

Abstract
Amplification of the EGFR gene and its truncation mutant EGFRvIII are hallmarks of glioblastoma. Although co-expression of EGFR and EGFRvIII confers a growth advantage, how EGFR and EGFRvIII influence the tumor microenvironment remains incompletely understood. Here we show that EGFR and EGFRvIII cooperate to induce macrophage infiltration via upregulation of the chemokine CCL2. EGFRvIII was significantly enriched in glioblastoma patient samples with high CCL2, and knockout of CCL2 in tumors co-expressing EGFR and EGFRvIII led to decreased infiltration of macrophages. KRAS was a critical signaling intermediate for EGFR and EGFRvIII-induced expression of CCL2. Our results illustrate how EGFR and EGFRvIII direct the microenvironment in glioblastoma.
Introduction
Glioblastoma is the most common primary brain tumor and among the most aggressive of all cancers (1). Average survival after diagnosis is only one year, even in the setting of extensive chemotherapy and radiotherapy (1). Amplification of the Epidermal Growth Factor Receptor (EGFR) is identified in more than half of tumors. One third of EGFR-amplified tumors also co-amplify EGFRvIII, a truncation mutant of EGFR (2). Amplification of EGFRvIII always occurs in tumors that also amplify EGFR, suggesting that co-amplification of those molecules confers a growth advantage (3). Our lab previously showed that: 1). EGFRvIII and EGFR cooperated to promote glioblastoma growth both in vitro and in vivo. 2). EGFRvIII was a substrate of EGFR, and 3). Phosphorylation of EGFRvIII by EGFR was critical for tumorigenesis and growth (3). EGFR and EGFRvIII cooperated to promote tumor growth more dramatically in vivo than in vitro, suggesting that tumor-extrinsic factors contribute to tumor growth (3).
The tumor microenvironment is a critical factor in initiation and progression of glioblastoma (4,5). EGFRvIII potentiates the expression of IL-6, IL-8 and LIF, indicating that EGFRvIII signals to the glioma microenvironment through upregulating specific cytokines (6-8). Interestingly, EGFRvIII promotes the growth of cells expressing wild-type EGFR via a paracrine IL-6 and/or LIF-dependent manner (8,9). However, how EGFR and EGFRvIII converge on cytokines to regulate immune cell infiltration remains incompletely understood.
Cytokines contribute to infiltration of immune cells including tumor-associated macrophages (TAMs). Macrophages represent up to half of the glioblastoma tumor mass, with the C-C chemokine CCL2 important for macrophage infiltration (10,11). Levels of CCL2 are elevated in glioblastoma, and drive progression by promoting immunosuppression (12,13). In this study, we demonstrate that EGFR and EGFRvIII cooperate through KRAS, to upregulate CCL2, promoting infiltration of macrophages.
Materials and Methods
Cell culture
U87 cells expressing control vector, EGFR, EGFRvIII and EGFR/EGFRvIII were described previously (3). Cells were maintained in DMEM supplemented with 10% FBS and 1× Penicillin-Streptomycin. A172 cells were purchased in 2017 (ATCC) and authenticated by ATCC using STR profiling. Cells were certified mycoplasma-free by ATCC. Cells were transduced with EGFR and EGFRvIII lentiviruses as described previously (3). A172 cells expressing control vector, EGFR, EGFRvIII and EGFR/EGFRvIII were maintained in DMEM supplemented with 10% FBS and 1× Penicillin-Streptomycin. The cells were passaged less than 15 times after thawing. GBM6 and GBM39 were from Dr. Jann Sarkaria at Mayo Clinic. The xenografted tumors were dissociated as described (14). The cells were grown in DMEM:F12 with 20ng/ml EGF, 20ng/ml FGF, 1×B27 supplement and 1× Penicillin-Streptomycin. For differentiation, the cells were grown in DMEM supplemented with 10% FBS and 1× Penicillin-Streptomycin. The cells were passaged less than 5 times after thawing. For cytokine-related assays, cells were grown for 24 hours in DMEM:F12 with 20ng/ml EGF, 20ng/ml FGF, 1×B27 supplement and 1x Penicillin-Streptomycin. MV-4–11 cells were purchased in 2015 (ATCC) and authenticated by ATCC using STR profiling. Cells were certified mycoplasma-free by ATCC. These cells were cultivated in IMDM supplemented with 10% FBS at 37°C and 5% CO2. The cells were passaged less than 5 times after thawing. Mycoplasma status was monitored monthly in the lab using the HEK-BlueTM detection kit (InvivoGen).
Transwell macrophage migration assay
The assay was carried out using QCMTM Chemotaxis 5μm 24-Well Cell Migration Assay kit (Millipore) following manufacturer’s instructions. Specifically, 1 × 106 macrophage cells (MV-4–11) per well were plated on transwell insert in 250 μL IMDM without FBS. Bottom chamber was filled with conditioned media (filtered through 0.45μm filters) from equal numbers of indicated cells. After 4 hrs, cells in the bottom chambers were lysed and data obtained by reading fluorescence 480/520nm as in the manufacturer’s protocol. Results were normalized to medium-only controls.
CRISPR knockout or siRNA knockdown of indicated genes
To generate CCL2 or KRAS CRISPR knockout, a pool of 3 different gRNA plasmids were used (Santa Cruz). The sequences for the CCL2 gRNAs were: 5’−3’: TACCTGGCTGAGCGAGCCCT; 5’−3’: TTTGGGTTTGCTTGTCCAGG; 5’−3’: TCAGGATTCCATGGACCACC. The sequences for the KRAS gRNAs were: 5’−3’: TCTCGACACAGCAGGTCAAG; 5’−3’: CAATGAGGGACCAGTACATG; 5’−3’: TTCTCGAACTAATGTATAGA. The corresponding HDR plasmids were co-transfected following manufacturer’s instructions (Santa Cruz). The control siRNA and KRAS siRNA were purchased from Dharmacon.
In vivo studies
Indicated cell lines were injected into the brain of 4–6 weeks old female BALB/C nu/nu mice. Specifically, mice were anesthetized using ketamine and xylazine. 3×105 cells were injected intracranially at 2 mm anterior and 1.5 mm lateral of the right hemisphere relative to bregma at a depth of 3 mm. Each group contains 10 mice. All mouse experiments were performed following protocols approved by UCSF’s Institutional Animal Care and Use Committee (IACUC).
Immunohistochemistry (IHC) staining
Excised tumors from mice were fixed with 10% neutral buffered formalin for 24 hours and changed to 70% ethanol. Tumors were paraffin-embedded and sectioned by the Brain Tumor Research Center at UCSF. For IHC, slides were deparaffinized and antigen retrieval was performed using a pressure cooker. The Mouse on Mouse (M.O.M.™) basic kit (Vector laboratories) was used for masking endogenous mouse antigen. The VECTASTAIN® ABC reagent (Vector laboratories) was used for signal detection. The CD45 antibody (05–1410) was purchased from EMD Millipore and was used at a concentration of 1:50. The CD38 (ab216343), CD68 (ab955) and TMEM119 (ab209064) antibodies were purchased from Abcam and were used at a concentration of 1:100. The CD206 antibody (AF2535SP) was purchased from R&D Systems and was used at a concentration of 1:50. Images were taken using a Zeiss AxioImager M1 microscope. Cells were quantified using Image J.
Immunofluorescent staining
Formalin-fixed paraffin sections from primary grade IV human glioblastoma tumor tissues were deparaffinized in xylene and rehydrated over a graded ethanol series, followed by treatment in 10 mM citrate buffer at pH 6.0 for 20 minutes in a steamer for antigen retrieval. Tissue sections were equilibrated in TBST and then blocked for 10 min with Ultravision Protein Block (Thermo Scientific) and incubated overnight at 4°C with primary antibodies against EGFR (mouse monoclonal DAK-H1-WT, Dako, 1:50), EGFRvIII (rabbit polyclonal 6549, Celldex, 1:100), CD45 (rat monoclonal SM1744P, Acris, 1:100), diluted in Dako REAL Antibody Diluent. Three washing steps in TBST were followed by incubation with secondary antibodies at room temperature for 1 hr diluted in Dako REAL Antibody Diluent (anti-mouse AF488, anti-rabbit AF594, anti-rat AF647, Thermo Fisher, 1:1000). Three washing steps in TBST were followed by incubation of the slides with DAPI diluted in PBS to a final concentration of 1 μg/ml for 5 minutes at room temperature. Following repeated washing, stained sections were mounted in ProLong® Gold antifade reagent (Thermo Fisher) and sealed using nail polish. Fluorescent images were taken using a Leica TCS SP8 Sted microscope with either a 100× oil immersion objective or a 40× water immersion objective. Taken images were further processed using the Leica Las X software, Adobe Photoshop and Adobe InDesign.
Real-time PCR
RNA was extracted using Quick-RNA MiniPrep kit according to the manufacturer’s instructions (ZYMO Research). cDNA was produced using iScript cDNA synthesis kit (Biorad). Real-time PCR was performed using SYBR FAST ABI Prism qPCR kit (KAPA Biosystems) on an AB7900HT real-time PCR machine (Applied Biosystems). The primers for CCL2: 5’−3’ CAGCCAGATGCAATCAATGCC; 5’−3’ TGGAATCCTGAACCCACTTCT. IL1B: 5’−3’ AATCTGTACCTGTCCTGCGTGTT; 5’−3’ TGGGTAATTTTTGGGATCTACACTCT. Actin: 5’−3’ CATGTACGTTGCTATCCAGGC; 5’−3’ CTCCTTAATGTCACGCACGAT. CXCR4: 5’−3’ GATCAGCATCGACTCCTTCA; 5’−3’ GGCTCCAAGGAAAGCATAGA. CXCL12: 5’−3’ ATGCCCATGCCGATTCTTCG; 5’−3’ GCCGGGCTACAATCTGAAGG. LIF: 5’−3’ CAGTGCCAATGCCCTCTTTAT; 5’−3’ GGCCACATAGCTTGTCCAGG. CD90/THY1: 5’−3’ CGCTCTCCTGCTAACAGTCTT; 5’−3’ CAGGCTGAACTCGTACTGGA. SOX1: 5’−3’ GCGGAAAGCGTTTTCTTG; 5’−3’ TAATCTGACTTCTCCTCCC.
Western blotting
Cells were lysed using RIPA buffer with cOmplete™ Mini protease inhibitor cocktail (Roche) and PhosSTOP (Roche). The IκB (9242) and p-IκB (9246) antibodies were purchased from Cell Signaling and used at a concentration of 1:500. The Actin antibody (sc-1615) was purchased form Santa Cruz and used at a concentration of 1:1000. The KRAS antibody (SAB2101297) was purchased from Sigma and used at a concentration of 1:500.
Cell surface EGFR/EGFRvIII staining and FACS sorting
Cells were dissociated using Accutase (Innovative Cell Technologies), washed with cell staining buffer with sodium azide (BioLegend), and stained with an Alexa Fluor® 488 anti-human EGFR antibody (AY13, BioLegend) for 20 minutes. Cells were sorted using a FACSAria3 sorter (BD) at the UCSF Laboratory for Cell Analysis core facility. Approximately 20,000 cells were sorted for each population.
Cytokine array
Cytokine levels in conditioned medium from equal number of indicated cell lines were measured using Human Cytokine Array Panel A (R&D systems #ARY005) following manufacturer’s instructions.
RNA-sequencing (RNA-seq)
RNA was extracted using Quick-RNA MiniPrep kit according to the manufacturer’s instructions (ZYMO Research). RNA-seq was performed with Illumina HiSeq4000 at University of California, Berkeley. Results were analyzed using Galaxy (https://usegalaxy.org/). The raw data was deposited to GEO (accession number: GSE117838). Heat map was generated using GENE-E. KEGG pathway analysis was performed using CPDB (http://cpdb.molgen.mpg.de/). Gene set enrichment was performed using GSEA oncogenic signatures (http://software.broadinstitute.org/gsea/index.jsp).
TCGA data analysis
EGFRvIII data was obtained from (2). CCL2 expression levels were downloaded from http://gliovis.bioinfo.cnio.es/ (TCGA, platform HG-133A). Three cases with unknown EGFR status and one case with EGFRvIII and EGFR euploid were excluded from analysis.
Statistical analysis
Unless otherwise noted, one-way ANOVA was performed and p<0.05 was considered statistically significant.
Results
EGFRvIII cooperates with EGFR to induce infiltration of CD45+ cells
To further clarify how EGFR and EGFRvIII promote tumor growth, we tested whether EGFR and EGFRvIII influenced infiltration of immune cells. We analyzed EGFRvIII positive sections from primary human glioblastoma, for expression of EGFR, EGFRvIII and CD45, a pan hematopoietic lineage marker (Figure 1A). Cells double-positive for EGFR and EGFRvIII were surrounded by CD45-positive cells (Figure 1A). Glioblastoma tumors are heavily infiltrated by immune cells, with infiltrating macrophages representing up to half of the tumor mass (11). To test whether inhibition of EGFR and EGFRvIII decreased macrophage attraction, we analyzed short-term cultures from two primary patient-derived xenograft (PDX) lines (GBM6 and GBM39--both EGFR and EGFRvIII co-amplified) (15), treated with DMSO or the EGFR inhibitor lapatinib. Conditioned media from equal numbers of DMSO-treated GBM6 and GBM39 cells attracted more CD45+ macrophages than media from lapatinib-treated GBM6 cells (Figure 1B and1C).
Figure 1.
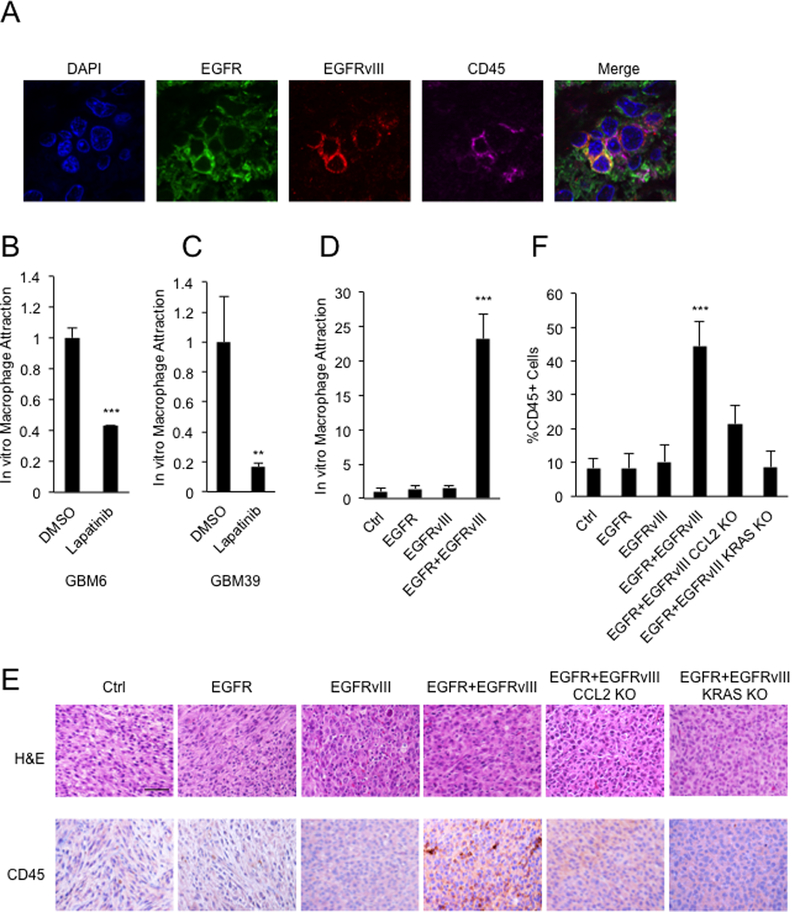
Co-expression of EGFR and EGFRvIII leads to infiltration of CD45+ cells. A, Cells double positive for EGFR and EGFRvIII colocalize with CD45+ cells in sections from primary EGFR/EGFRvIII+ human glioblastoma. B and C, Transwell migration assay showing attraction of macrophages (macrophage line: MV-4–11) in response to conditioned media from GBM6 and GBM39 cells treated with DMSO or 5μM lapatinib for 24 hours. D, Transwell migration assay showing attraction of macrophages (macrophage line: MV-4–11) in response to conditioned media from U87 cells co-expressing EGFR and EGFRvIII; E, H&E and IHC staining validating increased numbers of CD45 positive cells in intracranial xenografts driven by EGFR and EGFRvIII; and decreased numbers of CD45 positive cells in xenografts deleted for CCL2 and KRAS; F, quantification. Macrophage attraction levels were normalized to the control cell line. Results were reproduced in three independent experiments with triplicate samples. **, p<0.01, ***, p < 0.001. Scale bar: 50μM.
To test the function of EGFR and EGFRvIII in attracting macrophages in an isogenic system, we used our published cell lines, which stably express EGFR and EGFRvIII singly or together in U87:MG cells (3). Conditioned media from cells co-expressing EGFR and EGFRvIII attracted significantly more CD45+ macrophages than conditioned media from cells expressing EGFR or EGFRvIII alone (Figure 1D). Intracranial xenografted tumors co-expressing EGFR and EGFRvIII also showed significantly more CD45+ cells, compared to tumors expressing either EGFR or EGFRvIII alone (Figure 1E and1F). Together, these results show that co-expression of EGFR and EGFRvIII increases infiltration of immune cells in glioblastoma tumors.
Infiltration of both M1 and M2 macrophages in response to EGFR and EGFRvIIII
TAMs can polarize towards M1 pro-inflammatory and anti-inflammatory M2 phenotypes, respectively (16). However, M1 and M2 macrophages may coexist (17), depending on the balance of activating and inhibitory signals and their interplay with the tumor microenvironment (17). We stained intracranial xenografted tumors with M1 macrophage marker CD38 and M2 macrophage marker CD206. Tumors co-expressing EGFR and EGFRvIII showed significantly more CD38+ cells and CD206+ cells, compared to tumors expressing either EGFR or EGFRvIII singly (Figure 2A - C). Thus, in these xenografted tumors, co-expression of EGFR and EGFRvIII increases infiltration of both M1 and M2 macrophages.
Figure 2.
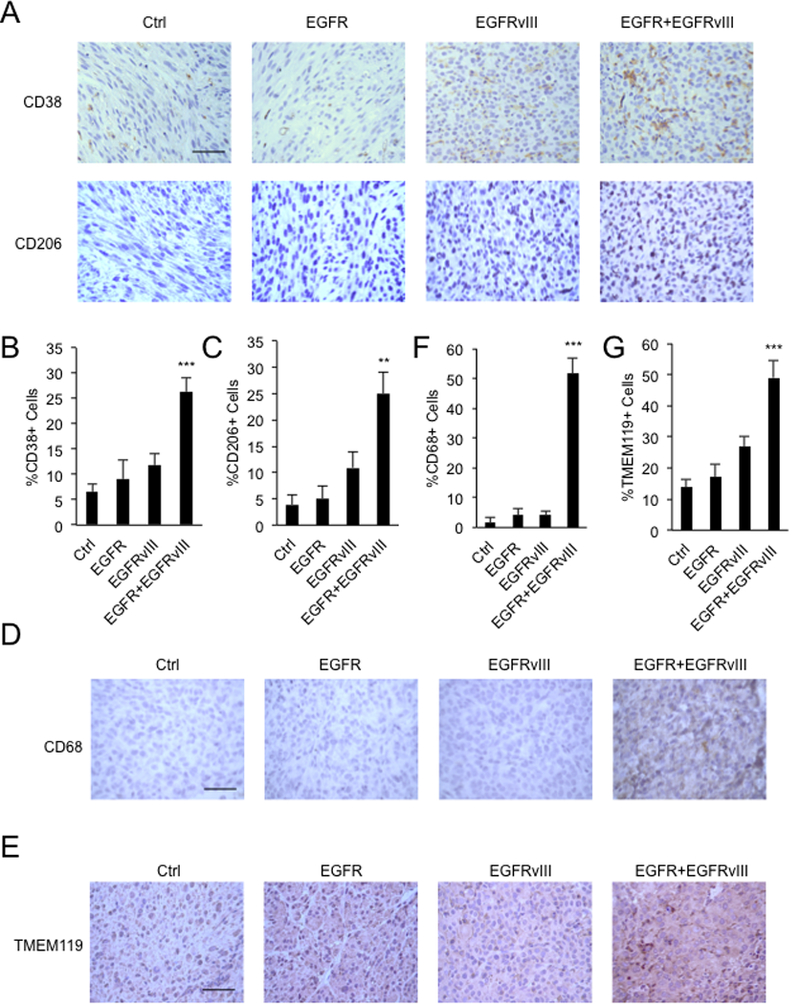
Co-expression of EGFR and EGFRvIII leads to infiltration of M1, M2 macrophages and microglia. A, IHC staining validating increased numbers of CD38 and CD206 positive cells in intracranial xenografts driven by EGFR and EGFRvIII; B and C, quantification. D, IHC staining validating increased numbers of CD68 positive cells in flank xenografts driven by EGFR and EGFRvIII; E, IHC staining validating increased numbers of TMEM119 positive cells in intracranial xenografts driven by EGFR and EGFRvIII; F and G, quantification.**, p<0.01, ***, p < 0.001. Scale bar: 50μM.
EGFRvIII cooperates with EGFR to induce infiltration of macrophages and microglia
To separately assess recruitment of macrophages from recruitment of brain-resident microglia, we injected tumor cells into flank regions of nude mice, a region free of microglia. Established tumors co-expressing EGFR and EGFRvIII showed significantly more infiltration of CD68+ monocyte lineage cells, compared to tumors expressing either EGFR or EGFRvIII alone (Figure 2D). Microglia, a brain-resident macrophage population, constitutes 5–10% of total brain cells (18). We therefore also analyzed intracranial xenografts, staining with TMEM119, a marker of microglia (19). Tumors co-expressing EGFR and EGFRvIII also showed significantly more TMEM119+ cells, compared to tumors expressing either EGFR or EGFRvIII alone (Figure 2E). Quantification further validated the results (Figure 2F and 2G). Together, these results suggest that EGFR and EGFRvIII cooperate to attract both peripheral macrophages and microglia into tumors.
Induction of cytokines and chemokines in response to EGFRvIII and EGFR
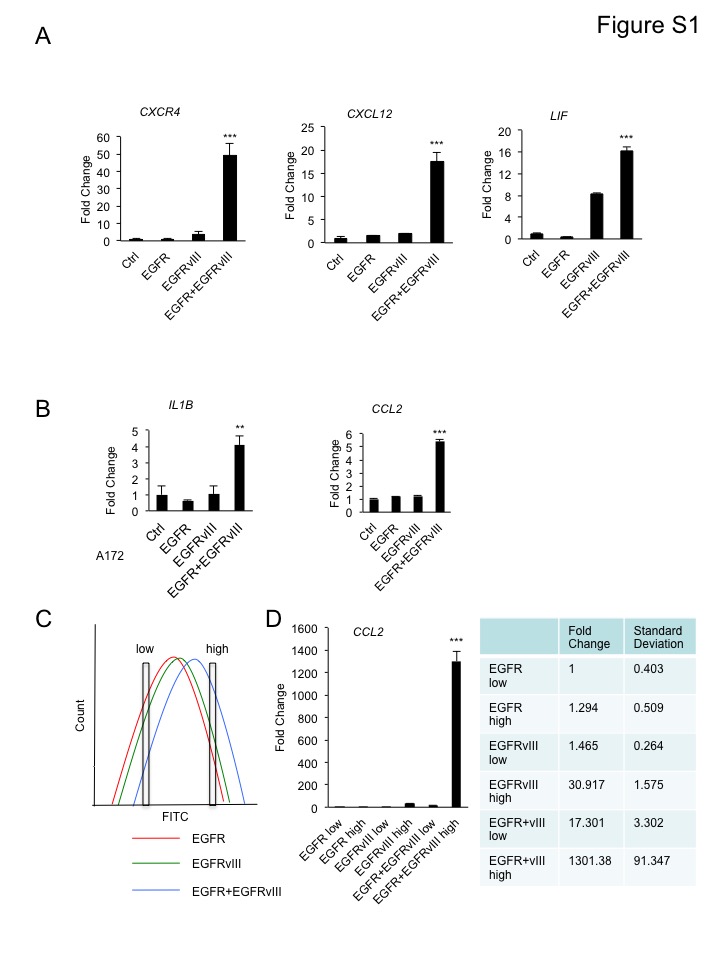
Next, we performed RNA-seq to determine whether co-expression of EGFR and EGFRvIII influenced the abundance of cytokines, to promote infiltration of CD45+ cells. Glioblastoma cells co-expressing EGFR and EGFRvIII upregulated the transcription of a large group of cytokines and chemokines, compared to cells expressing vector, EGFR or EGFRvIII alone. Top hits included IL6 and IL8, previously reported as regulated by EGFRvIII (20), and new candidates including IL1RN, IL33, IL11 and CCL2 (Figure 3A). Pathway enrichment analysis demonstrated that genes in cytokine-cytokine receptor interaction and chemokine signaling pathways were upregulated cooperatively by EGFR and EGFRvIII (Figure 3B). Using real-time PCR, we verified that EGFR and EGFRvIII upregulated transcription of IL1B, CCL2, LIF, CXCR4 and CXCL12 (Figure 3C, 3D and S1A).
Figure 3.
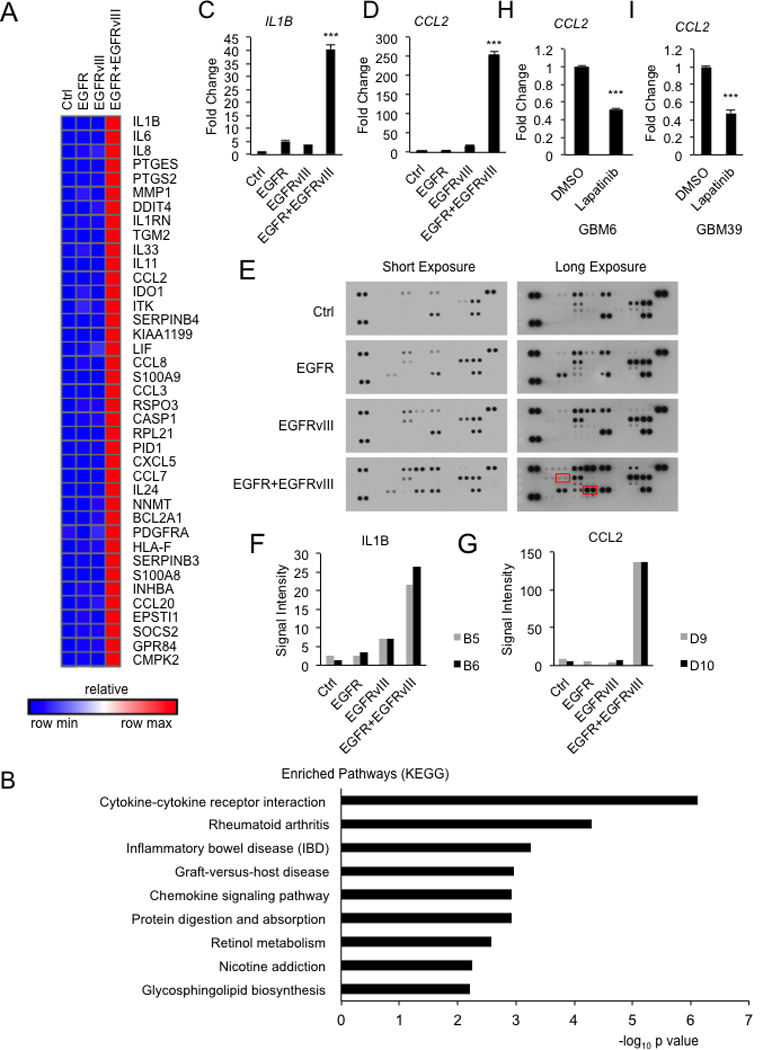
Co-expression of EGFR and EGFRvIII induces multiple cytokines. A, Heat map of mRNA levels for top 40 genes (listed from top to bottom according to expression levels) induced by EGFR and EGFRvIII. B, Pathway enrichment of genes induced by EGFR and EGFRvIII. C and D, Real-time PCR of IL1B and CCL2 from U87 cells expressing indicated genes. E-G, cytokine array (IL1B and CCL2 highlighted in red boxes) and quantification confirming that EGFR and EGFRvIII cooperate to drive expression of IL1B and CCL2 in U87 cells expressing indicated constructs. H and I, Real-time PCR of CCL2 from GBM6 and GBM39 cells treated with DMSO or 5μM lapatinib for 24 hours. Real-time PCR results were normalized to the control cell line Results were reproduced in at least three independent experiments with triplicate samples. ***, p < 0.001.
We verified these results in a second line, A172 glioblastoma cells, demonstrating that co-expression of EGFR and EGFRvIII also led to increased transcription of CCL2 and IL1B (Figure S1B). Next, we ruled out the possibility that increased receptor density on the surface of cells co-expressing EGFR and EGFRvIII accounted for induction of CCL2. We demonstrated that cells expressing EGFR or EGFRvIII alone, at cell surface receptor density comparable to that of cells co-expressing EGFR and EGFRvIII failed to induce significant amounts of CCL2. We labeled cells with Alexa Fluor® 488-conjugated EGFR antibody (recognizes both EGFR and EGFRvIII), and sorted cells with comparable EGFR/EGFRvIII levels (Figure S1C). For each cell line, an EGFR/EGFRvIII-high and an EGFR/EGFRvIII-low population were sorted (~10 fold difference in signal intensity). Within each sorted population, CCL2 mRNA expression was analyzed by real-time PCR (Figure S1C and S1D). Cells expressing EGFR or EGFRvIII alone, at comparable cell surface receptor level as cells co-expressing EGFR and EGFRvIII, expressed significantly less CCL2 (Figure S1D). Cells with a lower cell surface EGFR/EGFRvIII density expressed significantly less CCL2 than those cells with a higher cell surface EGFR/EGFRvIII density (Figure S1D). These results indicated that EGFR and EGFRvIII cooperate to induce CCL2 expression, and that receptor density affected the induction level.
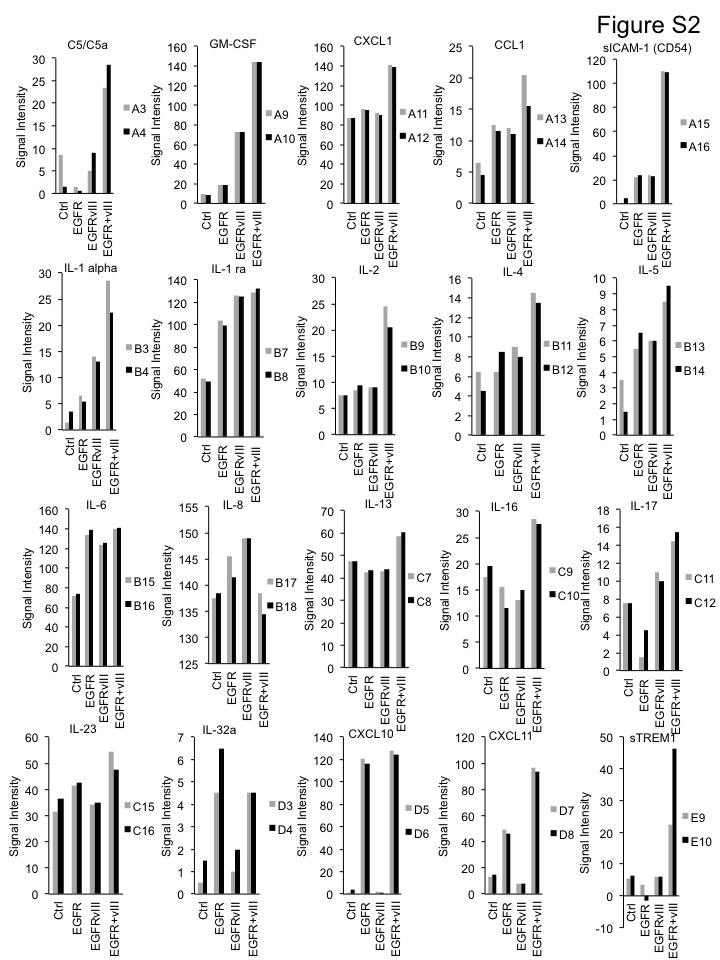
We went onto analyze expression of proteins using cytokine arrays. Consistent with the RNA-seq data, co-expression of EGFR and EGFRvIII also upregulated IL1B, CCL2 and other cytokine at the protein level (Figure 3E, 3F, 3G and S2). In addition, we treated short-term cultures of GBM6 and GBM39 (EGFR/EGFRvIII co-amplified) with lapatinib. This treatment led to significantly less production of CCL2 compared to control cells (Figure 3H and3I). Collectively, these data suggest that EGFR and EGFRvIII cooperate to drive expression of cytokines and chemokines.
EGFR and EGFRvIII signal through CCL2 to recruit macrophages
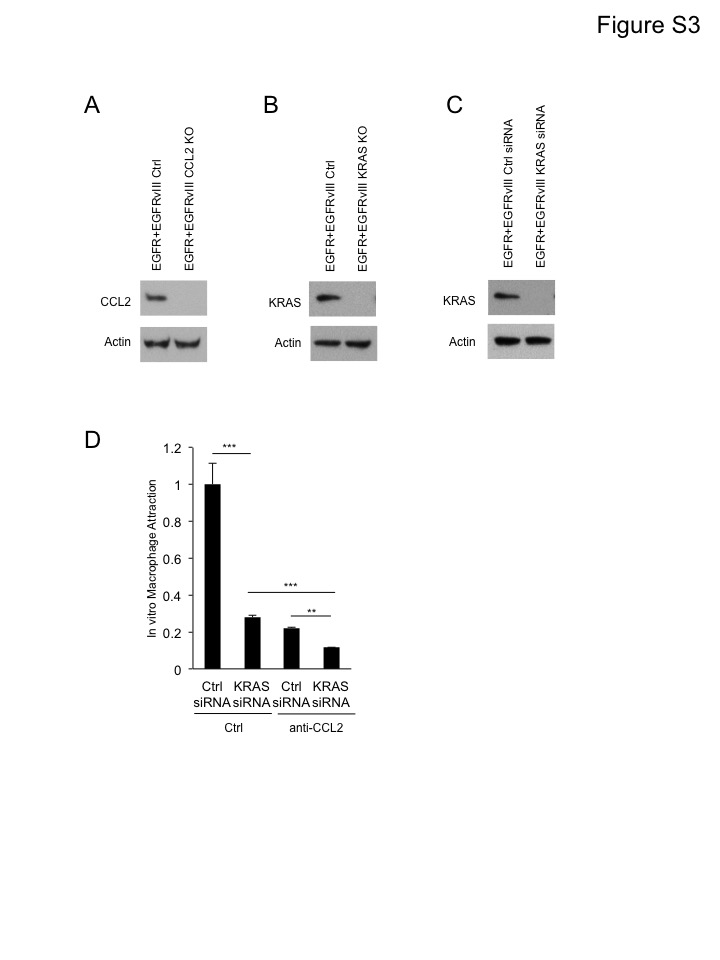
CCL2, among the most prominent chemokines for macrophage attraction, was induced by EGFR and EGFRvIII (Figure 3). Incubation of conditioned media from cells co-expressing EGFR and EGFRvIII, with a CCL2 neutralizing antibody, significantly reduced macrophage attraction (Figure 4A). Conditioned media from cells co-expressing EGFR and EGFRvIII, after CRISPR/Cas9 mediated deletion of CCL2, also led to decreased macrophage infiltration (Figure 4B and S3A). Knockout of CCL2 in intracranial xenografted tumors co-expressing EGFR and EGFRvIII led to significant decreases in the abundance of infiltrating CD45+ cells (Figure 1E and 1F) and modestly enhanced survival (Figure 4C). When we sacrificed animals at end point, tumors were localized to forebrains, where we injected the tumor cells. To extend this result, we analyzed genomic data from human glioblastoma. In TCGA datasets, EGFRvIII amplification was enriched significantly in samples with high expression of CCL2 mRNA (Figure 4D).
Figure 4.
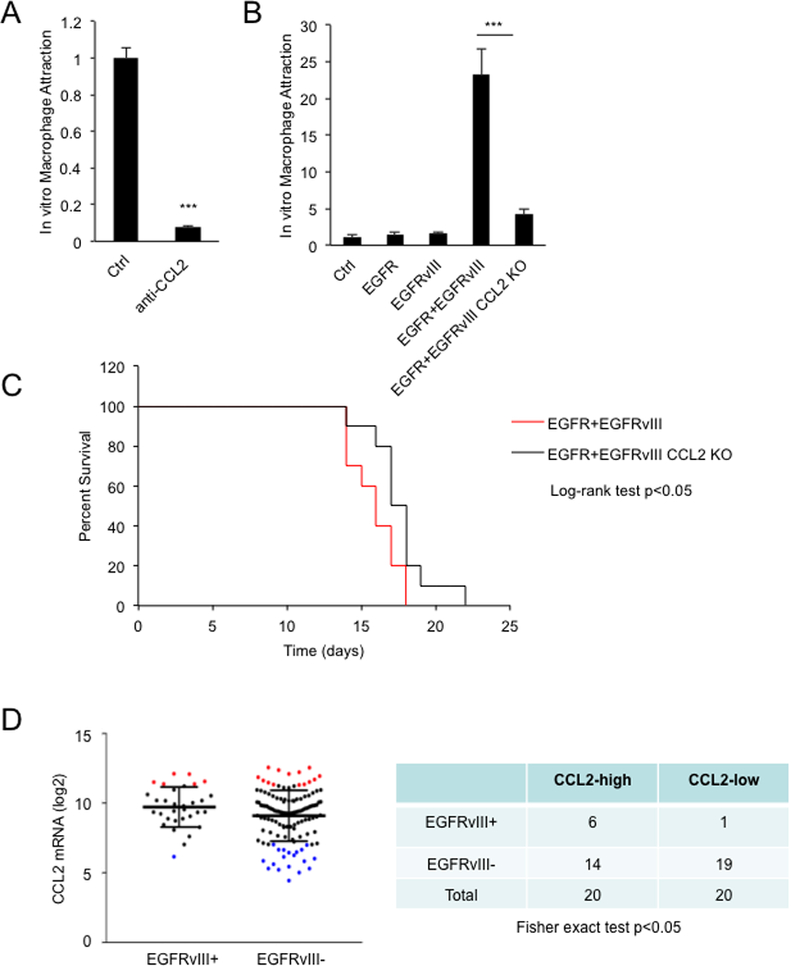
EGFR and EGFRvIII induce macrophage attraction through upregulating CCL2. A, Depletion of CCL2 in conditioned media from U87 cells expressing EGFR and EGFRvIII decreased attraction of MV-4–11 (antibody concentration: 1μg/ml). B, Knockout of CCL2 decreased attraction of MV-4–11. C, Kaplan-Meier curve of nude mice bearing intracranial xenografts of indicated cell lines. D, CCL2 expression levels were plotted according to EGFRvIII status in glioblastoma patient samples (TCGA). Red dots: 20 samples with highest expression of CCL2 mRNA. Blue dots: 20 samples with lowest expression of CCL2 mRNA. Macrophage attraction levels were normalized to the control cell line. Results were reproduced in three independent experiments with triplicate samples. ***, p< 0.001.
EGFR and EGFRvIII signal through KRAS to upregulate expression of cytokines
To determine how EGFR and EGFRvIII activate cytokines and chemokines, we looked for oncogenic signatures in gene sets enriched by EGFR and EGFRvIII (GSEA analysis). The top five gene sets were all related to KRAS (Figure 5A). Knockout of KRAS in cells co-expressing EGFR and EGFRvIII decreased expression of cytokines and chemokines including CCL2 (Figure 5B, 5C and S3B). Consistently, conditioned media from cells co-expressing EGFR and EGFRvIII, and knocked out for KRAS, attracted fewer macrophages than conditioned media from the same cells with intact KRAS (Figure 5D). Conditioned media from cells transfected with KRAS siRNA attracted significantly fewer macrophages compared to conditioned media from cells transfected with control siRNA (Figure S3C and S3D). In response to treatment with CCL2 neutralizing antibody, conditioned media from KRAS siRNA-transfected cells attracted fewer macrophages compared to conditioned media from control siRNA-transfected cells (Figure S3D). These results indicate that although CCL2 is a major chemokine for EGFR/EGFRvIII/KRAS-induced macrophage attraction, other KRAS-dependent cytokines also play minor roles. Intracranial tumors co-expressing EGFR and EGFRvIII and knocked out for KRAS also showed decreased infiltration by CD45+ cells (Figure 1E and 1F). These data indicate that EGFR and EGFRvIII signal through KRAS to upregulate expression of CCL2, and to promote infiltration by tumor-associated macrophages.
Figure 5.
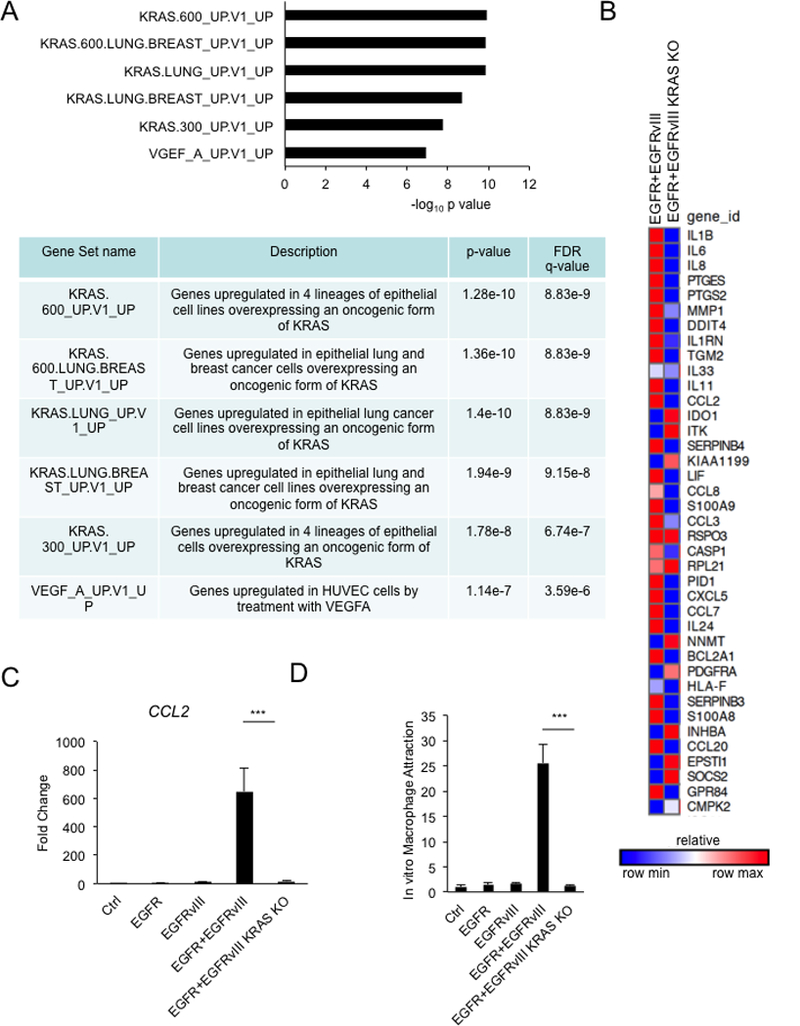
EGFR and EGFRvIII signal through KRAS to induce cytokine expression and macrophage attraction. A, Gene set enrichment analysis of genes upregulated by EGFR and EGFRvIII. B, Heat map of top 40 genes induced by EGFR and EGFRvIII in KRAS knockout U87 cells co-expressing EGFR and EGFRvIII. C, Real-time PCR. D, Knockout of KRAS decreased attraction of MV-4–11. Real-time PCR and macrophage attraction results were normalized to the control cell line. Results were reproduced in three independent experiments with triplicate samples.***, p < 0.001.
The NFκB pathway is an important pathway regulating inflammation. We tested IκB phosphorylation, a commonly used readout for NFκB activation. Under the same condition as our RNA-seq and cytokine array, we failed to observe increased phosphorylation of IκB (Figure S4). This result suggests that other pathways might be important in regulating EGFR/EGFRvIII/KRAS-induced cytokine expression.
Our RNA-seq results show that co-expression of EGFR and EGFRvIII upregulated CCL2, as well as stem cell markers such as CD90/THY1 and SOX1 (21,22) (Figure S5A). Expression of these stem cell markers was dependent on KRAS (Figure S5A). We also cultured GBM6 and GBM39 in stem cell medium or differentiated them with DMEM and FBS. Consistently, GBM6 and GBM39 expressed less CCL2, CD90/THY1 and SOX1 in differentiated media comparing to stem cell media (Figure S5B). These observations suggest that EGFR and EGFRvIII drive expression of CCL2 preferentially in glioblastoma stem cells.
Discussion
Amplification of EGFR and EGFRvIII represents hallmark genetic changes in glioblastoma. Amplification of EGFRvIII occurs only in tumors that amplify EGFR, suggesting that those two molecules could cooperate to promote tumor growth. Based on this co-amplification, we and others have previously studied EGFR and EGFRvIII in human cancers, to assess cooperation (3,8,9,23,24). We previously demonstrated co-expression of EGFR and EGFRvIII proteins in rare individual cells within glioblastoma, in all eleven of eleven primary human glioblastoma samples analyzed (3). To address whether this co-expression was coupled to cooperation between EGFR and EGFRvIII, we went on to show that EGFR and EGFRvIII cooperated in transformation in vitro and in vivo, that EGFR phosphorylated EGFRvIII, and that these two kinases converged on STAT3 (3). These published data illustrate clearly that EGFR and EGFRvIII cooperated through a cell-intrinsic mechanism. In this manuscript, we demonstrate that this cooperation extends to cell-extrinsic signaling. We show that EGFR and EGFRvIII promote cytokine expression in vitro leading to in vivo macrophage attraction through the KRAS pathway. These observations provide new insights, that cooperation of those two molecules can modulate the tumor microenvironment.
Glioblastoma tumors are heavily infiltrated by immune cells including macrophages, myeloid-derived suppressor cells and regulatory T-cells (25), among which infiltrating macrophages can represent half of the tumor mass (11). Here, we provide evidence that EGFR and EGFRvIII cooperatively upregulate CCL2 and promote infiltration of macrophages. Although CCL2 is a major chemokine recruiting macrophages, knockout of CCL2 in cells co-expressing EGFR and EGFRvIII did not completely block EGFR and EGFRvIII-induced macrophage attraction in vitro, indicating that other chemokines also contribute to macrophage recruitment. Our RNA-seq results suggest CCL8 as an additional candidate macrophage-recruiting chemokine (26), as CCL8 was also upregulated in cells co-expressing EGFR and EGFRvIII.
Within a glioblastoma tumor, the majority of cells express predominantly EGFR or EGFRvIII, with only rare cells co-expressing both EGFR and EGFRvIII (3). These observations suggest that EGFR and EGFRvIII may signal in glioma stem cells, with subsequent mitoses leading them to segregate independently to more differentiated daughter cells. Our results are consistent with observations that unlike their differentiated progeny, glioma stem cells show an increased capacity for active chemoattraction and recruitment of macrophages, through upregulation of cytokines (4). Our results suggest a possible link between KRAS-mediated cytokine expression and glioblastoma stemness.
Macrophages can be divided into tumor suppressive and tumor promoting classes (27). CCL2 can polarize macrophages toward a tumor promoting, immunosuppressive phenotype (28). Our results showed that co-expression of EGFR and EGFRvIII promoted infiltration of both tumor promoting (M2) and tumor suppressive macrophages (M1). This may be one reason why knockout of CCL2 in cells co-expressing EGFR and EGFRvIII only modestly impacted survival. Another issue is that macrophages induced by EGFR and EGFRvIII cannot in-turn influence the abundance or cellular activity of lymphocytes, which are absent in the nude mouse models used in this study. Notably, it was reported that B cells and T cells in the immune system, which nude mice lack, drove macrophage polarization to M2 (29,30). Our human cell-based model systems pose real limitations (31). While immunocompetent mouse models are better than nude mice for studying the tumor microenvironment,, we are unaware of immunocompetent models for glioblastoma in which EGFR and EGFRvIII are clear drivers. Such a model, which we are actively trying to develop, is essential to further delineate functionally how EGFR and EGFRvIII modulate the microenvironment in glioblastoma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
Z. An received support from Alex’s Lemonade Stand Foundation (CA-0091339), American Brain Tumor Association in honor of Juliana Schafer and Team Hope (BRF1800011), NIH T32CA108462 and Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation (7028233). C. Knobbe-Thomsen was supported by the German Cancer Aid (DKH #110663) and by the BMBF (#01ZX1401B). Q. Fan was supported by NIH grant R01CA221969. W. Weiss was supported by NIH grants R01CA221969, R01NS091620, P50CA097257, U01CA217864, and P30CA82103; the Samuel G. Waxman Cancer Research Foundation (A128431); and the Evelyn and Mattie Anderson Chair. We thank Drs Hideho Okada and Erin Simonds for useful discussions, Dr. Miller Huang and Lauren McHenry for critical review, and the UCSF Brain Tumor Core for help on intracranial injection.
Footnotes
Disclosure of Potential Conflicts of Interest: W. Weiss is a co-founder of StemSynergy Therapeutics. G. Reifenberger is a consultant for AbbVie.
References:
- 1.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annual review of pathology 2014;9:1–25. [DOI] [PubMed] [Google Scholar]
- 2.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer cell 2013;24(4):438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audia A, Conroy S, Glass R, Bhat KPL. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cells. Frontiers in oncology 2017;7:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mondal A, Kumari Singh D, Panda S, Shiras A. Extracellular Vesicles As Modulators of Tumor Microenvironment and Disease Progression in Glioma. Frontiers in oncology 2017;7:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurgis FM, Yeung YT, Tang MX, Heng B, Buckland M, Ammit AJ, et al. The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1beta-driven IL-6 secretion in glioblastoma cells. Oncogene 2015;34(22):2934–42. [DOI] [PubMed] [Google Scholar]
- 7.Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 2012;31(36):4054–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes & development 2010;24(16):1731–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zanca C, Villa GR, Benitez JA, Thorne AH, Koga T, D’Antonio M, et al. Glioblastoma cellular cross-talk converges on NF-kappaB to attenuate EGFR inhibitor sensitivity. Genes & development 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta neuropathologica 1997;93(5):518–27. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer research 2017;77(9):2266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer research 2016;76(19):5671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Annals of neurology 2003;54(3):388–92. [DOI] [PubMed] [Google Scholar]
- 14.Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Current protocols in pharmacology 2011;Chapter 14:Unit 14 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson H, Del Rosario AM, Bryson BD, Schroeder MA, Sarkaria JN, White FM. Molecular characterization of EGFR and EGFRvIII signaling networks in human glioblastoma tumor xenografts. Molecular & cellular proteomics : MCP 2012;11(12):1724–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. Journal of hematology & oncology 2017;10(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nature reviews Immunology 2018;18(4):225–42. [DOI] [PubMed] [Google Scholar]
- 19.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America 2016;113(12):E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 2018;37(12):1561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaikh MV, Kala M, Nivsarkar M. CD90 a potential cancer stem cell marker and a therapeutic target. Cancer biomarkers : section A of Disease markers 2016;16(3):301–7. [DOI] [PubMed] [Google Scholar]
- 22.Venere M, Han YG, Bell R, Song JS, Alvarez-Buylla A, Blelloch R. Sox1 marks an activated neural stem/progenitor cell in the hippocampus. Development 2012;139(21):3938–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng H, Hu B, Jarzynka MJ, Li Y, Keezer S, Johns TG, et al. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 2012;109(8):3018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Chakraborty S, Yang CR, Hatanpaa KJ, Cipher DJ, Puliyappadamba VT, et al. An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII. Oncogene 2014;33(33):4253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampson JH, Maus MV, June CH. Immunotherapy for Brain Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35(21):2450–56. [DOI] [PubMed] [Google Scholar]
- 26.Huma ZE, Sanchez J, Lim HD, Bridgford JL, Huang C, Parker BJ, et al. Key determinants of selective binding and activation by the monocyte chemoattractant proteins at the chemokine receptor CCR2. Science signaling 2017;10(480). [DOI] [PubMed] [Google Scholar]
- 27.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of clinical investigation 2012;122(3):787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. Journal of immunology 2014;192(8):3858–67. [DOI] [PubMed] [Google Scholar]
- 29.Wong SC, Puaux AL, Chittezhath M, Shalova I, Kajiji TS, Wang X, et al. Macrophage polarization to a unique phenotype driven by B cells. European journal of immunology 2010;40(8):2296–307. [DOI] [PubMed] [Google Scholar]
- 30.Chan T, Pek EA, Huth K, Ashkar AA. CD4(+) T-cells are important in regulating macrophage polarization in C57BL/6 wild-type mice. Cellular immunology 2011;266(2):180–6. [DOI] [PubMed] [Google Scholar]
- 31.Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: Good news and bad news. Science translational medicine 2016;8(354):354re3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.