

Abstract
Purpose:
Elevation of L-2-hydroxylgutarate (L-2-HG) in renal cell carcinoma (RCC) is due in part to reduced expression of L-2-HG dehydrogenase (L2HGDH). However, the contribution of L-2-HG to renal carcinogenesis and insight into the biochemistry and targets of this small molecule remains to be elucidated.
Experimental Design:
Genetic and pharmacologic approaches to modulate L-2-HG levels were assessed for effects on in vitro and in vivo phenotypes. Metabolomics was used to dissect the biochemical mechanisms that promote L-2-HG accumulation in RCC cells. Transcriptomic analysis was utilized to identify relevant targets of L-2-HG. Finally, bioinformatic and metabolomic analyses were used to assess the L-2-HG/L2HGDH axis as a function of patient outcome and cancer progression.
Results:
L2HGDH suppresses both in vitro cell migration and in vivo tumor growth and these effects are mediated by L2HGDH’s catalytic activity. Biochemical studies indicate that glutamine is the predominant carbon source for L-2-HG via the activity of malate dehydrogenase 2 (MDH2). Inhibition of the glutamine-MDH2 axis suppresses in vitro phenotypes in a L-2-HG dependent manner. Moreover, in vivo growth of RCC cells with basal elevation of L-2-HG is suppressed by glutaminase inhibition. Transcriptomic and functional analyses demonstrate that the histone demethylase KDM6A is a target of L-2-HG in RCC. Finally, increased L-2-HG levels, L2HGDH copy loss, and lower L2HGDH expression are associated with tumor progression and/or worsened prognosis in RCC patients.
Conclusion:
Collectively, our studies provide biochemical and mechanistic insight into the biology of this small molecule and provide new opportunities for treating L-2-HG driven kidney cancers.
Keywords: Kidney cancer, metabolism, oncometabolite, glutamine, epigenetics
Introduction
Cancer-associated mutations in metabolic enzymes have led to the identification of oncometabolites including fumarate, succinate and D-2-hydroxyglutarate (D-2-HG)(1–7). A unifying theme amongst these oncometabolites is their ability to competitively inhibit α-ketoglutarate (α-KG) dependent dioxygenases (αKGDs)(8–15). Members of the αKGDs include the TET (ten eleven translocation) enzymes (TETs 1–3) that hydroxylate 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) which can promote DNA demethylation(16), the Jumonji family of histone lysine demethylases, and prolyl hydroxylases (PHDs 1–3) which regulate Hypoxia Inducible Factors-1α and 2α (HIF-1α, HIF-2α). In leukemia, bioinformatic and functional studies indicate that a relevant target of D-2-HG is TET2(12, 17). However, the relevant target(s) of D-2-HG, fumarate, and succinate in other malignancies as well as the contribution to tumorigenesis remains less well characterized. For example, the in vivo efficacy of mutant IDH inhibitors for glioma has yielded conflicting results(18–20).
Elevations of the L-enantiomer of 2-HG (L-2-HG) are present in clear cell renal cell carcinoma (ccRCC), the most common histology(21, 22). Recent studies have also demonstrated increases of L-2-HG in the setting of hypoxia, mitochondrial dysfunction, and acidic conditions(23–29). Similar to D-2-HG, L-2-HG can competitively inhibit dioxygenases due to structural similarity to α-KG. Correspondingly, increased DNA and/or histone methylation has been found in the setting of raised L-2-HG(28–30). However, the underlying biochemistry, relevant targets, and contribution to tumor growth of this small molecule in renal cancer remain poorly understood. Elevations of L-2-HG in RCC are due to reduced expression of L-2-HG dehydrogenase (L2HGDH). L2HGDH is considered an enzyme of metabolite repair to counter the “off-target” activity of intermediary metabolism enzymes including lactate dehydrogenase (LDH) and malate dehydrogenases (MDH), particularly under acidic conditions (23, 26, 31–33). Inheritance of biallelic mutant L2HGDH results in a rare inborn error of metabolism referred to as L-2-HG aciduria that has been linked with brain tumors(34–39). The concept that small molecules can promote malignancy has now presented novel therapeutic opportunities. In the case of IDH gain of function mutations in leukemia, which result in raised D-2-HG levels, specific inhibitors that selectively inhibit mutant IDH (as opposed to wild-type IDH) are now approved for use. Based on these data, we determined the contribution of L-2-HG to the malignant phenotype of RCC. We also outline the biochemical axis that promotes L-2-HG accumulation as well as epigenetic targets. Collectively, our data demonstrate that L-2-HG promotes tumor growth in RCC, and therefore, identifies potential strategies for RCCs with raised levels of this small molecule.
Materials and Methods
Cell Culture
Renal cell lines were acquired from ATCC except RXF-393 (NCI) and OSRC-2 (Riken). Cells acquired from ATCC, NCI, and Riken were characterized via short tandem repeat (STR) profiling. As cells were passaged for less than 3 months after resuscitation and were periodically screened for Mycoplasma using a PCR based assay, no further authentication was performed. HEK293T and HK-2 cells were maintained with DMEM containing 10% FBS and penicillin/streptomycin. A498 cells were cultured in MEM containing 10% FBS and penicillin/streptomycin. RXF-393, OS-RC-2 and 769-P cells were maintained in RPMI containing 10% FBS and penicillin/streptomycin. For L-2-HG isotopologue analysis, cells were incubated in media without L-glutamine for 4 hrs and then incubated for 6 hrs in media containing 2mM L-Glutamine-13C5 (Sigma).
Glutaminase/MDH inhibitors and L-2-HG ester treatments
Glutaminase inhibitor (CB-839, Calithera Biosciences) was dissolved in DMSO and cells were treated with CB-839 (1 µM) for 48–72 hrs with replacement of CB-839 for every 24 hours. MDH inhibitor, a derivative of benzylpaullones (4k) was dissolved in DMSO and cells were treated with 1 µM for 48 hrs (40). L-2-HG ester was generated as previously reported (21). For effects on phenotypes in HK-2 renal epithelial cells, cells were continuously cultured in the indicated concentration of ester for 7–10 days followed by wound healing assays.
Lentivirus-based generation of stable cell lines
L2HGDH and L2HGDH A241G cDNA was inserted into LV2606 (kindly provided by John Kappes, UAB). For L2HGDH or MDH knockdown, the following shRNAs in pLKO.1 vector (Sigma) were used: shL2HGDH (sh3, TRCN0000064323), shL2HGD (sh4, TRCN0000064324) and shL2HGDH (sh5, TRCN0000064325), shMDH1 (TRCN0000275199), shMDH1 (TRCN0000028484) and shMDH2 (TRCN0000028485). For EZH2 knockdown, shRNA lentiviral vectors for human EZH2 gene (EZH2 shRNA) and control shRNA were kindly provided by Soory Varambally (UAB). To generate stable cell lines, lentiviral plasmids were transfected with packaging vectors into HEK293T cells using calcium phosphate method. Supernatants from transfected HEK293T cells were collected after 72h, filtered, and applied to cells. Viral transduced cells were selected in culture medium containing puromycin. All transduced cells represent polyclonal populations.
siRNA-mediated knockdown of KDM6A
A498 cells stably expressing L2HGDH were treated with 30 nM of control siRNA (#D-001800–01), KDM6A siRNA#1 (#J-014140–10) and KDM6A siRNA#2 (# J-014140–12) ON-TARGET plus siRNA (Dharmacon) using Lipofectamine® RNAiMAX (Invitrogen, cat# 13778030) transfection reagent for 72 hrs. Cells were again treated with 30 nM of control siRNA, KDM6A siRNA#1 and KDM6A siRNA#2 for 48 hrs.
2-HG Measurements
2-HG enantiomer analysis (i.e. D- and L-2-HG quantification) of samples was performed as previously described(21). For total 2-HG (D-2-HG + L-2-HG) measurement of samples from in vitro studies, cell pellets were washed in 1x cold PBS three times. Metabolites were extracted with 10% cold trichloroacetic acid (TCA) and the precipitate was removed by centrifugation. TCA in the supernatant was removed by vortexing with 4 volumes of 1,1,2-trichlorotrifluoroethane (FREON)-trioctylamine (Sigma) mixture and the upper aqueous layer was collected for analysis after centrifugation. Samples were analyzed by ion chromatography coupled with negative electrospray mass spectrometry (RFIC-MS) (Dionex) and 2-HG was determined by selected ion monitoring (SIM 147.1). 2-HG in cell extracts was quantified by using a calibration curve of 2-HG and normalized to protein content.
Animal studies
Immune-deficient nude (Nu/Nu) mice were obtained from Jackson Laboratory or Charles River and fed with standard chow diet. Cells were mixed with equal volume of Matrigel Matrix (Corning, #354230) and injected (RXF-393= 1 × 106, A498= 2 × 106 and OSRC-2= 1.5 × 106 cells/injection) subcutaneously in the flanks of 5–6 weeks-old nude (Nu/Nu) mice. Caliper measurements of growing tumors were taken periodically and the tumor volume was calculated. For glutaminase inhibition study, Caki-1 human clear cell renal cell carcinoma cells were mixed 1:1 with Matrigel and implanted subcutaneously in the flanks into female scid/bg mice (Charles River) (2.5 × 106 cells/mouse). On day 16 post-implant, mice were randomized to the following two groups (n=10 mice per group): 1) vehicle (25% hydroxyl-propyl-β-cyclodextrin) orally twice daily by gavage; or 2) CB-839 at 200 mg/kg orally twice daily by gavage. For all in vivo studies, tumors were measured with calipers and tumor volume was calculated using the formula = (a x b2/2) where ‘b’ is the smallest diameter and ‘a’ is the largest perpendicular diameter.
RNA extraction and Quantitative RT-PCR
Total RNA from cultured cells were extracted using Trizol reagent (Invitrogen). cDNA was synthesized using QuantiTect reverse transcription kit (Qiagen). Real-time qPCR was done using TaqMan expression assay probes (Invitrogen) for human L2HGDH (Hs00227575), SPOCK2 (Hs00360339) and SHISA2 (Hs01590823) in QuantStudio 6K Flex real-time PCR system (Applied Biosystem). RPLP0 probe (Hs99999902) was used as internal control, and ΔΔCt method was used to calculate relative mRNA levels.
Western blot analysis
α-L2HGDH (Proteintech, #15707–1-AP, GeneTex, #GTX32695), α−5hmc (Active Motif, #39791), α-KDM6A (GeneTex, #GTX120873), α-MDH2 (Abcam, #ab96193), α-H3K27Me3 (Millipore, #07–449), α-H3 (CST, #9715), α-EZH2 (CST, #5246) and β-actin (Abcam, #ab20272) were used as per manufacturers’ instructions.
Dot blot analysis of 5-hydroxymethylation (5-hmC)
Blotting of genomic DNA was performed as previously described(21). Membranes then blocked with 5% milk in TBST for 30 min and were incubated with anti-5-hmC antibody (Active Motif) overnight at 4° C and HRP-conjugated anti-rabbit IgG secondary antibody for 1 hr at room temperature. After washing three times with TBST, the membrane was treated with ECL and scanned.
Wound healing assay
Confluent cell monolayers in 6 well culture dishes were wounded by making a scratch using a pipette tip. Phase-contrast/bright field pictures were taken of the wounded area at day 0 and wound healing on the time point as mentioned in the respective figures. Distances between the wounded area was measured using ImageJ software and percentage migration/wound healing was calculated with a percentage of distance coverage by migrating cells into the scratch wound area.
Boyden chamber cell migration assay
Cells were harvested by trypsinization, washed with PBS and resuspended into serum free culture medium. 5–10 × 103 cells/100–200 µl in serum free medium was loaded into moistened upper chamber and 600 µl of complete culture medium containing 10% FBS was loaded into bottom chamber of Transwell (Corning) plate. Cells were incubated for 16 hrs. Non-migrated cells (upper chamber) were scrapped off using cotton swab and the migrated cells trapped into the membrane were stained with Diff-Quik Stain kit (Siemens). Bright field images of the migrated cells were capture and quantified per mm2 of the membrane.
Microarray analysis
Total RNA was isolated from A498 cells stably expressing control vector and L2HGDH and purity was assessed by gel electrophoresis (Agilent 2100 Bioanalyzer). Transcriptional profiling experiments were then carried out using the Illumina HumanHT-12 V4 Array. Data were analyzed using GeneSpring GX software. Differences between treatment groups were determined by an unpaired t-test and the multiple testing correction method of Benjamini Hochberg. A corrected P-value (q-value) was then calculated to correct for false-positive discoveries. Heatmaps were created with R using the gplot heatmap.2 package.
TCGA Gene expression, correlation and outcome analysis
Clinical and RNASeq (V2) raw data for normal and tumor samples (Kidney renal clear cell carcinoma) were downloaded from TCGA website (https://tcga-data.nci.nih.gov). In house developed parser was used to process and extract clinical information and gene expression levels of L2HGDH (http://ualcan.path.uab.edu/cgi-bin/ualcan-res.pl)(41). For survival analysis, patients were ordered by expression level and split into top 50% and bottom 50%. Using this information, a two condition (high expression vs. low expression) Kaplan-Meier survivability plot was generated using the survival library in R.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism6 software. Comparisons between groups for statistical significance were performed with a two-tailed unpaired t-test. A P-value of <0.05 was considered statistically significant in all cases.
Study Approval
All animal studies were approved by institutional animal care and use committee (IACUC).
RESULTS
L2HGDH Knockdown Promotes In Vitro Tumor Phenotypes in Renal Epithelial Cells
Prior studies demonstrating an increased prevalence of brain tumors in the context of L-2-HG aciduria suggest that L2HGDH may have tumor suppressor properties. We therefore examined the effects of reduced L2HGDH expression in HK-2 immortalized renal epithelial cell which have relatively high basal L2HGDH expression compared with RCC cells. Using shRNA, we created 3 distinct knockdown pools which demonstrated reduced expression of L2HGDH at the mRNA and protein levels (Fig. 1A and Supplementary Fig. S1A). Consistent with the role of L2HGDH in L-2-HG metabolism, knockdown clones demonstrated elevated cellular levels of 2-HG as determined by GC-MS (Supplementary Fig. S1B). As a functional readout of raised 2-HG levels, we assessed the effects of L2HGDH knockdown on DNA 5-hyroxymethylcytosine (5-hmC) levels as prior studies demonstrate that L-2-HG can competitively inhibit α-KG dioxygenases including the TET enzymes. Consistent with these findings, L2HGDH knockdown led to reduced DNA 5-hmC levels in renal epithelial cells (Supplementary Fig.S1C). L2HGDH knockdown resulted in a modest increase in proliferation in HK-2 cells (Supplementary Fig. S1D). We investigated additional in vitro phenotypes and found that knockdown of L2HGDH promoted a migratory phenotype as assessed by a wound healing scratch assay (Fig.1B-C). To assess if these effects were related to raised cellular levels of L-2-HG, we treated HK-2 cells with exogenous L-2-HG octyl ester which expectedly raised cellular levels of 2-HG (Supplementary Fig. S1E). L-2-HG ester treatment also promoted a migratory phenotype in HK-2 cells phenocopying the results of L2HGDH knockdown (Fig. 1D and Supplementary Fig. S1F).
Figure 1. The L2HGDH/L-2-HG axis regulates migratory phenotypes.
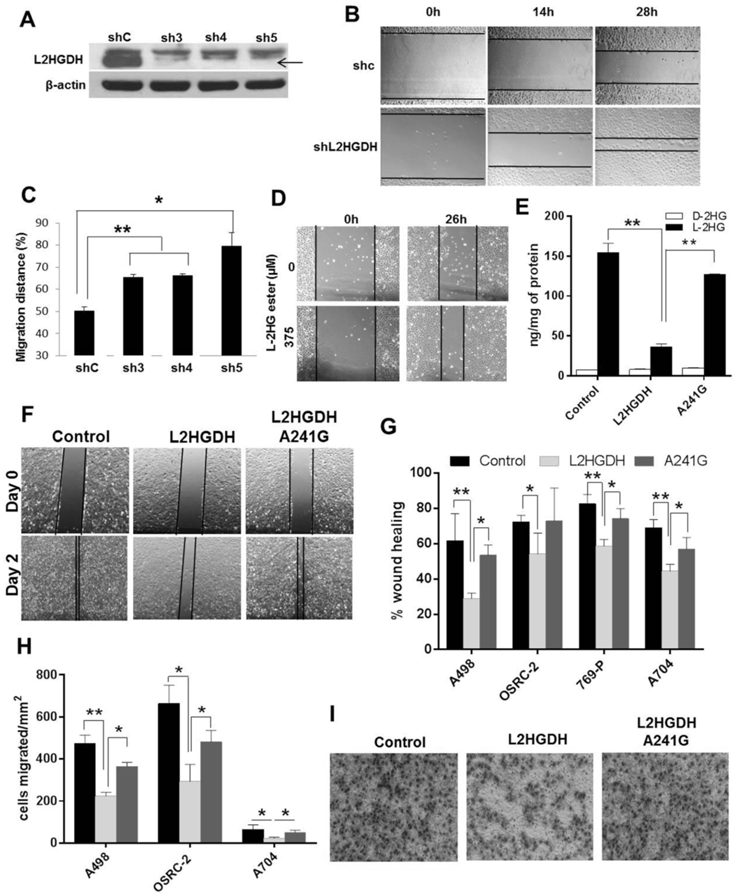
(A) HK-2 cells were transduced with control shRNA or three shRNAs targeting L2HGDH (sh3, sh4 and sh5) and puromycin resistant cells were selected to generate pooled stable cell lines. Validation of L2HGDH knockdown by immunoblotting (black arrow). (B and C) Wound healing of shL2HGDH stable cell lines seen using time-lapse phase contrast photography. Migration distance (%) calculated at 28 hr post-wound healing. Data are representative of two independent experiments (n=3/group). (D) Representative bright field images captured 26 hr post-wound creation in HK-2 cells treated with L-2-HG octyl ester. Data are representative of two independent experiments. (E) LC-MS analysis of L-2-HG and D-2-HG levels in control vector, WT L2HGDH, A241G expressing A498 cells. (F) Representative bright field images of A498 cells at day 0 and day 2 post-wound creation. (G) Relative wound healing of A498, OSRC-2, 769-P, A704 stably expressing control, L2HGDH and A241G vectors at 48, 36, 24 and 120 hrs post-wound creation, respectively. Data shown are the means ± SEM of two independent experiments (n=3/group). (H) Quantification of RCC cell migration (A498, OSRC-2, and A704) expressing control, L2HGDH and A241G. Data shown are the means ± SEM of two independent experiments (n=3/group). (I) Representative images of cell migration of A498 cells stably expressing control, L2HGDH and A241G vectors after 16 hrs incubation. (* indicates p < 0.05, ** indicates p < 0.01).
L2HGDH Suppresses Migratory Phenotypes in RCC Cells
Based on these data, we assessed the effects of L2HGDH reconstitution on RCC migratory phenotypes. Consistent with prior data from our group(21), we identified multiple RCC lines with reduced L2HGDH expression and raised L-2-HG levels relative to HK-2 cells (Fig. S2). We stably transduced RCC cells with WT L2HGDH cDNA or control vector. As an additional control, we stably transduced RCC cells with a L2HGDH point mutant (A241G) which results in a substitution of lysine with glutamate (K81E) previously reported in patients with L-2-HG aciduria and found to lack catalytic activity(39, 42). Protein expression was confirmed by immunoblotting (Supplementary Fig. S3A). In multiple lines tested, WT L2HGDH was able to significantly lower L-2-HG levels whereas only slight reductions in L-2-HG were noted with the A241G mutant (Fig. 1E and Supplemental Fig. S3B-C). Consistent with increased TET activity due to reduced L-2-HG, 5-hmC levels were increased in WT cells relative to control vector or A241G transduced cells (Supplementary Fig. S3D-E). Similar to our loss of function studies, L2HGDH restoration in RCC cells reduced migration as determined by scratch assay (Fig. 1F-G and Supplemental Fig. S4A-C). As an orthogonal assay, we assessed the effects of L2HGDH on migration via a Boyden chamber assay. In multiple RCC lines tested, WT L2HGDH suppressed Boyden chamber migration relative to either control or A241 mutant cells (Fig. 1H-I and Supplemental Fig. S4D-E). These data demonstrate that L2HGDH’s catalytic activity can suppress RCC migration.
L2HGDH Suppresses RCC Growth In vivo
These data prompted us to examine the effects of L2HGDH in vivo. We stably expressed L2HGDH in RXF-393 RCC cells which we previously demonstrated to express low levels of L2HGDH and had high levels of L-2-HG. Notably, L2HGDH restoration suppressed tumor growth (Fig. 2A-B). End of study analysis of tumor explants confirmed continued expression of L2HGDH in RCC cells (Supplemental Fig. S5). We next assessed if L2HGDH’s catalytic activity was required for effects on growth suppression. In both OSRC-2 and A498 RCC lines, tumors with WT L2HGDH grew more slowly than tumors expressing the A241G mutant (Fig. 2C-E). End of study tumors for WT tumors were less weight than A241G mutant tumors (Fig. 2F-G). Collectively, these data demonstrate that L2HGDH suppression of in vivo tumor growth is dependent on catalytic activity.
Figure 2. L2HGDH re-expression suppresses in vivo tumor growth of RCC cells.

(A-B) RXF-393 cells stably expressing control vector and wildtype L2HGDH were subcutaneously injected in 6 week old nude mice (n=10 per experimental group). Caliper measurements of the tumor were taken on the indicated days and the tumor volume at end of study was calculated. (C and D) Growth curves for OSRC-2 and A498 cells stably expressing wildtype L2HGDH and catalytic mutant A241G following subcutaneous injection in 6 week old nude mice (n=10 per experimental group). (E) Representative images of nude mice showing tumor growth of A498 cells stably expressing WT L2HGDH (red arrow) and A241G (blue arrow). Representative images (F) and average weights (G) of the harvested tumors of OSRC2 and A498 cells (L2HGDH and 241G) at the end of study. Data shown are the means ± SEM. (* indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001).
Biochemical Analysis of the Carbon Source for L-2-HG in RCC Cells
These data prompted us to delineate the biochemical mechanisms that promote L-2-HG accumulation in the setting of reduced L2HGDH expression. Prior studies demonstrate that α-ketoglutarate (α-KG) is the source for L-2-HG(31, 43). Two potential exogenous sources for α-KG include glucose and glutamine. Glutamine has previously been shown to be the predominant source of L-2-HG in hypoxic cancer cells (SF188 gliobastoma line) as well as activated T cells(23, 28). Culture of RCC cells with elevated basal L-2-HG levels in the absence of glucose resulted in a modest reduction in total 2-HG levels (Fig. 3A). In contrast, culture of cells in the absence of glutamine resulted in a dramatic reduction of total 2-HG levels (Fig. 3A). Enantiomer resolution of 2-HG demonstrated that glutamine deprivation markedly reduced L-2-HG levels in RCC cells (Supplemental Fig. S6A-B). Reductions of D-2-HG were also evident in cells cultured in the absence of glutamine. We next determined if glutamine is a direct carbon source for L-2-HG in RCC cells. Isotopologue studies with glutamine fully labeled with C13 (U-13C5) was used to determine if and how many carbons from glutamine are incorporated into L-2-HG. In both A498 and RXF-393 RCC cells, the predominant form of L-2-HG derived from U-13C5 glutamine is the m+5 fraction (C13 label incorporated into all 5 molecules of L-2-HG) (Fig. 3B and Supplemental Fig. S6C). These data are consistent with the working model that L-2-HG is generated by the direct reduction of α-KG to L-2-HG. The next most predominant isotopologue identified in A498 RCC lines is the m+3 fraction (C13 label incorporation into 3 carbons). These data would be consistent with α-KG going through a round of the TCA cycle which would result in the loss of 2 labeled carbons. L2HGDH re-expression in A498 cells led to reduced accumulation of labeled L-2-HG following incubation with U-13C5 as demonstrated by reduced levels of m+5 and m+3 L-2-HG isotopologues (Fig. 3C).
Figure 3. The role of glutamine in L-2-HG metabolism.
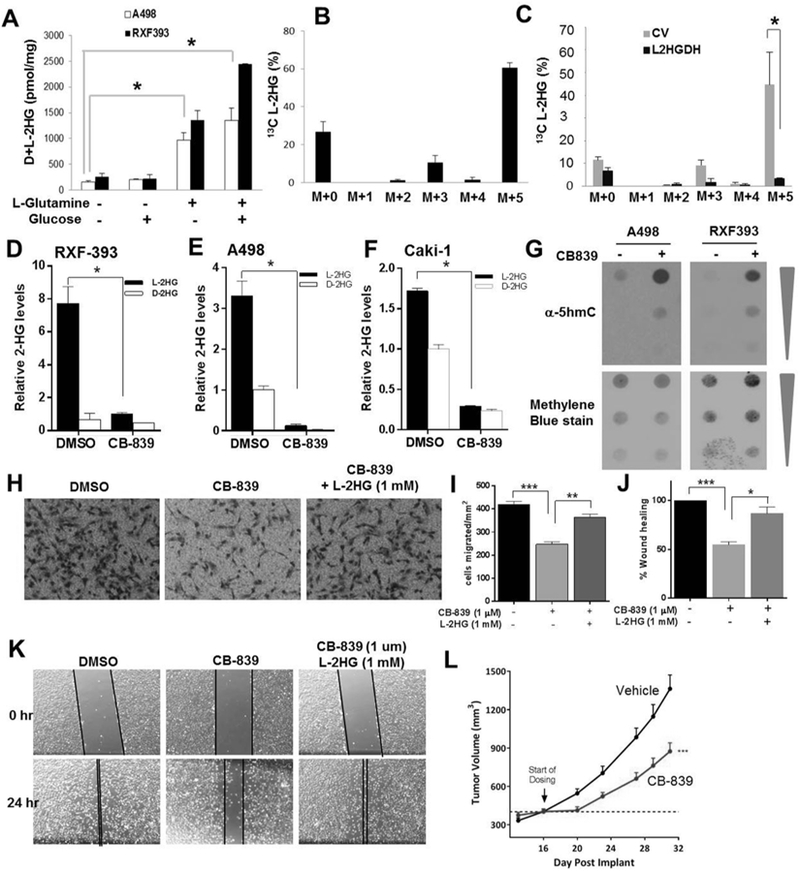
(A) A498 and RXF-393 cells were incubated in media containing either 5mM Glucose and/or 2mM L-Glutamine for 24 h. Total 2-HG level was measured. (B) L-2-HG isotopologue analysis showing abundance of 13C labelled L-2-HG in A498 cells starved for glutamine for 4 hrs and treated with 2mM 13C5 L-glutamine for 6 hrs. (C) L-2-HG isotopologue analysis showing abundance of 13C labelled L-2-HG in A498 cells (control and L2HGDH) starved for glutamine for 4 hrs and then incubated with 2mM 13C5 L-glutamine for 6 h. (D-F) Relative L-2-HG and D-2-HG levels of A498 (D), RXF-393 (E), and Caki-1 (F) RCC cells treated with glutaminase inhibitor CB-839 (1 µM) for 72 hrs. (G) Dot blot analysis of 5hmc levels in A498 and RXF-393 cells treated with CB-839 (1 µM) for 72 hrs. Upper panel: immunoblot for 5hmc. Bottom panel: methylene blue (MB) staining for total gDNA. (H-K) Caki-1 cells were treated with CB-839 (1 µM) for 48 hrs with or without L-2-HG (1 mM). Cells were harvested and then assessed for migration via Boyden chamber assay (panels H,I) and scratch assay (panels J,K). Data shown are the means ± SEM of two independent experiments (n=3/groups). (L) Growth curve of Caki-1 tumor xenografts in nude mice treated with vehicle or CB-839.
The contribution of glutamine to L-2-HG in RCC cells led us to consider if inhibition of glutamine metabolism could impact L-2-HG levels in RCC cells. A key step in glutamine utilization by cells is the conversion to glutamate by glutaminase. Recent studies demonstrate that glutaminase can be pharmacologically targeted (44). We therefore examined the effects of the glutaminase inhibitor CB-839 on L-2-HG metabolism in RCC cells. CB-839 treatment of RCC cells under glutamine containing conditions led to a significant reduction of cellular glutamate consistent with glutaminase inhibition (Supplemental Fig. S6D). Glutaminase inhibition led to a significant reduction of L-2-HG levels in multiple RCC lines (Fig. 3D-F). Small molecules including both D- and L-2-HG have been shown to competitively inhibit αKG dioxygenase including the TETs (1–3) which catalyze the conversion of DNA 5-methylC to 5-hydroxymethyl cytosine (5-hmC). Given the significant reductions of L-2-HG following glutaminase inhibition, we assessed for effects on DNA 5-hmC. In both A498 and RXF-393 cells, glutaminase inhibition resulted in increased DNA 5-hmC levels as determined via dot blot assay indicating that lowering of L-2-HG levels results in activation of TET activity (Fig. 3G). Given that CB-839 effectively lowers L-2-HG levels in RCC cells, we next assessed for effects on cellular migration. In Caki-1 RCC cells, CB-839 reduced cellular migration as determined by transwell migration assay (Fig. 3H-I) as well as scratch assay (Fig. 3J-K). Similar results on migration as determined by scratch assay were found in OSCR-2 and RXF-393 cells (Supplemental Fig. S7A-D). Moreover, for both assays evaluated, L-2-HG ester treatment was able to rescue the effects of glutaminase inhibition indicating that the effects of CB-839 on migration were in part mediated by lowering L-2-HG levels. Furthermore, glutaminase inhibition was able to suppress in vivo tumor growth in RCC cells (Fig. 3L). Collectively, these data demonstrate that glutamine metabolism promotes L-2-HG accumulation and that inhibition of this pathway can suppress tumor phenotypes.
MDH Promotes L-2-HG Accumulation in RCC Cells
Prior studies indicate that L-2-HG is produced by the promiscuous activity of dehydrogenases including malate dehydrogenase (MDH1 and 2)(31). We identified shRNA constructs that knockdown expression of both cytosolic MDH (MDH1) and mitochondrial MDH (MDH2) (Fig. 4A and Supplemental Fig. S8A). While knockdown of MDH1 reduced L-2-HG levels in RXF-393 cells (Supplemental Fig. S8B), MDH2 knockdown had a more pronounced effect on L-2-HG levels (Fig. 4B). In line with these data, pharmacologic MDH inhibition with a recently described inhibitor referred to as 4K also reduced 2-HG levels in RCC cells (Fig. 4C and Supplemental Fig. S8C). Similarly, MDH2 knockdown significantly reduced L-2-HG levels in OSRC-2 cells (Fig. 4D). We also observed that MDH2 knockdown reduced proliferation in OSRC-2 cells (Fig. 4E). Based on prior studies, we assessed effects of MDH2 knockdown on migration. We assessed migration at 16 hours to exclude effects on proliferation (Fig. 4F-G). Similar to prior manipulations that lowered L-2-HG levels, MDH2 knockdown reduced cell migration. However, the reduced migration could be rescued via treatment with L-2-HG ester. Collectively, these data demonstrate that MDH2 promotes L-2-HG accumulation and that inhibition of MDH2 can suppress in vitro tumor phenotypes in RCC cells with raised L-2-HG.
Figure 4. Knockdown of MDH lowers L-2-HG and suppresses in vitro tumor phenotypes in RCC cells.
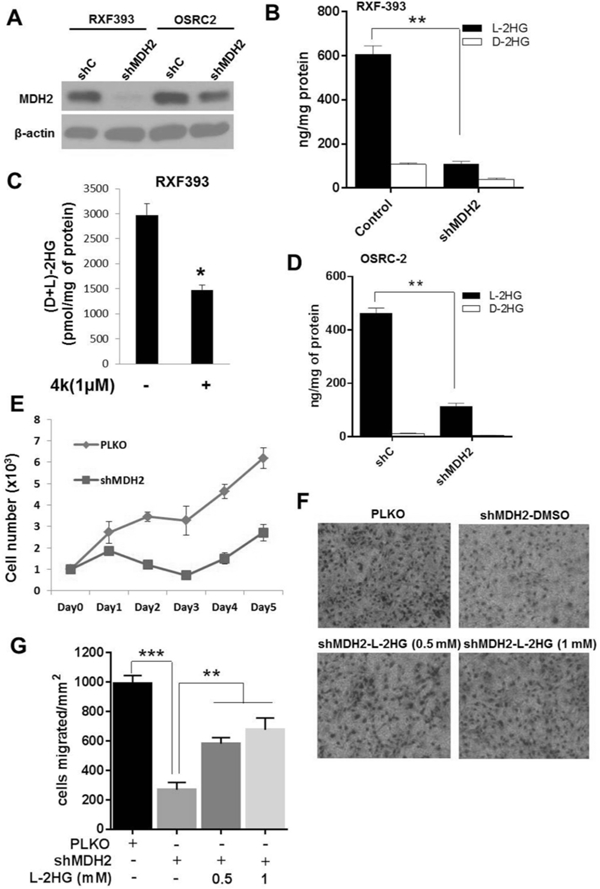
OSRC-2 and RXF-393 cells were transduced with PLKO control and shMDH2 vectors. (A) Western blot analysis of MDH2 knockdown in RXF-393 and OSRC-2 cells. (B) Intracellular L-2-HG and D-2-HG level in shMDH2 transduced RXF-393 cells. (C) RXF-393 cells were treated with MDH inhibitor (4k, 1uM) for 48 h, harvested, and assayed for total 2-HG levels. (D) Intracellular L-2-HG and D-2-HG levels in PLKO and shMDH2 transduced OSRC-2 cells. (E) Proliferation of OSRC-2 cells transduced with control PLKO and shMDH2 vector. Data shown are the means ± SEM of two independent experiments (n=3/group). (F, G) OSRC-2 cells transduced with shMDH2 were treated with or without L-2-HG ester (0.5 and 1 mM) for 48 hrs and allowed to migrate in Boyden’s chamber for 16 hrs. (F) Representative images of OSRC-2 cells migrated in Boyden’s chamber. (G) Quantification of OSRC-2 cells migrated in Boyden’s chamber. Data shown are the means ± SEM of two independent experiments (n=3/group). (* indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001).
L-2-HG Suppresses Targets of H3K27 Trimethylation (H3K27me3)
Small molecules including both D- and L-2-HG as well as fumarate and succinate have been shown to inhibit enzymes involved in histone and DNA methylation and therefore could impact gene expression. For deeper insight into L-2-HG’s effects, we performed comparative studies between A498/CV (high L-2-HG cells) and A498/L2HGDH (low L-2-HG cells). Gene expression array analysis comparing A498/CV and A498/L2HGDH cells reveals that genes re-expressed upon lowering L-2-HG cells are enriched for targets of polycomb repressor complex 2 (PRC2) and/or H3K27me3 target genes (Fig. 5A-B). PRC2 lays down the repressive H3K27me3 mark. Consistent with these data, WT L2HGDH lowered H3K27me3 levels in RCC cells whereas the A241G mutant did not (Fig. 5C). We validated that WT L2HGDH could increase expression of these target genes relative to control vector or A241G transduced cells (Fig. 5D). Given that L-2-HG can competitively inhibit lysine histone demethylases (KDMs), these data led us to consider KDMs that demethylate H3K27me3. Recurring mutations of KDM6A (also referred to as UTX) which encodes a H3K27 demethylase have been identified in renal cancer(45, 46). We therefore utilized siRNA to knock down the expression of KDM6A in A498 cells transduced with WT L2HGDH (A498/L2HGDH) which have reduced migration relative to either control or A241G mutant cells (Fig. 1G-H). Immunoblotting confirmed knockdown of KDM6A at the protein level (Fig. 5E). KDM6A knockdown in A498/L2HGDH cells enhanced migration and therefore phenocopied the enhanced migration of high L-2-HG cells (Fig. 5F-G). Based on these data, we assessed the effects of PRC2 inhibition in parental A498 cells which have high basal levels of L-2-HG. The catalytic subunit of PRC2, EZH2 (enhancer of zeste homologue 2), is required for H3K27 methyltransferase activity. Knockdown of EZH2 in A498 cells, confirmed by immunoblotting, resulted in reduced H3K27me3 levels (Fig. 5H). Notably, EZH2 knockdown resulted in reduced cell migration as determined by transwell chamber assay (Fig. 5I-J).
Figure 5. High L-2-HG inhibits activity of histone lysine demethylase to promote H3K27 trimethylation in RCC cells.
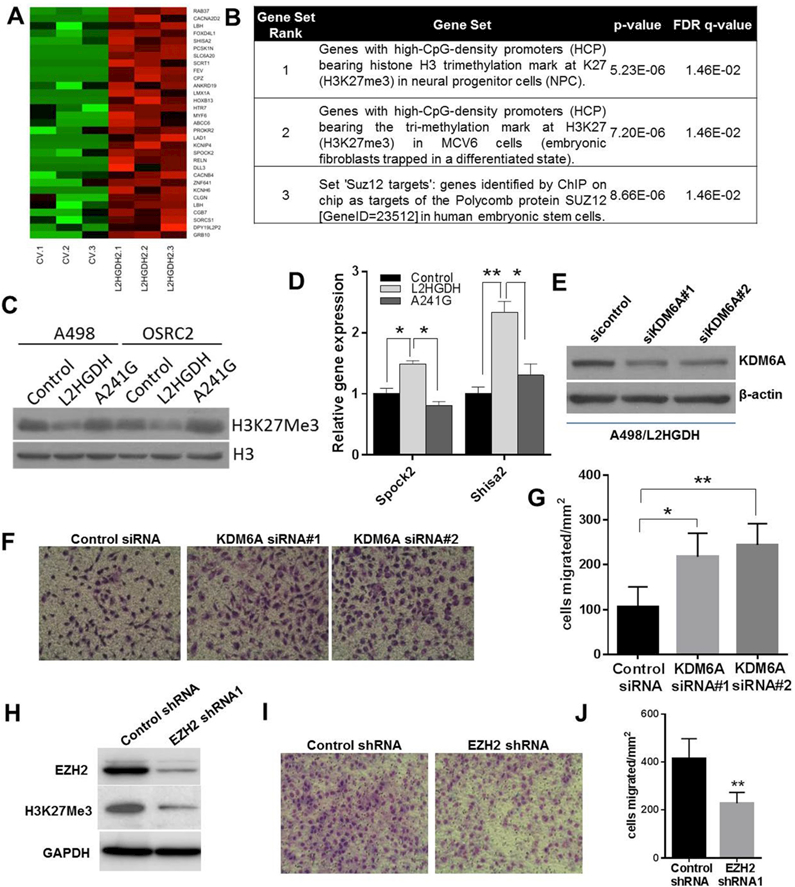
(A) Heat map of PRC2/H3K27me3 target genes with increased expression upon L2HGDH restoration (n=3/group). (B) GSEA of genes with increased expression upon L2HGDH restoration. (C) Immunoblots of H3K27Me3 levels in A498 and OSRC-2 cells expressing control, WT L2HGDH, and L2HGDH A241G. (D) Relative mRNA levels of SPOCK2 and SHISA2 in A498 cells stably expressing control, WT L2HGDH, and L2HGDH mutant A241G measured using RT-qPCR. (E) A498 cells expressing L2HGDH were treated with the indicated siRNA and then assessed by immunoblotting for KDM6A protein levels. (F) Representative images of A498/L2HGDH cells treated with the indicated siRNA migrated through a transwell insert. (G) Quantification of migration of A498/L2HGDH cells treated with the indicated siRNA. Data shown are the means ± SEM of two independent experiments (n=3/group). (H) Immunoblot for EZH2 and H3K27me3 in A498 cells transduced with control or EZH2 shRNA. (I) Representative images of A498 cells transduced with the indicated shRNA migrated through a transwell insert. (J) Quantification of migration of A498 cells transduced with the indicated shRNA. Data shown are the means ± SEM of two independent experiments (n=3/group). (* indicates p < 0.05, ** indicates p < 0.01).
Translational Analysis of the L-2-HG/L2HGDH Axis in RCC
Given these data, we next examined the translational significance of the L-2-HG/ L2HGDH axis in the context of RCC. Using the TCGA data set we examined the outcomes of RCC patients as a function of L2HGDH expression. Notably, patient with tumors in the lower 50% of L2HGDH expression had reduced survival (Fig. 6A). Correspondingly, lower L2HGDH mRNA expression was associated with higher tumor stage and grade (Fig. 6B-C). A primary mechanism by which L2HGDH expression is reduced is via copy loss of L2HGDH. We previously demonstrated using the TCGA data set that copy loss was associated with reduced expression(21). The L2HGDH gene is located at 14q, a commonly deleted region in ccRCC. Chromosomal losses of 14q resulting in loss of heterozygosity (LOH) has previously been shown to be associated with worsened prognosis in ccRCC patients(47, 48). We therefore analyzed the relationship between 14q LOH and L2HGDH expression based on a recent data set that examined both the genomic and transcriptomic landscape of 100 ccRCCs (49). 14q LOH was identified in 42/100 ccRCC tumors (supplemental table 1S). Consistent with prior studies, deletion was the primary mechanism for LOH (as opposed to alternate mechanism such as copy neutral LOH). LOH at the L2HGDH locus was present in 38 of these 42 cases (supplemental table 1S). Moreover, 14q LOH tumors demonstrate reduced L2HGDH expression relative to tumors without 14q copy number alterations (Fig. 6D). These data further support copy loss as a mechanism of reduced L2HGDH expression and that the L2HGDH locus is commonly included in 14q losses present in ccRCC. Consistent with these data, LOH at the L2HGDH locus was associated with worsened survival in the TCGA data set (Fig. 6E). Death from ccRCC is most commonly due to progression of disease that results in metastasis. We performed unbiased metabolomic profiling in normal kidney, primary tumors, and metastatic RCC tissues (manuscript in preparation). Consistent with prior studies, we identified elevations of total 2-HG levels in primary tumors relative to normal kidney (Fig. 6F). However, metastatic tissues had even higher total 2-HG levels compared with either primary tumors or normal kidney (Fig. 6F). We confirmed that the predominant enantiomer contributing to the total 2-HG pool in metastatic tissues was the L(S) enantiomer (Fig. 6G). Collectively, our translational studies support the biological relevance of the L-2-HG/L2HGDH axis to renal carcinogenesis identified by the in vitro and in vivo data presented.
Figure 6. Prognostic Significance of L2HGDH expression in RCC patients.
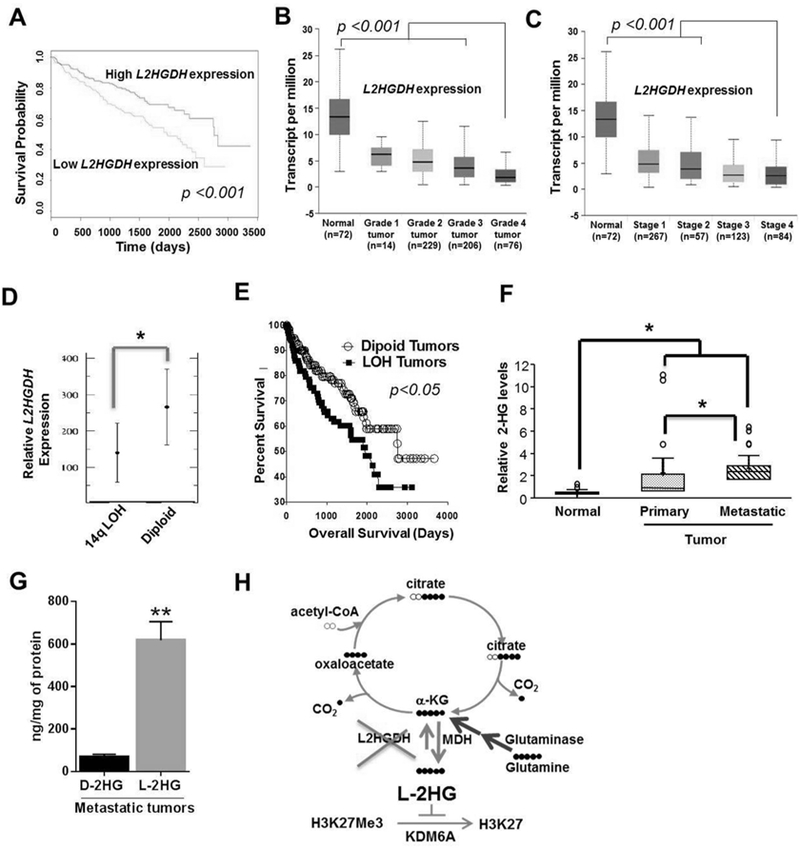
(A) Kaplan-Meier survival curve analysis in patients from TCGA data set with tumors expressing low L2HGDH mRNA expression (bottom 50%) relative to patients with tumors with high L2HGDH expression (upper 50%). Expression of L2HGDH (transcript per million) in increasing grades (Grade 1–4) (B) and stages (Stage 1–4) (C) of kidney tumors of patients from TCGA data set. (D) Relative L2HGDH mRNA expression as a function of 14q LOH. Data extracted from Sato et al. (E) Percentage survival curve of patients from TCGA data set as a function of L2HGDH copy number. (F) Relative total (D+L)-2-HG levels in normal, primary and metastatic kidney RCC deposits. (G) D-2-HG and L-2-HG levels in metastatic tumor deposits (n=3). (H) Graphical representation of biochemical axis of L-2-HG accumulation in RCC and therapeutic potential of glutaminase and MDH inhibitors to lower L-2-HG. (* indicates p <0.05 and ** indicates p <0.01).
Discussion
Here we provide clear evidence that the L-2-HG/L2HGDH axis has biological significance to renal carcinogenesis. In particular, our in vivo studies demonstrate that restoration of L2HGDH can suppress tumor growth. Moreover, our studies demonstrate that suppression of tumor growth is related to L2HGDH’s catalytic activity thereby bolstering the notion that L-2-HG is a bona fide oncometabolite that contributes to renal tumorigenesis. These data are in line with data from patients with L-2-HG aciduria in which L-2-HG levels are markedly elevated. A notable finding from this rare disease are multiple reports of patients with brain tumors suggesting that L2HGDH has tumor suppressor functions consistent with our data in RCC(34, 37, 38, 50, 51). In addition to brain tumors, a Wilms’ tumor of the kidney has also been reported in a L-2-HG aciduria patient(52). Wilms’ tumor is a common renal tumor affecting individuals in the pediatric population. To date, clear cell renal tumors have not been reported in these patients. However, patients with this disease process have significant neurological sequelae that may shorten lifespan relative to patients typically affected by RCC who are usually diagnosed in the seventh decade and beyond.
Although the concept of oncometabolites is well established, we would like to highlight that these are among the only data to demonstrate that lowering levels of a metabolite via a genetic approach (i.e. L2HGDH restoration) can impact tumor growth in vivo. In vivo studies on the role of fumarate and succinate are lacking. Rohle et al. reported that IDH1 knockdown via shRNA in IDH1 mutant (heterozygous) TS603 glioma cells could suppress tumor growth(18). One caveat to this study is that the IDH knockdown approach could not exclude effects on tumor growth mediated by the wild type IDH1 allele present in these cells. This is particularly relevant as recent studies by Clavert et al. demonstrate that non-mutated IDH1 is overexpressed in glioblastoma and that knockdown of wild type IDH1 can suppress glioma growth in vivo. Studies on the use of IDH inhibitors for glioma have yielded conflicting results in vivo(18, 19, 53). Moreover, recent transgenic models have demonstrated conflicting results on the role of mutant IDH in glioma progression(54, 55). In contrast, preclinical and clinical studies, particularly in leukemia, have provided clear rationale for the use of mutant IDH enzyme inhibitors to the point that they are now approved for use in the setting of IDH mutation(56–58). However, the effects are likely related to differentiation as in vitro studies in leukemia demonstrate that pharmacologic inhibition of mutant IDH may actually increase proliferation(58, 59). Our data are also the first to demonstrate that raised L-2-HG can promote a migratory phenotype. Prior studies indicate that D-2-HG can promote mesenchymal phenotypes in breast and colon cancer cells(60–62). However, L-2-HG failed to induce this behavior in colon cancer cells(61). Collectively, these data indicate that context is important with regard to the effects of either 2-HG enantiomer on tumor biology/phenotypes.
Bioinformatic analyses demonstrate that copy loss of L2HGDH is linked with 14q losses in ccRCC. Loss of 14q is associated with worsened outcomes in RCC patients(48, 63). Despite these data, the genes on 14q whose loss contributes to this clinical phenotype remain to be characterized. Our data indicated that L2HGDH is one of the relevant genes. Collectively, our data support a model in which 14q loss results in a cellular milieu that leads to elevation of L-2-HG in RCC. Our studies demonstrate possible strategies to lower L-2-HG levels in RCC including targeting the source of L-2-HG in RCC via the glutamine axis or via inhibition of MDH (Fig. 6H).
To date, studies on both 2-HGs have demonstrated that multiple histone marks can be impacted. However, a major challenge has been identifying the relevant histone demethylases. Our transcriptomic studies indicate that KDM6A is a target of L-2-HG in RCC and are in agreement with mounting evidence demonstrating the importance of the H3K27 methylation axis in renal cancer. As noted before, mutations of KDM6A (also referred to as UTX), which encodes the enzyme that demethylates H3K27, have been reported in renal cancer(45, 46). Notably, multiple studies have now reported that KDMs are HIF target genes which is particularly relevant in ccRCC in which inactivating mutations of VHL and ensuing HIF stabilization is a common event(64–68). A recent study by Chakraborty et al. indicated that VHL-defective RCC cells are dependent on the H3K27 methyltransferase activity of EZH1 for cell survival to counter the HIF-mediated induction of KDMs(69). Multiple studies indicate the importance of EZH2 in RCC phenotypes including in vivo tumor growth and therapy resistance(70, 71). Our data, therefore, add another layer of regulation of this axis in RCC via metabolism due to L-2-HG’s ability to act as a competitive inhibitor.
Our data may have biomarker implications for ccRCC, a major gap in the kidney cancer field. As previously noted, the L2HGDH gene is located at 14q, a region whose loss is associated with poorer outcomes. We previously established the connection between reduced L2HGDH expression and elevated L-2-HG levels in ccRCC. Given that reduced L2HGDH expression is associated with worsened outcomes and increasing stage/grade, L-2-HG levels in a tumor could serve as a biomarker that could dictate therapy and/or render prognostic information.
Finally, our data add to the growing body of evidence that inhibition of glutamine metabolism may have therapeutic relevance in renal cancer. These data have particular relevance given our data demonstrating the role of glutamine metabolism in maintaining L-2-HG levels in multiple RCC lines tested as well as the in vivo efficacy of a glutaminase inhibitor in RCC cells with elevated L-2-HG levels. Recent studies in the context of clinical trials indicate that glutaminase inhibition may have efficacy in a subset of RCC patients(72). Efficacy of glutaminase inhibition in RCC in vivo has been linked with PARP sensitivity and oxidative stress(73, 74). Therefore, despite our rescue experiments with esterified L-2-HG in vitro, we cannot exclude effects on these alternate pathways in vivo. Nevertheless, these data warrant future studies to explore whether L-2-HG could be utilized as a biomarker for therapeutics that target glutamine metabolism.
In summary, our studies demonstrate that L2HGDH has tumor suppressive effects and therefore support the concept of L-2-HG as an oncometabolite in kidney cancer. By delineating the metabolic events that promote L-2-HG accumulation and the downstream sequelae of this buildup, we provide new opportunities for therapeutic intervention in patients with L-2-HG driven kidney tumors.
Supplementary Material
Statement of Translational Relevance:
The small molecule L-2-hydroxyglutarate (L-2-HG) is elevated in clear cell renal cell carcinoma, the most common histology of kidney cancer, due to loss of the enzyme L-2-HG dehydrogenase (L2HGDH). Either pharmacologic or genetic approaches to raise intracellular levels of L-2-HG promote in vitro tumor phenotypes in nontransformed renal epithelial cells. In turn, re-expression of L2HGDH in kidney cancer cells reduced in vivo tumor growth. Biochemical studies indicate that the glutamine/ malate dehydrogenase 2 (MDH2) axis promotes L-2-HG elevation. Transcriptomic and functional analyses demonstrate that the histone demethylase KDM6A is a target of L-2-HG in RCC. Additionally, our studies indicate that alterations of the L-2-HG/L2HGDH are associated with patient outcomes and tumor progression. Collectively, our data demonstrate that L-2-HG is a bona fide oncometabolite in kidney cancer thereby providing opportunities for novel approaches to this malignancy.
Acknowledgments
Financial Support: The research reported in this article was supported by R01CA200653, 5I01BX002930, the UAB-HudsonAlpha Center for Genomic Medicine (to S.S.) and in part by the UAB Comprehensive Cancer Center (P30CA013148).
Footnotes
Conflicts of Interest: E. Emberley, A. Pan, J. Cheng, and T. Huang are employees of Calithera Biosciences. All other authors have declared no conflict of interest exist.
REFERENCES
- 1.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. The Journal of pathology 2011;224:334–43. [DOI] [PubMed] [Google Scholar]
- 3.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine 2009;361:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. The oncologist 2012;17:72–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005;8:143–53. [DOI] [PubMed] [Google Scholar]
- 9.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005;7:77–85. [DOI] [PubMed] [Google Scholar]
- 10.Smith EH, Janknecht R, Maher LJ, 3rd Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Human molecular genetics 2007;16:3136–48. [DOI] [PubMed] [Google Scholar]
- 11.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011;12:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & development 2012;26:1326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nature reviews 2010;11:607–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015;28:773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopinja J, Sevilla RS, Levitan D, Dai D, Vanko A, Spooner E, et al. A Brain Penetrant Mutant IDH1 Inhibitor Provides In Vivo Survival Benefit. Scientific reports 2017;7:13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 2014;4:1290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell research 2016;26:103–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism 2015;22:304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell reports 2014;7:1679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism 2015;22:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J Biol Chem 2016;291:20188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, et al. Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1alpha Stability in Aerobic Conditions. Cell metabolism 2016;24:740–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 2016;540:236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nature cell biology 2017;19:614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma S, Sun R, Jiang B, Gao J, Deng W, Liu P, et al. L2hgdh Deficiency Accumulates l-2-Hydroxyglutarate with Progressive Leukoencephalopathy and Neurodegeneration. Mol Cell Biol 2017;37. [DOI] [PMC free article] [PubMed]
- 31.Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis 2007;30:681–9. [DOI] [PubMed] [Google Scholar]
- 32.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 2017;13:494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Chawla G, Hurlburt AJ, Sterrett MC, Zaslaver O, Cox J, et al. Drosophila larvae synthesize the putative oncometabolite L-2-hydroxyglutarate during normal developmental growth. Proceedings of the National Academy of Sciences of the United States of America 2017;114:1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aghili M, Zahedi F, Rafiee E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J Neurooncol 2009;91:233–6. [DOI] [PubMed] [Google Scholar]
- 35.Barbot C, Fineza I, Diogo L, Maia M, Melo J, Guimaraes A, et al. L-2-Hydroxyglutaric aciduria: clinical, biochemical and magnetic resonance imaging in six Portuguese pediatric patients. Brain Dev 1997;19:268–73. [DOI] [PubMed] [Google Scholar]
- 36.Haliloglu G, Jobard F, Oguz KK, Anlar B, Akalan N, Coskun T, et al. L-2-hydroxyglutaric aciduria and brain tumors in children with mutations in the L2HGDH gene: neuroimaging findings. Neuropediatrics 2008;39:119–22. [DOI] [PubMed] [Google Scholar]
- 37.Moroni I, Bugiani M, D’Incerti L, Maccagnano C, Rimoldi M, Bissola L, et al. L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology 2004;62:1882–4. [DOI] [PubMed] [Google Scholar]
- 38.Ozisik PA, Akalan N, Palaoglu S, Topcu M. Medulloblastoma in a child with the metabolic disease L-2-hydroxyglutaric aciduria. Pediatr Neurosurg 2002;37:22–6. [DOI] [PubMed] [Google Scholar]
- 39.Rzem R, Veiga-da-Cunha M, Noel G, Goffette S, Nassogne MC, Tabarki B, et al. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proceedings of the National Academy of Sciences of the United States of America 2004;101:16849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker A, Kohfeld S, Lader A, Preu L, Pies T, Wieking K, et al. Development of 5-benzylpaullones and paullone-9-carboxylic acid alkyl esters as selective inhibitors of mitochondrial malate dehydrogenase (mMDH). European journal of medicinal chemistry 2010;45:335–42. [DOI] [PubMed] [Google Scholar]
- 41.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (New York, NY 2017;19:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rzem R, Van Schaftingen E, Veiga-da-Cunha M. The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie 2006;88:113–6. [DOI] [PubMed] [Google Scholar]
- 43.Struys EA, Gibson KM, Jakobs C. Novel insights into L-2-hydroxyglutaric aciduria: mass isotopomer studies reveal 2-oxoglutaric acid as the metabolic precursor of L-2-hydroxyglutaric acid. J Inherit Metab Dis 2007;30:690–3. [DOI] [PubMed] [Google Scholar]
- 44.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Molecular cancer therapeutics 2014;13:890–901. [DOI] [PubMed] [Google Scholar]
- 45.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010;463:360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature genetics 2009;41:521–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroeger N, Klatte T, Chamie K, Rao PN, Birkhauser FD, Sonn GA, et al. Deletions of chromosomes 3p and 14q molecularly subclassify clear cell renal cell carcinoma. Cancer 2013;119:1547–54. [DOI] [PubMed] [Google Scholar]
- 48.Monzon FA, Alvarez K, Peterson L, Truong L, Amato RJ, Hernandez-McClain J, et al. Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2011;24:1470–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nature genetics 2013;45:860–7. [DOI] [PubMed] [Google Scholar]
- 50.Patay Z, Orr BA, Shulkin BL, Hwang SN, Ying Y, Broniscer A, et al. Successive distinct high-grade gliomas in L-2-hydroxyglutaric aciduria. J Inherit Metab Dis 2015;38:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patay Z, Mills JC, Lobel U, Lambert A, Sablauer A, Ellison DW. Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR Am J Neuroradiol 2012;33:940–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogers RE, Deberardinis RJ, Klesse LJ, Boriack RL, Margraf LR, Rakheja D. Wilms tumor in a child with L-2-hydroxyglutaric aciduria. Pediatr Dev Pathol 2010;13:408–11. [DOI] [PubMed] [Google Scholar]
- 53.Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol 2017;133:629–44. [DOI] [PubMed] [Google Scholar]
- 54.Waitkus MS, Pirozzi CJ, Moure CJ, Diplas BH, Hansen LJ, Carpenter AB, et al. Adaptive Evolution of the GDH2 Allosteric Domain Promotes Gliomagenesis by Resolving IDH1(R132H)-Induced Metabolic Liabilities. Cancer Res 2018;78:36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Philip B, Yu DX, Silvis MR, Shin CH, Robinson JP, Robinson GL, et al. Mutant IDH1 Promotes Glioma Formation In Vivo. Cell reports 2018;23:1553–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kats LM, Vervoort SJ, Cole R, Rogers AJ, Gregory GP, Vidacs E, et al. A pharmacogenomic approach validates AG-221 as an effective and on-target therapy in IDH2 mutant AML. Leukemia 2017;31:1466–70. [DOI] [PubMed] [Google Scholar]
- 57.Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov 2017;7:478–93. [DOI] [PubMed] [Google Scholar]
- 58.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013;340:622–6. [DOI] [PubMed] [Google Scholar]
- 59.Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes & development 2013;27:1974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mishra P, Tang W, Putluri V, Dorsey TH, Jin F, Wang F, et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J Clin Invest 2018;128:323–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Scientific reports 2016;6:36289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J Biol Chem 2012;287:42180–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klatte T, Rao PN, de Martino M, LaRochelle J, Shuch B, Zomorodian N, et al. Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma. J Clin Oncol 2009;27:746–53. [DOI] [PubMed] [Google Scholar]
- 64.Lee HY, Choi K, Oh H, Park YK, Park H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol Cells 2014;37:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochemical and biophysical research communications 2008;372:892–7. [DOI] [PubMed] [Google Scholar]
- 66.Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 2010;30:344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 2008;283:36542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proceedings of the National Academy of Sciences of the United States of America 2009;106:4260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chakraborty AA, Nakamura E, Qi J, Creech A, Jaffe JD, Paulk J, et al. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Science translational medicine 2017;9. [DOI] [PMC free article] [PubMed]
- 70.Liu L, Xu Z, Zhong L, Wang H, Jiang S, Long Q, et al. EZH2 promotes tumor cell migration and invasion via epigenetic repression of E-cadherin in renal cell carcinoma. BJU international 2014. [DOI] [PubMed]
- 71.Adelaiye-Ogala R, Budka J, Damayanti NP, Arrington J, Ferris M, Hsu CC, et al. EZH2 Modifies Sunitinib Resistance in Renal Cell Carcinoma by Kinome Reprogramming. Cancer Res 2017;77:6651–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Funda Meric-Bernstam NMT, Mier James Walter, DeMichele Angela, Telli Melinda L., Fan Alice C.. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS), alone and in combination with everolimus (E) in patients (pts) with renal cell cancer (RCC). Journal of Clinical Oncology 34, no 15_suppl (May 2016) 4568–4568. 2016. [Google Scholar]
- 73.Abu Aboud O, Habib SL, Trott J, Stewart B, Liang S, Chaudhari AJ, et al. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res 2017;77:6746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okazaki A, Gameiro PA, Christodoulou D, Laviollette L, Schneider M, Chaves F, et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J Clin Invest 2017;127:1631–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.