

Not unlike Tolstoy’s remark about happy and unhappy families, current wisdom in vascular biology holds that healthy blood vessels are (excluding their tissue-specific characteristics) mostly similar, whereas vessels in different vascular diseases are mostly different. Re-evaluation of the literature, however, leads us to observe that unresolved vascular remodeling is a key element of virtually all vascular diseases. This commonality not only suggests the possibility of unifying principles that govern these processes but raises the possibility that methods to restore normal remodeling or promote resolution could effective treatments for a multiplicity of disease states.
Postnatal physiological vascular remodeling encompasses sprouting of existing blood vessels to form new ones (angiogenesis), and growth and remodeling of small vessels into larger arteries that support higher blood flow (arteriogenesis). Importantly, both of these processes are self-limited. Angiogenesis is stimulated by tissue demand for oxygen and nutrients due to tissue growth, exercise, or wound healing1. VEGF secretion by cells in ischemic tissue is the main driver but once blood flow is established in the new vessels, oxygen and nutrient transport into the tissue decreases VEGF expression, shuts down angiogenesis and restores vascular stability. Arteriogenesis is triggered mainly by increased blood flow and hence fluid shear stress through small vessels, which induces a local inflammatory response leading to VEGF production2. The resultant outward remodeling, vessel enlargement and de novo formation of arterial vasculature restore normal levels of shear stress, resolving vascular wall inflammation and completing the repair. These processes are of relatively short duration, on the order of days to weeks in mouse models, and fully restore normalcy.
In contrast, prolonged or chronic stimulation results in pathological remodeling and leads to formation of morphologically and functionally abnormal vessels. One example is an atherosclerotic plaque which form due to converging biomechanical (disturbed flow and high wall stress), metabolic (hyperlipidemia, hyperglycemia), inflammatory and thrombotic factors3,4. Plaque initiation and progression appear to be driven by altered endothelial phenotype, including elevated permeability that allows lipoproteins to enter the vessel wall, increased expression of leukocyte attractants that mediate recruitment of monocytes and other immune cells, and increased migration and growth of smooth muscle cells, which then form a neointima that narrows the vessel lumen. Further, vascular wall inflammation increases sensitivity of endothelial cells to TGFβ, leading to endothelial-mesenchymal transition (EndMT)5. The resultant endothelial-derived mesenchymal cells drive further increases in permeability, altered ECM and inflammation, thus perpetuating the incomplete repair state.
A similar process occurs in vascular malformations caused by mutations in CCM and HHT genes 6,7. In inherited form of these diseases, patients have a heterozygous loss of function mutation in a key gene (CCM1,2 or 3 for CCM; or Alk1, endoglin or Smad4 for HHT). A “second hit” mutation plus angiogenic or inflammatory stimuli leads to clonal expansion and formation of lesions with poorly formed vessel walls that are prone to rupture and bleeding. Once again, the pattern of elevated permeability, abnormal matrix, inflammation and EndMT can form positive feedback circuits that contribute to lesion progression. Yet another example is chronic inflammation due to persistent infection, injury or autoimmune processes. In these conditions, angiogenic blood vessels remain immature with increased permeability, matrix remodeling and expression of leukocyte adhesion molecules that perpetuate inflammation 8.
There are several well-conserved features that distinguish stable from remodeling vasculature, (Table 1). In stable vessels, the endothelium rests on a basement membrane comprised mainly of laminins and type IV collagen with associated glycoproteins but low levels of provisional matrix proteins such as fibronectin and fibrin 4. By contrast, all forms of instability are associated with metalloprotease secretion, matrix degradation assembly of provisional matrices, and alterations in key endothelial signaling inputs. In stable vessels, angiopoetin 1, which binds its receptor Tie1 on the endothelium to stabilize the vessels, is high, while its antagonist, angiopoetin 2, is low 9. Junctional permeability is low in stable vessels and elevated during inflammation and remodeling 8. This rule has exceptions since fenestrated endothelium allows passage of fluids, and high endothelial venules allow passage of cells, but these forms of permeability have distinct mechanisms. All forms of remodeling are also associated with a shift toward a more oxidative state, or in the case of pathologies, to oxidative Stress10
While stable vessels exhibit normal endothelial cell fate markers, vascular instability is associated with EndMT, a process characterized by variable loss of endothelial markers and the appearance of mesenchymal markers 11. Functional consequences include increased permeability, increased expression of leukocyte adhesion receptors, enhanced cell migration and expression and deposition of provisional matrix. EndMT has been observed in physiological and pathological angiogenesis, and in atherosclerosis, chronic inflammation and vascular malformations.
How do physiological and pathological remodeling differ? In physiological remodeling, the initiating stimulus disappears once adaptation completed, leading to restoration of normalcy. By contrast, in pathological remodeling, the initial triggers generally persist. Furthermore, perhaps due to the prolonged period of instability and remodeling, even when initiating triggers are removed, the vasculature does not return to its normal state. Such persistence involves positive feedback loops that maintain the endothelium in the remodeling/disease state, leading to disease progression. Several examples illustrate this point.
In atherosclerosis, statins, for example, slow progression but do not stop or reverse the disease even when plasma lipids are drastically lowered; endothelial cells overlying atherosclerotic plaques remain activated. Some relevant positive feedback loops involve increased vascular permeability and expression of leukocyte adhesion molecules. Entry of circulating matrix proteins such as fibronectin and fibrinogen into the vessel wall contributes to deposition of pro-inflammatory provisional matrix. Leaky endothelium also allows entry of lipoproteins into inflamed vessel wall under oxidative stress, resulting in production of pro-inflammatory oxidized lipoproteins that further increase lipid uptake and inflammatory mediators. Activation of adaptive immune responses to damaged proteins within the plaque and enhanced entrance of circulating inflammatory cells due to increased expression of adhesion molecules accelerate disease3. Similar processes take place in transplant arteriopathy characterized by low-grade persistent vascular inflammation and incomplete repair 12. CCMs also exhibit this constellation of increased permeability, altered matrix, inflammatory activation and EndMT that very likely mediate disease progression 7.
These findings imply that persistent instability is a central feature of multiple vascular disorders (Fig 1). EndMT is very likely an important component due to conversion of ECs to migratory mesenchymal cells that cause irreversible changes in tissue architecture. EndMT is induced by inflammation-driven decreases in protective endothelial FGF signaling, which sensitizes ECs to TGFβ 11. Thus transformed, endothelial cells express high levels of leukocyte adhesion molecules and altered ECM, and have high vascular permeability, further driving inflammation. These events establish a positive feedback loop in which EndMT drives inflammation that leads to further EndMT. This inflammation/TGFβ circuit also promotes fibrosis, which is largely irreversible.
Figure 1. Persistent instability and disease.
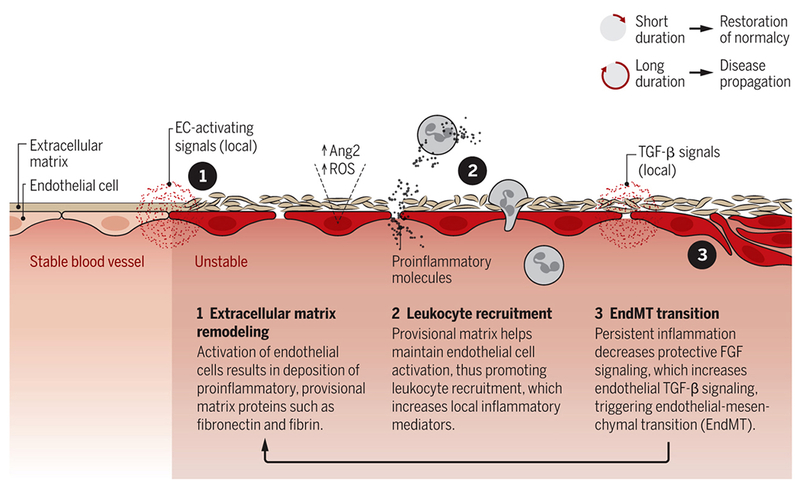
Local signals activate blood vessel endothelial cells (ECs), triggering events that transform ECs and create further instability. This is a feedforward cycle, as instability leads to matrix remodeling, leukocyte recruitment, inflammation, and EC transformation, which accelerates matrix remodeling and so on. Long duration of this process may lead to numerous pathologies. ROS, reactive oxygen species.
This hypothesis implies that interventions to restore vascular stability might improve outcomes in multiple disease states. This might be achieved via agonists for Tie2, FGF receptors or other pathways that promote stability, or antagonists for inflammatory or endothelial TGFβ pathways that promote instability. The key requirement is to break the feedback loops that maintain the remodeling state and thereby restore tissue homeostasis. While this notion flies in the face of current trends toward personalized medicine, the search for unifying principles that govern health and disease is as old as medicine. much remains to be done before we achieve the deep understanding of biological processes needed to design effective interventions in vascular disease, yet our current state of knowledge offers a glimpse of the path forward.
References
- 1.Adams RH & Alitalo K Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8, 464–78 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Simons M & Eichmann A Molecular controls of arterial morphogenesis. Circ Res 116, 1712–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tabas I, Garcia-Cardena G & Owens GK Recent insights into the cellular biology of atherosclerosis. J Cell Biol 209, 13–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr., Finney AC, Woolard MD & Orr AW The arterial microenvironment: the where and why of atherosclerosis. Biochem J 473, 1281–95 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen PY et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 125, 4514–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tual-Chalot S, Oh SP & Arthur HM Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front Genet 6, 25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lampugnani MG, Malinverno M, Dejana E & Rudini N Endothelial cell disease: emerging knowledge from cerebral cavernous malformations. Curr Opin Hematol 24, 256–264 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Nagy JA, Benjamin L, Zeng H, Dvorak AM & Dvorak HF Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11, 109–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parikh SM Angiopoietins and Tie2 in vascular inflammation. Curr Opin Hematol 24, 432–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santilli F, D’Ardes D & Davi G Oxidative stress in chronic vascular disease: From prediction to prevention. Vascul Pharmacol 74, 23–37 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Dejana E, Hirschi KK & Simons M The molecular basis of endothelial cell plasticity. Nat Commun 8, 14361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell RN Allograft arteriopathy: pathogenesis update. Cardiovasc Pathol 13, 33–40 (2004). [DOI] [PubMed] [Google Scholar]