surface. (F and F’) Representative TEM images showing that myelin wraps are precociously formed in cortical layer I of TFEB cKO at P14 (red arrows in F’). Red dash lines delineate the brain surface. n=3 animals for both genotypes. (G) Quantification of normalized myelin proteins from P8 control and TFEB cKO whole brain lysates. (H) Quantification of the averaged MBP fluorescent intensity in control and TFEB cKO cortices. (I-I’) Double immunofluorescent labeling using antibodies directed against CC1 (green) and PLP (red) shows that PLP and differentiated OL cell bodies are present in cortical layer I of TFEB cKO mice at P21. (J) Quantification of CC1+ cell density through all cortical layers at P21. (K-L’) In situ hybridization with ASPA probes showing that in 2-month-old control mice only a few ASPA+ OL are present in cortical layer I (red arrowhead in L). In contrast, TFEB cKO mice harbor increased numbers of ASPA+ OLs (red arrowheads in L’). (L) and (L’) represent the inset in (K) and (K’), respectively. (M) Quantification of ASPA+ cell density in 2-month-old control and TFEB cKO cortices. (N-O) Characterization and quantification of MBP+ OLs in 15-month-old control and TFEB cKO mice. White vertical bars delineate cortical layer I. (P-Q) Representative confocal images and quantification showing that genetic deletion of TFEB in OL lineage cells from P4 causes aberrant MBP expression in cortical layer I. Error bars represent SEM. Scale bars: 1 mm in (A’) for (A) and (A’); 100 μm in (D’) for (C)-(D’); 100 μm in (E’) for and (E’); 1 μm in (F’) for (F) and (F’); 10 μm in (I’) for (I) and (I’); 100 μm in (K’) for (K) and (K’); 100 μm in (L’) for (L) and (L’); and 100 μm in (P’) for (N), (N’), (P), and (P’).
We next addressed whether TFEB cKO mice contain altered OL number in adulthood. At P21, TFEB cKO mice have significantly increased numbers of CC1+ OLs in the cortex (Figures 3I–J). This phenotype is observed in both 2-month-old (Figures 3K–M and Figures S3G–H) and in 15-month-old TFEB cKO mouse brains throughout all cortical layers (Figures 3N–O and Figure S3H), and is found in diverse brain regions including hippocampus and cerebellum (Figures S3F–H and Figures S2G–H). In particular, both 2- and 15-month-old TFEB cKO mice exhibit an approximately 5-fold increase of the total OL number in cortical layer I (quantified in Figures 3M and 3O, respectively). The increased OL number is not due to misregulation of OPC migration or proliferation, as OPC spatial patterns and numbers are not different between TFEB cKO and littermate controls (Figures S3I–J’; quantified in Figure S3K). This is further supported by our observation showing that PDGFRa-CreER; TFEBF/F mutant mice exhibit ectopic MBP immunofluorescence in cortical layer I when TFEB is deleted in OPCs from P4 (Figures 3P–Q and S3N–N‘), a developmental stage when OPC migration has ceased (Kessaris et al., 2006). Thus, TFEB controls OL number and is required for proper myelination timing in diverse brain regions.
TFEB cKO Mice Have Thicker Myelin Sheaths
The extended expression of TFEB in OLs from P14 to adulthood suggests that it may participate in myelin sheath formation and maintenance (Figure S1) (Auer et al., 2018; Snaidero et al., 2014). We characterized the myelination pattern in the corpus callosum (CC), a white matter brain region enriched with TFEB-expressing OLs and commissural axons projecting from both brain hemispheres (Figure 1). In control mice at P21, only a small subset of axons are myelinated (Figure 4A; quantified in Figure 4B). In contrast, TFEB cKO mice exhibit a significantly increased density and percentage of myelinated axons, whereas the total axon number remains the same as compared to controls (Figure 4A’; quantified in Figure 4B). In addition, the thickness of myelin sheaths is significantly increased in the absence of TFEB, measured by g-ratios (the ratio of inner axonal diameter to the total outer diameter; Figures 4C–C‘; quantified in Figures 4D–E). Importantly, the average diameter of myelinated axons and the percentage of myelinated axons with different diameters are largely unchanged in TFEB cKO mice (Figure S4A and Figures S4B–C, respectively), showing that the increased myelin sheath thickness is unlikely to be a consequence of altered axon calibers.
Figure 4. TFEB cKO Mice Harbor Thicker Myelin Sheaths and are Precociously Myelinated in White Matter.

(A and A’) Representative TEM micrographs from P21 control (TFEBF/F) and TFEB cKO (Olig2-Cre; TFEBF/F) corpus callosum showing that myelinated axon density is increased in the absence of TFEB. (B) Quantification of myelinated axon number (left), total axon number (middle), and myelinated axon percentage (right) in the corpus callosum from TFEBF/F (Flox), Olig2-Cre; TFEBF/+ (Het) and Olig2-Cre; TFEBF/F (cKO) mice. (C and C’) Representative high-magnification TEM micrographs from P21 control and TFEB cKO corpus callosum. (D and E) Quantification of average g-ratios of myelinated axons (E), and as a function of axon diameter (D), in P21 TFEB cKO mice (red) compared with littermate controls (TFEBF/F in black and Olig2-Cre; TFEBF/+ in blue). n•180 axons from 3 animals per genotype. (F and F’) Representative high-magnification TEM micrographs from 6-month-old control and TFEB cKO corpus callosum. (G and H) Quantification of average g-ratios of myelinated axons (H), and as a function of axon diameter (G), in 6-month-old TFEB cKO mice (red) as compared with littermate controls. n≥180 axons from 3 animals per genotype. Error bars represent SEM. Scale bars: 5 m in (A’) for (A) and (A’); and 0.5 m in (F’) for (C)-(F’).
Myelination continues throughout adulthood (Hill et al., 2018; Hughes et al., 2018; Young et al., 2013). Therefore, we next characterized 6-month-old CC and found that TFEB cKO mice exhibit thicker myelin sheaths (compare Figure 4F’ to 4F; see also Figure S4K; quantified in Figures 4G–H) and increased myelin wrap numbers (Figures S4I–J). Consistent with previous findings that a subset of axons in the CC remain unmyelinated in adulthood (Sturrock, 1980), we found that approximately only 40% of axons are myelinated in the CC of 6 month-old littermate controls (Figure S4K; quantified in Figure S4L). The percentage of unmyelinated axons, average unmyelinated axon diameter, as well as the distribution of unmyelinated axons with different diameters remain unchanged in TFEB cKO mice (Figure S4K; quantified in Figures S4L–M), suggesting that separate mechanisms prevent this subset of axons being myelinated in the CC. Taken together, TFEB controls myelination timing and is required to limit the myelin sheath thickness in developing and adult white matter.
TFEB Functions Cell-autonomously to Promote OL Apoptosis and Inhibit OL Maturation
To determine TFEB cell-autonomous functions, we acutely purified OPCs from P12 control or TFEB cKO brains and differentiated them in vitro in the presence of all known trophic factors for OLs but in the absence of neurons or other cell types (Emery and Dugas, 2013). We characterized OL differentiation by static staining (Figures 5B–G) and by live imaging (Figures 5H–I). Like rat OLs (Zuchero et al., 2015), mouse OLs differentiate in a stereotypic manner in vitro (Figures 5B, 5C, S5E, and S5G, summarized in Figure 5A; see also Movie S1). Remarkably, at differentiation day 4 approximately 40% of control OLs exhibit fragmented cellular processes (red arrows in Figure 5C; quantified in Figure 5F) and most of these cells are immunopositive for cleaved Caspase-3 (red arrows in Figure 5C’; quantified in Figure 5G). The cells with fragmented processes are negative for CalceinAM (live cell marker) but positive for ethidium homodimer 1 (EtHD-1; dead cell marker; Figure S5A), confirming that they die during differentiation. In contrast, TFEB cKO OLs exhibit significantly decreased cell death events during differentiation (Figures 5D–E’ and Figure S5A’; quantified in Figures 5F–G). These results are confirmed by a live imaging assay using the cell death marker Annexin V to visualize OL programmed cell death in real time (Dumont et al., 2001)(Figure 5H and Figures S5B–C’; quantified in Figures 5I and S5D; see also Movies S1 and S2). Additional, overexpressing TFEB in wildtype OPCs promotes OL cell death during differentiation (Figures 5J–K’ and Figure S5K; quantified in Figure 5L). Thus, TFEB acts cell-autonomously in OLs to facilitate programmed cell death during differentiation.
Figure 5. TFEB Mediates OL Programmed Cell Death during Normal Differentiation.

(A) Diagram showing OL morphological changes and cell death during in vitro differentiation. (B-E’) Representative micrographs of control OLs (B-C’) and TFEB cKO OLs (D-E’) that have differentiated for 2 days (B and D) and 4 days (C, C’, E, and E’) in culture. A subset of control OLs undergo programmed cell death at day 4 (C and C’), as evidenced by their fragmented cellular processes (red arrows in C) and the presence of cleaved Caspase-3 immunofluorescent signals (red arrows in C’). In contrast, TFEB cKO OLs display reduced cell death (compare E to C, and E’ to C’; quantified in F and G). (F and G) Quantification of live cell ratio (F) and cleaved Caspase-3+ cell ratio (G) in TFEB cKO and littermate control OLs. n≥6 biological replicates. (H) Representative live-imaging micrographs of littermate control (top panels) and TFEB cKO OLs (bottom panels) throughout OL differentiation (left three columns), labeled by Annexin V (red). Note that a subset of control OLs became Annexin V+ at 72 hr post differentiation, a stage when the OPCs have just differentiated into the pre-OL stage but not yet become fully mature. (I) Quantification of Annexin V+ cell area throughout OL in vitro differentiation. n=4 independent experiments with 2–12 replicates per experiment. (J-K’) Representative micrographs of OLs transfected with plasmids expressing GFP alone (J and K) or GFP-TFEB (J’ and K’) at day 1 (J and J’) and day 7 (K and K’) after transfection followed with differentiation. (L) Quantification of the ratio of GFP+ cells to the total cells following the transfection with the plasmid expressing GFP or GFP-TFEB. n ≥6 biological replicates for each condition. Error bars represent SEM. Scale bars: 100 μm in (E’) for (B)-(E’); 100 μm in (H); and 100 μm in (K’) for (J)-(K’).
To address whether TFEB controls premature OL differentiation and maturation, we characterized myelin protein expression in differentiating OLs. We found that at day 2 and day 4, the percentages of MBP+ cells are the same in control and TFEB cKO OLs (Figures S5E–F), and that the mRNAs of several myelin genes are not changed in the absence of TFEB (Figure S5J). At day 6 following differentiation, TFEB cKO OLs exhibit increased cell surface areas (quantified in Figure S5I) and begin expressing elevated levels of mature OL markers, including MOG (Figures S5G–G’; quantified in Figure S5H), MOBP, and PLP1 (Figure S5J). These results demonstrate that loss of TFEB does not prevent OL differentiation but rather causes a small but significant acceleration of OL maturation in culture. Conversely, overexpressing TFEB in wildtype OLs strongly blocks cell differentiation (Figures S5L–M), together showing that TFEB is necessary and sufficient to inhibit OL maturation.
TFEB Promotes Subsets of Endoplasmic Reticulum (ER) Stress Gene Expression and Induces Pro-Apoptotic Factor PUMA in Pre-OLs
To address the molecular mechanisms underlying TFEB-dependent OL development and myelination, we employed an unbiased, whole-genome RNA sequencing approach with acutely purified pre-OLs, the differentiation stage when TFEB is most highly enriched (Figure 1) and playing crucial roles (Figures 2–5). We first validated sample purity by analyzing a panel of molecular markers that each define a specific brain cell type (Figure S6A). We then performed differential gene expression analysis between TFEB cKO and heterozygous (Het) pre-OLs using DESeq2 (Love et al., 2014) (see also STAR Methods). We found 1895 significantly downregulated genes and 1760 significantly upregulated genes in TFEB cKO pre-OLs (Figures 6A and 6B; see the entire list of genes in Table S1). We analyzed lysosomal and autophagy genes in TFEB cKO pre-OLs, as TFEB is best known for its function in lysosomal biogenesis and autophagy (Sardiello et al., 2009; Settembre et al., 2011). Many lysosomal and autophagy genes are not significantly changed in the absence of TFEB (61/89 lysosomal genes and 38/51 autophagy genes are not significantly changed, see Figures S6B–C). Furthermore, TFEB cKO OLs contain the same number of Lamp1+ puncta (Figure S6D–E’; quantified in Figure S6F). We also found that TFE3, another master regulator for lysosome biogenesis (Martina et al., 2014), is expressed and its mRNA level remains unchanged in TFEB cKO OLs (Figure S6G). These results suggest that TFEB might function cooperatively or redundantly with TFE3 in regulating lysosome biogenesis in OLs, similarly to what has been observed in the immune system (Pastore et al., 2016). Because TFEB has been shown to mainly function as a transcriptional activator (Sardiello et al., 2009), we focused on the downregulated genes in the absence of TFEB and utilized Gene Ontology (GO) term analysis to help identify molecular pathways that may be involved in its function. Our GO term analysis of downregulated genes identified hits in pathways involving mitochondrial membrane permeability (GO:0035795; Figure 6C and Table S2) and intrinsic apoptotic signaling pathways in response to endoplasmic reticulum (ER) stress (GO:0070059; Figure 6C and Table S3). In particular, the expression of a pro-apoptotic gene Bbc3/PUMA is among the most-downregulated genes in TFEB cKO pre-OLs (Figure 6A; −1.679 Log2 fold change; see also Figure S6G). We also discovered that Ddit3/CHOP (Figure 6A; see also Figure S6G), a critical gene for ER stress response (Lin and Popko, 2009), as well as genes involved in DNA damage repair (Gadd45b and Ddit4; Figure 6A) are significantly downregulated in TFEB cKO pre-OLs. These results suggest that TFEB may activate ER stress and/or apoptosis pathways to control OL survival.
Figure 6. TFEB Promotes Subsets of Endoplasmic Reticulum (ER) Stress Gene Expression and Induces Pro-Apoptotic Factor PUMA in Pre-OLs.

(A) Volcano plot showing differentially expressed genes in pre-OLs acutely purified from P12 TFEB cKO (Olig2-Cre; TFEBF/F) as compared to those purified from littermate control mice (Olig2-Cre; TFEBF/+). Each dot represents a gene. Blue color dots are genes up- or down-regulated greater than two-fold. Four selected genes are denoted and highlighted in red. (B) Heatmap showing expression levels (in z scores) of genes up- or down-regulated greater than two-fold in TFEB cKO pre-OLs by RNAseq. Replicates of TFEB heterozygous control cells (Het) and KO cells form distinct clusters are shown by the dendrogram. Each row corresponds to a gene, and rows are ordered by fold change values from the highest to the lowest. See Table S1 for the full gene list. (C) Gene ontology (GO) term analysis (PANTHER Overrepresentation Test) by using genes down-regulated more than 2-fold as input. Top 10 most enriched biological process (BP) terms are shown. Only the narrowest term from each hierarchical group in the analysis is plotted. The full list of enriched terms can be found in Table S2. (D) qRT-PCR of PUMA(Bbc3) mRNA expression in differentiating OLs from TFEB cKO mice and littermate controls. n=3 biological replicates for both genotypes and for each time points. Error bars represent SEM.
In mouse embryonic fibroblast cells, TFE3 can directly bind to the PUMA E-Box region, and overexpressing TFEB or TFE3 upregulates PUMA expression (Martina et al., 2016). To test if TFEB promotes PUMA transcriptional expression in differentiating OLs, we validated our RNAseq results in which PUMA mRNA was downregulated in acutely purified, TFEB cKO pre-OLs. PUMA mRNAs are the same in control and TFEB cKO OLs at day 2 during differentiation (Figure 6D), a time point when OL cell death has not yet occurred (Figures 5B and 5D). By day 4 when OL programmed cell death begins, PUMA mRNA significantly increases in control OLs but is largely downregulated in TFEB cKO OLs (Figure 6D). Importantly, TFEB-dependent upregulation of PUMA mRNA is not via p53, a putative transcriptional activator for PUMA in some cellular contexts, as OL-conditional deletion of p53 does not cause OL ectopic survival or aberrant myelination (Figures S7C–D’; quantified in Figure S7E).
TFEB Functions Through the PUMA-Bax/Bak Axis to Regulate the Location and Timing of CNS Myelination
PUMA acts as a pro-apoptotic factor by binding to anti-apoptotic Bcl-2 family members to induce Bax/Bak activation, mitochondrial dysfunction, and Caspase activation (Hikisz and Kilianska, 2012; Simon et al., 2016). We next set out to determine if PUMA functions downstream of TFEB to promote OL programmed cell death. We found that indeed PUMA mRNA is expressed by differentiating OLs in vitro (Figure 6D) and in vivo (Figures S7A–B; 21/36 ENPP6+ cells from 4 animals co-express PUMA in the cerebellar molecular layer). PUMA−/− mutants show a significant reduction in OL cell death in culture (Figure 7A; quantified in Figures 7B–C) and a significant increase in OL number in the cortex (Figures S7F–F“; quantified in Figure S7G). Compared to wildtype animals, PUMA−/− mutants exhibit fully-penetrant (4/4 animals), ectopic presence of myelin proteins in the cerebellar ML and cortical layer I at P14, phenocoyping TFEB cKO mice (Figures 7D’ and 7F’, respectively; quantified in Figures 7E and 7G). Interestingly, PUMA−/− mutants show normal thickness of myelin sheaths in the corpus callosum compared to littermate controls (Figures S7H–I), and significantly increased g-ratios in comparison to those in TFEB cKO mice (Figure S7J). These results suggest that TFEB exerts additional functions in regulating myelination besides its roles in mediating pre-OL cell death.
Figure 7. TFEB-PUMA-Bax/Bak Axis Controls the Location and Timing of CNS Myelination.

(A) Representative live imaging micrographs of wildtype (WT; left) and PUMA−/− OLs (right) labeled by CaleinAM (green; live cells) and Annexin V (red; dead cells) 7 days after in vitro differentiation. Note that a subset of WT OLs died (yellow arrows in the left panel) whereas most PUMA−/− OLs survived (right panel). (B) Quantification of the ratio of the total live cell area (CalceinAM+) to the total cell area (CalceinAM+ and Annexin V+) in WT and PUMA−/− OLs. (C) Quantification of the total Annexin V+ cell area throughout OL differentiation. n=3 independent experiments with more than 5 replicates per experiment. (D-D” and F-F”) Representative confocal micrographs of the cerebellar ML (D-D”) and upper cortical layers (F-F”) from P14 WT (D and F), PUMA−/− (D’ and F’), and CNP-Cre; BaxF/F; Bak−/− mice (D” and F”), showing that PUMA−/− and CNP-Cre; BaxF/F; Bak−/− mutant mice exhibit ectopic MBP immunofluorescence in the cerebellar ML (D’ and D”, respectively) and cortical layer I (F’ and F”, respectively; white vertical bars demarcate layer I). (H-H”) A representative P7 sagittal brain section showing that MBP immunofluorescent signals are primarily located in the corpus callosum and deep cortical layers in WT (H; see also Figure 3A). In contrast, PUMA−/− (H’) and CNP-Cre; BaxF/F; Bak−/− mice (H”) display widespread MBP fluorescent signals precociously in many brain regions including upper cortical layers, fornix, internal capsule, and the thalamus. (E, G, and I) Quantification of the averaged MBP fluorescent intensity in P14 cerebellar ML (E), P14 cortical layer I (G), and P7 forebrain (I) from WT (black circles), PUMA−/− (red circles), and CNP-Cre; BaxF/F; Bak−/− mice (blue circles). (J) Performance of littermate control (TFEBF/F, n=9) and TFEB cKO mice (CNP-Cre; TFEBF/F, n=9) in an open-field test, showing the total distance traveled and averaged velocity through the entire 10-minute session. Left two panels show the representative movement paths of a littermate control and a TFEB cKO mouse through the entire session, respectively. (K) Performance of littermate control (TFEBF/F, n=7) and TFEB cKO (CNP-Cre; TFEBF/F, n=5) on an accelerating rotarod (4–40rpm). The averaged latency to fall off for individual trials (left) and the averaged latency to fall off for a given day (right) are plotted. *P<0.05, two-way ANOVA followed by Bonferroni’s multiple comparisons test. (L) The TFEB-PUMA-Bax/Bak axis controls the location and timing of CNS myelination. TFEB is highly expressed by pre-OLs and it facilitates programmed cell death of a subset of pre-OLs during development. TFEB induces PUMA mRNA expression, which encodes a pro-apoptotic factor that triggers Bax/Bak-dependent programmed cell death. The continuous expression of TFEB in myelinating OLs acts as a brake on OL maturation and myelination. Error bars represent SEM. Scale bars: 100 μm in (A); 100 μm in (F”) for (D)-(F”); and 1 mm in (H”) for (H)-(H”).
To further investigate if PUMA functions through Bax/Bak-dependent programmed cell death, we analyzed Bax−/− mutants and found that they exhibit normal myelination patterns in the cerebellum (compare Figure S7K’ to S7K). However, Bax can function redundantly with Bak in promoting OL programmed cell death (Kawai et al., 2009). Therefore, we next generated OL-specific, Bax conditional knockout mice in a Bak mutant background (CNP-Cre; BaxF/F; Bak−/−). CNP-Cre; BaxF/F; Bak−/− mutants show fully-penetrant (4/4 animals), ectopic myelin presence in both the cerebellar ML and cortical layer I (Figures 7D” and 7F”, respectively; quantified in Figures 7E and 7G), again phenocopying TFEB cKO and PUMA−/− mutants. To further address whether OL programmed cell death determines the timing of myelination, we analyzed early postnatal PUMA−/− and CNP-Cre; BaxF/F; Bak−/− mutants in which programmed OL cell death is genetically blocked. In wildtype forebrain, differentiated MBP+ OLs are primarily located in the corpus callosum and deep cortical layers, which are among the earliest myelinated brain regions (Figure 7H). Similar to TFEB cKO mice (Figure 3A’), P7 PUMA−/− mutants and CNP-Cre; BaxF/F; Bak−/− mutants exhibit numerous differentiated OLs “precociously” in brain regions that are typically myelinated at much later developmental stages (Figures 7H’ and 7H”, respectively; quantified in Figure 7I). These results together show that the TFEB-PUMA-Bax/Bak axis determines the location and timing of CNS myelination.
To understand the physiological consequences of precocious and ectopic myelination, we next performed open-field tests to assess gross locomotor activity in 4-month-old TFEB cKO mice. We found no difference in total distance traveled and average velocity between TFEB cKO mice and littermate controls (Figure 7J). We then evaluated sensory-motor coordination with accelerating rotarod tests. Intriguingly, TFEB cKO mice exhibit enhanced performance at the first trial on day 1 (Figure 7K; average time to fall off is 89.7±38.8 seconds for control and is 211.4±23.8 seconds for TFEB cKO mice, mean±SEM, n•5 animals per genotype), but show normal performance for the remainder of trials in comparison to littermate controls (Figure 7K). These results suggest that disrupted myelination timing and location might alter certain aspects of sensory-motor function.
DISCUSSION
Over a century ago, Fleschig described that myelination occurs precisely in time and space in humans (Flechsig Of Leipsic). Little is known, however, about the cellular and molecular mechanisms that govern the temporal and regional specificity of myelination. Previous work showed that ectopically inducing OL apoptosis delays developmental myelination (Caprariello et al., 2015). In this study we revealed intrinsic mechanisms underlying pre-OL programmed cell death, which in turn control the location and timing of CNS myelination. We found that TFEB is highly expressed by OL lineage cells and is enriched in pre-OLs, a transient but vulnerable stage during OL development and myelination. TFEB functions cell-autonomously to promote the expression of ER stress genes and PUMA, which encodes a pro-apoptotic factor that induces Bax/Bak-dependent programmed cell death. Selective OL elimination mediated by the TFEB-PUMA-Bax/Bak pathway ensures the spatial and temporal specificity of developmental myelination and certain sensory-motor functions (Figure 7L).
Oligogenesis and myelin remodeling continues throughout adulthood, with nearly half of OLs generated after 4 months of age in rodents (Hughes et al., 2018). Recent work suggests that subsets of pre-OLs still undergo elimination after being differentiated in the adult brain (Hill et al., 2018; Hughes et al., 2018), however the underlying molecular mechanisms remain largely unknown. The continuous expression of TFEB in OL lineage cells into adulthood, as well as the increased OL cell numbers in 15-month-old TFEB cKO mice suggest that TFEB may regulate survival of adult-born OLs. Recent work also demonstrates that axons in the superficial cortical layers have intermittent myelin patterns along their length, and that gaps between adjacent myelin sheaths are very slowly occupied over time (Hill et al., 2018; Hughes et al., 2018). It will be important to address if ectopically survived OLs in TFEB cKO mice can precociously myelinate axon segments within these gaps, and if the extent of myelination by individual OLs, including the sheath number and sheath length, is controlled by TFEB.
What are the upstream signaling events converging onto TFEB in OLs? Previous work demonstrated that ectopically enlarging axon calibers of cerebellar granule cells led to increased OPC proliferation and differentiation, as well as de novo myelination in the cerebellar ML (Goebbels et al., 2017). This finding suggests that neuronal trophic cues act upstream of OL differentiation and survival. Indeed, cerebellar granule cell axons with enlarged diameter exhibit elevated expression of many secreted proteins, including Bdnf, Vegfc, and Tmsb4x, all of which are associated with OL development (Goebbels et al., 2017). Therefore, it will be of great interest to determine whether axonal signals regulate TFEB, and if so what the identities of these signals are.
How does TFEB act as the brake on OL maturation and myelination? Our whole genome transcriptome analysis revealed several candidate pathways possibly regulated by TFEB, including Wnt signaling pathway and PTEN-AKT-mTOR signaling pathway (Figure 6 and Tables S1 and S2), all of which have been previously demonstrated to regulate OL differentiation and myelination (Fancy et al., 2009; Figlia et al., 2018; Wood et al., 2013). In cancer and liver cells the mTOR complex directly regulates the activity and localization of TFEB, and TFEB in turn modulates mTOR signaling activity (Di Malta et al., 2017; Settembre et al., 2013). It remains to be determined with respect to the molecular mechanisms through which TFEB regulates OL maturation and myelination.
The precocious and ectopic myelination phenotypes in TFEB cKO mice provide a useful model to begin addressing the physiological consequences of disrupted myelination timing and location. TFEB cKO mice exhibit altered performance in accelerating rotarod tests, suggesting that aberrant myelination might influence circuitry-specific neural functions. Ectopic myelination on subsets of parallel fibers could potentially change their conduction velocity and therefore alter the synchronization of nerve pulse propagation along these fibers (Wyatt et al., 2005). Further electrophysiological studies and comprehensive behavioral tests will begin to address the underlying neural mechanisms. Additionally, TFEB cKO mice are precociously myelinated during early development. It will be important to investigate if this precocious myelination compromises cognitive functions, especially in the late-myelinating prefrontal cortex (PFC), which integrates neural circuits critical for emotional regulation, memory storage, and attention (Flechsig Of Leipsic). Finally, TFEB cKO adult mice harbor significantly increased OL numbers and possibly saturated myelination, allowing for further investigations on their impacts upon adaptive myelination capacity and related neural functions (Monje, 2018).
In conclusion, the TFEB-PUMA-Bax/Bak pathway regulates critical aspects of OL development and controls the spatiotemporal pattern of CNS myelination. Our observations provide a foundation for future work to identify the upstream signaling pathways and intracellular events through which TFEB regulates myelination. It will be of great interest to determine whether the same pathway facilities OL cell death under demyelinating insults, and whether modulating this pathway promotes myelin repair.
STAR METHODS TEXT
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lu O. Sun (lsun2@stanford.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals.
Experiments involving animals followed a protocol approved by the Administrative Panel on Laboratory Animal Care (APLAC) of Stanford University in accordance with NIH guidelines. Mice were housed in plastic cages with disposable bedding on normal light-dark cycles (8:30am-8:30pm with light on, and 8:30 pm-8:30am with light off) with food and water ad libitum. All mice appeared healthy, received regular monitoring from veterinary and animal care staff, and were not involved in prior procedures or testing. The day of birth in this study was designated postnatal day 0 (P0) and all the developmental stages were specified in figures and figure legends. All mice used in the behavior assays were test naïve for individual behavioral paradigms. Male and female mice were used for all experiments except for accelerating rotarod tests where only male mice were utilized. We do not have evidence showing that male and female mice differ significantly in oligodendrocyte cell survival. TFEB Knock-in (TFEBKI) mice were generated by the Knockout Mice Project (KOMP; project number: CSD35261), TFEBLacZ/+ and TFEBFlox/+ (TFEBF/+) mice were generated from TFEBKI mice and maintained in the C57BL/6 background (see details in Figure S1C). TFEBLacZ/+ mice were genotyped using PCR reactions with two pairs of primers to amplify the LacZ allele (5’-gggatctcatgctggagttcttcg-3’ and 5’-atccactgagccatctctccaacc-3’). TFEBF/+ mice were genotyped using PCR reactions with two pairs of primers to amplify the WT and the Flox allele (5’-caagtagaactgagtcaaggcatactgg-3’ and 5’-atccactgagccatctctccaacc-3’). The sizes of PCR products of WT, LacZ, and Flox allele are 352bp, 492bp, and 604bp, respectively. Olig2-Cre (Jackson Laboratory, Olig2tm2(TVA,cre)Rth/J, Stock#011103), PDGFRa-CreER (Jackson Laboratory, B6N.Cg-Tg(Pdgfra-cre/ERT)467Dbe/J, Stock #018280), and PUMA−/− mice (Jackson Laboratory, C57BL/6-Bbc3tm1Ast/J, Stock#011067) were described previously (Kang et al., 2010) (Simon et al., 2016). CNP-Cre mice were generously provided by Prof. Klaus-Armin Nave (Lappe-Siefke et al., 2003). Sox2-Cre (Edil3Tg(Sox2-cre)1Amc/J, Stock#004783), Actb-Flp1 (B6; SJL-Tg(ACTFLPe)9205Dym/J, Stock#003800), Ai14 (B6; 129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, Stock#007908), p53 F/F (B6.129P2-Trp53tm1Brn/J, Stock#008462), Bax−/− (B6.129X1-Baxtm1Sjk/J, Stock#002994), and BaxF/F; Bak−/− (B6;129-Baxtm2Sjk Bak1tm1Thsn/J, Stock#006329) mice were obtained from the Jackson Laboratory.
Culture for mouse oligodendrocyte lineage cells and HEK293T cells.
Primary oligodendrocyte precursor cells (OPCs) purified from individual P12 male or female mouse brains were cultured in the “proliferation medium” containing DMEM-SATO Base Growth Medium (Emery and Dugas, 2013) supplemented with 4.2 μg/ml forskolin (Sigma-Aldrich, Cat#F6886), 10 ng/ml PDGF (Peprotech, Cat#100–13A), 10 ng/ml CNTF (Peprotech, Cat#450–02), and 1 ng/ml neurotrophin-3 (NT-3; Peprotech, Cat#450–03) in a 37°C, 10% CO2 humidified incubator. Oligodendrocytes (OLs) were cultured in the “differentiation medium” containing DMEM-SATO Base Growth Medium(Emery and Dugas, 2013) supplemented with 4.2 μg/ml forskolin (Sigma-Aldrich, Cat#F6886), 10 ng/ml CNTF (Peprotech, Cat#450–02), and 40 ng/ml thyroid hormone (T3; Sigma-Aldrich, Cat#T6397). OPCs and OLs were authenticated by their stereotypical morphologies and distinct molecular makers (see Figures 1J and S6A). HEK293T cells were obtained from Clontech (Cat#632273) without the prior knowledge regarding the sex of this cell line. HEK293 cells were cultured in DMEM (Gibco, Cat#11960044) supplemented with 10% fetal bovine serum (FBS; Gibco, Cat#16000044), 2mM L-glutamine (Gibco, Cat#25030081), 1mM sodium pyruvate (Gibco, Cat#11360070), and 1,000 U/ml Penicillin-Streptomycin (Gibco, Cat#15140148) in a 37°C humidified incubator containing 5% CO2. HEK293T cells were not authenticated after purchase.
METHOD DETAILS
In situ hybridization.
In situ hybridization was performed on fresh frozen sagittal brain sections (12 μm) using RNAscope® Fluorescent Multiplex Assay as described by vendor’s protocol (ACDbio, Cat#320850). In situ probes include: TFEB (ACDbio, Cat#434701), Ddap (RNAscope®3-plex negative control probe, Cat#320871), Olig2 (Cat#447091-C2), ENPP6 (Cat#511021-C2), ASPA (Cat#425891), PDGFRa (Cat#480661-C2), MBP (Cat#451491 and Cat#451491-C2), and Bbc3 (PUMA; Cat#423721). Fluorescent images were taken using a Zeiss Axio Imager M1 and Axiovision software, or using a Zeiss 710 inverted confocal microscope and Zen software.
Immunopanning for oligodendrocyte lineage cells and in vitro differentiation.
Immunopanning for oligodendrocyte lineage cells was performed as previously described (Emery and Dugas, 2013). Briefly, the animals were quickly decapitated and the brains were dissected out and diced into ~1-mm3 chunks in DPBS without Ca2+/Mg2+ at room temperature. The brain tissues were dissociated in a papain buffer containing Earle’s Balanced Salt Solution (EBSS; Invitrogen, Cat#14155–063) supplemented with 1 mM MgSO4, 0.46% glucose, 2 mM EGTA, 26 mM NaHCO3, 20 units/ml of papain (Worthington Biochemical, Cat#LS003126), and 250 units/ml of DNase I (Worthington Biochemical, Cat#LS002007). The tissues were digested in the papain buffer under 5% CO2/95% O2 gas flow at 34°C for 90 minutes to make a single-cell suspension. After digestion and sequential trituration, the single-cell suspension was incubated onto two BSL-1-coated plates (10 minutes each; Vector Laboratories, Cat#L1100) to deplete endothelial cells and microglia. The cell suspension was then transfer for 20 minutes on a plate coated with rat anti-mouse CD45 (1.25 μg in 12 ml of DPBS/0.2%BSA; BD Pharmingen, Cat#550539) to deplete remaining microglia. The remaining cell suspension was incubated for minutes on a rat anti-PDGFRa (rat anti-mouse CD140A, 10 μg in 12 ml of DPBS/0.2% BSA; BD Pharmingen, Cat#558774) coated plate to isolate OPCs. To collect premyelinating and myelinating OLs, the remaining cell suspension was first incubated on a plate coated with mouse A2B5 monoclonal antibody ascites to deplete remaining OPCs. The cell suspension was next incubated on an anti-myelin oligodendrocyte glycoprotein (MOG) hybridoma-coated plate for 30 minutes to harvest myelinating oligodendrocytes, followed by an additional anti-MOG hybridoma-coated plate for 30 minutes to deplete any remaining myelinating OLs. Finally, the cell suspension was incubated on an anti-galactocerebroside (GalC) hybridoma-coated plate for minutes to harvest premyelinating OLs. To differentiate OPCs in vitro, purified OPCs were plated at a density of 5,000–20,000 cell/well in 24-well plates and cultured in the “proliferation medium” (see details in “Culture for mouse oligodendrocyte lineage cells”) for 24 hours before the medium was completely replaced with the “differentiation medium” (see details in “Culture for mouse oligodendrocyte lineage cells”). The cells were cultured for additional 6 to 10 days in the “differentiation medium” with half of the medium being replaced by fresh medium every two days.
4-hydroxytamoxifen (4-HT) injections.
4HT (Sigma-Aldrich, Cat#H7409) was dissolved in sunflower oil (Sigma-Aldrich, Cat#W530285–5KG-K) and was delivered by a single intraperitoneal injection from P4 to P7 with a dose of 100 μg per animal. The brains from these animals were processed and analyzed at P21.
Immunohistochemistry.
Mouse brains were perfused with the chilled 4% PFA/PBS solution and post-fixed in the same solution for 3 hours at 4°C before being cryopreserved with a 30% sucrose/PBS solution at 4°C overnight. Cultured cells were briefly fixed with a chilled 4% PFA/PBS solution for 5 minutes and rinsed 3X with PBS on ice. For immunostaining, 16–18 μm sagittal brain sections or coverslips with cultured cells were first rinsed in the blocking solution containing PBS supplemented with 0.1% TritonX-100 (PBST; Sigma-Aldrich, Cat#T8787) and 10% normal goat serum (Gibco, Cat#PCN5000) for 10 minutes at room temperature. The sections or coverslips were then incubated with the primary antibodies in the staining solution (PBST supplemented with 1% normal goat serum) at 4°C overnight. Primary antibodies used in this study include: chicken anti-LacZ (Aves lab, 1:1000, Cat#BGL-1040), rabbit anti-DsRed (Takara Clontech, 1:1000, Cat#632496), rabbit anti-NG2 (EMD Millipore, 1:500, Cat#ab5320), rat anti-MBP (Abcam, 1:100, Cat#ab7349), rabbit anti-PLP (Abcam, 1:200, Cat#ab105784), rabbit anti-cleaved Caspase-3 (Asp175) (Cell Signaling Technology, 1:200, Cat#9661S), mouse anti-CC1 (EMD Millipore, 1:200, Cat#OP80–100UG), rabbit anti-PDGFRa (Santa Cruz Biotechnology, 1:200, Cat#sc-338), rat anti-Ctip2 (Abcam, 1:500, Cat#ab18465), Cux1 (CDP; Santa Cruz Biotechnology, 1:200, Cat#sc-13024), rabbit anti-Calbindin (Swant, 1:2000, Cat#CB38), mouse anti-MOG hybridoma (1:50) (Emery et al., 2009), and rabbit anti-GFP (Thermo Fisher Scientific, 1:1000, Cat#A-11122). The brain sections or coverslips were rinsed 30 minutes for 4 times in PBST at room temperature, and further incubated with appropriate Alexa Fluor 488 or 594 secondary antibodies (Thermo Fisher Scientific, 1:1000, diluted in PBST containing 1% normal goat serum) for 1 hour at room temperature. DAPI (Thermo Fisher Scientific, 5 μg/mL, Cat#D1306) and HCS CellMask™ Blue Stain (Thermo Fisher Scientific, 1:1000, Cat#H32720) were used for counterstaining for brain sections and cultured cells, respectively. Fluorescent images were taken using a Zeiss Axio Imager M1 and Axiovision software, or using a Zeiss 710 inverted confocal microscope and Zen software.
X-gal staining.
Mouse brains were perfused with a chilled 4% PFA/PBS solution and post-fixed in the same solution for 1 hour at 4°C before being cryopreserved with 30% sucrose/PBS solution at 4 °C overnight. Brain sections (18 μm) were collected on slides with a cryostat and rinsed 2X in PBS before staining. Brain sections were then incubated with the X-gal staining solution containing 5 mM potassium ferricyanide (Sigma-Aldrich, Cat#702587), 5 mM potassium ferrocyanide (Sigma-Aldrich, Cat#31254), 2mM MgCl2 (Sigma-Aldrich, Cat#M8266) and 1 mg/ml X-gal (Sigma-Aldrich, Cat#10651745001) in a humified chamber for 24 hours at 37°C. Tissue sections were rinsed in PBS 3x and bright-field images were taken using a Zeiss Axio Scan.Z1 scope.
Measurement and quantification of the averaged MBP fluorescent intensity.
For the measurement of MBP fluorescent intensity, the micrograph was first converted to a binary image with the ImagJ “Make Binary” tool. The area of interest was then selected on the binary image by the ImageJ “Freehand Selections” tool. The mean MBP fluorescent intensity value was directly measured by the ImageJ “Measure” tool, using the readout of “mean grey value”. For the quantification of averaged MBP fluorescent intensity, the mean intensity values in 3-to-5 adjacent brain sections from one animal were measured and the averaged MBP fluorescent intensity from those brain sections was determined for further statistical analysis. At least 3 animals per genotype were analyzed and the N number was specified in the Figures and Figure Legends.
Brain lysate extraction.
The brain was dissected on ice and placed into a chilled PYREX Dounce Tissue Grinder (Thermo Fisher Scientific, Cat#7722–7), filled with 7 ml of chilled homogenization buffer containing 0.32M sucrose (Sigma-Aldrich, Cat#S9318), 10mM HEPES (pH=7.4; Sigma-Aldrich, Cat#H3375), 2mM EDTA (Sigma-Aldrich, Cat#1233508), and protease inhibitors (Sigma-Aldrich, Cat#11697498001). Brains were homogenized with 10–15 strokes on ice and spin down at 3000 rpm at 4°C for 15 minutes to remove pelleted fraction. The supernatant was then collected and protein concentrations were determined by a BCA assay (Thermo Fisher Scientific, Cat#23225).
Western immunoblotting and quantification of protein expression.
For cultured cells, each well of 6-well plates was rinsed with room-temperature DPBS then lysed in chilled RIPA Buffer (Thermo Fisher Scientific, Cat#89900) supplemented with 1X cOmplete protease inhibitor (Sigma-Aldrich, Cat#5892791001) for 10 minutes on ice. Cell lysate was then collected with a cell scraper and frozen at −80°C until further processing. Protein concentration was measured with a BCA assay (Thermo Fisher Scientific, Cat#23225) and protein samples were denatured with 5X Sample Buffer for 5 minutes at 95°C. 0.2–2 μg of total proteins was loaded onto a Trigly 4–12% gradient polyacrylamide gels (Thermo Fisher Scientific, Cat#NP0336BOX) and run at 180V for 0.5–1hr for separation. Proteins were then transferred to 0.45 nM PVDF membrane (EMD-Millipore, Cat#IPVH00010) at 100V for 1–2 hours on ice. Blots were washed in solution containing 0.1% Tween-20 in PBS (PBST) and blocked at room-temperature for 1 hr in Odyssey blocking buffer (LI-COR, Cat#92740000). Primary antibodies were incubated overnight at 4°C on a rocker in Odyssey blocking buffer. Primary antibodies include: rabbit anti-TFEB (Bthyl Laboratories, 1:1000, Cat# A303–673A), rabbit anti-PDGFRα (Santa Cruz Biotechnology, 1:200, Cat#sc338), mouse anti-β actin (Santa Cruz Biotechnology, 1:500, Cat#sc-47778), rat anti-MBP (Abcam, 1:500, Cat#ab7349), mouse anti-CNPase (EMD Millipore, 1:1000, Cat#MAB326), rabbit anti-GAPDH (ProSci Inc., 1:500, Cat#3781), and mouse anti-GAPDH (Santa Cruz Biotechnology, 1:1000, Cat#365062). Blots were washed with PBST 3x for 20 minutes and incubated at room-temperature for 1 hour with HRP or IRDye secondary antibodies in Odyssey blocking buffer. Blots were then washed in PBST 3x for 20 minutes each. HRP blots were developed with WesternBright Sirius-HRP Substrate (Advansta, Cat#K-12043-D10) for 2 minutes and imaged with X-Ray films. Blots incubated in IRDye secondary antibodies were imaged on a LI-COR Odyssey CLx. For protein quantification, film blots were scanned and densitometry was performed with ImageJ and normalized to GAPDH. For LI-COR Odyssey CLx imaged blots, densitometry was performed on the LI-COR software and normalized to GAPDH.
Molecular cloning and OPC transfection.
GFP-TFEB expression construct was generated by subcloning mouse TFEB cDNA into a pEGFP-N1-Flag vector, which was a gift from Patrick Calsou (Addgene plasmid, #60360) (Britton et al., 2014). Briefly, TFEB cDNA was amplified by PCR reactions from a TFEB (NM_011549) mouse tagged ORF clone (Origene Technologies, Cat#MR223016) with primers 5’-tctcgagctcaagcttgccaccatggctcagctcgctcagt-3’ and 5’-gatcccgggcccgcggtaccttcagaacatcaccctcctccat-3’. pEGFP-N1-Flag vector was linearized with restriction enzymes KpnI (New England Biotechnology, Cat#R0142S) and HindIII (New England Biotechnology, Cat#R0104S). Amplified TFEB cDNA was then subcloned into pEGFP-N1-Flag vector via KpnI and HindIII sites by recombination-mediated cloning (Infusion-HD cloning kit; Clontech, Cat#639648). The expression of GFP-TFEB was further validated by transfecting HEK293T cells with Lipofectamine 3000 (Thermo Fisher Scientific, Cat#L3000008) for 24 hours and by analyzing cell lysate with Western immunoblotting (see Figure S5N). mouse OPC transfection was performed as previously described (Zuchero et al., 2015). Briefly, 1 μg DNA was electroporated into 106 purified mouse OPCs using Lonza/Amaxa nucleofector kit (Lonza, Cat#VPI-1006). After the transfection, cells were briefly re-suspended and plated onto PDL-coated plastic coverslips with a density of 30,000/well in 24-well plates. Cells were allowed to recover in the “proliferation medium” for 24 hours before the medium was completely replaced by the “differentiation medium”.
Transmission Electron Microscopy (TEM).
Brains were processed for TEM analysis as previously described (Zuchero et al., 2015). Briefly, animals were perfused with the chilled Karlsson-Schltz Fixative containing 13mM NaH2PO4, 87mM Na2HPO4, 85.6mM NaCl, 2.5% glutaraldehyde (Electron Microscopy Sciences, Cat#16320), and 4% paraformaldehyde (Electron Microscopy Sciences, Cat#15710), and post-fixed in the same fixative solution at 4°C for 24 hours before further dissection. For the TEM analysis, the superficial layer of motor or somatosensory cortex was carefully dissected out for further EM sectioning. The pia surface of the brain was kept intact for the orientation of the EM sections. For TEM analysis on the corpus callosum, sagittal brain sections near the midline were obtained and only the medial region of the corpus callosum were further sectioned and analyzed. For TEM analysis on the cerebellum, coronal sections were obtained from a micro-dissected region covering the cerebellar molecular layer, Purkinje cell layer, and the granule cell layer. The pia surface of the cerebellum was kept intact for TEM section orientation. All tissues were serially dehydrated, embedded, and sectioned by Stanford Cell Science Imaging Facility as previously described (Zuchero et al., 2015). Images were acquired with a JEOL 1400 TEM.
Live imaging and data analysis.
Live imaging of OPCs and differentiating OLs was described previously with minor modifications (Zuchero et al., 2015). Briefly, OPCs purified from P12 mouse brains were allowed to recover and proliferate for 24 hours in the “proliferation medium” in 24-well plates (Thermo Fisher Scientific, Cat#08–772-1H) before the medium was completely replaced by the “differentiation medium”. The “differentiation medium” was supplemented with Annexin V Red Reagent for Apoptosis (Essen Bioscience, 1:200, Cat#4641). The plates were transferred into IncuCuyte ZOOM Live Cell Imaging System (Essen Bioscience) and imaged continuously for 6 to 11 days at 37°C and 10% CO2, with 2-hour frame rates. Half of the differentiation medium was replaced with fresh medium supplemented with Annexin V Red Reagent every two days during the entire imaging course. The Annexin V+ cell areas were measured by IncuCyte software provided by the vendor. For some experiments, 30 minutes before the end of live imaging CalceinAM (Thermo Fisher Scientific, 1:1000, Cat#C3100MP) and/or ethidium homodimer-1 (EthD-1, Thermo Fisher Scientific, 1:1000, Cat#E1169) were added into the medium to label live cells and dead cells, respectively.
Quantitative RT-PCR.
50,000 to 100,000 cells were rinsed with room-temperature DPBS, lysed in chilled RLT buffer (QIAGEN, Cat# 79216) supplemented with 2-β mercaptoethanol (Sigma-Aldrich, 1:100, Cat#M6250), and immediately frozen at −80°C until further processing. RNA was isolated using the RNeasy Micro Kit (QIAGEN, Cat#74004) with on-column DNase treatment. RNA was reverse-transcribed using a High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Cat#4387406) and used as a template for PowerUp SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat#A25741) in 20 μL reactions for cycling in a QuantStudio 3 QPCR machine (Thermo Fisher Scientific). Primers were designed to bridge splice-junctions using Primer-BLAST-NCBI or previously published sequences and used at 200 nM. Primer efficiency was measured for each primer set and was only used for experiments if efficiency was between 90% and 115% (r2 > 0.98). Primer sequences for mouse genes include: Cnp-f, tccacgagtgcaagacgctattca and Cnp-r, tgtaagcatcagcggacaccatct; Mobp-f, tccacaggaacctttcacaac and Mobp-r, gctcggagaacttctggttc; Plp1-f, gcactgttctgtggatgtgg and Plp1-r, gtggtgtagaagccctcagc; Bbc3 (PUMA)-f, agcacttagagtcgcccgtg and Bbc3 (PUMA)-r, tgctgctcttcttgtctccgc; and Eef1a-f, gtcctgattgttgctgctgg and Eef1a-r, tctgactgtatggtggctcg. CT values were registered using noiseband thresholding, and melting curves were checked to ensure formation of a single product within the expected size range. All CT values were detected before completion of the 38th cycle. CT values were converted to absolute values based on standard curves for each primer set and normalized to the averaged CT values of Eef1a.
Whole-genome RNA sequencing and analysis.
Total RNA from acutely purified premyelinating OLs was extracted using the RNeasy Micro Kit (QIAGEN, Cat#74004) with on-column DNase treatment. RNA quality was accessed by Bioanalyzer and samples with high RNA integrity number (>9) were used for library construction. TruSeq RNA Sample Prep Kit (Illumina, Cat#RS-122–2001 and RS-122–2002) was used to construct poly(A) selected paired-end sequencing library. Sequencing was conducted with the Illumina HiSeq 4000 Sequencer at the Stanford Functional Genomics Center. Read mapping, transcript assembly, and expression estimation were performed on usegalaxy.org. Briefly, mapping of Illumina 75 bp paired-end reads to the mouse reference genome (University of California, Santa Cruz Genome Browser version mm10) was performed using TopHat (version: 2.1.0) and Bowtie software (version: 2.2.5.0). After read mapping, Htseq-count followed by DESeq2 (version: 2.11.38) was applied to determine the raw read counts and normalized counts in order to compare gene expression levels between TFEB knockout and heterozygous pre-OLs (Love et al., 2014).
Gene Ontology (GO) term analysis was performed using the online service of PATHER Classification System as previously described (Mi et al., 2010). Genes with padj < 0.01 and log2FoldChange < −1 were used as the input for “Statistical overrepresentation test” with default settings. Fold enrichment values for the top 10 most enriched biological process (BP) terms were plotted. Only the narrowest term from each hierarchical group in the analysis was shown. False discovery rate (FDR) was calculated by Fisher’s Exact with FDR multiple test correction from the program.
Behavioral assays.
All behavioral experiments were performed independently and blindly without knowing mouse genotypes by Stanford Behavioral and Functional Neuroscience Core. Before starting the behavioral tests, mice were allowed to adapt to reversed light-dark cycles for a week. The reversed light-dark cycle is 8:30am-8:30pm with light off, and 8:30 pm-8:30am with light on. All the behavioral tests were conducted under dim yellow light illuminance during the daytime. For the open-field assay, assessment took place in an arena made of white squared box (length × width × height: 76cm × 76cm × 50cm). A 4-month-old animal (male or female) was placed in a corner of the open field arena and allowed to freely move about for 10 minutes while being tracked by an automated tracking system Ethovision XT (Noldus Information Technology, Wageningen, the Netherlands) via a camera mounted on the ceiling. At the end of each trial the surface of the arena is cleaned with 1% virkon solution. Distance moved, velocity, and time spent in each pre- pre-defined zones are recorded. For the accelerating rotarod test (model#ENV-575M, Med Associate Inc.), male mice were placed on a stationary rotarod for 10 seconds before the rod gradually accelerated from 4rpm to 40 rpm. The latency before falling off the rod was recorded and maximum time for testing is 300 seconds. Animals were tested for three trials per day with 15-minute inter-trial breaks, and they were tested for three days with one-day break in between (day 1, 3, and 5).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data analyses except for the RNAseq analysis (see Method Details) were performed using GraphPad Prism 7.0. Data are shown as mean±SEM. The “n” numbers for each experiment are specified in the text and figure legends. For in vivo experiments including Figures 1, 2, 3, 4, 6, 7, S1, S2, S3, S4, S7, each data point represents an individual animal. For in vitro experiments including Figures 5, 6D, 7A–C, and S5, each data point represents a biological replicate. For Figures S5H–I, n≥200 cells from 3 animals per genotype. For Figures S6D–F, n≥11 cells from 2 animals per genotype. For the comparison between two groups, statistical significance was determined using two-tailed Student’s t-test. For multiple comparisons, one-way or two-way ANOVA followed by Tukey’s multiple comparisons test or Bonferroni’s multiple comparisons test were performed. The criterion for statistical significance was set at P<0.05.
DATA AND SOFTWARE AVAILABILITY
All raw sequencing data are publicly available at National Center for Biotechnology Information (NCBI) BioProject, www://www.ncbi.nlm.nih.gov/bioproject. The accession number for the data reported in this paper is BioProject: PRJNA483757.
Supplementary Material
HIGHLIGHTS.
Transcription Factor EB (TFEB) is highly expressed by premyelinating oligodendrocytes
TFEB functions cell-autonomously to eliminate OLs in unmyelinated brain regions
TFEB activate the PUMA-Bax/Bak pathway to control myelination location and timing
TFEB induces ER stress gene expression in OLs and sets a brake on CNS myelination
ACKNOWLEDGEMENTS
We thank Drs. Liqun Luo, J. Bradley Zuchero, Steven A. Sloan, and Christopher J. Bohlen for helpful comments on the manuscript. We thank Dr. Onkar Dhande for technical assistance, and members of Barres and Luo laboratories for assistance and discussions. This work was supported by Helen Hay Whitney Foundation (L.O.S.), National Multiple Sclerosis Society Career Transition Award (L.O.S., TA-1705–27634), NIH Pathway to Independence Award (L.O.S., 1K99EY029330), Vincent J. Coates Research Foundation (B.A.B.), Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (B.A.B), the Myra Reinhard Family Foundation (B.A.B), NIH RO1 NS089786–05 (M.T-L), Stanford Neuroscience Microscopy Service (NIH NS069375), Stanford Cell Sciences Imaging Facility (NCRR 1S10RR026780), and Stanford Functional Genomic Facility (NIH, S10OD018220). L.O.S. is a Helen Hay Whitney postdoctoral fellow.
Footnotes
This paper is dedicated to the memory of our wonderful mentor and colleague, Dr. Ben A. Barres, who recently passed away.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
L.O.S. has applied for patents related to this paper.
REFERENCES
- Auer F, Vagionitis S, and Czopka T (2018). Evidence for Myelin Sheath Remodeling in the CNS Revealed by In Vivo Imaging. Curr Biol 28, 549–559 e543. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, and Raff MC (1992). Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70, 31–46. [DOI] [PubMed] [Google Scholar]
- Barres BA, and Raff MC (1994). Control of oligodendrocyte number in the developing rat optic nerve. Neuron 12, 935–942. [DOI] [PubMed] [Google Scholar]
- Bergles DE, and Richardson WD (2015). Oligodendrocyte Development and Plasticity. Cold Spring Harb Perspect Biol 8, a020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Kokkosis AG, and Aguirre A (2017). Oligodendroglia-lineage cells in brain plasticity, homeostasis and psychiatric disorders. Curr Opin Neurobiol 47, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, Frit P, and Calsou P (2014). DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res 42, 9047–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprariello AV, Batt CE, Zippe I, Romito-DiGiacomo RR, Karl M, and Miller RH (2015). Apoptosis of Oligodendrocytes during Early Development Delays Myelination and Impairs Subsequent Responses to Demyelination. J Neurosci 35, 14031–14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, et al. (2017). Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont EA, Reutelingsperger CP, Smits JF, Daemen MJ, Doevendans PA, Wellens HJ, and Hofstra L (2001). Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat Med 7, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, and Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, and Dugas JC (2013). Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb Protoc 2013, 854–868. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, and Rowitch DH (2009). Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev 23, 1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlia G, Gerber D, and Suter U (2018). Myelination and mTOR. Glia 66, 693–707. Flechsig Of Leipsic, P. DEVELOPMENTAL (MYELOGENETIC) LOCALISATION OF THE CEREBRAL CORTEX IN THE HUMAN SUBJECT. The Lancet 158, 1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, et al. (2017). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat Neurosci 20, 10–15. [DOI] [PubMed] [Google Scholar]
- Hikisz P, and Kilianska ZM (2012). PUMA, a critical mediator of cell death--one decade on from its discovery. Cell Mol Biol Lett 17, 646–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Li AM, and Grutzendler J (2018). Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Orthmann-Murphy JL, Langseth AJ, and Bergles DE (2018). Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, and Bergles DE (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K, Itoh T, Itoh A, Horiuchi M, Wakayama K, Bannerman P, Garbern JY, Pleasure D, and Lindsten T (2009). Maintenance of the relative proportion of oligodendrocytes to axons even in the absence of BAX and BAK. Eur J Neurosci 30, 2030–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, and Richardson WD (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingseisen A, and Lyons DA (2018). Axonal Regulation of Central Nervous System Myelination: Structure and Function. Neuroscientist 24, 7–21. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33, 366–374. [DOI] [PubMed] [Google Scholar]
- Lin SC, Huck JH, Roberts JD, Macklin WB, Somogyi P, and Bergles DE (2005). Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron 46, 773–785. [DOI] [PubMed] [Google Scholar]
- Lin W, and Popko B (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci 12, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, and Jacobowitz DM (2004). Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol 472, 318–329. [DOI] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, and Puertollano R (2016). TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35, 479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, and Puertollano R (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 7, ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, and Thomas PD (2010). PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 38, D204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M (2018). Myelin Plasticity and Nervous System Function. Annu Rev Neurosci 41, 61–76. [DOI] [PubMed] [Google Scholar]
- Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, Lim JA, Zare H, Raben N, Ballabio A, et al. (2016). TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, and Jacobson MD (1993). Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262, 695–700. [DOI] [PubMed] [Google Scholar]
- Saab AS, and Nave KA (2017). Myelin dynamics: protecting and shaping neuronal functions. Curr Opin Neurobiol 47, 104–112. [DOI] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, and Ballabio A (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14 , 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tesic Mark M, Molina H, and Tessier-Lavigne M (2016). Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell 164, 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaidero N, Mobius W, Czopka T, Hekking LH, Mathisen C, Verkleij D, Goebbels S, Edgar J, Merkler D, Lyons DA, et al. (2014). Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturrock RR (1980). Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol 6, 415–420. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, and Arlotta P (2014). Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, and Macklin W (1997). Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol 137, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TL, Bercury KK, Cifelli SE, Mursch LE, Min J, Dai J, and Macklin WB (2013). mTOR: a link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neuro 5, e00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt KD, Tanapat P, and Wang SS (2005). Speed limits in the cerebellum: constraints from myelinated and unmyelinated parallel fibers. Eur J Neurosci 21, 2285–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, and Richardson WD (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci 19, 1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HJ, Vainshtein A, Maik-Rachline G, and Peles E (2016). G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat Commun 7, 10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, and Richardson WD (2013). Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, and Barres BA (2013). Intrinsic and extrinsic control of oligodendrocyte development. Curr Opin Neurobiol 23, 914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, Fu MM, Sloan SA, Ibrahim A, Olson A, Zaremba A, Dugas JC, Wienbar S, Caprariello AV, Kantor C, et al. (2015). CNS myelin wrapping is driven by actin disassembly. Dev Cell 34, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data are publicly available at National Center for Biotechnology Information (NCBI) BioProject, www://www.ncbi.nlm.nih.gov/bioproject. The accession number for the data reported in this paper is BioProject: PRJNA483757.