

Abstract
Purpose:
Testosterone suppression in prostate cancer (PC) is limited by serious side effects and resistance via restoration of androgen receptor (AR) functionality. ELK1 is required for AR-dependent growth in various hormone-dependent and castration resistant PC models. The amino terminal domain of AR docks at two sites on ELK1 to co-activate essential growth genes. This study explores the ability of small molecules to disrupt the ELK1-AR interaction in the spectrum of PC, inhibiting AR activity in a manner that would predict functional tumor selectivity.
Experimental design:
Small molecule drug discovery and extensive biological characterization of a lead compound.
Results:
We have discovered a lead molecule (KCI807) that selectively disrupts ELK1-dependent promoter activation by wild-type and variant ARs without interfering with ELK1 activation by ERK. KCI807 has an obligatory flavone scaffold and functional hydroxyl groups on C5 and C3’. KCI807 binds to AR, blocking ELK1 binding, and selectively blocks recruitment of AR to chromatin by ELK1. KCI807 primarily affects a subset of AR target growth genes selectively suppressing AR-dependent growth of PC cell lines with a better inhibitory profile than enzalutamide. KCI807 also inhibits in vivo growth of castration/enzalutamide-resistant cell line-derived and patient-derived tumor xenografts. In the rodent model, KCI807 has a plasma half-life of 6h and maintenance of its antitumor effect is limited by self-induced metabolism at its 3’-hydroxyl.
Conclusions:
The results offer a mechanism-based therapeutic paradigm for disrupting the AR growth-promoting axis in the spectrum of prostate tumors while reducing global suppression of testosterone actions. KCI807 offers a good lead molecule for drug development.
Keywords: Prostate cancer, Castration resistance, Androgen receptor, ELK1, Drug discovery
INTRODUCTION
Prostate cancer (PC) predominantly afflicts older men and is relatively slow-growing and immune-resistant. Hence the most effective treatment strategy following surgical resection or radiation is chronic management to suppress tumor growth rather than acute cytotoxic therapies. A unique feature of prostate oncogenesis is its dependence on androgen, which acts by binding to and activating transcriptional signaling by the androgen receptor (AR). Both early stage and advanced prostate tumors are generally dependent on AR for growth (1, 2). Residual or recurrent PC is commonly treated by suppressing testicular androgen synthesis, typically by disruption of the hypothalamic-pituitary-gonadal axis (chemical castration) (3, 4). In addition, AR antagonists or an inhibitor of intra-tumor testosterone synthesis may be used (5). Unfortunately, the initial responders to androgen deprivation therapy (ADT) tend to develop hormone refractory disease, referred to as castration resistant PC (CRPC), which nevertheless continues to depend on AR. Growth signaling may be sustained in CRPC largely through amplification of AR (6) or expression of its splice variants, which lack the ligand binding domain (7). AR splice variants are frequently co-expressed with full length AR with which they heterodimerize and translocate to the nuclear compartment in a ligand-independent manner (8). AR splice variants, in collaboration with full length AR, confer optimal hormone-independent growth and insensitivity to anti-androgens and their expression is both functionally and clinically linked to tumor progression (9). Resistance mechanisms also include hormone-independent cross-talk between AR and certain signaling pathways, alterations in the AR co-regulator complement or mutation of AR (6).
Adverse effects associated with ADT are both acute (fatigue, hot flashes) and long-term (hyperlipidemia, insulin resistance, cardiovascular disease, anemia, osteoporosis, sexual dysfunction and cognitive defects) and include loss of the feeling of well-being (3, 10, 11). The high affinity androgen antagonist enzalutamide, used to treat CRPC, inactivates AR by blocking its nuclear entry and impairing its transcriptional activity (12) but it necessarily also abrogates AR signaling in normal tissues and is ineffective against AR splice variants or highly overexpressed AR (13). The recurrent and metastatic disease is then treated by chemotherapy, which is typically non-curative and has adverse side effects. Newer types of chemotherapy, immunotherapy and radiation therapy offer valuable, but limited, improvements (14–17). Experimental drugs that block coactivator protein binding to AR have been developed through high throughput small molecule screening and they are effective against AR splice variants (18) but they may also be expected to impair AR function in normal tissues.
Thus the current clinical paradigm for long-term treatment of advanced PC is ubiquitous suppression of AR signaling. However, these approaches have two major limitations: 1. ineffectiveness against advanced tumors in which functional AR has been restored through any of the aforementioned mechanisms and 2. the need to deprive the patient of androgen or AR function in all tissues and the consequent multiple side effects noted above. A strategic approach to addressing the dual limitation of ADT is to identify and disrupt a functional arm of AR that (i) is preserved as a crucial mechanism for supporting growth in CRPC and (ii) is necessary for tumor growth, but not for the physiological role of androgen in differentiated normal tissues. Mechanisms of growth signaling by AR that are tumor-specific could potentially offer a highly sensitive point of attack, even in cells that have acquired resistance to ADT and anti-androgen therapies.
As androgen plays a major role in all physiological aspects of the normal prostate including development, differentiation, maintenance and function (19), malignant prostate cells must selectively support mechanisms that redirect androgen/AR signaling to strongly support growth. In the classical model of gene regulation by AR the receptor requires bound ligand to homo-dimerize, enter the nucleus and bind to DNA at well-characterized androgen response elements (AREs) associated with target genes (20, 21). When the bound ligand is an agonist, AR then recruits coactivators; in contrast, when bound to antagonists, co-repressors are preferentially recruited (20). AR contains sites of co-regulator binding that are either ligand-dependent or -independent. However, in PC cells that are adapted to grow in the absence of hormone, the AR apo-protein is localized in the nucleus, where it is transcriptionally active even in the absence of hormone (7, 22, 23). AR localized in the nucleus cannot optimally bind to AREs without androgen; nevertheless, both ligand-bound and -unbound AR will still activate a large set of growth supporting genes and support growth through associations with chromatin via putative tethering proteins (22). We have previously reported that ELK1 is an AR tethering protein that is obligatory for androgen/AR-dependent malignant growth in a variety of well-established PC/CRPC models (24, 25).
ELK1 is a downstream effector of the MAPK signaling pathway and belongs to the ternary complex factor (TCF) sub-family of the ETS family of transcription factors. ELK1 characteristically binds to purine-rich GGA core sequences (26) and is in a repressive or passive association with many cell proliferation genes. Phosphorylation by ERK transiently hyper-stimulates ELK1 to activate its target genes including association with serum response factor (SRF) for activation of immediate early genes (26–28). Chromatin sites of AR binding are highly enriched for ELK1 binding DNA cis-elements (29). ELK1 was at least partially required for a substantial proportion (~ 27 percent) of all gene activation by androgen in PC cells (24). Tethering of AR by ELK1 in PC/CRPC cells enables constitutive activation of a crucial set of growth genes by AR and this is not associated with ELK1 phosphorylation and does not require the transactivation domain of ELK1 (24, 25). AR binds to ELK1 (Kd = 2×10−8 M) by utilizing the two ERK docking sites in ELK1, through its amino-terminal A/B domain, which lacks the ligand binding site. The AR docking is essential for growth as demonstrated by the dominant-negative effect of a docking site mutant of ELK1 on growth of PC cells that are insensitive to MEK inhibition (25). The A/B domain is adequate for ELK1-dependent gene activation, as are splice variants of AR (24, 25).
The goal of this study is to explore development of a new class of small molecule drugs that could suppress growth signaling by AR or its variants required by PC/CRPC with potentially decreased effects on other actions of the receptor. To accomplish this, we undertook an unbiased cell-based screen to search for small molecules that could block the association of AR with ELK1 and further derived a lead molecule from an initial hit. Here we report on this small molecule discovery including a detailed evaluation of its mechanism of action and selectivity and efficacy as an inhibitor of the growth of PC/CRPC cells and tumors.
MATERIALS AND METHODS
Cell Culture and Reagents
293FT cells were from Invitrogen. All other cell lines were from the American Type Culture Collection (Manassas, VA). As the cell lines purchased from ATCC were all expanded and frozen at low passage (< passage 5), they were not further authenticated or tested for mycoplasma. These frozen batches of cells were not maintained beyond 10 passages in the course of the experiments. All cell growth conditions have been previously described (24, 25). Antibodies sc-7305 and sc-47724 were from Santa Cruz Biotechnology (Santa Cruz, CA) and antibody ab32106 was from Abcam (Cambridge, MA). Testosterone was from Sigma-Aldrich. Lipofectamine ™ 2000 was from Thermo Scientific (product number 78410). Flavonoid compounds were from Selleckchem (S2320), BOC Sciences (480–23-9), INDOFINE Chemical Company (021111S, T-406, D-409, D-412, H-025, H-410, D-123), Extrasynthase (1223, 1362S, 1204, 1104S, 1342, 1204), Cayman Chemical (10010275, 18649), Sigma-Aldrich (CDS06791) and Sana Cruz (sc-267859). shRNAs targeting AR and ELK1 and non-targeting control shRNA in the lentiviral expression vector pLKO.1-puro were from Sigma-Aldrich. The pLVX-AR-V7 plasmid, pLVX control plasmid and plasmids for RLuc8.6-AR and Turbo635FP-AR were a kind gift from Dr.Yan Dong, Tulane University (New Orleans, LA). To construct Turbo-ELK1, AR coding sequences in Turbo635FP-AR were replaced with coding sequences for ELK1.
Generation of recombinant cell lines for high throughput screening and counter screening of small molecule libraries
The recombinant cells used for primary screening were generated from HeLa HLR cells, kindly provided by Dr. Johann Hofman (Innsbruck Medical University), which were originally designed to serve as a cell-based assay system to measure modulation of MAPK activity. HeLa HLR cells have a stably integrated minimal promoter-luciferase reporter containing five upstream Gal4 elements (Gal4-TATA-Luc) and also constitutively express a Gal4-ELK1fusion protein in which the Gal4 DNA binding domain is substituted for the ETS DNA binding domain of ELK1. We stably transduced these cells with a vector expressing the full-length AR. The full length AR was subcloned from the pCMV expression vector (Origene) into the pCDH-CMV-MCS-EF1-Puro cDNA Cloning and Expression Vector (System Biosciences) at NheI (upstream) and BamHI (downstream) sites. The lentiviral vector expressing full length AR was then packaged in lentivirus and the HeLa HLR cells were infected as described below in the sub-section ‘Lentivirus-mediated-Transduction’. After 72h of infection, 2 ug/mL of puromycin was added to the culture media to select for the transduced cells. The cells were plated at low density for colony formation (20 −40 colonies) in a 100 mm dish. Clonal cells were isolated using cloning cylinders from CORNING (Cat. #3166–8). The selected clones were further expanded and then tested for luciferase induction by testosterone. The clone that gave the greatest luciferase signal to noise ratio in response to testosterone treatment was then chosen for use in the primary screening assay for high throughput small molecule screening.
The cells generated for counter screening comprise HeLa cells stably transduced with a lentiviral plasmid construct containing a minimal promoter-luciferase reporter and an upstream androgen response element (ARE) sequence. The cells were also transduced with a lentiviral expression plasmid for the full-length AR. These constructs were made as follows. Custom synthesized PCR primers were used to amplify and clone the ARE sequence from a pG5luc plasmid construct containing an ARE sequence element (25) into the pGreenFire1TM -mCMV-EF1-Neo (Plasmid) at SpeI (upstream) and BamHI (downstream) sites. The lentiviral vector expressing ARE-luciferase reporter was then packaged in lentivirus and parental HeLa cells were infected as described below under the sub-section ‘Lentivirus-mediated-Transduction’. After 72h of infection, 400 ug/mL of Geneticin was added to the culture media to select for the transduced cells. These cells were then infected with the lentivirus containing the full length AR expression plasmid described above. After 72h of infection, 2 ug/mL of Puromycin was added to the culture media to select for the transduced cells. Clonal cells harboring both the ARE-promoter-luciferase reporter and also stably expressing AR were then isolated using cloning cylinders as described above. The selected clones were further expanded and then tested for luciferase induction by testosterone. The clone that gave the greatest luciferase signal to noise ratio in response to testosterone treatment was then chosen for use in the counter screening assay for high throughput small molecule screening. All of the plasmid constructs generated above were sent to either the Plant-Microbe Genomics Facility for DNA Sequencing at The Ohio State University (Columbus, OH) or to Genewiz (South Plainfield, NJ) to verify DNA sequences before the constructs were used in the studies.
The recombinant HeLa cells generated above were routinely grown in DMEM supplemented with 10% FBS and 100 units/ml penicillin, 100µg/ml streptomycin, 2mM L-glutamine mixture (Invitrogen) and the appropriate selection antibiotics. The antibiotics used in the culture media for the primary screening cells included 100µg/ml Hygromycin (Invitrogen) (to maintain Gal4-ELK1), 100µg /ml Geneticin (Invitrogen) (to maintain Gal4-TATA-Luc) and 2 µg /ml Puromycin (Sigma-Aldrich) (to maintain AR). The antibiotics used in the culture media for the counter screening cells included 400ug/ml Geneticin (Invitrogen) (to maintain ARE-TATA-Luc) and 2 µg /ml Puromycin (Sigma-Aldrich) (to maintain AR).
High Throughput Screening
The high throughput screening was conducted at University of Michigan’s Center for Chemical Genomics Screening facility. Recombinant primary screening cells were first depleted of hormone by growing them for 24h in media in which the serum used was heat-inactivated and charcoal-stripped. The cells were then plated in 384-well white flat bottom plates (5,000 cells/well) (Corning Product #3570) using a Multidrop (Thermo Fisher Scientific, Waltham, MA). The plates were then incubated for 24h prior to adding compounds. The following day compounds from the LOPAC, Prestwick, or Maybridge Hitfinder libraries were added using a high-density replication (pintool) in a 0.2 µL volume in the test wells using a Biomek FX liquid handler (Beckman Coutler, Indianapolis, IN) to achieve final media concentration of 10 µM of each compound. Using the Multidrop (ThermoLab Systems, Helsinki, Findland), testosterone was added in addition to the compounds to achieve a final media concentration of 10 nM. As the compounds were re-constituted from powder stocks using dimethyl sulfoxide (DMSO) as the solvent, the final media concentration of DMSO was 0.4% v/v. For the assay negative control on each plate, one row of wells on each plate contained 10 nM testosterone and 0.4% v/v of DMSO. For the assay positive control on each plate, one row of wells on each plate contained 10 nM testosterone and 10 uM enzalutamide dissolved in DMSO (0.4% v/v of DMSO in the wells). The plates were incubated for 24h at 37°C in 5% CO2. The medium was then aspirated leaving a residual volume of 10µl using an ELx 405-plate washer (BioTek U.S.). Then, 10 uL of the assay reagent Bright-Glo (Promega Corp., Madison WI) was added to each well. Luciferase activities in the wells were then measured using a PHEROStar plate reader (BMG Labtech, Ortenburg, Germany). A total of 18,270 compounds were tested in the primary screen. A ‘hit’ was initially defined using relatively low stringency criteria as a compound able to reduce luciferase activity in the test well ≥ 3 standard deviations below the negative control wells or to a level ≥ 40% of the enzalutamide control wells. For the primary assay this definition produced 1613 ”hits” for an overall hit rate of 8.8%. The 1613 compounds were then tested again in the primary screening assay in parallel with the counter screening assay in triplicate at 10uM, with compound additions to plates using Mosquito X1 (Hertfordshire, UK). A hit was defined as a compound able to reduce luciferase activity in the test wells ≥ 3 standard deviations below the negative control wells and that was unable to reduce luciferase activity ≥ 50% in the counter screen. By this definition, 92 ”hits” were obtained. Compounds were further prioritized based on their ability to inhibit in the primary screen by ≥ 80% and produced no inhibition in the counter screen. One of the top hits was prioritized for this study.
BRET Assay
HEK293T cells were transfected with DNA/calcium phosphate co-precipitates. Two days post-transfection, 1 × 105 cells per well were re-seeded into white-walled, 96-well plates. After 2 h coelenterazine was added to the cells and BRET emissions were measured at 535 nm (RLuc) and 635 nm (Turbo) using a CLARIOstar (BMG Labtech) dual plate reader. For the inhibitor experiments, KCI807 or DMSO vehicle control were added to cells 24 h post-transfection. Data are presented as 635 nm/535 nm ratios normalized to the ratios obtained from cells transfected with RLuc constructs alone.
Purified Proteins
Purified human AR was purchased and recombinant his tagged ELK1 was purified as previously described (25).
Surface Plasmon Resonance
Immobilization of protein to the CM5 sensor chip by an amine coupling reaction and evaluation of binding of the analyte have previously been described in (25).
Competition binding experiments were executed on the Biacore 3000 system at a flow rate of 5–10 µl/min in HBS-N buffer (30, 31). A fixed concentration (200 nM) of AR in the presence of increasing concentrations of KCI807 or enzalutamide was passed over a covalently stabilized ELK1 sensor surface for 5 min at 50 µl min-1. The sensor surface was regenerated between experiments by dissociating any formed complex in HBS-N buffer for 30 min, followed by a further 30-min stabilization period. After regeneration, the SPR signal returned to the original level (baseline). In all cases, baseline was established in the presence of the vehicle used for the compounds (DMSO) appropriately diluted in HBS-N buffer. The binding curves were analyzed using the heterogeneous analyte competition model. The kinetic curves were analyzed for a one-to-one Langmuir fitting model provided with the Biacore 3000 instrument software.
mRNA expression profiling
mRNA expression profiling was performed at RUCDR Infinite Biologics, Piscataway, NJ using Clariom D Arrays. The samples were amplified using Ovation Pico WTA System Version 2 kits from Nugen (P/N: 3302–96) following the procedure detailed in the User Guide from an original input of 40ng of RNA. The amplification was automated on the Caliper Sciclone liquid handling workstation from Perkin Elmer (P/N: 124901). The amplified cDNA underwent a QC check using the Trinean Dropsense96 to confirm that a sufficient amount was generated and that it was of an appropriate quality to be used for the next step. Amplified cDNA was then fragmented and labeled using the Encore Biotin Module from Nugen (P/N: 4200–96) following the procedure detailed in the User Guide. Following fragmentation, 2uL of the fragmented product was used for a QC check on the Caliper LabChip GX from Perkin Elmer to confirm that the samples were adequately fragmented and were within the correct size range. Fragmented and labeled cDNA was hybridized to the Affymetrix Clariom D cartridge array (P/N: 902922) using the GeneChip Hybridization, Wash, and Stain Kit (P/N: 900720) and following the procedure detailed in the User Guide. The cartridges were placed in a rotisserie hybridization oven at 45°C for 16 h to hybridize. The hybridized cartridges were then washed and stained on the GeneChip Fluidics Station 450 (P/N: 00–0079) from Affymetrix following the procedure detailed in the User Guide. They were then immediately scanned on the GeneChip Scanner 3000 7G (P/N: 00–00212) from Affymetrix. After the scans were complete, the Cel data files were run through a QC check using Affymetrix Expression Console software (now part of Transcriptome Analysis Console (TAC) Software). The PM means were checked to ensure consistency across the processed samples and to meet a minimum threshold. Hybridization controls were graphed to ensure consistent hybridization within the samples. Feature intensity and probeset means were also graphed and checked.
Other in vitro methodologies
Chromatin immunoprecipitation (ChIP) assays, transfections and reporter luciferase assays, lentivirus-mediated-transduction, colony growth assays, cell monolayer growth assay, and RNA isolation, reverse transcription, and real time PCR have all been previously described in (24, 25). Customized PCR primers were designed using the Agilent Technologies’ Primer Design tool, along with the QuickChange II XL Site-Directed Mutagenesis Kit (#200521) to synthesize the AR-F876L mutant from the lentiviral vector expressing full length AR.
Determination of serum levels of KCI807 and identification of its metabolites by LC-MS/MS
Determination of KCI807 serum levels. The concentrations of KCI807 in mouse serum samples were quantitatively determined by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). In brief, 50 μL of mouse serum was acidified by adding 50 μL 1% formic acid, followed by extraction with 0.5 mL ethyl acetate. The top layer was transferred to a new tube and evaporated to dryness under a stream of nitrogen in a water bath at 37oC. The residual was reconstituted in 50 μL mobile phase, and the supernatant was subjected to the LC-MS/MS analysis. LC-MS/MS analyses were performed on an AB SCIEX (Foster City, CA) QTRAP 6500 LC-MS/MS system, which consists of a SHIMADZU (Kyoto, Japan) Nexera ultra high performance liquid chromatography system coupled with a hybrid triple quadrupole / linear ion trap mass spectrometer. Chromatographic separation was performed on a Waters Xterra C18 column (50 × 2.1 mm, 3.5µm) under a gradient elution consisting of mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile), at the flow rate of 0.3 mL/min. KCI807 was monitored using the multiple reaction monitoring mode under positive electrospray ionization. Mass spectrometer parameters were optimized to obtain the most sensitive and specific mass transitions for KCI807 by direct infusion 0.5 µM of the standard solution into the ion source with a syringe pump. The Turbo ion-spray voltage was set at 4500 V and the source temperature was set at 500 °C. Collision gas was optimized at medium level with curtain gas, ion source gas 1 and ion source gas 2 delivered at 20, 30 and 30 psi, respectively. The dwell time was set for 50 ms. KCI807 was monitored at the most sensitive and specific mass transition of m/z, 254.9>137.0. The linear calibration curve was established at KCI807 concentration range of 5 – 1000 nM in mouse serum. The intra- and inter-day precision and accuracy of the quality control samples were within the generally acceptable criteria for bioanalytical methods (< 15%).
Metabolite identification.
To identify potential metabolites of KCI807 in mice, serum and liver samples collected from the control (untreated) and KCI807-treated mice were subjected to the LC-MS/MS analysis. Sample preparation and chromatographic separation were the same as that described above. Column eluents of the control and drug-treated samples were surveyed using different scan modes including full scan, product scan, and multiple reaction monitoring scan. Due to the lack of adequate sensitivity in the full scan and product scan modes, the multiple reaction monitoring scan mode was selected to detect potential metabolites of KCI807, including those involved in methylation, oxidation, reduction, glucuronidation, glycosylation, acetylation, sulfation, as well as double-modifications (e.g., glucuronide-glucuronide, glucuronide-sulfate, glucuronide-glycosylate, and sulfate-glycosylate double conjugates). The LC-MS/MS was operated under the optimized condition as for KCI807, and hypothesized theoretical mass transitions for potential metabolites were monitored (Supplementary Tables 4 and 5). If a chromatographic peak monitored at the theoretical transition(s) corresponding to a particular metabolite was identified in the drug-treated samples but not in the control (untreated) samples, this metabolite was tentatively determined as a metabolite of KCI807 (Supplementary Figure 9)
Tumor xenograft model studies
All of the animal model studies had prior approval of The Institutional Animal Care and Use Committee (IACUC) of Wayne State University.
The 22Rv1 human CRPC xenograft model was established by bilateral subcutaneous implant (SC) of 22Rv1 cells and serial passaging of the tumors in male SCID mice. KCI807 was administered intraperitoneally (IP) in a volume of 0.2 mL/20 g mouse with the following formulation: 5% DMSO (v/v) with 0.5% NaHCO3 (v/v), in 1% carboxymethylcellulose (CMC). For preliminary dose determinations, mice were given a single dose of the compound daily x 3 days, with escalation if no symptoms were noted e.g. 100, 150 and 250 mg/kg body weight. Mice were then observed for immediate and peri-acute post-injection toxicity by monitoring weight and body condition for 7 days. As the mice were asymptomatic post all doses given, the highest dose range of 250–260 mg/kg body weight was used for anti-tumor efficacy studies of the compound. Enzalutamide was administered orally by a well-established regimen of 50mg/kg daily (32). Male SCID mice were implanted bilaterally SC with 30–50 mg tumor fragments by 12 gauge trocar, and randomly distributed to various treatment and vehicle control groups (5 mice per group). Treatment typically began 3 days post-implant to determine antitumor efficacies and to further evaluate potential cumulative toxicities. Tumors were measured with a caliper 3 times/week and tumor masses (in mg) estimated by the formula, mg = (a x b2)/2, where “a” and “b” are tumor length and width in mm, respectively. Mice were sacrificed when cumulative tumor burdens reached 5–10% of body weight (1–2g) in the control group. In a parallel experiment, groups of 5 mice were administered KCI807 as described above but were sacrificed on Days 3, 11 and 19 at 6h following the last injection to monitor plasma levels of unmetabolized KCI807.
PDX-PR011 is a prostate tumor xenograft derived from an initial bone biopsy donated by a CRPC patient at Karmanos Cancer Institute. The biopsy was implanted sc into a SCID male mouse and ultimately metastasized to the lungs. The metastatic lungs were harvested and implanted into a fresh mouse, which then subsequently formed a sc tumor. The tumor xenograft retained AR expression (Supplementary Figure 14c). Tumor implantation in mice (5 mice per group), the KCI807 treatment regimen and tumor growth measurements were conducted as described above with the exception that treatment with KCI807 was initiated within 1 day of tumor implantation because of the relatively more aggressive growth rate of the tumors.
Statistical methods
All of the in vitro experiments were repeated at least three times. The error bars in all graphs represent the standard deviation, unless otherwise stated. Statistical analysis was performed using two-sample t-test or one-way ANOVA with post-hoc LSD (Least Square Differences).
In the gene expression analysis using the mRNA profiling data, the differentially expressed genes were identified using the following cutoff definitions: 2-fold change and unadjusted p-value (by a 2-sided two-sample t-test) < 0.05. These genes were analyzed by Venn diagram and the hypergeometric test was used to assess whether the intersection of separately identified genes with elevated expression was statistically significant. The canonical pathway analyses were further carried out for these genes by IPA (Ingenuity Pathway Analysis, http://www.ingenuity.com) with the following parameters: genes only reference set, direct and indirect relationships, experimentally observed confidence, and human species. The detected canonical pathways were statistically tested by a hypergeometric test at a 5% significance level and the ratios of molecules present in the dataset out of all the function related molecules were calculated for each of detected canonical pathways.
In the mouse xenograft studies, a total of 5 mice were used for each of three treatment groups, resulting in a total of 15 mice, and each mouse had two tumors, implanted in their left and right flanks. The tumor growth rates were statistically compared among five treatment groups using a linear mixed-effects model and, in particular, in order to consider flank side-specific variation, the flank side was introduced as another level for each group in addition to the mouse-specific level. Non-zero tumor volumes were log-transformed to meet the normality assumption before linear mixed-effects modeling. The reported p-values were not adjusted for multiple comparisons. The tumor volume curves were depicted by median and an interval of semi-interquartile range on the basis of the raw tumor volume values.
For the drug concentration-time profile, groups of 5 mice were used for each of three time points at Day 3, 11, and 19, resulting in a total of 15 mice, and the measurements of drug concentrations at each time point were repeated 4 times. The comparisons between two time points (Day 3 vs. Day 11 and Day 11 vs. Day 19) were performed using a linear mixed-effects model by considering within- and between-mouse variations. All concentrations were log-transformed before linear mixed-effects modeling and the p-values were adjusted by Bonferroni correction. The drug concentration-time profile was depicted by median and an interval of semi-interquartile range on the basis of the raw concentration values.
RESULTS
Discovery of the lead compound
For initial discovery of small molecules that selectively blocked the binding of ELK1 and AR, we developed a stringent and methodical system for high throughput screening (HTS) of a high diversity compound library. We expected a relatively high probability of success because we were screening for molecules that could bind at any one of a minimum of four target sites, considering the two AR docking sites on ELK1 and the corresponding binding sites in AR. Therefore, we screened a diversity library of ~ 20,000 small molecules based on experience at our facility that the primary hit rate for a single target in cell-based HTS by moderately stringent criteria is 1–2 %.
For the primary screen, we used recombinant AR+ HeLa cells harboring a TATA-dependent promoter-luciferase reporter construct with an upstream cluster of Gal4 elements (Gal4-TATA-Luc) as well as a Gal4-ELK1 fusion protein gene. When these cells are treated with testosterone AR is translocated to the nucleus where it binds to Gal4-ELK1 and activates the reporter gene. The cells for counter screening were identical to the primary screening cells with the exception that an androgen response element (ARE) sequence replaced the Gal4 elements in the promoter and Gal4-ELK1 was absent. Compounds of interest should only suppress the signal in the primary screening assay, as the only difference between these two assays is AR recruitment to the promoter via ELK1 binding vs. direct DNA binding (Illustrative schematic in Supplementary Figure 1). The Z-factor for the primary screening assay was 0.734 and for the counter screening assay it was 0.711. In both assays enzalutamide, which does not allow nuclear translocation of AR, completely suppressed the signal (Supplementary Figure 2a, 2b) and hence served as the positive control for the HTS. We initially screened two pilot sets of compound libraries, LOPAC and Prestwick and then the Maybridge Hit Finder library, which is a diversity set, at a compound concentration of 10 µM. By the criteria described under Materials and Methods, the primary screen produced 1613 hits. Elimination of false positives by counter-screening resulted in 92 compounds with variable potencies (40% - 100% inhibition) in the primary screen. A further stringent triage yielded 15 hits from which we chose 5,7,3’,4’-tetrahydroxyflavone (Hit 1), based on potency of inhibition in the primary screen (80% - 100% in < 6 hours) and virtual absence of an effect in the counter screen (Figure 1a). This hit was also prioritized for further studies because it belongs to a large class of well-studied natural products and could potentially be introduced relatively rapidly in the clinic in an appropriately modified form.
Figure 1: Scaffold specificity and selectivity of Hit1 (5,7,3’,4’-Tetrahydroxyflavone) as an inhibitor of ELK1-dependent promoter activation by AR.

Dose response curve for (a) 5,7,3’,4’-Tetrahydroxyflavone (b) 5,7,3’,4’-Tetrahydroxyisoflavone (c) 5,7,3’,4’-Tetrahydroxyflavanone for inhibition of promoter activation by testosterone in the primary screening assay (ELK1-dependent promoter activation by AR) compared with the counter screening assay (ARE-dependent promoter activation by AR). The cells were simultaneously treated with compound and testosterone for 6h and promoter activity was measured as reporter luciferase activity.
Hit 1 is itself highly unstable (easily oxidized) in vitro and rapidly metabolized in vivo because of its multiple phenolic hydroxyl groups including hydroxyls on adjacent carbon atoms (33, 34); hence its reported anti-inflammatory, neuroprotective and other physiological effects (35, 36) may be related to its metabolites rather than its original structure. In order to identify the essential structural elements required for selective activity against the ELK1-AR complex, we conducted structure-activity analysis using the same in vitro assay as in the primary screening. First, we tested the effect of substituting the flavone scaffold in Hit 1 with the closely related flavanone and isoflavone scaffolds. Hit 1 was unable to affect either ELK1-dependent or ARE-dependent promoter activation by AR upon scaffold substitution (Figure 1b, 1c). Further structure-activity analysis, using derivatives of Hit 1 with individual or combinatorial substitution of hydroxyl groups by hydrogen, indicated that in the A ring, only the 5-hydroxyl group is necessary for activity but by itself it confers weak activity at best (Table 1). A second hydroxyl substitution on the B ring enhances activity, optimally at the 3’ position and sub-optimally at the 4’ position (Table 1). Simultaneous substitution of all four hydroxyl groups with methoxy groups or substitution at the 3’ position alone with a methoxy group abolished activity (Table 1). Further, methoxy substitutions on carbons at positions 6, 7 and 4’ were moderately tolerated (Table 1). Additionally, methoxy substitution on the carbon at position 3 was not tolerated (Table 1), predicting possible steric hindrance from any bulky substitutions at this position.
Table 1:
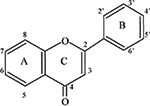
Structure-activity analysis of compound Hit 1
| Structurea | Flavonoid | –R3 | –R5 | –R6 | –R7 | –R3’ | –R4’ | IC50(μM)b |
|---|---|---|---|---|---|---|---|---|
 |
5,7,3’,4’-Tetrahydroxyflavone | H | OH | H | OH | OH | OH | 1.01 |
| 5,3’,4’-Trihydroxyflavone | H | OH | H | H | OH | OH | 1.01 | |
| 5,7,4’-Trihydroxyflavone | H | OH | H | OH | H | OH | No effect | |
| 7,3’,4’-Trihydroxyflavone | H | H | H | OH | OH | OH | No effect | |
| 5.7-Dihydroxyflavone | H | OH | H | OH | H | H | No effect | |
| 3’.4’-Dihydroxyflavone | H | H | H | H | OH | OH | No effect | |
| 7,3’-Dihydroxyflavone | H | H | H | OH | OH | H | No effect | |
| 5,4’-Dihydroxyflavone | H | OH | H | H | H | OH | 5.48 | |
| 5,3’-Dihydroxyflavone | H | OH | H | H | OH | H | 0.53 | |
| 7,4’-Dihydroxyflavone | H | H | H | OH | H | OH | No effect | |
| 5-Hydroxyflavone | H | OH | H | H | H | H | >10 | |
| 3’-Hydroxyflavone | H | H | H | H | OH | H | No effect | |
| 5,3’-Dihydroxy-6,7,4’-trimethoxyflavone | H | OH | OCH3 | OCH3 | OH | OCH3 | 4.17 | |
| 5,7,4’-Trihydroxy-3’-methoxyflavone | H | OH | H | OH | OCH3 | OH | No effect | |
| 5,7,3’-Trihydroxy-4’-methoxyflavone | H | OH | H | OH | OH | OCH3 | 1.37 | |
| 5,7,3’,4’-Tetrahydroxy-3-methoxyflavone | OCH3 | OH | H | OH | OH | OH | No effect | |
| 5,7,3’,4’-Tetramethoxyflavone | H | OCH3 | H | OCH3 | OCH3 | OCH3 | No effect |
R8, R2’, R5’ and R6’ comprise H in all cases
IC50 for inhibition of ELK1-dependent promoter activation by AR. The primary screening assay (ELK1-dependent promoter activation by AR) was used to determine the IC50 values for Hit1 and its various derivatives, using a compound dose range of 1-10μM.
As a secondary test of target selectivity, we examined the effect of 5,3’-dihydroxyflavone on activation of ELK1 by MEK/ERK using HeLa cells co-transfected with the Gal4-TATA-Luc construct, the expression plasmid for the Gal4-ELK1 fusion protein and either an expression plasmid for a constitutively active MEK1 protein (caMEK1) or vector control. Activation of the luciferase reporter by MEK1 was completely inhibited by the MEK inhibitor trametinib but 5,3’-dihydroxyflavone did not inhibit the promoter activation (Supplementary Figure 3).
To conclude, 5,3’-dihydroxyflavone, which is predictably more stable than Hit 1, is the minimal structure that is fully and selectively active against the target ELK1-AR interaction. We name this lead compound KCI807.
Disruption of the ELK1-AR binary complex and selective inhibition of chromatin recruitment by binding of KCI807 to AR
We also developed a BRET assay of the association of ELK1 with AR to demonstrate the ability of KCI807 to inhibit the in situ association of AR with ELK1 (Supplementary Figure 4). BRET coupling between ELK1 and AR fusions (Supplementary Figures 4a and 4b) expressed in HEK293T cells increased hyperbolically with increasing acceptor/donor ratios, indicative of specific interactions between AR and ELK1 (Supplementary Figure 4c). Pre-incubation of cells with KCI807 inhibited BRET coupling between ELK1 and AR (Supplementary Figure 4d). These data further support the inhibitory mechanism proposed for KCI807.
Next, it was determined by surface plasmon resonance (SPR) that KCI807 binds to purified immobilized AR with a dissociation constant of 7×10−8 M (Figure 2a). In contrast, KCI807 did not bind to immobilized ELK1 (Figure 2b). The functional relevance of the binding data is also supported by the fact that the binding of AR to either 5-hydroxyflavone or 3’-hydroxyflavone was extremely weak (with dissociation constants of 5.9×10−6 M and 1.43×10−6 M, respectively) (Supplementary Figure 5), consistent with their weak or absent activity compared to KCI807 in Table 1. SPR analysis further demonstrated that KCI807 blocked binding of purified AR (used as analyte) to purified immobilized ELK1 progressively with increasing molar ratios relative to AR (Figure 2b). In contrast, although SPR analysis could demonstrate the binding of enzalutamide to AR with a dissociation constant of 1.7×10−9 M (Figure 2c), enzalutamide was unable to block the binding of AR to ELK1 (Figure 2d).
Figure 2: Binding of KCI807 to AR and disruption of its binary and chromatin complexes with ELK1.

(a) Surface plasmon resonance (SPR) kinetic curves for quantitative analyses of KCI807 binding to AR. AR was immobilized and KCI807 (analyte) was diluted in a series (0, 10, 20, 40, 80, 160, and 320nM). The calculated kon,koff and Kd values are 3.67 × 104 M−1 s−1 , 2.54 × 10−3 s−1 and 6.92 × 10−8 M respectively. (b) SPR kinetic curves for quantitative analysis of inhibition of binding of AR (used as analyte) to immobilized ELK1 by KCI807. A fixed concentration of AR (200 nM) was combined with KCI807 diluted in a series (0, 50, 100, 200, 400, 800, and 1600 nM). (c) SPR kinetic curves for quantitative analysis of enzalutamide binding to AR. AR was immobilized and enzalutamide (analyte) was diluted in a series (0, 6.25, 12.5, 25, 50, 100, 200nM). The calculated kon koff and Kd values are 3.4 × 105 M−1 s−1, 5.8 × 10−4 s−1 and 1.70 × 10−9 M respectively. (d) SPR kinetic curves to test for inhibition of binding of AR (used as analyte) to immobilized ELK1 by enzalutamide. AR (200 nM) was combined with enzalutamide (0 uM or 1 uM). (e and f) LNCaP cells plated in charcoal stripped serum were treated with R1881 (10nM), R1881 (10nM) + KCI807 (20uM), or vehicle for 2h. The cells were fixed and subjected to the chromatin immunoprecipitation (ChIP) assay using antibody to AR. ChIP signals from established sites of AR recruitment by ELK1 (e) or from canonical ARE enhancer sites associated with the KLK3 and TMPRSS2 genes (f) are plotted as percent of input DNA. In e and f, genomic coordinates are shown for the hg18 human genome.
We then tested the ability of KCI807 to selectively block recruitment of AR by ELK1 to chromatin in situ. Chromatin immunoprecipitation (ChIP) assays showed that in LNCaP prostate cancer cells, KCI807 prevented association of AR with previously (24) established sites in the chromatin at which ELK1 recruits AR (Figure 2e). In contrast, KCI807 did not impede AR recruitment at the well-established canonical ARE enhancer sites associated with the KLK3 (PSA) and TMPRSS2 genes (Figure 2f).
The set of complementary results described above establish that KCI807 directly binds to AR and selectively blocks its physical association with ELK1, inhibiting ELK1-dependent transcriptional activity of AR.
Narrow genotropic effects of KCI807 in CRPC cells
KCI807 also inhibited hormone-independent promoter activation by the splice variant AR-V7 (Supplementary Figure 6). The relatively higher concentrations of the compound required for this inhibition is likely because AR-V7 was highly overexpressed in the assay system.
To test the selectivity of KCI807 for ELK1-dependent gene activation by AR vs. other target genes of the receptor, we examined 22Rv1 CRPC cells, which are dependent on both full-length AR and AR-V7 for hormone-independent growth. In these cells, depletion of full length AR using lentiviral shRNA (Figure 3a, left panels) led to reduction in mRNA levels of representative AR target genes previously (24) shown to be activated by AR in either an ELK1-dependent manner (Figure 3b, genes left of dashed line) or ELK1-independent manner (Figure 3b, genes right of dashed line). Depletion of ELK1 using lentiviral shRNA (Figure 3a, right panels) caused reduction only in the mRNAs for genes previously reported to be ELK1-dependent for regulation by AR (Figure 3c, genes left of dashed line). Treatment of the cells with KCI807 decreased expression of only the genes known to be supported synergistically by ELK1 and AR (Figure 3d, genes left of dashed line). Similar results were also obtained for representative genes using LNCaP and VCaP cells (Supplementary Figure 7).
Figure 3: Transcriptional targets of KCI807.

(a) 22Rv1 cells were infected with lentivirus expressing shRNA selective for full length AR or ELK1 shRNA or control shRNA. The cell lysates were analyzed by western blot to confirm knockdown of full length AR (top left panel) or ELK1 (top right panel). Real time RT-PCR was used to confirm depletion of mRNA for full length AR (bottom left panel) or ELK1 mRNA (bottom right panel). (b) In the 22Rv1 cells in which full length AR was depleted, the mRNAs for the indicated panel of genes were measured by real time RT-PCR. (c) In the 22Rv1 cells in which ELK1 was depleted, the mRNAs for the same panel of genes were measured by real time RT-PCR. (d) 22Rv1 cells were treated with 10 µM of KCI807 for 72h and real time RT-PCR was used to measure mRNAs for the indicated panel of genes tested in b and c. Genes on the left of the dashed line in panels b-d are those that require ELK1 for activation by AR and those on the right of the dashed line are ELK1-independent AR target genes. (e) Venn diagram showing overlaps among genes showing ≥ 2 fold elevated expression in the control treated 22Rv1 cells compared to cells treated with AR shRNA, ELK1shRNA or KCI807 (10 uM). The p value was based on the hypergeometric test.
To examine the global genotropic effects of KCI807 in 22Rv1 cells, Affymetrix DNA microarray analysis was used for comparative transcriptome profiling of decreases in mRNA levels due to drug treatment, depletion of AR or depletion of ELK1 (Venn diagram in Figure 3e, annotated gene lists provided in Supplementary Tables 1–3). Consistent with our published data on other PC cells (24), AR was at least partially dependent on ELK1 for activation of about a third of its target genes and these genes were functionally strongly and primarily enriched for cell cycle progression and mitosis (Supplementary Figure 8a, 8b). The AR-ELK1 activated genes inhibited by KCI807 were similarly primarily enriched for cell proliferation genes (Supplementary Figure 8a, 8c) although the compound did affect a smaller subset of ELK1-independent AR target genes with weak functional clusters (Supplementary Figure 8a, 8d), possibly by interfering with binding of AR to one or more unidentified proteins. The limited number of genes affected by KCI807 that were not activated by AR did not show ontological clustering of high significance (Supplementary Figure 8a, 8e).
The above results comprehensively demonstrate that the target gene set of KCI807 is principally associated with functional roles in cell cycle progression and mitosis within the AR signaling axis, and this functional clustering is virtually exclusively associated with synergistic activation by ELK1 and AR.
Selective in vitro growth inhibition by KCI807 and comparison with enzalutamide
KCI807 inhibited both androgen-dependent and androgen-independent in vitro growth of standard AR-dependent PC/CRPC cell line models. After initiation of colony formation of the enzalutamide-resistant (37) 22Rv1 CRPC cells, further colony growth was virtually completely inhibited by KCI807 beyond 125nM compound, with an IC50 of 33.12 nM (Figure 4a and 4b). With respect to dose response of the cell growth inhibition by the MTT viability assay, KCI807 was more effective than enzalutamide in 22Rv1 (androgen-independent cells) (Figure 4c and 4d) and LNCaP (androgen-dependent cells) (Figure 4e and 4f) cells; moreover, in these cell lines KCI807 completely inhibited cell growth whereas enzalutamide only showed partial effects even at a concentration of 20 uM (Figure 4c – 4f). VCaP cells (androgen-dependent) were the most sensitive to KCI807 as well as enzalutamide at comparable doses (Figure 4g and 4h). KCI807 but not enzalutamide inhibited the growth of LNCaP cells transfected with the F876L mutant of AR, which is known to confer enzalutamide resistance (Supplementary Figure 9). Among the above PC/CRPC cell lines, in no case did KCI807 treatment alter the protein level of AR or AR-V7 (Supplementary Figure 10). KCI807 did not appreciably affect the growth of AR-negative cell lines including DU145 (prostate cancer cells), HeLa (cervical cancer cells), HEK293 (adenovirus transformed kidney fibroblasts) and H1650 (lung adenocarcinoma cells) (Supplementary Figure 11). As an additional control, tetrahydroxyisoflavone and tetrahydroxyflavonone, both of which deviate minimally in structure from KCI807, did not inhibit growth of 22Rv1, LNCaP or VCaP cells (Supplementary Figure 12), confirming the specific compound structure dependence for PC cell growth inhibition by KCI807.
Figure 4: Inhibition of AR-dependent PC/CRPC clonogenic survival and cell growth by KCI807 and comparison with enzalutamide.

(a) 22Rv1 cells were seeded in phenol red-free growth media. Treatment with the indicated concentrations of KCI807 began 48h later, replenishing the treatments every 48h. Colonies were stained with crystal violet 10 days after plating. Treatments were conducted in triplicate. Representative images are shown. (b) Data from replicate colony formation assays described in Panel a are plotted as a histogram. Each bar represents the average number of colonies for each triplicate treatment. (c-h) The growth inhibitory effects of KCI807 were simultaneously compared with the effect of enzalutamide using the MTT assay. Twenty four hours after plating the cells, they were treated with the indicated concentrations of each compound. The data are shown in the following order: 22Rv1 Cells + KCI807 (c) 22Rv1 Cells + enzalutamide (d) LNCaP Cells + KCI807 (e) LNCaP Cells + enzalutamide (f) VCaP cells + KCI807 (g) VCaP cells + enzalutamide (h).
The growth inhibitory effect of KCI807 is thus selective for PC cells that are dependent on AR and/or AR-V7 or mutant AR. Further, this compound shows a better growth inhibitory profile than enzalutamide in well-established prostate cancer cell line models without decreasing the expression level of AR or AR-V7.
Suppression of CRPC growth in vivo by KCI807
The in vivo anti-tumor efficacy of KCI807 was tested using two types of model tumor xenografts in male SCID mice. The first was generated from the enzalutamide-resistant 22Rv1 CRPC cell line and the second was a patient-derived tumor from an enzalutamide-resistant bone metastatic CRPC (patient-derived tumor xenograft, PDX-PR011). The tumors were xenografted on day zero.
In the case of the 22Rv1 CRPC xenograft model, treatment was initiated on day 3 (representing early stage disease where implanted tumors have established blood flow) when either KCI807 was administered intraperitoneally (IP) at a dose of 250–300 mg/kg, every other day or enzalutamide was given orally (PO) at 50–60 mg/kg, daily on a standard regimen (38). The experiment was terminated when the tumor burden in the placebo group reached 5–10% of body weight. Tumor growth was inhibited by KCI807 but not by enzalutamide (Figure 5a). A delayed recovery of tumor growth in mice treated with KCI807 followed a sharp decline in plasma level of unmetabolized compound (median 70 × 10−9 M on Day 19, compared to 3.87 × 10−7 M on Day 11) suggesting self-induced enhancement of clearance rate of the compound in the rodent model during the chronic dosing schedule of the compound. The major plasma metabolites of KCI807 comprised glucuronidation and other modifications at the 3’ position (Supplementary Figure 13). The mice treated with KCI807 were asymptomatic for the duration of the study and did not show appreciable weight loss (Supplementary Figure 14a).
Figure 5: Inhibition of in vivo CRPC tumor growth by KCI807.

(a) Tumor xenografts of 22Rv1 cells were implanted sc into male SCID mice (5 mice per group) in both flanks. Drugs were administered beginning on Day 3 of implantation. The treatments include 250mg/Kg of KCI807 injected intraperitoneally, on alternate days or vehicle control. In parallel, groups of 5 mice similarly administered KCI807 were sacrificed precisely 6h after the last injection of KCI807 on Day 3, Day 11 and Day 19 for analysis of plasma levels of unmetabolized KCI807. For comparison of anti-tumor efficacy, enzalutamide was administered to a separate group of mice following the standard regimen of daily oral administration of 50mg/kg. Tumor volume curves and plasma concentration-time profiles are plotted as median and an interval of semi-interquartile range on the basis of their raw values. The p-values were obtained using a linear mixed-effects model after a log-transformation of the raw values. (b) PDX-PR011 tumor xenografts were implanted sc into male SCID mice (5 mice per group) in both flanks. The vehicle control and KCI807 were administered as described for Panel a, with the exception that treatment was begun on Day 1 after tumor implantation. Tumor volumes were monitored as described above.
In the case of the patient-derived PDX-PR011 tumor xenograft, treatment was initiated one day after tumor implantation because of the extremely aggressive growth of this tumor, which required termination of the experiment within 12 days due to the heavy tumor burden in the control mice. Despite such aggressive tumor growth and despite the self-induced clearance of KCI807 in rodents noted above, tumor growth was significantly inhibited by KCI 807 (Figure 5b). Again, the treated mice were asymptomatic and did not show appreciable weight loss (Supplementary Figure 14b). As expected, the PDX-PR011 tumor xenograft was insensitive to enzalutamide (Supplementary Figure 15).
DISCUSSION
Current and experimental modalities of therapeutic targeting of the AR signaling axis in prostate cancer, including suppression or inhibition of testosterone synthesis and antagonists that bind to either the ligand binding pocket of AR or to the amino-terminal hormone-independent activation functions of AR, entail global disruption of androgen/AR actions (39). This study supports the concept that a critical growth signaling arm of AR could be selectively disrupted by small molecules in a manner that would not only spare most other transcriptional activities of AR but also overcome mechanisms that restore AR function, causing resistance to current treatments. Using an unbiased HTS approach combined with SAR studies, we have discovered and characterized a flavonoid molecule (KCI807) that directly binds to AR, blocks binding of ELK1 to AR and also prevents recruitment of AR to chromatin by ELK1. As a result, KCI807 selectively inhibits transcriptional activity of AR mediated by ELK1 binding DNA elements vs. canonical AREs. This is reflected by selective inhibition by KCI807 of ELK1-dependent activation of endogenous genes by AR. Target genes of ELK1-dependent transcriptional activation by AR are critical for cell proliferation with gene ontology analysis showing enrichment primarily for cell cycle and mitosis genes, entirely consistent with previous reports on the androgen-independent transcriptional program supported by AR and AR-V7 (40, 41). KCI807 does not inhibit all the ELK1-dependent target genes of AR and this is theoretically attributable to promoter context-dependent proteins associated with the ELK1-AR complex. Nevertheless, the compound does inhibit a large ELK1-AR target gene set that is primarily enriched for cell cycle progression and mitosis genes. Accordingly, KCI807 selectively inhibits AR-dependent prostate cancer cell growth in vitro and tumor growth in vivo including both cell line-derived and primary patient-derived tumors that are enzalutamide resistant. As may be expected from the fact that the A/B domain of AR is entirely adequate for the role of AR as a co-activator of ELK1, KCI807 is also able to inhibit ELK1-dependent transcriptional activity of AR-V7 and growth of AR-V7 expressing prostate cancer cells and tumors that are enzalutamide resistant by virtue of AR-V7 overexpression. As the A/B domain of AR alone was previously shown to be capable of activating ELK1-dependent genes and partially supporting growth of LNCaP cells (25), the effect of KCI807 on growth signaling by wtAR may therefore also extend to AR-V7. KCI807 also inhibits PC cell growth supported by the F876L mutant of AR which is insensitive to enzalutamide. KCI807 does not affect the expression of either AR or AR-V7 proteins. The structure of KCI807 only retains the minimal features of the initial hit from our HTS that are required for optimally disrupting the functional interaction of AR with ELK1, i.e., a flavone scaffold and two hydroxyl groups at specific positions (5 and 3’). Comparative SPR binding data on the mono-hydroxylated derivatives demonstrates that the 5 and 3’ hydroxyl groups are both essential for optimal binding of KCI807 to AR. Notably, KCI807 lacks hydroxyl groups at positions 7 and 4’ that destabilize its parent compound.
Similar to other nuclear receptors, including steroid receptors, the amino-terminal domain of AR is intrinsically disordered (42). This is a major obstacle for structure-based design of small molecule drugs targeting functional motifs within this region of the receptor. Nevertheless, it has been previously demonstrated by the empirical approach of small molecule screening and structure-activity studies that small molecules including certain bis-phenols (EPI) (43) and Sintokamide A (44) can bind selectively to this domain of AR with profound inhibitory effects on coactivator binding. However, as ARE-mediated transcriptional activity of AR is unaffected by KCI807, this compound clearly does not interfere with coactivator binding to AR. For binding to ELK1, the amino-terminal domain of AR only requires the two ERK docking sites in ELK1 (25). KCI807 binds to AR with a dissociation constant that is only ~ 3 times that for the binding of ELK1 to AR (25) possibly by binding to a substructure required for the formation of the recognition site for one of the two AR docking sites on ELK1. Nevertheless, KCI807 did not interfere with the activation of ELK1 by ERK. Indeed, it has not been possible to identify structures within the amino-terminal region of AR that are similar to the two ELK1 docking site recognition sites in ERK previously identified using substrate peptides for hydrogen exchange mass spectrometry and X-ray crystallography (45, 46). Therefore, there appears to be some degree of conformational flexibility that enables AR to bind to ELK1. By extension of this principle, it may be possible to identify other small molecules that selectively disrupt the AR-ELK1 complex by binding to other sites on AR or to either one of its two docking sites on ELK1.
From pharmacological and clinical perspectives, the results of studies of KCI807 establish that the ELK1-AR interaction is a drugable target that is highly desirable. Notwithstanding its ability to disrupt activation of critical growth genes supported by the ELK-AR synergy, there appears to be a possibility that KCI807 may affect a minor subset of AR target genes by interfering with binding of AR to a yet unidentified protein(s), although this would likely have a much narrower range of effects than systemic testosterone suppression. Indeed this and other off-target genotropic effects of KCI807 did not reveal strong ontological trends. These results are consistent with the fact that ELK1 primarily associates with growth genes and we currently do not have any evidence of a role for ELK1 in AR function in normal differentiated tissues. Nevertheless, the relative impact of this mode of targeting AR signaling on non-growth related functions of AR in various normal tissues can only be evaluated through future clinical studies. KCI807 showed a better inhibition profile of PC/CRPC cell growth in vitro when compared with enzalutamide in the same dose range using standard cell line models and also inhibited growth of PC cells resistant to enzalutamide by virtue of expression of AR-V7 or AR F876L. KCI807 did not appreciably affect the growth of a variety of non-malignant and malignant AR-negative cell lines and showed no apparent toxicity in mouse tumor xenograft models while inhibiting the growth of aggressively growing, enzalutamide-resistant and AR-V7 positive tumors and also aggressively growing enzalutamide-resistant patient-derived tumors. Regrowth of the tumors coincided with apparently drug-induced clearance of KCI807, demonstrating reversibility of tumor growth inhibition by KCI807, consistent with selective targeting of growth signaling rather than differentiation or survival pathways. The delayed clearance mechanisms for KCI807 induced in the rodent model appear to be centered on modification of the 3’ hydroxyl group. This self-induced metabolism of the compound may be host-species related but in any event, it could be addressed in humans through the development of next generation molecules and/or optimal treatment regimens. Additionally, future drug development efforts could explore other small molecules targeting the ELK1-AR complex by binding to either AR or to ELK1.
In conclusion, our studies establish a new mechanism-based paradigm for treatment of PC by demonstrating that the ELK1-dependent arm of androgen/AR signaling is a drugable and potentially functionally tumor selective target. This mode of targeting AR is likely to be effective in the spectrum of prostate tumors including CRPC that is resistant to enzalutamide and abiraterone and may obviate the need for systemic testosterone suppression.
Supplementary Material
Intellectual Property: Candidate drug molecules described in this report and their derivatives as well as the drug discovery methods described herein are covered by pending patents to Wayne State University.
Translational Relevance
The mainstay treatment for advanced prostate cancer is testosterone suppression via castration and the use of new generation androgen antagonists. These treatments have devastating side effects due to deprivation of testosterone or functional androgen receptor in a variety of normal tissues, severely compromising quality of life. Moreover, the tumors typically become resistant to the treatment by restoring androgen receptor function through different mechanisms including expression of androgen receptor splice variants and mutants. This manuscript establishes a novel mechanistic paradigm that systematically addresses both these problems by means of small molecule drug discovery. The studies report on the anti-tumor efficacy and target selectivity of a lead compound, KCI807. This compound represents a promising class of small molecules for drug development to suppress tumor growth in the spectrum of prostate cancer without the need for testosterone suppression.
ACKNOWLEDGEMENTS
We thank the Biospecimen Core at Karmanos Cancer Institute and Dr. Elisabeth Heath for sharing the prostate cancer PDX model. We also thank Dr. Sijana Dzinic and Juiwanna Kushner for assistance with the animal model experiments. We are grateful to Curtis Krier at RUCDR Infinite Biologics for his help in facilitating the Affymetrix DNA microarray studies.
Financial Support: This work was supported by DoD grant W81XWH-17–1-0242 to M.R., and W81XWH-17–1-0243 to P.S., NIH 5T32CA009531–29 NRSA T32 Fellowships to R.R. and T.M
Footnotes
The authors declare that they have no conflict of interest
REFERENCES
- 1.Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(12):3876–83. [DOI] [PubMed] [Google Scholar]
- 2.Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(27):3651–8. [DOI] [PubMed] [Google Scholar]
- 3.Dunn MW, Kazer MW. Prostate cancer overview. Seminars in oncology nursing. 2011;27(4):241–50. [DOI] [PubMed] [Google Scholar]
- 4.Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(12):1596–605. [DOI] [PubMed] [Google Scholar]
- 5.Yin L, Hu Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nature reviews Urology. 2014;11(1):32–42. [DOI] [PubMed] [Google Scholar]
- 6.Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Molecular endocrinology (Baltimore, Md). 2011;25(6):897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. The Journal of biological chemistry. 2012;287(23):19736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5(6):1646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sprenger CC, Plymate SR. The link between androgen receptor splice variants and castration-resistant prostate cancer. Hormones & cancer. 2014;5(4):207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myklak K, Wilson S. An update on the changing indications for androgen deprivation therapy for prostate cancer. Prostate cancer. 2011;2011:419174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holzbeierlein JM, McLaughlin MD, Thrasher JB. Complications of androgen deprivation therapy for prostate cancer. Current opinion in urology. 2004;14(3):177–83. [DOI] [PubMed] [Google Scholar]
- 12.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakazawa M, Antonarakis ES, Luo J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Hormones & cancer. 2014;5(5):265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. The New England journal of medicine. 2010;363(5):411–22. [DOI] [PubMed] [Google Scholar]
- 16.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–54. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson S, Franzen L, Parker C, Tyrrell C, Blom R, Tennvall J, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. The lancet oncology. 2007;8(7):587–94. [DOI] [PubMed] [Google Scholar]
- 18.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. The Journal of clinical investigation. 2013;123(7):2948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayward SW, Cunha GR. The prostate: development and physiology. Radiologic clinics of North America. 2000;38(1):1–14. [DOI] [PubMed] [Google Scholar]
- 20.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465–74. [DOI] [PubMed] [Google Scholar]
- 21.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonit M, Zhang J, Salazar M, Cui H, Shatnawi A, Trumbly R, et al. Hormone depletion-insensitivity of prostate cancer cells is supported by the AR without binding to classical response elements. Molecular endocrinology (Baltimore, Md). 2011;25(4):621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Gonit M, Salazar MD, Shatnawi A, Shemshedini L, Trumbly R, et al. C/EBPalpha redirects androgen receptor signaling through a unique bimodal interaction. Oncogene. 2010;29(5):723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patki M, Chari V, Sivakumaran S, Gonit M, Trumbly R, Ratnam M. The ETS domain transcription factor ELK1 directs a critical component of growth signaling by the androgen receptor in prostate cancer cells. The Journal of biological chemistry. 2013;288(16):11047–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosati R, Patki M, Chari V, Dakshnamurthy S, McFall T, Saxton J, et al. The Amino-terminal Domain of the Androgen Receptor Co-opts Extracellular Signal-regulated Kinase (ERK) Docking Sites in ELK1 Protein to Induce Sustained Gene Activation That Supports Prostate Cancer Cell Growth. Journal of Biological Chemistry. 2016;291(50):25983-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaw PE, Saxton J. Ternary complex factors: prime nuclear targets for mitogen-activated protein kinases. The international journal of biochemistry & cell biology. 2003;35(8):1210–26. [DOI] [PubMed] [Google Scholar]
- 27.Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358(6385):414–7. [DOI] [PubMed] [Google Scholar]
- 28.Zhang HM, Li L, Papadopoulou N, Hodgson G, Evans E, Galbraith M, et al. Mitogen-induced recruitment of ERK and MSK to SRE promoter complexes by ternary complex factor Elk-1. Nucleic acids research. 2008;36(8):2594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer cell. 2010;17(5):443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, Li X, Guo L-H. Development of a label-free competitive ligand binding assay with human serum albumin on a molecularly engineered surface plasmon resonance sensor chip. Analytical Methods. 2012;4(11):3718–23. [Google Scholar]
- 31.Bahloul A, Michel V, Hardelin JP, Nouaille S, Hoos S, Houdusse A, et al. Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Human molecular genetics. 2010;19(18):3557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guerrero J, Alfaro IE, Gomez F, Protter AA, Bernales S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. The Prostate. 2013;73(12):1291–305. [DOI] [PubMed] [Google Scholar]
- 33.Gao S, Hu M. Bioavailability challenges associated with development of anti-cancer phenolics. Mini reviews in medicinal chemistry. 2010;10(6):550–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramešová Š, Sokolová R, Tarábek J, Degano I. The oxidation of luteolin, the natural flavonoid dye. Electrochimica Acta. 2013;110:646–54. [Google Scholar]
- 35.Nabavi SF, Braidy N, Gortzi O, Sobarzo-Sanchez E, Daglia M, Skalicka-Woźniak K, et al. Luteolin as an anti-inflammatory and neuroprotective agent: a brief review. Brain research bulletin. 2015;119:1–11. [DOI] [PubMed] [Google Scholar]
- 36.Ravishankar D, Watson KA, Boateng SY, Green RJ, Greco F, Osborn HM. Exploring quercetin and luteolin derivatives as antiangiogenic agents. European journal of medicinal chemistry. 2015;97:259–74. [DOI] [PubMed] [Google Scholar]
- 37.Patki M, Huang Y, Ratnam M. Restoration of the cellular secretory milieu overrides androgen dependence of in vivo generated castration resistant prostate cancer cells overexpressing the androgen receptor. Biochemical and biophysical research communications. 2016;476(2):69–74. [DOI] [PubMed] [Google Scholar]
- 38.Zhan Y, Cao B, Qi Y, Liu S, Zhang Q, Zhou W, et al. Methylselenol prodrug enhances MDV3100 efficacy for treatment of castration-resistant prostate cancer. International journal of cancer. 2013;133(9):2225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crona DJ, Milowsky MI, Whang YE. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clinical pharmacology and therapeutics. 2015;98(6):582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research. 2012;72(14):3457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McEwan IJ. Intrinsic disorder in the androgen receptor: identification, characterisation and drugability. Molecular bioSystems. 2012;8(1):82–90. [DOI] [PubMed] [Google Scholar]
- 43.Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, et al. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(17):4466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banuelos CA, Tavakoli I, Tien AH, Caley DP, Mawji NR, Li Z, et al. Sintokamide A Is a Novel Antagonist of Androgen Receptor That Uniquely Binds Activation Function-1 in Its Amino-terminal Domain. The Journal of biological chemistry. 2016;291(42):22231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Molecular cell. 2002;9(6):1241–9. [DOI] [PubMed] [Google Scholar]
- 46.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, et al. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Molecular cell. 2004;14(1):43–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.