

Abstract
Immunotherapy has clinical activity in certain virally-associated cancers. The tumor antigens targeted in successful treatments are not well defined and have important implications for the study and design of novel immunotherapies. We used a personalized immunogenomic approach to elucidate the global landscape of anti-tumor T-cell responses in complete regression of human papillomavirus-associated metastatic cervical cancer after tumor-infiltrating adoptive T-cell therapy. Remarkably, the immunodominant T-cell reactivity was directed against mutated neoantigens or a cancer-germline antigen, rather than the canonical viral antigens underlying the disease. T cells targeting viral tumor antigens did not display preferential in vivo expansion during cancer regression. T-cell clonotypes specific for both viral and non-viral tumor antigens resided predominantly in the programmed cell death 1 (PD-1) expressing T-cell compartment before treatment suggesting that PD-1 blockade may unleash diverse anti-tumor T-cell reactivities. These findings suggest a new paradigm of targeting non-viral antigens in immunotherapy of virally-associated cancers.
Immunotherapy can induce the regression of certain virally-associated epithelial malignancies such as human papillomavirus (HPV)-induced cervical (1), head and neck (2), and anal (3) cancers. However, the tumor antigens involved in T-cell-mediated regression of these malignancies remain poorly defined. The viral oncoproteins expressed by HPV+ tumors are conspicuous potential candidate tumor regression antigens as they are immunologically foreign and constitutively expressed by the cancers (4). However, evidence for the importance of these antigens in immunotherapy-mediated tumor regression is limited. Efforts to induce tumor regression by targeting HPV-oncoproteins with specific immunotherapy, such as therapeutic cancer vaccines, have not been effective in the treatment of invasive cancers (5, 6). Combining therapeutic vaccination with chemotherapy, which eliminates elevated levels of myeloid-derived suppressor cells, has demonstrated augmented immunogenicity, but whether this approach will result in tumor regression requires further study (7). It is also intriguing that in early-phase clinical trials, response rates to programmed cell death 1 (PD-1) immune checkpoint blockade appear to be similar in patients with virus-positive and -negative carcinomas of the head and neck (2). T cells targeting the protein products of somatic mutations (cancer neoantigens) (8–11) and epigenetically dysregulated genes (cancer-germline antigens) (12, 13) have been implicated in immunotherapy-induced regression of certain non-viral cancers. Thus, one unexplored explanation unifying these observations may be that non-viral tumor antigens are targeted in regression of HPV+ cancers. To explore this hypothesis, we performed a global landscape analysis of the viral and non-viral antigens targeted by T cells in patients successfully treated with immunotherapy for a virally-associated epithelial cancer.
We studied two patients with HPV+ metastatic cervical carcinoma who experienced complete cancer regression that is ongoing 44 (patient 3775 with HPV16+ squamous cell carcinoma) and 37 (patient 3853 with HPV18+ adenocarcinoma) months after adoptive transfer of tumor-infiltrating lymphocytes (TIL) (1). The infused cells, hereafter referred to as TIL-3775 and TIL-3853, consisted of T cells expanded from TIL cultures selected for reactivity against the HPV-E6 and/or -E7 oncoproteins (1). However, these cultures also contained T cells with initially uncharacterized antigen specificities. To fully define the spectrum of antigens targeted by the therapeutic T cells, we combined next-generation sequencing with functional immunological assays (fig. S1). T-cell reactivity was examined against three classes of potentially immunogenic tumor antigens: HPV-encoded antigens, mutated neoantigens, and cancer-germline antigens (fig. S1). Briefly, constructs encoding full-length versions of HPV-encoded genes and cancer-germline genes expressed by the patient’s metastatic tumor were generated (fig. S1 and table S1) (14). Further, putative somatic mutations identified by whole-exome sequencing of patients’ tumors were incorporated into tandem minigene (TMG) constructs (14, 15). Minigenes encoding each somatic mutation flanked bilaterally by 12 amino acids from the wild-type (WT) sequence (mutant 25-mer) were concatenated to yield a TMG (14, 15). Subsequently, autologous dendritic cells (DCs) were electroporated with in vitro transcribed RNA from gene constructs and used as targets for TIL in immunological assays (14). The secretion of the T-cell effector cytokine interferon-γ (IFN-γ) measured by enzyme-linked immunospot (ELISPOT) assay and upregulation of the T-cell activation marker CD137 by flow cytometry were evaluated. Given limitations in the ability to reliably grow tumor cell lines from metastatic cervical cancers, the personalized immunogenomic approach used here enabled screening for tumor-specific antigens without the requirement for autologous tumor cell lines.
We first investigated whether the infused TIL contained T-cell reactivity against the HPV-encoded proteins, L1, L2, E1, E2, E4, E5, E6 and E7. Consistent with prior results, T cells specific for the E6 and/or E7 antigens were detected in both patients (Fig. 1A and B) (1). Reactivity against other HPV proteins was not found (Fig. 1A and B). In TIL-3775, the response against E6 was CD8+ T-cell-mediated whereas CD4+ and CD8+ T cells recognized E7 (Fig. 1C). The T-cell response against E7 in TIL-3853 was mediated by CD4+ T cells (Fig. 1D).
Fig. 1. Therapeutic TIL used for successful treatment of patients with metastatic HPV+ cervical cancer targeted viral and non-viral tumor antigens.
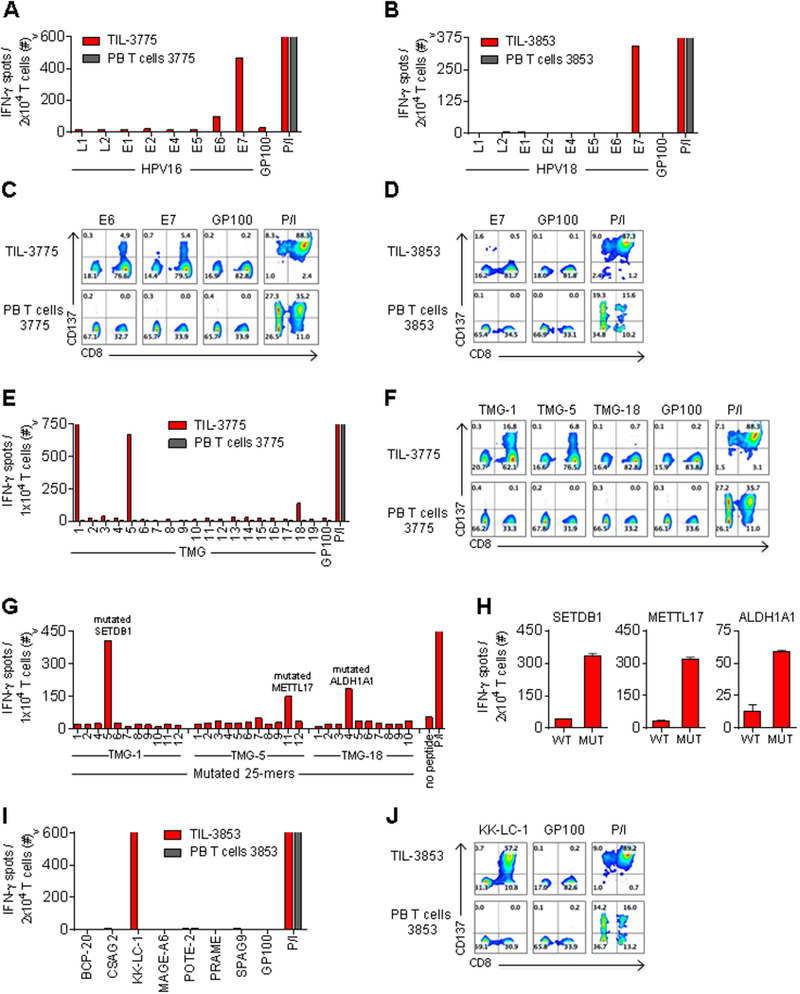
(A and B) IFN-γ ELISPOT assay of (A) TIL-3775 and (B) TIL-3853, compared with pre-treatment PB T cells from these patients after co-culture with autologous DCs electroporated with RNA encoding HPV type-specific antigens or glycoprotein 100 (GP100, negative control). (C and D) Flow cytometric analysis (FCA) of CD137 expression on (C) TIL-3775 and (D) TIL-3853, and PB T cells, co-cultured with DCs electroporated with RNA encoding (C) HPV16-E6, HPV16-E7 or GP100, and (D) HPV18-E7 or GP100. (E) IFN-γ ELISPOT assay of TIL-3775 and PB T cells co-cultured with DCs electroporated with TMG-1 to TMG-19, or GP100 RNA. (F) FCA of CD137 expression on TIL-3775 and PB T cells upon co-culture with DCs electroporated with TMG-1, TMG-5, TMG-18 or GP100 RNA. (G) IFN-γ ELISPOT assay of TIL-3775 co-cultured with APCs pulsed with individual mutated 25-mer peptides encoded in the indicated TMG (table S2). Mutated 25-mers 3 and 10 encoded in TMG-1 and TMG-5, respectively, could not be synthesized. (H) Reactivity of TIL-3775 to the mutated (MUT) 25-mer peptides encoded by TMG-1 (SETDB1E>D), TMG-5 (METTL17E>K), and TMG-18 (ALDH1A1N>I) or their wild-type (WT) counterparts pulsed on DCs. Error bars represent standard deviation of duplicate wells. (I) IFN-γ ELISPOT assay of TIL-3853 and PB T cells to DCs electroporated with the indicated cancer-germline antigens or GP100 RNA. (J) FCA of CD137 expression on TIL-3853 and PB T cells upon co-culture with DCs electroporated with KK-LC-1 or GP100 RNA. Phorbol 12-Myristate 13-Acetate and Ionomycin (P/I) stimulation was used as a positive control. T-cell reactivity was measured at 20–24 hours in all co-culture assays. “>” denotes off-scale values. All FCA’s are pre-gated on live+ CD3+ T cells. Experiments were performed as single determinations unless stated otherwise. All data, except E and G, are representative of ≥ two independent experiments.
We next examined whether the infused TIL contained reactivity against mutated neoantigens. TIL-3775 was screened for reactivity against 19 TMG constructs that together encoded 222 identified somatic mutations (14). Specific reactivity was detected to TMG-1, TMG-5, and TMG-18 (Fig. 1E). The T-cell responses against all three TMGs were CD8+ T-cell-mediated (Fig. 1F). To identify the specific neoantigen(s) recognized within TMGs, individual mutant 25-mer peptides encoded by each of the recognized TMGs were screened (table S2). Reactivity to mutated peptides derived from SET domain bifurcated 1 (SETDB1), methyltransferase like 17 (METTL17) and aldehyde dehydrogenase 1 family member A1 (ALDH1A1) was detected in TIL-3775 (Fig. 1G and fig. S2A). In each case, T cells preferentially recognized the mutant 25-mer peptide compared to the WT counterparts (Fig. 1H and fig. S2B). Using HLA binding prediction algorithms, the minimal epitopes were determined for SETDB1E>D and METTL17E>K mutation-reactive CD8+ T cells (fig. S2C). The epitope for ALDH1A1N>I mutation-reactive CD8+ T cells could not be elucidated at this stage (data not shown). TIL-3853 was also screened for reactivity against 72 identified somatic mutations (14), but no T-cell responses were detected (data not shown).
Next, the TIL from each patient were examined for reactivity against cancer-germline antigens (14). TIL-3775 did not demonstrate reactivity against any of seven antigens tested (table S1). TIL-3853 demonstrated reactivity against a single cancer-germline antigen, Kita-kyushu lung cancer antigen 1 (KK-LC-1) (Fig. 1I and table S1). This reactivity was CD8+ T-cell mediated (Fig. 1F). Thus, in addition to T cells targeting the HPV-oncoproteins, T cells with reactivity against mutated neoantigens or a cancer-germline antigen contributed to the pool of infused T cells in each of the effective treatments.
To characterize the polyclonality of T-cell responses targeting tumor antigens, we identified antigen-specific T-cell receptor (TCR) clonotypes in the infused TIL of each patient. The dominant TCR beta-chain (TCRB) sequences identified in the T-cell populations enriched for specific tumor antigen reactivity (HPV-E6, HPV-E7, TMG-1, TMG-5, TMG-18 for TIL-3775; HPV-E7 and KK-LC-1 for TIL-3853) were paired with the corresponding TCR alpha-chain sequences to reconstruct functional TCRs using single-cell RT-PCR (16) and/or pairSEQ (17) (fig. S3 and table S3) (14). Subsequently, these TCRs were cloned into retroviral expression vectors and used to genetically engineer autologous peripheral blood (PB) T cells (14, 16). For patient 3775, one CD8+ T-cell derived TCR demonstrated specific HPV-E6 recognition (Fig. 2A). An additional four TCRs, two from the CD4+ and two from the CD8+ T-cell population, displayed specific HPV-E7 reactivity (Fig. 2A). Five, one, and two CD8+ T-cell derived TCRs demonstrated specific recognition of the mutant SETDB1E>D, METTL17E>K, and ALDH1A1N>I peptides, respectively (Fig. 2A). For patient 3853, one CD4+ T-cell derived TCR conferred specific HPV-E7 reactivity, and one CD8+ T-cell derived TCR recognized KK-LC-1 (Fig. 2B). Thus, T-cell responses targeting viral and non-viral tumor antigens in the infused TIL of patients were mono- or oligoclonal.
Fig. 2. Immunodominant anti-tumor T-cell responses in therapeutic TIL were directed against mutated neoantigens and the cancer-germline antigen KK-LC-1, rather than HPV antigens.
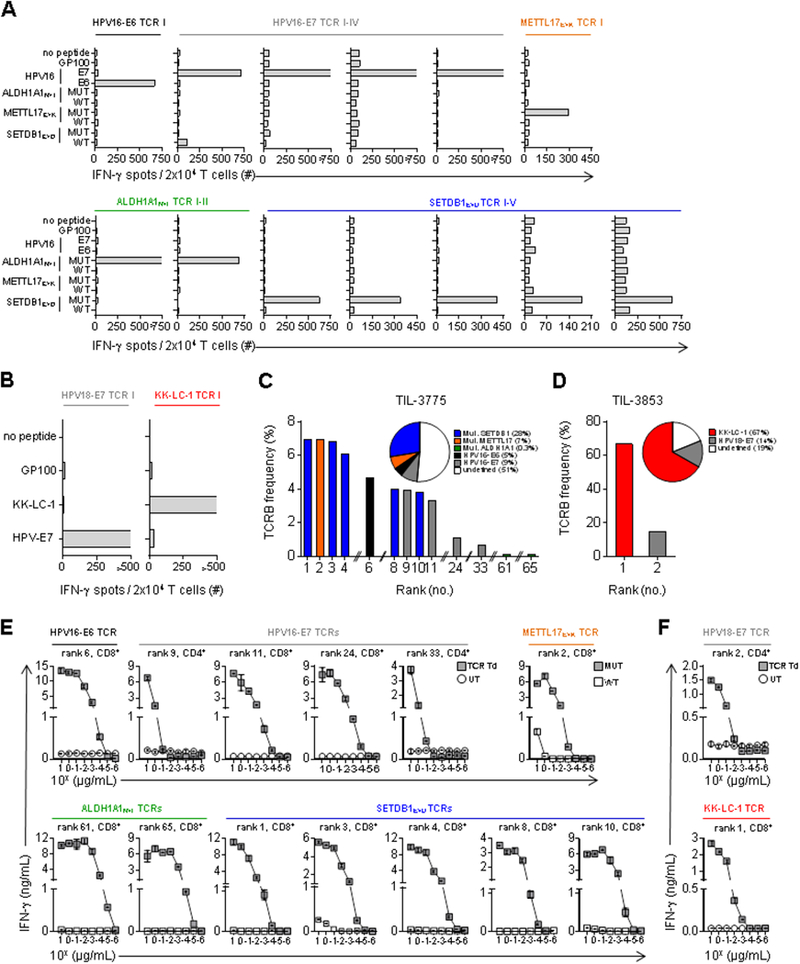
(A and B) IFN-γ ELISPOT assay of autologous pre-treatment PB T cells retrovirally transduced with TCRs identified from infused TIL of patients. (A) Reactivity of PB T cells from patient 3775 transduced with the 13 identified TCRs to peptide pools of HPV16-E6, HPV16-E7 and GP100 (negative control), and wild-type (WT) and mutated (MUT) SETDB1E>D, METTL17E>K, and ALDH1A1N>I 25-mer peptides pulsed on autologous DCs. (B) Reactivity of PB T cells from patient 3853 transduced with the 2 identified TCRs stimulated with autologous DCs pulsed with peptide pools of HPV18-E7, KK-LC-1 and GP100 (negative control). T-cell reactivity was measured at 20–24 hours in co-culture assays. The TCR transduction efficiencies were >40%, as determined by anti-mouse TCRB constant region flow cytometry (data not shown). P/I stimulation was used as a positive control in all co-culture assays. “>” denotes off-scale values. Experiments were performed as single determinations. Data are representative of two independent experiments. (C and D) Frequency and rank of the individual tumor antigen-specific TCRB clonotypes identified in infused TIL as determined by TCRB deep sequencing analysis. (C) In TIL-3775, CD8+ clonotypes were all mutation-specific (rank 1, 2, 3, 4, 8, 10, 61 and 65), the HPV16-E6-specific (rank 6) and two HPV16-E7-specific (rank 11 and 24) clonotypes. CD4+ clonotypes were two HPV16-E7-specific (rank 9 and 33) clonotypes. (D) In TIL-3853, the KK-LC-1-specific clonotype (rank 1) was CD8+ and the HPV18-E7-specific clonotype (rank 2) was CD4+. Pie charts display the sum of the frequencies of individual TCRB clonotypes with indicated tumor antigen specificity in the infused TIL. White color indicates the frequency of TCRB clonotypes with undefined specificity. (E and F) Functional avidity assay testing by IFN-γ enzyme-linked immunospot assay (ELISA) of autologous PB T cells transduced with the indicated tumor-antigen specific TCRs co-cultured with autologous antigen-presenting cells pulsed with titrated concentrations of cognate peptides (fig. S4). (E and F) Functional avidity of (E) 13 identified TCRs from patient 3775 and (F) two TCRs from patient 3853. Reactivity of TCR transduced T cells (TCR Td) and untransduced T cells (UT) (control T cells) are shown for HPV-specific TCRs (E and F) and KK-LC-1-specific TCR (F). Reactivity of TCR transduced T cells against mutated (MUT) and wild-type (WT) cognate epitopes are shown for mutation-specific TCRs (E). HLA-class I restricted CD8+-T-cell derived TCRs (rank 1, 2, 3, 4, 6, 8, 10, 11, 24, 61 and 65 in TIL-3775 (Figure 2C); rank 1 in TIL-3853 (Figure 2D)) were transduced into PB CD8+ T cells, and CD4+-T-cell derived TCRs (rank 9 and 33 in TIL-3775 (Figure 2C); rank 2 in TIL-3853 (Figure 2D)) were transduced into PB CD4+ T cells. TCR transduced T cells were enriched by anti-mouse TCRB constant region to yield >90% CD8+ or CD4+ TCR transduced T cells in each case for use in co-culture assays (data not shown). Error bars represent standard deviation of duplicate wells. Data are representative of two independent experiments.
Having identified the clonotypes that targeted each class of tumor antigens, we next determined their relative frequencies in the infused TIL using TCRB deep sequencing. In TIL-3775, the five HPV-specific clonotypes were found at variable frequencies (range 0.7–4.7%) and rank (range 6–33) among all clonotypes (Fig. 2C). Similarly, the eight mutation-specific clonotypes were also represented at variable frequencies (range 0.12–6.94%) and rank (range 1–65) (Fig. 2C). The cumulative frequency of mutation-specific clonotypes accounted for 35% of TIL-3775, while the HPV-specific clonotypes represented 14% (Fig. 2C). TIL-3853 displayed a highly skewed T-cell repertoire (Fig. 2D). The HPV-E7-specific clonotype was the second most frequent and constituted 14% of TIL-3853, while the KK-LC-1-specific clonotype was the most frequent at 67% (Fig. 2D). Thus, both patients who experienced complete tumor regression received TIL that contained a relatively low frequency of HPV-targeted T cells compared to the frequency of non-viral tumor antigen-targeted T cells.
To directly compare the functional avidities of TCRs targeting the viral and non-viral tumor antigens identified in the infused TIL, we first determined the minimal epitopes targeted by each of the identified tumor-reactive TCRs (fig. S4 and table S4). To account for the potential influence of co-receptor usage on TCR avidity, TCRs that recognized HLA-class I restricted antigens (fig. S4A and B) were transduced into autologous peripheral blood CD8-enriched T cells, whereas CD4+ T-cell derived TCRs were transduced into CD4-enriched T cells (table S4). In all cases, TCR transduced cells were enriched for TCR expression and CD8+ or CD4+ phenotype to >90% purity prior to use in co-culture assays. Next, we measured the functional avidity of TCR transduced T cells using autologous antigen-presenting cells pulsed with titrated concentrations of cognate peptide. For patient 3775, the three HPV-specific CD8+ T-cell derived TCRs showed recognition of target cells pulsed with cognate peptide concentrations as low as 10−4 μg/ml, while the two HPV-specific CD4+ T-cell derived TCRs required 10−1 μg/ml cognate peptide concentration to provoke recognition (Fig. 2E). The eight mutated neoantigen-specific CD8+ T-cell derived TCRs exhibited a range of functional avidities spanning from 10−3 to 10−5 μg/ml in cognate peptide concentrations (10−3 μg/ml for the METTL17E>K–specific TCR, 10−4 μg/ml for the five SETDB1E>D-specific TCRs, and 10−5 μg/ml for the two ALDH1A1N>I-specific TCRs). For patient 3853, the HPV-E7 specific CD4+ T-cell derived TCR recognized as little as 10−2 μg/ml of peptide while the KK-LC-1 specific CD8+ T-cell derived TCR displayed recognition down to a concentration of 10−3 μg/ml cognate peptide (Fig. 2F). Thus, both viral and non-viral tumor antigen-targeting TCRs displayed a range of functional avidities in both patients.
To evaluate the function and persistence of infused tumor antigen-specific T cells after adoptive transfer, we first examined the tumor antigen reactivity in the circulation. Post-treatment PB T cells from both patients displayed specific reactivity against all identified viral and non-viral tumor antigens, while minimal to no reactivity was detected pre-treatment (Fig. 3A and B). Next, we longitudinally tracked tumor antigen-specific T-cell clonotypes using TCRB deep sequencing. In patient 3775, infused tumor antigen-specific T cells represented 27% and 29% of the total circulating T cells at 1 and 4.2 months post-treatment during tumor regression, and 2% at 13 months during remission. Pre-treatment, they accounted for only 0.06% of all T cells (Fig. 3C). The relative proportion of HPV- to neoantigen-specific T cells was similar during tumor regression and remission (Fig. 3C). At the clonotypic level, fluctuations in the relative frequencies were observed among and between clonotypes targeting the viral and non-viral tumor antigen classes, without strict preservation of the numerical hierarchy measured in the infused TIL (Fig. 3C). The dynamic nature of clonotype persistence appeared not to be associated with differences in TCR avidity (fig. S4). In patient 3853, infused tumor antigen-specific T cells accounted for 12%, 3%, and 3% of total circulating T cells at 1 (tumor regression), 5.6 and 10.6 (remission) months post-treatment, respectively, while they represented only 0.003% of pre-treatment T cells (Fig. 3D). The KK-LC-1-specific clonotype remained dominant over the HPV-specific clonotype at ≥10-fold higher frequencies, and was among the most dominant clonotypes in PB (rank 1–5) across all time-points measured (Fig. 3D). Taken together, adoptively transferred viral and non-viral tumor antigen-reactive T cells in both patients remained functional and persisted at elevated levels in the circulation for months during tumor regression and ongoing remission.
Fig. 3. Repopulation of peripheral blood (PB) by infused T cells targeting viral and non-viral tumor antigens throughout cancer regression and remission.
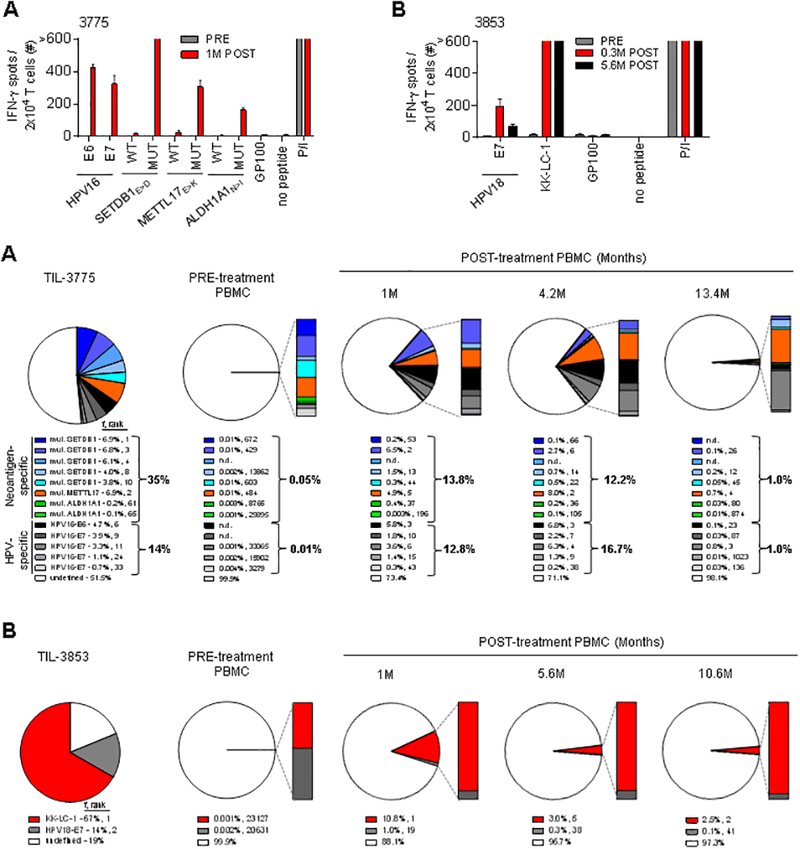
(A and B) Reactivity of PB T cells from before (PRE) and after (POST) treatment was assessed by IFN-γ ELISPOT assay against peptides from tumor antigens identified in patient’s infused TIL. (A) Reactivity of PB T cells from patient 3775 from before and 1 month (M) after treatment to peptide pools of HPV16-E6, HPV16-E7 and GP100 (negative control), and mutated (MUT) and wild-type (WT) SETDB1E>D, METTL17E>K, and ALDH1A1N>I 25-mer peptides pulsed on autologous DCs. (B) Reactivity of PB T cells from patient 3853 from before, 0.3 and 5.6 M after treatment to peptide pools of HPV18-E7, KK-LC-1 and GP100 (negative control) pulsed on autologous DCs. T-cell reactivity was measured at 20–24 hours in co-culture assays. Error bars represent standard deviation of duplicate wells. “>” denotes off-scale values. P/I stimulation was used as a positive control in all co-culture experiment. (C and D) Frequency (f) and rank of individual tumor antigen-specific TCRB clonotypes as identified within (C) TIL-3775 and (D) TIL-3853 among PB mononuclear cells (PBMC) pre- and post-treatment (at the indicated months (M)), as determined by TCRB deep sequencing. Pie charts display the sum of the frequencies of individual TCRB clonotypes with indicated tumor antigen specificity in the infused TIL.
Cell surface expression of PD-1 on PB CD8+ T cells functions as a biomarker to identify tumor antigen-specific T cells in metastatic melanoma patients (18), but whether it identifies these T cells in patients with epithelial cancers is unknown. We therefore tested whether expression of PD-1 can identify both viral and non-viral tumor antigen-specific T cells in the PB of the two patients we studied. In both cases, a minor population representing ≤10% of CD4+ and CD8+ T cells co-expressed PD-1 in pre-treatment PB (Fig. 4A). Next, we used flow cytometric sorting followed by TCRB deep sequencing to isolate and identify circulating PD-1+ and PD-1− populations. Twelve of 13 tumor antigen-specific T-cell clonotypes, including those associated with HPV and neoantigen reactivities, were uniquely detected in the PD-1+ but not the PD-1− T-cell populations of patient 3775 (Fig. 4B). For patient 3853, frequencies of both the CD8+ KK-LC-1- and the CD4+ HPV-specific T-cell clonotypes were enriched in the PD-1+ compared to the PD-1− T-cell populations (Fig. 4C). These findings demonstrate that PD-1 expression identifies both viral and non-viral tumor antigen-specific T cells in PB of cervical cancer patients, suggesting that T cells targeting both tumor antigen classes may be unleashed by anti-PD-1 blockade therapy.
Fig. 4. Programmed cell death 1 (PD-1) expression identifies both viral and non-viral tumor antigen-specific T cells in the circulation of patients with metastatic cervical carcinoma before treatment.
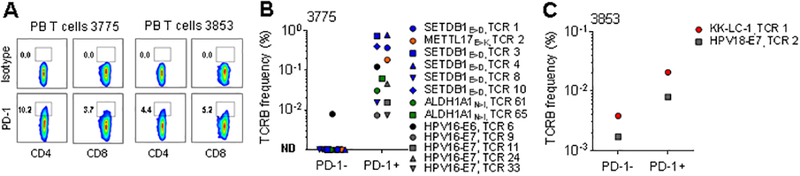
(A) Expression of PD-1 on CD4+ and CD8+ T cells from PB before treatment for (left) patient 3775 and (right) patient 3853. (B and C) Frequency of individual tumor antigen-specific TCRB clonotypes as identified for (B) patient 3775 in TIL-3775 and for (C) patient 3853 in TIL-3853 (Fig. 2D) within sorted PD-1− and PD-1+ PB CD4+ and CD8+ T-cell subsets was determined by TCRB deep sequencing analysis. Graphs display CD4+ and CD8+ clonotypes. The specificity of each TCRB clonotype and its rank in the infused TIL is indicated next to the symbol. ND in (B) indicates clonotypes not detected (<0.001%) in sorted PD-1− population (symbols shown on the x-axis).
This study reveals the targeting of both viral and non-viral tumor antigens by adoptively transferred TIL that resulted in complete regression of metastatic HPV+ cervical cancer. Remarkably, the immunodominant T-cell reactivity in infused TIL was directed against mutated neoantigens in one patient (35% of TIL) and the cancer-germline antigen KK-LC-1 in another patient (67% of TIL). HPV-antigen-targeted T cells represented a subdominant population of administered tumor-specific T cells in both cases (14% of TIL). Given that both patients’ infused TIL contained T cells specific for both viral and non-viral tumor antigens, as well as T cells with undefined specificity, the T-cell component(s) responsible for tumor eradication cannot be precisely determined. The relatively high frequency of infused viral and non-viral tumor antigen-specific T cells coupled with their persistence in the circulation following adoptive transfer suggests that they might have contributed to the complete cancer regressions. Furthermore, the finding that T cells targeting both viral and non-viral tumor antigens resided preferentially in the PD-1+ PB T-cell compartment suggests that anti-PD-1 therapy may act through targeting of a broad variety of tumor antigen-specific T cells in HPV+ cancers. These results have important implications for the development and immune-monitoring of future immunotherapy trials for cervical cancer and possibly other virally-induced cancers.
The immunogenicity of somatic mutations and of the cancer-germline antigen KK-LC-1 in cervical cancer revealed in this study might be exploited for therapeutic benefit. Cervical cancers harbor a variable number of somatic mutations (19, 20). Inactivation of the tumor suppressor proteins p53 and retinoblastoma, by the high-risk HPV E6 and E7 oncoproteins, respectively, impedes DNA damage repair in HPV-transformed cells (21, 22). Consequently, these cells can accumulate genomic alternations at an accelerated rate over time, including non-synonymous somatic mutations which might be expressed as neoantigens (21, 22). Tumors from patients 3775 and 3853 contained 222 and 72 mutations, respectively. In the case of patient 3375, we identified CD8+ T-cell responses against three different mutated neoantigens. How frequently somatic mutations elicit T-cell reactivity in these cancers merits further study. All identified mutations were unique or rare passenger mutations by examining the catalogue of somatic mutations in the COSMIC database. As such, targeting this class of tumor antigens requires personalized therapy that is unique to each patient (23). By contrast, infused TIL from patient 3853 recognized the cancer-germline antigen KK-LC-1, but not the mutated neoantigens. KK-LC-1 is a shared tumor antigen expressed in 40% (12/30) of metastatic cervical cancers tested using real-time qPCR (data not shown). Additionally, this antigen is reported to be expressed in gastric (81%) (24), triple-negative breast (53%) (25) and non-small cell lung (40%) (26) cancers. Importantly, KK-LC-1 is not expressed in normal tissues, except for the testis (25), suggesting it may be safely targeted by T cells. Indeed, patient 3853 received ~50 billion KK-LC-1-specific CD8+ T cells which persisted long-term at levels >2.5% without causing discernable autoimmune toxicities. Analysis of TIL grown from eight other metastatic cervical cancers that expressed KK-LC-1 demonstrated KK-LC-1-reactive TIL in two cases (data not shown), suggesting that a TCR gene transfer approach targeting this antigen may provide a strategy to circumvent the limitations of the endogenous T-cell repertoire. The KK-LC-1-specific TCR we identified recognized its cognate epitope in the context of HLA-A*01:01, a common allele expressed in ~28% of the Caucasian population (fig. S4B and F), and conferred recognition of peptide-pulsed target cells and antigen+ cancer cell lines as evidenced by IFN-γ production (fig. S5B and C). In addition, KK-LC-1-specific TCR transduced T cells as well as the KK-LC-1-reactive TIL infused into the patient specifically produced effector cytokine TNF-α and mobilized the cytotoxic degranulation marker CD107a (LAMP-1) (fig. S5D), and exerted specific cytolytic activity (fig. S5E), against cancer cell lines in an antigen- and HLA-dependent manner. Thus, these data demonstrate the potential for development of a new generation of immunotherapies based on a novel paradigm of targeting non-viral tumor antigens in cervical cancers and possibly other virally-associated tumors.
HPV-associated cancers harbor the cancer-driving E6 and E7 oncoproteins, historically the primary targets for antigen-specific immunotherapies in these malignancies (4). Despite their foreign nature, only 43% (23/54) of TIL from cervical cancers were shown to possess oncoprotein reactivity (27, 28). We found that the frequency of oncoprotein-reactive T cells infiltrating the metastatic tumors in our patients were similar to or less than the frequency of mutated neoantigen- or cancer-germline antigen-specific T cells (table S5). This may suggest that viral proteins are not necessarily more immunogenic than alternative classes of tumor antigens. In contrast to non-viral tumor antigens (29), HPV-oncoproteins have the desirable attributes of universal and constitutive expression by HPV+ cancers making them conceptually attractive antigens to target. Whether T cells solely targeting HPV-oncoproteins can cause regression of HPV+ cancers is currently under investigation using an avid TCR (30) gene therapy approach (NCT02280811 and NCT02858310).
In summary, this study reveals a previously unrecognized participation of T cells targeting non-viral tumor antigens in immunotherapy of HPV+ cervical cancer. Analysis of patients who have not responded to immunotherapy may provide additional insight into the importance of T cells targeting this class of tumor antigens for clinical effects. Our study provides an impetus for further research into the relative contribution of both viral and non-viral tumor antigen reactivities to the anti-tumor activity of adoptive T-cell therapies and immune checkpoint inhibitors for virally-associated malignancies.
Supplementary Material
Acknowledgments:
We thank the Division of Cancer Treatment and Diagnosis Tumor Repository at the NCI for providing cancer cell lines, A. Mixon, S. Farid and W. Telford for flow cytometry support, Y-C. Lu, M. El-Gamil, Y-F. Li, S.A. Feldman, S. Ilyas, A. Gros and J.R. Wunderlich for reagents, E. Tran, K. Hanada, L. Jia and L. Zhang for technical support, and the members of the Surgery Branch for helpful discussions. The data reported in this manuscript are tabulated in the main manuscript and in the supplementary materials. The raw sequence data are available through the National Center for Biotechnology Information Bioproject database, ID PRJNA342632. S.S. and C.S.H. have filed a patent application (U.S. application no. 62/327,529) that relates to the KK-LC-152–60-specific TCR. This research was supported by the Center for Cancer Research intramural research program of the NCI and the Milstein Family Foundation. B. Howie has employment and equity ownership with Adaptive Biotechnologies. H.S. Robins has consultancy, equity ownership, patents and royalties with Adaptive Biotechnologies. Other authors have no potential conflicts of interest.
Footnotes
References and Notes:
- 1.Stevanovic S et al. , Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol 33, 1543–1550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seiwert TY et al. , Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 17, 956–965 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Morris VK, Ciombor KK, Salem ME, A multi-institutional eETCTN phase II study of nivolumab in refractory metastatic squamous cell carcinoma of the anal canal (SCCA). J Clin Oncol 34, (2016). [Google Scholar]
- 4.van der Burg SH, Melief CJ, Therapeutic vaccination against human papilloma virus induced malignancies. Curr Opin Immunol 23, 252–257 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Kenter GG et al. , Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin Cancer Res 14, 169–177 (2008). [DOI] [PubMed] [Google Scholar]
- 6.van Poelgeest MI et al. , HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J Transl Med 11, 88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welters MJ et al. , Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med 8, 334ra352 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Rizvi NA et al. , Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran E et al. , Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Allen EM et al. , Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Rooij N et al. , Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31, e439–442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvistborg P et al. , TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology 1, 409–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins PF et al. , A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21, 1019–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Materials and methods are available as supplementary materials at the Science website.
- 15.Lu YC et al. , Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 20, 3401–3410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasetto A et al. , Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res 4, 734–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howie B et al. , High-throughput pairing of T cell receptor alpha and beta sequences. Sci Transl Med 7, 301ra131 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Gros A et al. , Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 22, 433–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexandrov LB et al. , Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojesina AI et al. , Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duensing S, Munger K, Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer 109, 157–162 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Moody CA, Laimins LA, Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10, 550–560 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Stronen E et al. , Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Shida A et al. , Frequent High Expression of Kita-Kyushu Lung Cancer Antigen-1 (KK-LC-1) in Gastric Cancer. Anticancer Res 35, 3575–3579 (2015). [PubMed] [Google Scholar]
- 25.Paret C et al. , CXorf61 is a target for T cell based immunotherapy of triple-negative breast cancer. Oncotarget 6, 25356–25367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuyama T et al. , Identification of a new cancer/germline gene, KK-LC-1, encoding an antigen recognized by autologous CTL induced on human lung adenocarcinoma. Cancer Res 66, 4922–4928 (2006). [DOI] [PubMed] [Google Scholar]
- 27.de Vos van Steenwijk PJ et al. , An unexpectedly large polyclonal repertoire of HPV-specific T cells is poised for action in patients with cervical cancer. Cancer Res 70, 2707–2717 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Piersma SJ et al. , Human papilloma virus specific T cells infiltrating cervical cancer and draining lymph nodes show remarkably frequent use of HLA-DQ and -DP as a restriction element. Int J Cancer 122, 486–494 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Kerkar SP et al. , MAGE-A is More Highly Expressed Than NY-ESO-1 in a Systematic Immunohistochemical Analysis of 3668Cases. J Immunother 39, 181–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Draper LM et al. , Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin Cancer Res 21, 4431–4439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones S et al. , Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran E et al. , Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C et al. , Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y et al. , Immune epitope database analysis resource. Nucleic Acids Res 40, W525–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang P et al. , A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol 4, e1000048 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peper JK et al. , An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J Immunol Methods 405, 192–198 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA, Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66, 8878–8886 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen CJ et al. , Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67, 3898–3903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haga-Friedman A, Horovitz-Fried M, Cohen CJ, Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J Immunol 188, 5538–5546 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Szymczak AL et al. , Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 22, 589–594 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.