

Abstract
Purpose:
Head and neck squamous cell carcinoma (HNSCC), a common cancer worldwide, is etiologically associated with tobacco use, high alcohol consumption and high risk human papillomaviruses (HPV). The Notch signaling pathway, which is involved in cell differentiation decisions with differential downstream targets and effects depending on tissue type and developmental stage, has been implicated in human HNSCC. Notch1 is among the most frequently mutated genes in both HPV-positive and HPV-negative HNSCC. These mutations are predicted to inactivate the function of Notch. Other studies have argued the opposite - that Notch signaling is increased in HNSCC.
Experimental Design:
To assess the role of Notch signaling in HPV-positive and HPV-negative HNSCC, we utilized genetically engineered mouse (GEM) models for conventional keratinizing HNSCC, in which either HPV16 E6 and E7 oncoproteins or a gain of function mutant p53 are expressed, and in which we inactivated canonical Notch signaling via expression of a dominant negative form of MAML1 (DNMAML1), a required transcriptional co-activator of Notch signaling.
Results:
Loss of canonical Notch signaling increased tumorigenesis in both contexts and also caused an increase in nuclear β-catenin, a marker for increased tumorigenic potential. When combined with loss of canonical Notch signaling, HPV oncogenes led to the highest frequency of cancers overall and the largest number of poorly differentiated (high grade) cancers.
Conclusions:
These findings inform on the contribution of loss of canonical Notch signaling in head and neck carcinogenesis.
Keywords: Notch, HNSCC, Cancer, HPV, p53
Introduction
Head and Neck Squamous Cell Carcinoma (HNSCC) is one of the most common cancers in the world, accounting for approximately 600,000 cases worldwide and 50,000 in the U.S annually [1]. These cancers have been broadly placed into two groups: HPV-negative cancers, which make up approximately 75% of the cancers; and HPV-positive cancers, which account for the remaining 25% and are most commonly found in the oropharynx [2, 3]. These groups are distinct both etiologically as well as genetically: HPV-negative HNSCCs are strongly associated with a history of tobacco and alcohol use [4] and frequently contain mutations in p53 as well as other cancer-associated genes [5, 6], whereas HPV-positive HNSCCs are characterized primarily by the presence of HPV and have very few mutations in cancer associated cellular genes [5, 6]. Differences are seen at many other levels. Morphologically, most HPV-positive HNSCCs are of non-keratinizing immature type resembling tonsillar crypt epithelium in contrast to the conventional keratinizing HPV-negative HNSCCs [7], have distinct gene expression profiles from HPV-negative HNSCCs that are more similar to those of HPV-positive cervical cancers [8], and while these tumors appear to be high-grade (poorly differentiated) histologically, patients with HPV-positive HNSCCs have much-improved outcomes compared to patients with HPV-negative HNSCC [7].
Despite the dissimilarities between these groups, chemoradiation remains the standard of care for both types of HNSCC. This therapy, however, is associated with severe morbidities consequent to the location of the primary tumor and high levels of radiation used, and overall 5-year survival rates are poor (approximately 60% overall, particularly for patients with HPV-negative HNSCC [7]. Identifying factors underlying development of HNSCC as well as further differentiating HPV-positive and HPV-negative HNSCC is an important step towards preventing new cases as well as creating better and more targeted treatment methods for the cancers that arise.
Recent exome-seq studies have pointed to a potential link between HNSCC and the Notch signaling pathway [5, 6]. This pathway is highly conserved among metazoans, having first been discovered through studies in Drosophila [9]. Despite being a relatively simple signaling pathway in cells, it can have differential effects depending on the tissue context and interactions with numerous additional pathways [10]. Briefly, a ligand from a signal-sending cell (JAG 1–2 and DLL1–3 in humans) binds to a Notch receptor on an adjacent cell (NOTCH1–4 in humans). The Notch receptor is then cleaved twice, first by ADAM and then by γ-SECRETASE, leading to a release of the Notch Intracellular domain (NICD). NICD then translocates to the nucleus, associates with RBP-Jκ (Recombination Signal Binding Protein for Immunoglobulin Kappa J Region) and MAML (Mastermind-like) proteins to induce transcription of target genes. Unlike many pathways, the Notch pathway does not have an amplification step, making it highly influenced by gene dosage [11]. In the epithelium, the Notch signaling pathway is an important factor in differentiation [12]. One important way in which Notch signaling affects this is via its target protein HES1, which promotes cells beyond the basal layer to begin the first steps of differentiation also known as the basal-to-spinous switch.
The first evidence linking the Notch signaling pathway to cancer was found in acute T-cell lymphoblastic leukemia [13]. Here the pathway acted as an oncogene; Notch1 contained a truncating mutation that resulted in a constitutively active NICD. However, it can also act as a tumor suppressor in other contexts, such as in the squamous cell carcinoma of the skin [14]. Its relation to HPV-mediated disease has yet to be fully described, with conflicting evidence regarding its role [15, 16]. However, the two, aforementioned exome-seq studies independently found that NOTCH1, and to a lesser extent, other Notch receptors were mutated at a high rate in both HPV-positive and HPV-negative HNSCC [5, 6]. More recently, TCGA data by the Cancer Genome Atlas Network has also found NOTCH1 to be frequently mutated in HNSCC [17]. In contrast to the mutations in acute T-cell lymphoblastic leukemia [13], the locations of the NOTCH1 truncating and missense mutations found in these studies were primarily thought to result in an inactive protein. Because HPV encodes its own viral oncogenes, HPV-associated cancers typically have fewer mutations in cancer-related genes compared to non-HPV-associated head and neck cancers [5, 6, 18]. Therefore, the high prevalence of a particular mutated gene indicates a potentially important interplay between that gene and HPV. These observations have raised the hypothesis that Notch can act as a tumor suppressor in head and neck carcinogenesis, although some experimental studies have provided evidence to the contrary [reviewed in [19]], including several recent studies demonstrating that: a) levels of expression of components of the Notch pathway and downstream targets are increased in some HNSCC while lower in others [20]; b) the levels of NICD vary greatly in HNSCC [21]; and c) the Notch suppressor, Dtx1, is epigenetically repressed in its expression in some HNSCC [22]. Collectively, these studies indicate that Notch signaling may be oncogenic in some HNSCC and tumor suppressive in others.
We decided to test specifically the hypothesis that loss of Notch signaling increases head and neck tumorigenesis, and that its effect is greater in conjunction with HPV as opposed to other factors. Our laboratory has previously developed a mouse model for head and neck carcinogenesis in which mice are treated with the carcinogen 4-Nitroquinoline 1-oxide (4NQO) in their drinking water, inducing squamous cell carcinoma in the squamous epithelium of the tongue and esophagus [23]. 4NQO induces head and neck cancers in mice expressing the HPV16 oncogenes E6 and E7 under the Keratin 14 transcriptional promoter [23], and E6 and E7 act synergistically in this process [24]. The phenotype of SCC arising in this setting is a conventional keratinizing squamous cell carcinoma. To examine the effect of loss of Notch signaling, we inhibited the pathway by expressing a dominant-negative form of MAML1 (DNMAML1) in mice [25]. We utilized this transgene in conjunction with either HPV16 E6 and E7 or the gain-of-function mutant p53R172H, which is the mouse homolog to the R175H mutation in human p53 commonly found in HPV-negative human HNSCC, to simulate HPV-positive or HPV-negative head and neck tumorigenesis, respectively. We found that loss of Notch signaling significantly increased tumor incidence in both the HPV-positive and HPV-negative contexts at least partially through increased expression of the cancer-associated biomarker β-CATENIN and not through increased cellular proliferation. Additionally, loss of Notch signaling synergized with HPV oncogenes, but not the gain-of-function mutant p53R172H, to induce high-grade carcinomas.
Materials and Methods
Mice for HPV-positive study
K14E7 transgenic mice [26] on an FVB genetic background were crossed with K14CreERTM mice from Jackson Laboratory on a CD1 background to generate K14CreERTM/K14E7 F1 mice. K14E6 mice [27] on an FVB background were crossed with DNMAML1 mice [25] on a BL6 background to generate DNMAML1/K14E6 F1 mice. Experimental mice (nontransgenic, DNMAML1/K14CreERTM, K14E6/K14E7, DNMAML1/K14CreERTM/K14E6/K14E7) were generated through intercrosses of the aforementioned mice. All experimental mice were on the same FVB/BL6/CD1 mixed genetic background and consisted of both male and female mice.
Mice for HPV-negative study
K14CreERTM transgenic mice (Jackson Laboratory) on a CD1 genetic background were crossed with either p53+/− mice (Jackson Laboratory) on an FVB background or p53R172H/+ mice [28] on an FVB background to generate p53+/−/K14CreERTM and p53R172H/+/K14CreERTM F1 mice, respectively. These mice were crossed to either DNMAML1 [25] or nontransgenic mice on a mixed FVB/BL6 background to generate experimental mice (p53+/−, p53R172H/+/K14CreERTM, p53+/−/K14CreERTM/DNMAML1 and p53R172H/+/K14CreERTM/DNMAML1). All experimental mice were on the same FVB/BL6/CD1 mixed genetic background.
To activate the K14CreERtam transgene and express DNMAML1 and p53R172H, mice were injected intraperitoneally at 6 weeks of age with 4 mg of tamoxifen dissolved in corn oil per day for 5 days (Sigma-Aldrich).
All mice for the study were housed in the Association for Assessment of Laboratory Animal Care-approved Wisconsin Institute for Medical Research Vivarium. All procedures were in accordance with an animal protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee.
4 Nitroquinoline 1-oxide treatment and Histologic Analysis
7 days following tamoxifen treatment, mice were treated with 10 μg/mL 4-Nitroquinoline 1-oxide in their drinking water for 8 weeks as previously described [29]. Mice were then given normal drinking water for 16 weeks before being sacrificed, or until they were moribund. The tongue and esophagus were first scored for the presence of grossly visible tumor nodules before being fixed with 4% paraformaldehyde in PBS, paraffin-embedded and sectioned for histology at 10 μm intervals. Every tenth serial section was stained with hematoxylin and eosin (H&E) and subjected to histopathological analysis by a pathologist (D.B.) to determine the presence and the severity of squamous dysplasia and invasive carcinoma. In this model, the squamous cell carcinomas in both HPV-positive and HPV-negative setting develop from the keratinizing surface (rather than tonsillar crypt) epithelia of the tongue and esophagus and phenotypically are of the conventional keratinizing type as opposed to nonkeratinizing carcinoma that is associated with origination from the tonsillar crypt.. For this reason, they were graded according to the conventional grading scheme as well-, moderately- and poorly-differentiated (grade I, II and III, respectively) and sarcomatoid (spindle-cell phenotype resembling high-grade soft tissue sarcoma). Squamous dysplasia was graded as mild, moderate and severe/carcinoma in situ. Tumor multiplicity was assessed as the number of discrete tumor foci separated by normal non-dysplastic epithelium.
Immunofluorescence/Immunohistochemistry
Immunohistochemistry and immunofluorescence was performed as previously described [30]. Primary Antibodies used were anti-BrdU (1:50 dilution, Calbiochem #NA61), anti-HES1 (1:5000 dilution, Cell Signaling #11988S), anti-MCM7 (1:200 dilution, NeoMarkers #47DC141), and anti β-catenin (1:100, Pharmingen #610154).
Levels of hyperplasia
A minimum of 3 mice per genotype were injected intraperitoneally with 0.3 mL of 5-Bromo Deoxyuridine (BrdU) at 12.5 mg/mL in PBS 1 hour prior to sacrifice. Immunohistochemistry was performed to detect BrdU, and 10 microscopic frames of tongue and esophageal tissue were captured at 200 times magnification for each mouse. Percentage of basal and suprabasal cells positive for BrdU was counted on ImageJ using a macro kindly provided by Dr. David Ornelles (Wake Forest).
Statistical Analysis
We used a 2-sided Fisher’s Exact Test to assess differences in gross tumor incidence, a 2-sided Student’s T-Test to assess differences in % BrdU+ cells and a 2-sided Wilcoxon Rank Sum Test to assess differences in tumor severity and tumor multiplicity. A power analysis was performed prior to the study to determine the minimum number of mice that would allow for a sufficient statistical power.
Results
DNMAML1 is expressed in the epithelia upon activation of CreER™
To examine the effect that loss of Notch signaling has on head and neck cancer, we used a ROSA26 knock-in mouse strain that conditionally expresses a dominant-negative acting, truncated MAML1 (DNMAML1) protein that blocks recruitment of co-activators by MAML proteins to complexes with ICN, thereby preventing transcriptional activation by all four Notch receptors [25]. Expression of DNMAML1 requires CRE/LOX-mediated excision of a strong polyadenylation signal positioned upstream of its transcript. DNMAML1 is fused to GFP, allowing one to monitor expression of DNMAML1. To assess the autonomous role of Notch signaling in head and neck carcinogenesis, we directed expression of CRE from the K14Cre transgene. DNMAML1/K14Cre mice displayed alopecia and spontaneous skin cancers (date not shown), similar to that previously observed in DNMAML1/SM22-Cre mice [14]. We therefore used K14CreERTM, allowing us to temporally control CRE activity using tamoxifen. Activation of CRE in DNMAML1/K14CreERTM (heretofore referred to as DNMAML1) mice at 6–8 weeks of age significantly reduced the above-described skin phenotypes, with only mild hair loss and low skin papilloma incidence noted. We still saw robust expression of DNMAML1 in this setting (Fig. 1A), allowing us to pursue long-term head and neck carcinogenesis studies.
Figure 1:
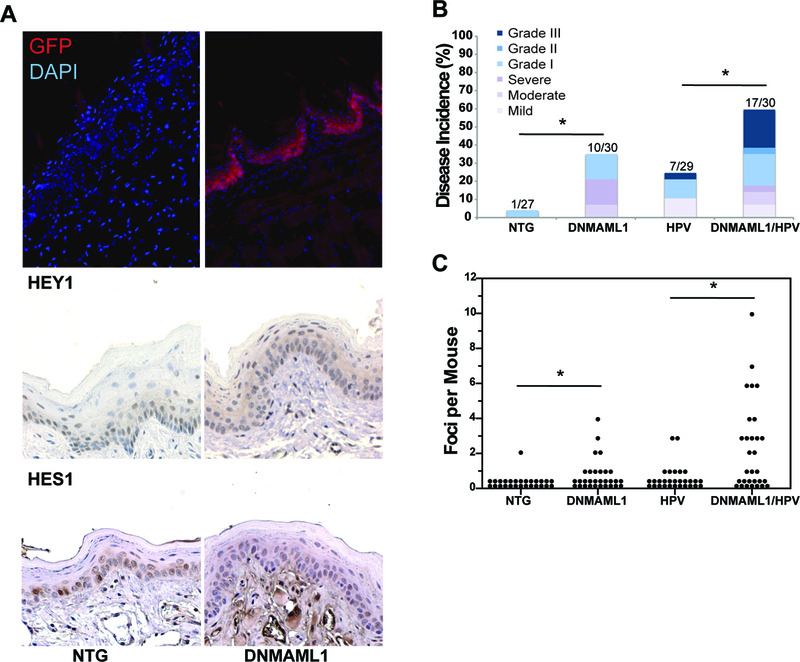
A) Analysis of DNMAML1 gene expression upon K14CreER™ activation. To confirm DNMAML1 expression, immunofluorescence was performed against GFP (red), which is fused to the DNMAML1 gene, with a DAPI counterstain (blue). Expression of HEY1 and HES1, two Notch-dependent proteins, are shown via immunohistochemistry as evidence of lost Notch signaling upon DNMAML1 expression. Representative images of the tongue are shown (200x). B) Gross Tumor Incidence in the tongue and esophagus separated by histological grade of disease (mild-severe dysplasia and grade I-III carcinoma) for all mice in the HPV-positive cohort. Following 4NQO treatment, mice were sacrificed, and the tongue and esophagus were scored for grossly visible lesions. Incidence p-value via Fisher’s Exact Test: (NTG versus DNMAML1) = 0.04, (NTG versus HPV) = 0.052, (DNMAML1 versus DNMAML1/HPV) = 0.119, (HPV versus DNMAML1/HPV) = 0.017. C) Tumor Multiplicity in the tongue and esophagus for mice in the HPV-positive cohort. Tumor multiplicity was assessed by pathologist blinded to treatment groups as the number of discrete tumor foci separated by normal tissue. p-value via Wilcoxon Rank Sum Test: (NTG versus DNMAML1) = 0.006, (NTG versus HPV) = 0.039, (DNMAML1 versus DNMAML1/HPV) = 0.012, (HPV versus DNMAML1/HPV) = 0.001.
Loss of Notch signaling increases tumor incidence and severity in the presence or absence of expression of HPV oncogenes, E6 and E7
We first compared the susceptibility of mice to head and neck cancer in the presence of absence of Notch signaling in both an HPV-positive and HPV-negative context. A well-validated approach to inducing head and neck cancers in mice involves treatment with the chemical carcinogen, 4-Nitroquinoline 1-Oxide (4NQO), placed in drinking water. Epithelial cells lining the oral cavity and esophagus absorb 4NQO, where it is metabolically activated, and forms DNA adducts that result in mutations [23, 31]. 4NQO has also been shown to upregulate the EGFR pathway, which is often activated in human HNSCCs [32]. 4NQO-induced carcinomas also progress through multiple stages of dysplasia and are histologically similar to their human counterparts [33]. For our study, we treated mice with 4NQO in the drinking water for 8 weeks, followed by 16 weeks of normal drinking water. We chose the 8 week treatment period because this induced tumors in around 30% of K14E6/K14E7 mice in a prior study [24]. At the 24 week endpoint, mice were sacrificed, the tongue and esophagus scored for grossly visible tumors, and tissues processed for histopathological assessment of neoplastic disease.
To examine the effect of loss of Notch signaling in the context of expression of HPV oncogenes, we monitored disease in cohorts consisting of nontransgenic mice, mice expressing HPV16 E6 and E7 (E6/E7), mice expressing DNMAML1, and mice expressing both E6/E7 and DNMAML1. Nontransgenic (heretofore referred to as NTG) mice exhibited a low rate of tumor incidence, with only 1 of 27 mice developing an overt tumor, consistent with our prior study [24]. In contrast, 33% of DNMAML1 mice (n=30) had grossly visible tumors, significantly higher than in the NTG group (p = 0.04) (Fig. 1B). K14E6/K14E7 (heretofore referred to as HPV) mice also had a higher gross tumor incidence (24%) compared to NTG mice (p = 0.059),.This difference was not quite statistically significant via a Fisher’s Exact Test before factoring in disease severity, but it is similar to our prior study [24]. Expression of DNMAML1 on the HPV16 transgenic mouse background (DNMAML1/K14CreERTM/K14E6/K14E7 - heretofore referred to as DNMAML1/HPV mice) led to the highest incidence of gross tumors (57%), which was significantly higher than in the HPV mice that do not express DNMAML1 (p = 0.017) (Fig. 1B).
Tongue and esophagus tissues were sectioned, stained with hematoxylin and eosin and every 10th section examined histologically for the presence and severity of squamous dysplasia and invasive carcinoma. Because SCC in this model are of the conventional keratinizing type, they are amenable to grading as grade I-III. Table 1 summarizes disease severity detected in each mouse. Table 1 summarizes the worst stage of disease detected in each mouse. The only neoplastic lesions found in the NTG group was a single grade I invasive carcinoma, whereas DNMAML1 mice had numerous grade I carcinomas as well as varying grades of squamous dysplasia (p = 0.006). HPV mice showed similar tumor severity to DNMAML1 mice. The DNMAML1/HPV mice displayed the worst severity of neoplastic disease. This group developed a high frequency of grade II and grade III squamous cell carcinoma, whereas HPV mice only had 1 grade III carcinoma. There were significant differences in severity of disease between either DNMAML1 (p = 0.029) or HPV (p = 0.010) mice and DNMAML1/HPV mice (Table 1).
Table 1.
Severity of disease in the tongue and esophagus*
| Genotype** | No disease | Mild dysplasia | Moderate dysplasia | Severe dysplasia | Grade I Carcinoma | Grade II Carcinoma | Grade III carcinoma | Sarcomatoid carcinoma | Total mice |
|---|---|---|---|---|---|---|---|---|---|
| NTG | 26 | 1 | 27 | ||||||
| DNMAML1 | 20 | 2 | 4 | 4 | 30 | ||||
| HPV | 22 | 3 | 3 | 1 | 29 | ||||
| DNMAML1/HPV | 13 | 2 | 2 | 1 | 5 | 1 | 6 | 30 | |
| p53het | 29 | 1 | 3 | 1 | 34 | ||||
| DNMAML1/p53het | 16 | 1 | 1 | 4 | 2 | 24 | |||
| p53mut | 28 | 3 | 4 | 1 | 1 | 37 | |||
| DNMAML1/p53mut | 8 | 1 | 1 | 3 | 5 | 2 | 1 | 21 |
Tissues were examined by a pathologist blinded to treatment groups and mice categorized based upon the worst stage of disease present in their tongue and esophagus. Disease states included dysplasia (mild, moderate, severe) and invasive carcinoma (I, II and III, and sarcomatoid carcinoma). p-values via Wilcoxon Rank Sum Test for comparing disease severity: (NTG versus DNMAML1) = 0.006, (NTG versus HPV) = 0.025, (DNMAML1 versus DNMAML1/HPV) = 0.029, (HPV versus DNMAML1/HPV) = 0.010, (p53mut versus DNMAML1/p53mut) = 0.007, (DNMAML1/p53het versus DNMAML1/p53mut) = 0.021.
abbreviations used in table for genotypes: NTG - nontransgenic; DNMAML1 - DNMAML1/K14CreERTM; HPV - K14E6/K14E7; DNMAML1/HPV - DNMAML1/K14CreERTM/K14E6/K14E7; p53het - p53+/−/K14CreERTM; DNAMAL1/p53het - p53+/−/K14CreERTM/DNMAML1; p53mut - p53R172H/K14CreERTM; DNAMAL1/p53mut - p53R172H/K14CreERTM/DNMAML1.
Loss of Notch signaling caused an increase in tumor multiplicity as well (Fig 1C). Again, this difference was most pronounced when comparing DNMAML1 (p = 0.012) or HPV (p = 0.001) mice to DNMAML1/HPV mice. The latter group most often had multifocal carcinoma, in many cases, more than four cancers per animal. There was also a significant increase in tumor multiplicity comparing nontransgenic mice to either DNMAML1 (p = 0.006) or HPV (p = 0.039) mice. These results also indicate that loss of Notch signaling as well as HPV oncogenes contribute to carcinoma tumorigenesis in vivo.
Loss of Notch Signaling Increased Tumor Incidence Most with a p53 Gain of Function Mutation
To model HPV-negative HNSCC, we utilized the gain-of-function mutant p53R172H as a tumor driver, p53+/− (heretofore referred to as p53het) mice as a control group, both in the presence or absence of expression of the DNMAML1 transgene. p53R172H/K14CreERTM (heretofore referred to as p53mut) mice exhibited a higher incidence of grossly visible tumors (24%) than p53het mice (15%), however this difference was not significant (p = 0.379) (Fig. 2A). There was also a non-significant increase in tumor incidence when DNMAML1 was expressed on the p53+/− background (i.e. p53+/−/DNMAML1/K14CreERTM mice, heretofore referred to as DNMAML1/p53het mice) (33% vs 15%) (p = 0.118). However, expression of DNMAML1 in conjunction with p53R172H (i.e. p53R172H/DNMAML1/K14CreERTM mice, heretofore referred to as DNMAML1/p53mut mice) led to a significant increase in tumor incidence (62% vs 24%) (p = 0.010).
Figure 2:

A) Gross Tumor Incidence in the tongue and esophagus separated by histological grade of disease for mice in the HPV-negative cohort (mild-severe dysplasia and grade I-sarcomatoid carcinoma). Incidence p-value via Fisher’s Exact Test: (p53mut versus DNMAML1/p53mut) = 0.010, (DNMAML1/p53het versus DNMAML1/p53mut) = 0.076. B) Tumor Multiplicity in the tongue and esophagus for all mice in the HPV-negative cohort. p-value via Wilcoxon Rank Sum Test: (p53mut versus DNMAML1/p53mut) = 0.002, (DNMAML1/p53het versus DNMAML1/p53mut) = 0.047. (C) Cancer Multiplicity for mice that developed cancer in the tongue and esophagus for HPV, DNMAML1/HPV, p53mut and DNMAML1/p53mut mice. Among the mice that developed cancer, DNMAML1/HPV mice had significantly more cancers than DNMAML1/p53mut mice (p value via Wilcoxon Rank Sum Test = 0.020), whereas there was no difference between HPV and p53mut mice.
While DNMAML1/p53mut mice had similar tumor multiplicities to DNMAML1/HPV mice, DNMAML1/p53mut mice did have increased tumor multiplicity in comparison to either p53mut mice (p = 0.002) or DNMAML1/p53het mice (p = 0.047) (Fig. 2B). Additionally, DNMAML1/p53mut had a statistically significantly higher disease severity than p53mut (p = 0.007) as well as DNMAML1/p53het mice (p = 0.021).
An important difference in the HPV-negative model studies was that, despite having a significantly higher overall disease severity compared to all other groups, DNMAML1/p53mut mice had very few high-grade carcinomas (Table 1). 1 out of 21 (4.8%) DNMAML1/p53mut mice developed a grade 3 carcinoma, whereas 6 out of 30 (20%) DNMAML1/HPV mice developed this grade of disease. Additionally, among the mice that developed cancer, DNMAML1/HPV mice had a significantly higher average multiplicity of cancers than did the DNMAML1/p53mut mice (average of 2.8 versus 1.5 cancers, p = 0.020) (Fig. 2C). Taken together, these results indicate that Notch deficiency synergized with HPV oncogenes in carcinogenesis to a greater extent than with p53R172H.
Loss of Notch signaling only modestly increases cellular proliferation
Previous studies have indicated that loss of Notch signaling in the skin of mice led to hyperplasia [14, 34], leading us to hypothesize that there could be similar effects in the oral mucosa. In order to determine rates of cellular proliferation in the tongue and esophagus upon 4NQO treatment, we injected mice intraperitoneally with BrdU 1 hour prior to sacrifice. We then performed immunohistochemistry against BrdU among all groups and measured basal and suprabasal proliferation (Fig. 3). DNMAML1 mice exhibited significantly higher basal cell proliferation compared to NTG mice in both the tongue (p = 0.042) and esophagus (p = 0.009) as well as higher suprabasal cell proliferation in the tongue (0.0003). However, this increase was overshadowed by the expression of HPV oncogenes E6 and E7, both of which, but particularly E7, have previously been reported to increase cellular proliferation [23], with no significant difference in basal or suprabasal proliferation between HPV mice and DNMAML1/HPV mice. DNMAML1 also did not increase proliferation on either a p53+/− or p53R172H genetic background. DNMAML1/p53mut mice did have slightly increased cellular proliferation compared to p53+/− mice with or with DNMAML1, but this difference was not significant. This analysis suggests that loss of Notch signaling has a modest effect on cellular proliferation in the absence of other transgenes, but this effect is not sufficient to account for increased tumorigenesis in either the HPV-positive (E6/E7) or HPV-negative (p53R172H) settings.
Figure 3:
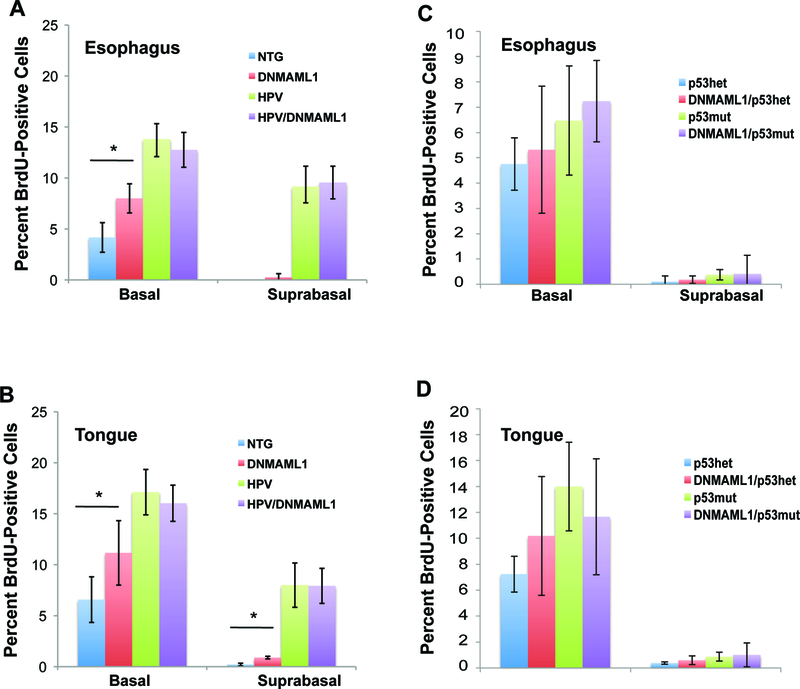
Analysis of cellular proliferation with loss of Notch signaling on an HPV-positive or HPV-negative background. To determine basal and suprabasal cellular proliferation in the tongue and esophagus, mice were injected intraperitoneally with BrdU prior to sacrifice. The tissue was then stained via immunohistochemistry against anti-BrdU. A minimum of 3 mice from each genotype (NTG, DNMAML1/K14CreER, E6/E7, DNMAML1/K14CreER/E6/E7) were stained, and 8 to 10 sections per mouse were captured at 200 times magnification. Each section was scored for percentage of basal and suprabasal proliferation using ImageJ. A) Average Proliferation for the esophagus for the HPV-positive cohort. p-value via Student’s T-Test for basal NTG versus DNMAML1 = 0.009. B) Average proliferation for the tongue for the HPV-positive cohort. p-value via Student’s T-Test for basal NTG versus DNMAML1 = 0.042, for suprabasal NTG versus DNMAML1 < 0.001. C) Average proliferation for the esophagus for the HPV-negative cohort. D) Average proliferation for the tongue for the HPV-negative cohort. p53mut mice exhibited slightly higher proliferation in the tongue and esophagus with or without DNMAML1, but this difference was not significant. DNMAML1 did not increase cellular proliferation in the tongue or esophagus.
DNMAML1-expressing mice exhibit increased expression of MCM7 and decreased expression of HES1
Notch signaling is first found to be active in the parabasal layer in epithelia. It is at this layer and more superficial layers where the Notch pathway is thought to promote differentiation by activating expression of HES1 [35, 36]. We looked for changes in expression of biomarkers for Notch signaling in the parabasal and suprabasal layer upon DNMAML1 expression. The most well-known target of the Notch signaling pathway in human is HES1, a protein important for proper differentiation of epithelia [12]. All groups of mice without DNMAML1 expressed HES1 in the epithelia (Fig. 4A). Conversely, all groups of mice that expressed DNMAML1 showed greatly decreased expression of HES1 in the epithelia. This was most apparent in tumors; tumors arising on HPV mice showed high HES1 staining throughout the tumor, tumors arising on DNMAML1/HPV mice had greatly decreased HES1 staining.
Figure 4:

Analysis of MCM7 and HES1 expression in normal and cancerous epithelia. A) Normal or Cancerous tongue sections stained with anti-HES1 (brown) with a hematoxylin counterstain (200x). B) Normal or Cancerous tongue sections stained with anti-MCM7 (brown) with a hematoxylin counterstain (200x).
We next looked at MCM7, an E2F-responsive gene. HPV-expressing mice express MCM7 throughout their epithelium, due to E7’s inactivation of pocket proteins including pRb, which regulate E2F activity [37] MCM7 is also a marker for cells undergoing proliferation, as it is normally expressed in the S phase of the cell cycle. DNMAML1-expressing mice expressed MCM7 in the basal and parabasal layer, whereas nontransgenic mice expressed MCM7 only in the basal layer (Fig. 4B). Results were similar in p53+/− and p53R172H/K14CreERTM mice with and without DNMAML1 (Supplemental Fig. 1). These results indicate that the basal-to-spinous switch is delayed in the DNMAML1-expressing epithelium, and this correlates with the increased proliferation in DNMAML1-expressing mice. Consistent with prior studies, MCM7 was up-regulated in its expression throughout the basal and suprabasal compartments in mice expressing the HPV oncogenes, E6 and E7. This is due to E7’s aforementioned effect of causing expression of MCM7 in all layers of the epithelium, which would mask any effect of DNMAML1 in this context [37] (Fig. 4B).
DNMAML1-expressing mice exhibit increased nuclear and cytoplasmic β-CATENIN expression
Inactive β-CATENIN normally accumulates at the plasma membrane in complex with E-CADHERIN [38]. However, when activated, β-CATENIN translocates to the cytoplasm and nucleus, driving expression of various genes in the Wnt signaling pathway. Because the Notch signaling pathway has been previously described to have crosstalk with the Wnt signaling pathway and has been seen to alter β-CATENIN expression in the skin [14, 39], we performed immunofluorescence against β-CATENIN (Fig. 5). We found that DNMAML1-expressing mice showed increased levels of nuclear β-CATENIN in the epithelium in comparison to those mice not expressing DNMAML1. Increased levels of β-CATENIN have been reported to be highly associated with various cancers including HNSCC [40], and it has also been described as a marker for more poorly differentiated cells in the epithelium [41]. Furthermore, previous studies have shown that elevated β-CATENIN in human oral SCC cells increased invasion/migration potential and promoted HNSCC tumor growth in nude mice [42]. Due to the strong correlation between increased β-CATENIN and HNSCC, we conclude that this is one mechanism for tumorigenesis induced through inactivation of Notch signaling that could be contributing to enhanced susceptibility to HNSCC.
Figure 5:
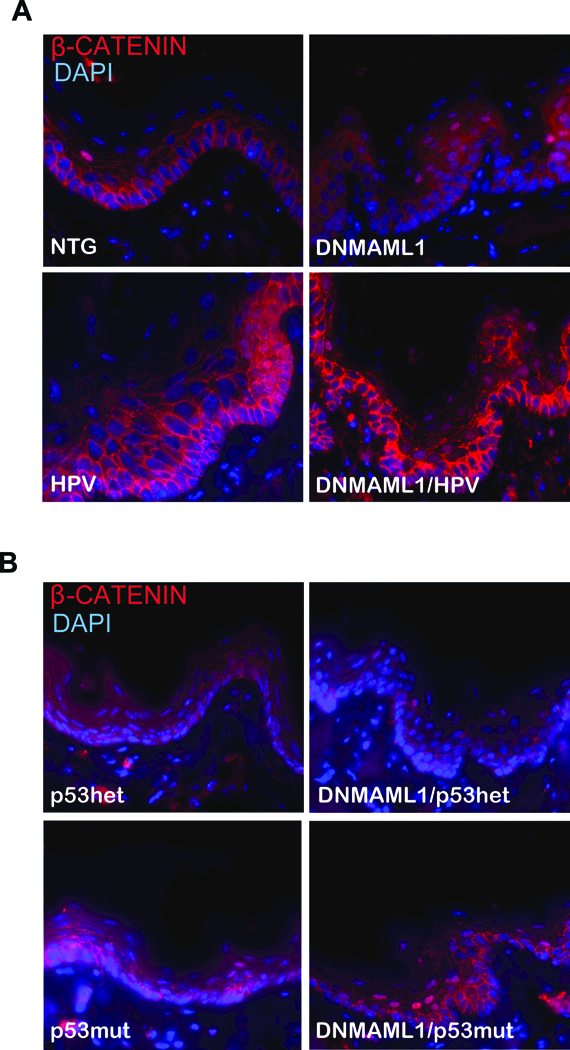
Analysis of β-CATENIN in the tongue epithelium. Immunofluorescence was performed against β-CATENIN (red) with a DAPI (blue) counterstain. Representative images of the tongue are shown for each genotype of A) the HPV-positive cohort and B) the HPV-negative cohort at a high magnification (400x).
Discussion
The Notch signaling pathway has been studied for over a century [9], yet there is still much about it that is unknown. A role of the Notch pathway in HNSCC has recently been raised based upon these cancers accumulating inactivating mutations in the pathway. This was observed in HPV-positive as well as HPV-negative HNSCC and led to the hypothesis that Notch is a tumor suppressor in many head and neck cancers. Other studies have argued quite the opposite - that Notch acts as an oncogene in HNSCC. Here, we demonstrate that, in mice, inhibition of canonical Notch signaling via expression of DNMAML1 increases the incidence of head and neck tumorigenesis in both HPV-positive and HPV-negative settings.
Inactivation of canonical Notch signaling together with HPV oncogene expression leads to highest frequency of cancers and percentage of mice with high-grade cancers.
Even though DNMAML1 expression significantly increased tumor incidence compared to nontransgenic mice, the tumors that arose were either benign or low-grade carcinomas, suggesting that DNMAML1 expression on its own has a relatively limited ability to cause high-grade cancers. This was also true when DNMAML1 was expressed on the p53+/− background. In contrast, 20% of DNMAML1/HPV mice developed high-grade carcinomas (Table 1), and these mice had the highest frequencies of cancers, significantly higher than observed with the DNMAML1/p53mut mice (Fig 2C). This effect cannot be explained simply by the HPV oncogenes being inherently more oncogenic than p53R172H because these two groups of mice without DNMAML1 (HPV and p53mut mice) exhibited almost identical disease spectrums (Table 1). These results point to a clear cooperativity between E6/E7 and loss of Notch signaling in inducing frequent and high-grade carcinomas.
In the case of high risk HPV-associated skin carcinomas, the E6 proteins of the cutaneous HPVs are able to bind to and inhibit MAML1 [43], an activity not shared with high risk mucosal HPV E6 proteins (such as HPV16 E6). The importance of Notch signaling in suppressing skin carcinogenesis was also evident in our study, as 4NQO-treated mice expressing DNMAML1 developed high rates of SCC originating from the perioral skin (Supplemental Fig. 2), likely due to contact with the 4NQO present in the drinking water. High risk mucosal HPV E6 proteins instead inhibit p53 [44].. Interestingly, a link between p53 and Notch signaling has been described [45]. Thus different HPVs may have evolved different means to the same end. That expression of DNMAML1 on the HPV16 transgenic background increased severity of disease indicates that modulation of Notch signaling by high-risk mucosal HPV oncogenes must be rate limiting.
Effect of the Notch signaling pathway on the epithelia
The Notch receptors primarily act through a canonical pathway, in which the cleaved Notch receptor translocates to the nucleus and interacts with MAML1 and RBP-J to affect transcription of downstream targets. However, the Notch receptors can also act in a RBP-J and MAML1-independent fashion to exert other effects. It is important to note that DNMAML1 only inhibits the canonical pathway and may affect the epithelia and tumorigenesis in different ways than inactivating mutations in 1 of more of the Notch receptors.
One area in which the canonical Notch signaling pathway affects the stratified squamous epithelia is in the basal-to-spinous switch [12, 36]. While repressed in the basal layer, the pathway is activated in the suprabasal layer. Here, it both activates spinous gene expression and represses basal gene expression. An important mediator of spinous gene expression is the Notch-dependent protein HES1. As evidenced in Figure 4, HES1 expression was lost upon expression of DNMAML1, indicating that the basal-to-spinous switch may be disrupted in the DNMAML1-expressing tissue. Increased tumor severity is highly associated with less well-differentiated cells, and interrupting the switch from the poorly differentiated basal layer to the more differentiated spinous layer may be an important factor in increased tumor severity.
A dysregulation of β-CATENIN was also observed. DNMAML1-expressing mice expressed increased activated (nuclear) β-CATENIN, and this was primarily seen in the spinous layer of the epithelium (Fig. 5). β-CATENIN activation has been linked to more poorly differentiated epithelial cells [41], and therefore its activation in suprabasal cells provides further evidence that differentiation to the spinous layer has been disrupted due to loss of Notch signaling, which exerts significant influence on the basal-to-spinous switch in the suprabasal cells of the epithelium.
How can Notch also be oncogenic in HNSCC?
While our study provides clear evidence that canonical Notch signaling suppresses tumorigenesis in mouse models for both HPV-positive and HPV-negative HNSCC, consistent with inactivation mutations being often found in Notch genes in human HNSCC, there is also a growing body of evidence supporting the opposing hypothesis that Notch is playing an oncogenic role in at least some HNSCCs (see Introduction). The simplest explanation is that the role of Notch is context dependent, and may reflect differences that lie in how an individual cancer arises. What are the differences in cancers, or the pre-cancers that give rise to them, that determine whether Notch is oncogenic, tumor suppressive or even neutral deserves further investigation.
Notch signaling as a therapeutic target in HNSCC
Because the Notch signaling pathway has been implicated in so many cancers, there have been many studies examining the therapeutic approaches towards targeting Notch signaling in cancer [46]. Many of these efforts have focused on inhibiting the pathway via γ-SECRETASE inhibitors or other small molecule inhibitors. However, there are also multiple possibilities for activating the Notch signaling pathway as a therapeutic target. A recent study found that N-methylhemeanthidine chloride (NMHC), an alkaloid isolated from an herbaceous plant, exhibits anti-tumor activity in Acute Myeloid Leukemia (where Notch is tumor suppressive [47] by promoting Notch1 processing and release of NICD [48]. There is also an ongoing clinical trial examining the efficacy of Notch pathway activation via resveratrol in preventing growth of low-grade neuroendocrine tumors (ClinicalTrials.gov identifier NCT01476592). Furthermore, Notch inhibition has also been shown to reduce cancer stem cell (CSC) populations in human HNSCC tissue both alone and in combination with conventional chemotherapy [49]. Studies such as these may provide valuable insight on the prospects of Notch activation to treat cancers.
Regardless of whether the Notch signaling pathway is being targeted for inhibition or activation, however, there are important caveats to take into consideration. This is due to the fact that the pathway can be both oncogenic and tumor suppressive and is sensitive to gene dosage effects [11]. One study found that a Notch-inhibiting drug caused gastrointestinal cells to move towards a secretory instead of epithelial fate, leading to an imbalance and gastrointestinal toxicity [50]. Activating the Notch signaling pathway in the head and neck could lead to the development of hematopoietic cancers, where the Notch signaling pathway is oncogenic. Additionally, as previously discussed, there is the hypothesis that hyperactive Notch signaling could also be oncogenic in the head and neck (see Introduction). Any treatment options developed, therefore, must be extremely sensitive to these issues.
The Notch signaling pathway in other HPV-associated cancers
The Notch signaling pathway has been studied in the context of cervical cancer for some time, with a variety of conflicting results. CaSki cells have something akin to an oncogene addiction to a cleaved and constitutively active NOTCH1, in that inhibiting NOTCH1 reduces cell viability [15]. Conversely, repressing Notch signaling via ΔNp63α promoted cellular proliferation in other cervical cancer cell lines [16]. These studies have been almost entirely based on cell culture work, with little study of Notch signaling in vivo. Studying the effect of loss of Notch signaling in cervical carcinogenesis utilizing an HPV-cervical cancer mouse model [37], therefore, may be of value.
Supplementary Material
Statement of Transcriptional Relevance.
Head and Neck Squamous Cell Carcinoma (HNSCC) is a common cancer worldwide with only a 60% 5-year survival following standard of care, which is associated with significant acute and long-term consequences. Human papillomaviruses (HPVs) cause a subset of HNSCC distinct from HPV-negative HNSCC at molecular, histopathological and clinical-response levels. Deep sequencing studies found loss-of-function mutations in the Notch signaling pathway in some HNSCCs; whereas, other studies found Notch signaling was increased in some HSCCCs. This has led to a controversy - is Notch a tumor suppressor, an oncogene, or both in HNSCC? We demonstrate that inactivation of canonical Notch signaling drives head and neck carcinogenesis in mouse models of keratinizing HNSCC, both in the presence and in the absence of HPV oncogenes. However, HPV oncogenes synergize with lost Notch signaling to induce more and higher grade cancers than seen in the HPV-negative mouse model.
Acknowledgements:
We acknowledge Warren Pear (University of Pennsylvania) for generating and providing us with the DNMAML1 mice and Professor David Ornelles (Wake Forest University) for sharing his Image J macro for counting BrdU-positive cells.
Financial Support:
Patrick Nyman: National Institute of Health Grants T32AI078985
Darya Buehler: no grants to disclose
PaulLambert: National Institute of Health Grants P01CA022443, P50DE026787, R35CA210807
Footnotes
The authors declare no potential conflicts of interest
References
- 1.Cancer Facts and Figures. In: American Cancer Society Edited by Society AC. Atlanta, GA; 2011. [Google Scholar]
- 2.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L et al. : Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. Journal of the National Cancer Institute 2000, 92(9):709–720. [DOI] [PubMed] [Google Scholar]
- 3.Stein AP, Saha S, Yu M, Kimple RJ, Lambert PF: Prevalence of human papillomavirus in oropharyngeal squamous cell carcinoma in the United States across time. Chemical research in toxicology 2014, 27(4):462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franceschi S, Talamini R, Barra S, Baron AE, Negri E, Bidoli E et al. : Smoking and drinking in relation to cancers of the oral cavity, pharynx, larynx, and esophagus in northern Italy. Cancer research 1990, 50(20):6502–6507. [PubMed] [Google Scholar]
- 5.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ et al. : Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333(6046):1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A et al. : The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333(6046):1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H et al. : Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. Journal of the National Cancer Institute 2008, 100(4):261–269. [DOI] [PubMed] [Google Scholar]
- 8.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ et al. : Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer research 2007, 67(10):4605–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohr OL: Character Changes Caused by Mutation of an Entire Region of a Chromosome in Drosophila. Genetics 1919, 4(3):275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artavanis-Tsakonas S, Rand MD, Lake RJ: Notch signaling: cell fate control and signal integration in development. Science 1999, 284(5415):770–776. [DOI] [PubMed] [Google Scholar]
- 11.Andersson ER, Sandberg R, Lendahl U: Notch signaling: simplicity in design, versatility in function. Development 2011, 138(17):3593–3612. [DOI] [PubMed] [Google Scholar]
- 12.Blanpain C, Lowry WE, Pasolli HA, Fuchs E: Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes & development 2006, 20(21):3022–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD et al. : TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66(4):649–661. [DOI] [PubMed] [Google Scholar]
- 14.Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J et al. : Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer research 2006, 66(15):7438–7444. [DOI] [PubMed] [Google Scholar]
- 15.Kuncharin Y, Sangphech N, Kueanjinda P, Bhattarakosol P, Palaga T: MAML1 regulates cell viability via the NF-kappaB pathway in cervical cancer cell lines. Experimental cell research 2011, 317(13):1830–1840. [DOI] [PubMed] [Google Scholar]
- 16.Yugawa T, Narisawa-Saito M, Yoshimatsu Y, Haga K, Ohno S, Egawa N et al. : DeltaNp63alpha repression of the Notch1 gene supports the proliferative capacity of normal human keratinocytes and cervical cancer cells. Cancer research 2010, 70(10):4034–4044. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas N: Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517(7536):576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klussmann JP, Mooren JJ, Lehnen M, Claessen SM, Stenner M, Huebbers CU et al. : Genetic signatures of HPV-related and unrelated oropharyngeal carcinoma and their prognostic implications. Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15(5):1779–1786. [DOI] [PubMed] [Google Scholar]
- 19.Egloff AM, Grandis JR: Molecular pathways: context-dependent approaches to Notch targeting as cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2012, 18(19):5188–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun W, Gaykalova DA, Ochs MF, Mambo E, Arnaoutakis D, Liu Y et al. : Activation of the NOTCH pathway in head and neck cancer. Cancer research 2014, 74(4):1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rettig EM, Chung CH, Bishop JA, Howard JD, Sharma R, Li RJ et al. : Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated with NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev Res (Phila) 2015, 8(4):287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaykalova DA, Zizkova V, Guo T, Tiscareno I, Wei Y, Vatapalli R et al. : Integrative computational analysis of transcriptional and epigenetic alterations implicates DTX1 as a putative tumor suppressor gene in HNSCC. Oncotarget 2017, 8(9):15349–15363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strati K, Pitot HC, Lambert PF: Identification of biomarkers that distinguish human papillomavirus (HPV)-positive versus HPV-negative head and neck cancers in a mouse model. Proceedings of the National Academy of Sciences of the United States of America 2006, 103(38):14152–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jabbar S, Strati K, Shin MK, Pitot HC, Lambert PF: Human papillomavirus type 16 E6 and E7 oncoproteins act synergistically to cause head and neck cancer in mice. Virology 2010, 407(1):60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I et al. : Notch signaling is an important regulator of type 2 immunity. The Journal of experimental medicine 2005, 202(8):1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herber R, Liem A, Pitot H, Lambert PF: Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. Journal of virology 1996, 70(3):1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song S, Pitot HC, Lambert PF: The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. Journal of virology 1999, 73(7):5887–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT et al. : Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119(6):847–860. [DOI] [PubMed] [Google Scholar]
- 29.Strati K, Lambert PF: Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer research 2007, 67(24):11585–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF: Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Molecular and cellular biology 2003, 23(24):9094–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramotar D, Belanger E, Brodeur I, Masson JY, Drobetsky EA: A yeast homologue of the human phosphotyrosyl phosphatase activator PTPA is implicated in protection against oxidative DNA damage induced by the model carcinogen 4-nitroquinoline 1-oxide. J Biol Chem 1998, 273(34):21489–21496. [DOI] [PubMed] [Google Scholar]
- 32.Kao YY, Tu HF, Kao SY, Chang KW, Lin SC: The increase of oncogenic miRNA expression in tongue carcinogenesis of a mouse model. Oral Oncol 2015, 51(12):1103–1112. [DOI] [PubMed] [Google Scholar]
- 33.Kanojia D, Vaidya MM: 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol 2006, 42(7):655–667. [DOI] [PubMed] [Google Scholar]
- 34.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M et al. : Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003, 33(3):416–421. [DOI] [PubMed] [Google Scholar]
- 35.Blanpain C, Horsley V, Fuchs E: Epithelial stem cells: turning over new leaves. Cell 2007, 128(3):445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuchs E: Finding one’s niche in the skin. Cell Stem Cell 2009, 4(6):499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM: Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer research 2003, 63(16):4862–4871. [PubMed] [Google Scholar]
- 38.Valenta T, Hausmann G, Basler K: The many faces and functions of beta-catenin. Embo j 2012, 31(12):2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naganuma S, Whelan KA, Natsuizaka M, Kagawa S, Kinugasa H, Chang S et al. : Notch receptor inhibition reveals the importance of cyclin D1 and Wnt signaling in invasive esophageal squamous cell carcinoma. Am J Cancer Res 2012, 2(4):459–475. [PMC free article] [PubMed] [Google Scholar]
- 40.Santoro A, Pannone G, Papagerakis S, McGuff HS, Cafarelli B, Lepore S et al. : Beta-catenin and epithelial tumors: a study based on 374 oropharyngeal cancers. Biomed Res Int 2014, 2014:948264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu AJ, Watt FM: beta-catenin signalling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development 1999, 126(10):2285–2298. [DOI] [PubMed] [Google Scholar]
- 42.Yang F, Zeng Q, Yu G, Li S, Wang CY: Wnt/beta-catenin signaling inhibits death receptor-mediated apoptosis and promotes invasive growth of HNSCC. Cellular signalling 2006, 18(5):679–687. [DOI] [PubMed] [Google Scholar]
- 43.Brimer N, Lyons C, Wallberg AE, Vande Pol SB: Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31(43):4639–4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM: The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63(6):1129–1136. [DOI] [PubMed] [Google Scholar]
- 45.Dotto GP: Crosstalk of Notch with p53 and p63 in cancer growth control. Nature reviews Cancer 2009, 9(8):587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purow B: Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol 2012, 727:305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S et al. : A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011, 473(7346):230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye Q, Jiang J, Zhan G, Yan W, Huang L, Hu Y et al. : Small molecule activation of NOTCH signaling inhibits acute myeloid leukemia. Sci Rep 2016, 6:26510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao ZL, Zhang L, Huang CF, Ma SR, Bu LL, Liu JF et al. : NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci Rep 2016, 6:24704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H et al. : Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005, 435(7044):959–963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.