

Abstract
Background:
Mutations in recombination-activating gene (RAG) 1 and RAG2 are associated with a broad range of clinical and immunologic phenotypes in human subjects.
Objective:
Using a flow cytometry–based assay, we aimed to measure the recombinase activity of naturally occurring RAG2 mutant proteins and to correlate our results with the severity of the clinical and immunologic phenotype.
Methods:
Abelson virus–transformed Rag2–/– pro-B cells engineered to contain an inverted green fluorescent protein (GFP) cassette flanked by recombination signal sequences were transduced with retroviruses encoding either wild-type or 41 naturally occurring RAG2 variants. Bicistronic vectors were used to introduce compound heterozygous RAG2 variants. The percentage of GFP-expressing cells was evaluated by using flow cytometry, and high-throughput sequencing was used to analyze rearrangements at the endogenous immunoglobulin heavy chain (Igh) locus.
Results:
The RAG2 variants showed a wide range of recombination activity. Mutations associated with severe combined immunodeficiency and Omenn syndrome had significantly lower activity than those detected in patients with less severe clinical presentations. Four variants (P253R, F386L, N474S, and M502V) previously thought to be pathogenic were found to have wild-type levels of activity. Use of bicistronic vectors permitted us to assess more carefully the effect of compound heterozygous mutations, with good correlation between GFP expression and the number and diversity of Igh rearrangements.
Conclusions:
Our data support genotype-phenotype correlation in the setting of RAG2 deficiency. The assay described can be used to define the possible disease-causing role of novel RAG2 variants and might help predict the severity of the clinical phenotype.
Keywords: Recombination-activating gene 2, VDJ recombination, severe combined immunodeficiency, Omenn syndrome, autoimmunity, genotype-phenotype correlation
GRAPHICAL ABSTRACT
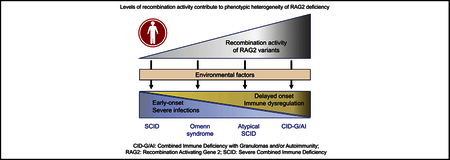
Recombination-activating gene (RAG) 1 and RAG2 proteins initiate the VDJ recombination process, allowing generation of T and B lymphocytes with a largely diversified repertoire of antigen receptor specificities (reviewed by Notarangelo et al1). In particular, RAG1 and RAG2 form a heterotetrameric complex that binds to the recombination signal sequences flanking each of the variable (V), diversity (D) and joining (J) gene segments and introduces double-strand DNA breaks that are sealed by hairpins. On opening of the hairpin by ARTEMIS, proteins of the nonhomologous end-joining machinery complete the VDJ recombination process by mediating joining of the coding ends and formation of signal joins.1
Mutations of the RAG1 and RAG2 genes in human subjects are associated with a broad range of clinical and immunologic phenotypes, including severe combined immunodeficiency (SCID) with absence of T and B lymphocytes (T−B− SCID),2 Omenn syndrome (OS),3 atypical severe combined immunodeficiency (AS),4 combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI),5 and other milder presentations.
We have reported previously on a flow cytometry–based assay in which Abelson virus (v-Abl)–transformed Rag1–/– pro-B cells engineered to contain an inverted green fluorescent protein (GFP) cassette flanked by recombination signal sequences (pMX-INV) were transduced with retroviruses encoding either a wild-type (WT) or a mutant human RAG1 protein.6 In this assay, because the Rag1−/− pro-B cells express both RAG2 and the components of the nonhomologous end-joining machinery, the proportion of GFP+ cells can be used to measure the recombinase activity of the introduced human RAG1 protein. The results obtained supported correlation between levels of recombination activity of the mutant RAG1 proteins and the severity of the clinical and immunologic phenotype observed in patients with RAG1 deficiency.6
Here, using a similar assay based on a v-Abl Rag2−/− pro–B-cell line containing a single pMX-INV integrated cassette, we report on the systematic analysis of recombination activity of 41 naturally occurring human RAG2 variant proteins (including compound heterozygous variants) identified in 58 patients with various clinical and immunologic phenotypes. Our results demonstrate that genotype-phenotype correlation also exists for patients with RAG2 deficiency. Therefore this assay might be useful in determining the pathogenicity of newly identified RAG2 mutations.
METHODS
Patients
Deidentified clinical, molecular, and immunologic data of patients with RAG2 mutations were collected both by means of literature search and through collaboration with referring clinicians. The study was approved by the Institutional Review Board at Boston Children’s Hospital and at local institutions of the referring physicians and was conducted according to protocol 16-I-N139 approved by the National Institutes of Health Institutional Review Board.
Analysis of recombination activity
The v-Abl Rag2−/− Em-Bcl2 pro–B-cell line with a pMX-INVand human truncated hCD4 cassette was generated by transducing v-Abl Rag2−/− pro-B cells with a retrovirus containing the pMX–INV–internal ribosome entry site (IRES)–hCD4 plasmid, as previously described.7,8 Cells with successful integration of the pMX-INV-IRES-hCD4 cassette express a truncated human CD4 surface protein, allowing for selection by using anti-hCD4 magnetic beads (MACS; Miltenyi Biotech, Bergisch Gladbach, Germany). The hCD4-enriched cells were further transduced with a retrovirus containing either the pBMN-WT-hRAG2-IRES-hCD2 or pBMN-Mut-hRAG2-IRES-hCD2 plasmid expressing WT or mutant (Mut) human RAG2 (hRAG2) protein, respectively. In parallel, Rag2−/− v-Abl pro-B cells were transduced with a retrovirus containing pBMN-empty-IRES-hCD2 construct as a negative control.
To analyze the recombination activity of 2 distinct RAG2 variants for which compound heterozygosity had been identified in patients, we developed bicistronic vectors (RAG2 [allele 1]-T2A-RAG2 [allele 2]-IRES-hCD2) using overlapping PCR (see the Methods section in this article’s Online Repository at www.jacionline.org). Successfully transduced cells were enriched with anti-hCD2 magnetic beads (MACS; Miltenyi Biotech). As a negative control, v-Abl Rag2−/− Eμ-Bcl2 pMX-INV-IRES-hCD4 pro-B cells, hereafter referred to as Rag2−/− Abl pro-B cells, were transduced with a retrovirus containing a pBMN-empty-IRES-hCD2 plasmid, allowing for expression of hCD2 but not hRAG2.
The Rag2−/− v-Abl pro-B cells transduced with the vectors indicated above were incubated with 3 μmol/L STI-571/imatinib (Novartis, Basel, Switzerland), an inhibitor of the Abl kinase, thereby maintaining cells in the G0-G1 phase of the cell cycle and allowing for RAG1 and RAG2 to initiate VDJ recombination more efficiently, whereas the presence of the Bcl2 transgene prevents cell death. After 96 hours, cells were harvested and GFP expression was analyzed by using flow cytometry on gating on cells expressing both hCD4 and hCD2. The relative recombination activity of the individual RAG2 variants was calculated as a percentage of WT-hRAG2 activity.
High-throughput sequencing analysis of rearrangements at the endogenous immunoglobulin heavy chain locus
Analysis of immunoglobulin heavy chain (Igh) locus rearrangements in transduced Rag2−/− v-Abl pro-B cells was performed by Adaptive Biotechnologies (Seattle, Wash), as described previously.9 Sequences were aligned to a reference genome, and Igh VDJ gene definitions were based on ImMuno-GeneTics system (see Table E1 in this article’s Online Repository at www.jacionline.org).10 These sequences are available in Table E1. Heat maps were generated to detail the frequency of individual V-J rearrangements within unique sequences of rearrangement products. The Shannon diversity index and Simpson index were calculated by using the PAST program, as described previously,11 and according to the following formulas:
where R indicates total templates, i indicates unique rearrangements, pi indicates the proportion of the total sequences belonging to the ith unique rearrangement, and D indicates dominance and unevenness.
In particular, the Simpson D index takes into account both richness and evenness. With this index, 0 represents infinite diversity, and 1 indicates no diversity.
Statistical analysis
Statistical analyses for 1-way ANOVA for nonparametric analysis (Kruskal-Wallis) and post hoc analysis of the unpaired, 1-tailed, nonparametric Mann-Whitney test were carried out with Prism 5 software (GraphPad, La Jolla, Calif).
RESULTS
Patients’ clinical and molecular features
We compiled a list of 58 RAG2-deficient patients from published manuscripts and unpublished sources. These patients were divided into 4 different groups based on their clinical and immunologic presentation and according to the diagnostic criteria of the Primary Immunodeficiency Treatment Consortium.12 In particular, 20 patients were given a diagnosis of T−B−SCID,2,4,13–21 28 patients were given a diagnosis of OS,2,4,14–19,22–27 7 patients were given a diagnosis of AS,26,28 and 3 patients were given a diagnosis of CID-G/AI (Table I).2,4,5,13–28
TABLE I.
Clinical and genetic information of patients with RAG2 deficiency
| Clinical phenotype | Autoimmunity/inflammation | Infections/malignancy | Patient no. | Mutation | Age | ALC (cells/μL) | CD3 (cells/μL) | CD19 (cells/μL) | CD16/56 (cells/μL) | References |
|---|---|---|---|---|---|---|---|---|---|---|
| CID-G/AI (n = 3) | Aplastic anemia; granulomas in spleen, lungs, and skin; ITP; neutropenia; splenomegaly | ARDS, disseminated and vaccine-associated varicella, meningitis, pneumonia, sinusitis, infections with Cryptococcus species, EBV, and RSV | P1 | a. T77N; b. G451A | 2 y | 769–1,554 | 538–1,057 | 54–202 | 131–355 | 5 |
| P2 | a. G451A; b. M459L | 9 mo | 480 | 138 | 12 | 86 | 28 | |||
| P3 | a-b. F62L | 5 y | 687 | 391 | 78 | 215 | 28 | |||
| AS (n = 7) | AIHA, alopecia areata, APS, biliary cirrhosis, granulomas in skin and bone marrow, hepatomegaly, IBD, ITP, neutropenia, splenomegaly, psoriasis, polyarthritis | Bronchiectasis, cholecystitis, chronic diarrhea, DIC, fungal nail infections, hyper-IgM syndrome, jaundice, Klebsiella species, meningitis, molluscum, oral thrush, otitis media, PJP, pneumonia, Pseudomonas species sepsis, rhinorrhea, skin rash-papules and abscesses, vaccine, associated varicella, RSV, CMV, and EBV viremia | P4 | a-b. M459L | 4 mo | NA | 691 | 173 | 657 | 26 |
| P5 | a. R73H; b. P180H | 13 mo | 1,479 | 401 | 524 | 654 | ||||
| P6 | a-b. G35A | 7 mo | 1,027 | 716 | 105 | 209 | 28 | |||
| P7 | a-b. G35A | 10 mo | 1,108 | 13 | 276 | |||||
| P8 | a-b. G35A | 12 mo | 2,700 | 717 | 131 | 898 | ||||
| P9 | a. G35A; b. E437K | 5 mo | 3,460 | 1,384 | 62 | 1,176 | ||||
| P10 | a-b. E407X | 18 y | 3,480 | 2,750 | 0 | 661 | 28 | |||
| OS (n = 28) | AIHA, eczema erythroderma, generalized edema, hepatomegaly, hepatosplenomegaly, lymphadenopathy, seborrhea-like dermatitis, splenomegaly | BCGitis, chronic diarrhea, CMV infection, failure to thrive, LAD, interstitial pneumonia, myocarditis, onychomycosis, recurrent chest infection and URTI, PJP, prolonged rotavirus infection, Pseudomonas pneumonia, Staphylococcus aureus skin infection, sepsis, severe infections | P11 | a.K440N; b.P253R | 7 mo | 792 | 103 | 8 | 657 | 17 |
| P12 | a. S160L; b. M502V | 3 wk | 9,064 | 7,250 | 0 | 1,360 | 17 | |||
| P13 | a-b. T215I | NA | 600 | 240 | 0 | 198 | 16 | |||
| P14 | a-b. M459L | 4 mo | 869 | 149 | 4 | 279 | 26 | |||
| P15 | a. C41W; b. M285R | 1 wk | 5,880 | 2,646 | 1 | 2,470 | 4 | |||
| P16 | a.C41W; b.M285R | 0 mo | 660 | 554 | <6 | 66 | ||||
| P17 | a-b. A456T | 0 mo | 45,000 | 41,000 | 0 | NA | 23 | |||
| P18 | a. Q278X; b. R73H | 0 mo | 8,339 | 7,071 | 0 | NA | 25 | |||
| P19 | a-b. R229W | 0 mo | 280 | 162 | 17 | 53 | 4 | |||
| P20 | a-b. R229W | 4 mo | 77 | 4 | 2 | 4 | ||||
| P21 | a-b. R229W | 4 mo | NA | 61 | 2 | 19 | 4 | |||
| P22 | a-b. R229W | NA | 287 | 46 | 6 | 184 | 16 | |||
| P23 | a-b. R229W | NA | 1,972 | 1,045 | 39 | 375 | 16 | |||
| P24 | a-b. R229W | 15 d | 889 | 595 | 4 | 231 | 17 | |||
| P25 | a. R229W; b.G95R | NA | 1,953 | 1,074 | 78 | 19 | 16 | |||
| P26 | a. G95R; b. W453R | <5 mo | 10,000 | 5,200 | 204 | NA | 22 | |||
| P27 | a-b. R229Q | 2 wk | 8,600 | 3,698 | <86 | 3,956 | 4, 24 | |||
| P28 | a.R39G; b.R229Q | 2 mo | 10,000 | 7,400 | 0 | NA | 14 | |||
| P29 | a. R229Q; b. locus del | 2.5 mo | 322 | 113 | 16 | 161 | 2, 4 | |||
| P30 | a-b. E480X | NA | 11,000 | 2,778 | 0 | 3,740 | 19 | |||
| P31 | a.G95R; b.E480X | 3 mo | 11,000 | 2,871 | 0 | 3,800 | 27 | |||
| P32 | a-b. I444M | 3 mo | 620 | 415 | <6 | 180 | 18 | |||
| P33 | a-b. W416L | 3 mo | 34,000 | 31,647 | 0 | 1,347 | 17 | |||
| P34 | a.-b. W453R | NA | NA | NA | NA | NA | 15 | |||
| P35 | a-b. G35V | NA | 1,850 | 537 | 130 | 370 | 16 | |||
| P36 | a-b. M443I | 1 mo | 5,700 | 2,354 | 11 | NA | 25 | |||
| P37 | a-b. G157V | NA | 5,600 | 4,592 | 56 | 504 | 19 | |||
| P38 | a-b. G35V | 4 mo | 1,320 | 488 | 0 | 500 | 27 | |||
| SCID (n = 20) | Eczema, erythroderma, lymphadenopathy, hepatosplenomegaly might be present in patients with maternal T-cell engraftment | BCGitis, diarrhea, failure to thrive, oral ulcers, oral thrush, oral candidiasis pneumonia, protracted diarrhea, respiratory distress | P39† | a-b. N474S | 1 wk | 1,120 | 22 | 1 | 784 | 4 |
| P40 | a-b. R229W | NA | 110 | 40 | 0 | 45 | 16 | |||
| P41 | a-b. R229W | 1 mo | 1,076 | 126 | 1 | 869 | ||||
| P42 | a-b. R229W | NA | NA | NA | NA | NA | 21 | |||
| P43 | a-b. R229W | NA | NA | NA | NA | NA | 21 | |||
| P44 | a-b.T215I & R229Q | 12 d | 684 | 5 | 7 | 382 | 20 | |||
| P45 | a. R39G; b. R229Q | NA | NA | 0 | 0 | NA | 13 | |||
| P46 | a-b. D65Y | NA | 400 | 16 | 8 | 228 | 19 | |||
| P47 | a-b. Q16X | NA | NA | NA | NA | NA | 15 | |||
| P48 | a-b. G35V | NA | 576 | 110 | 0 | 432 | 16 | |||
| P49 | a-b. G35V | NA | 290 | 9 | 0 | 159 | 16 | |||
| P50 | a-b. G35V | NA | NA | 0 | 0 | NA | 14 | |||
| P51 | a-b. G35V | 3 mo | NA | NA | NA | NA | 17 | |||
| P52 | a-b. R41W | 6 mo | NA | 5 | 1 | 804 | ||||
| P53 | a-b. C478Y | NA | NA | 0 | 0 | NA | 2 | |||
| P54 | a-b. W307X | 4 mo | 720 | <7 | <7 | 533 | 4 | |||
| P55 | a-b. C478Y | 0 mo | 2,000 | 80 | <20 | 820 | 4 | |||
| P56† | a-b. C478Y | 2 wk | 5,000 | 3,500* | <50 | 2,050 | 2, 4 | |||
| P57 | a-b. K127X | 5 mo | 810 | NA | <8 | 760 | 18 | |||
| P58 | a-b. K127X | 1 mo | 978 | <10 | <10 | 918 | 18 |
Age, Age at presentation; AIHA, autoimmune hemolytic anemia; ALC, absolute lymphocyte count; APS, antiphospholipid syndrome; ARDS, acute respiratory distress syndrome; BCGitis, systemic dissemination of the attenuated Mycobacterium bovis bacillus of the tuberculosis vaccine; CMV, cytomegalovirus; DIC, disseminated intravascular coagulation; IBD, inflammatory bowel disease; ITP, immune thrombocytopenic purpura; LAD, lymphadenopathy; NA, not available; N, normal; PJP, Pneumocystis jirovecii pneumonia; RSV, respiratory syncytial virus; URTI, urinary tract infection.
Maternal.
Maternal engraftment.
Analysis of the distribution of RAG2 mutations by disease phenotype revealed that some mutations were observed in multiple patients with the same phenotype, such as p.G35A, which was reported in 4 patients with AS, and the p.C478Y mutation, which was detected in 3 patients with SCID. Moreover, 6 other mutations (G95R, K127X, M285R, G451A, W453R, and E480X) were identified in 2 patients each, with the same clinical phenotype (Fig 1). Missense mutations at residues G35, T215, and R229 were identified in 24 patients who presented with either SCID or OS, indicating that these mutations are associated with a severe but somewhat variable phenotype. Furthermore, missense mutations at residues R73 and M459 were reported in patients with either OS or AS. By contrast, other mutations (F62L, T77N, and G451A) were reported only in patients with CID-G/AI (Fig 1).
FIG 1.

Distribution of RAG2 mutations. Graphic presentation of different RAG2 mutations according to their position in the RAG2 protein and abundance of the patients according to various clinical presentations. For 5 RAG2 mutations (M1T, M110L, Y195D, C446W, and H481P), complete genetic and clinical information for the patients is not available. PHD, Plant homology domain.
Distribution and recombination activity of mutant RAG2 proteins
As a preliminary step to analyze the recombination activity of the RAG2 variants identified in the patients, we transduced murine Rag2−/− Abl pro-B cells with a vector encoding for either human or mouse WT RAG2 (WT-hRAG2 and WT-mRAG2) or with an empty vector and compared levels of GFP expression on treatment with STI-571 (Fig 2, A). As shown in Fig 2, B, similar levels of GFP expression were induced by hRAG2 and mRAG2, whereas no GFP expression was observed in cells transduced with an empty vector (Fig 2, B). Altogether, these data indicate that hWT-RAG2 can cooperate efficiently with mWT-RAG1 in mediating recombination.
FIG 2.

Assay to measure recombination activity of human RAG2 variants. A, Schematic representation of the recombination assay. Rag2−/− v-Abl–transformed pro-B cells in which an inverted GFP cassette flanked by a recombination signal sequence (RSS) had been integrated stably in the genome (top) were transduced with a retroviral vector encoding either WT or mutant human RAG2 (hRAG2) and hCD2 as a reporter (middle). In this system GFP expression is measured by using fluorescence-activated cell sorting as a readout of successful recombination induced by the RAG2 protein in the presence of WT-mRag1 and the intact nonhomologous end-joining (NHEJ) pathway (bottom). LTR, Long terminal repeat. B, Representative fluorescence-activated cell-sorting dot plot of Rag2−/− v-Abl pro-B cells transduced with an empty vector (pBMN[mock]) or with retroviral vectors encoding either WT human RAG (WT-hRAG), WT mouse Rag2 (WT-mRAG2), or RAG2 mutants identified in patients with various clinical presentations (T−B−SCID, OS, and CID-G/AI).
Initially, Rag2−/− Abl pro-B cells were transduced with vectors encoding for various RAG2 mutant proteins (C478Y, A456T, W416L, and G451A) that had been identified in patients with SCID, OS, OS, and CID-G/AI, respectively. As shown in Fig 2, B, no detectable levels of GFP expression were induced by the C478Y mutant protein, whereas the G451A mutant protein supported significant levels of GFP expression, although levels were lower than in the presence of hWT-RAG2. Finally, low but detectable levels of GFP expression were supported by the A456Tand W416L mutant proteins associated with OS (Fig 2, B).
Based on these data, we performed a comprehensive analysis of the recombinase activity of 41 naturally occurring RAG2 mutations, including 31 for which no data of functional activity had been reported previously. Among the 41 mutations tested, 6 were nonsense mutations, and the remaining 35 were missense mutations. Both the nonsense and missense mutations were distributed broadly along the length of the hRAG2 protein (Fig 3, A, and Table II). A higher density of mutations was observed in the plant homology domain at the C-terminus of the molecule, where mutations affected 18% of the amino acids included in the region compared with 6% and 3% of the amino acids being targeted by mutations in the core and acidic regions of RAG2, respectively (Fig 3, B). Although the 6 nonsense mutations supported no or minimal levels of recombination (range, 0.1% to 2.9% of WT-hRAG2), variable levels of functional activity could be ascribed to the 35 missense mutations (mean, 25.2% [range, 0.2% to 109.1%] of WT-hRAG2; Fig 3, C). Overall, missense variants supported significantly greater recombination activity than nonsense variants (P < .01; Fig 3, C). Finally, no clear difference of functional activity was observed for mutations affecting the plant homology domain or other domains of the protein (Fig 3, D).
FIG 3.

Recombination activity of mutant RAG2 proteins. A, Graphic representation of the recombination activity of various RAG2 mutations according to their position in different domains of the RAG2 protein (core domain: amino acids 1–383; plant homology domain [PHD]: amino acids 414–487). Solid circles identify nonsense mutations, and triangles represent missense mutations. Means ± SEMs are shown to illustrate recombination activity. For each RAG2 variant, the assay was performed in triplicates. B, Frequency of pathogenic mutations (defined as recombination activity <80% WT-hRAG2) per amino acid length of various domains of the hRAG2 protein. C, Recombination activity (expressed as percentage of the activity of WT-hRAG2) of nonsense and missense hRAG2 mutations. Bars represent means ± SEMs. Statistical analysis was performed with the Mann-Whitney test. D, Recombination activity (expressed as percentage of the activity of WT-hRAG2) of missense mutations affecting the PHD and non-PHD domains of the hRAG2 protein. Bars represent means 6 SEMs. Statistical analysis was performed with the Mann-Whitney test. n.s., Not significant.
TABLE II.
Summary of RAG2 mutations and recombination activity levels
| No. | Amino acid position | Domain affected by the mutation | Mutation | Mean recombination activity (% of WT hRAG2) | SEM |
|---|---|---|---|---|---|
| 1 | 1 | Core | M1T | 65.3 | 2.2 |
| 2 | 16 | Core | Q16X | 1.7 | 0.4 |
| 3 | 35 | Core | G35A | 22.1 | 3.1 |
| 4 | 35 | Core | G35V | 0.4 | 0.3 |
| 5 | 39 | Core | R39G | 0.2 | 0.1 |
| 6 | 41 | Core | C41W | 0.2 | 0.4 |
| 7 | 62 | Core | F62L | 19.6 | 3.0 |
| 8 | 65 | Core | D65Y | 6.8 | 1.2 |
| 9 | 73 | Core | R73H | 12.4 | 1.4 |
| 10 | 77 | Core | T77N | 42.6 | 2.7 |
| 11 | 95 | Core | G95R | 0.3 | 0.2 |
| 12 | 110 | Core | M110L | 74.6 | 1.8 |
| 13 | 127 | Core | K127X | 0.1 | 0.0 |
| 14 | 157 | Core | G157V | 0.4 | 0.2 |
| 15 | 160 | Core | S160L | 5.8 | 0.6 |
| 16 | 180 | Core | P180H | 31.1 | 0.5 |
| 17 | 195 | Core | Y195D | 2.0 | 0.3 |
| 18 | 215 | Core | T215I | 67.2 | 1.0 |
| 19 | 229 | Core | R229Q | 8.9 | 1.0 |
| 20 | 229 | Core | R229W | 10.5 | 0.5 |
| 21 | 253 | Core | P253R | 95.4 | 2.3 |
| 22 | 278 | Core | Q278X | 0.1 | 0.1 |
| 23 | 285 | Core | M285R | 24.7 | 0.8 |
| 24 | 307 | Core | W307X | 0.2 | 0.2 |
| 25 | 386 | Acidic | F386L | 109.1 | 5.0 |
| 26 | 407 | Acidic | E407X | 2.9 | 0.4 |
| 27 | 416 | PHD | W416L | 1.4 | 0.2 |
| 28 | 437 | PHD | E437K | 0.9 | 0.2 |
| 29 | 440 | PHD | K440N | 26.7 | 2.4 |
| 30 | 443 | PHD | M443I | 0.4 | 0.2 |
| 31 | 444 | PHD | I444M | 2.7 | 0.3 |
| 32 | 446 | PHD | C446W | 2.9 | 0.1 |
| 33 | 451 | PHD | G451A | 66.3 | 4.8 |
| 34 | 453 | PHD | W453R | 0.6 | 0.1 |
| 35 | 456 | PHD | A456T | 16.0 | 2.9 |
| 36 | 459 | PHD | M459L | 30.8 | 0.6 |
| 37 | 474 | PHD | N474S | 97.5 | 5.9 |
| 38 | 478 | PHD | C478Y | 0.2 | 0.1 |
| 39 | 480 | PHD | E480X | 2.8 | 0.6 |
| 40 | 481 | PHD | H481P | 23.8 | 3.9 |
| 41 | 502 | — | M502V | 99.6 | 3.4 |
The activity of each RAG2 variant was determined by at least 3 independent experiments from which the mean value was calculated.
PHD, Plant homology domain.
Interestingly, among the 41 RAG2 variants tested, 4 (P253R, F386L, N474S, and M502V) showed levels of recombination activity comparable with that associated with hWT-RAG2 (Table II).
For all of the RAG2 variants tested, we reported their minor allele frequency (MAF) in the Genome Aggregation Consortium (gnomAD) database,29 which calculated the combined annotation-dependent depletion (CADD)–PHRED score30 to predict deleteriousness and incorporated these data and the results of functional analysis of recombination activity in a scheme to assess pathogenicity according to guidelines of the American College of Medical Genetics and Genomics.31 We reported in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) all the variants studied here (see Table E2 in this article’s Online Repository at www.jacionline.org).
Correlation of the recombination activity of mutant RAG2 proteins with clinical phenotype
On excluding 3 patients whose RAG2 mutant proteins (P253R, N474S, and M502V) supported WT levels of recombination activity, we aimed to determine whether the mean recombination activity of the mutant alleles identified in the remaining 55 patients correlated with the severity of the clinical phenotype. As shown in Fig 4, mutations associated with SCID and OS allowed similar levels of recombination activity, which were significantly lower than those identified in patients with the AS and CID-G/AI phenotypes. Although only a few patients with CID-G/AI were included in this study, these data support the notion that genotype-phenotype correlation exists also for human RAG2 deficiency, as previously found for RAG1 deficiency.6
FIG 4.

Correlation between RAG2 recombination activity and severity of clinical presentation. Representation of recombination activity of mutant RAG2 alleles according to the clinical phenotype in 55 patients with RAG2 deficiency. Patients with variants resulting in values of 100% were omitted from this analysis. In the case of patients with compound heterozygous mutations, recombination activity corresponding to the allele with the higher activity is shown. Bars represent means ± SEMs.
Analysis of recombination activity of compound heterozygous RAG2 variants
Among the 58 patients, 16 were compound heterozygous for 15 distinct combinations involving a total of 21 distinct RAG2 variants (Table I). In several of these cases, the 2 RAG2 alleles supported markedly different levels of recombination activity, making it difficult to predict the net effect of the compound heterozygous RAG2 variants on V(D)J recombination.
Furthermore, the RAG complex is expressed as a heterotetramer including 2 molecules each of RAG1 and RAG2, so that the simultaneous presence of 2 distinct RAG2 variants in the complex might have very different effects than 2 copies of each variant alone. To investigate this in greater detail, we generated bicistronic retroviral vectors expressing equimolar amounts of 2 distinct RAG2 cDNA variants and did this for 5 cases of compound heterozygous variants that had been identified in patients P2, P9, P12, P44, and P45 included in this study. In parallel, a bicistronic vector expressing 2 copies of hWT-RAG2 was also generated. Analysis of the recombination activity supported by the bicistronic vectors produced results that were not easily predictable based on the activity of each individual variant (Fig 5, A). For example, P44 was compound heterozygous for 2 variants (T251I and R229Q) that supported 67.2% and 8.9% WT recombination activity, respectively. The bicistronic vector including both variants allowed levels of recombination activity (63%) that were very similar to those of the allele with higher residual function. By contrast, P9 was compound heterozygous for the G35A and E437K variants, which supported 22.1% and 0.9% recombination activity, respectively, whereas the bicistronic vector expressing both of these variants yielded levels of recombination activity (3.9%) that were similar to the less functional allele.
FIG 5.

Recombination activity of compound heterozygous RAG2 variants. A, Analysis of the recombination activity supported by bicistronic vectors simultaneously expressing 2 RAG2 variants and comparison with recombination activity of single variants. Experiments were done in triplicates. B, Pearson correlation analysis between recombination activity supported by bicistronic vectors versus number of unique sequences. C, Graphic representation of Igh repertoire diversity. D, Quantitative measurement of diversity and unevenness of the Igh repertoire.
Next, we studied the richness and diversity of V-D-J rearrange ments introduced by the bicistronic vectors at the endogenous Igh locus in Rag2−/− v-Abl pro-B cells. As shown in Fig 5, B, there was a robust correlation between levels of recombination activity (as measured based on GFP expression) and the number of unique reads of Igh rearrangements obtained on introduction of bicistronic vectors expressing 2 copies of WTor mutant RAG2 protein. more detailed analysis of V-J rearrangements (Fig 5, C, and see Fig E1 in this article’s Online Repository at www.jacionline.org) showed that introduction of 2 copies of hWT-RAG2 into Rag2−/− v-Abl pro-B cells generated a very polyclonal pattern of rearrangements, and similar results were obtained with 2 other bicistronic vectors that contained either the M502Vor T215I variants. By contrast, a more restricted pattern of V-D-J rearrangements was detected on transduction with the other 3 bicistronic vectors (G451A-M459L, R39G-R229Q, and G35A-E437K). Furthermore, the repertoire of Igh rearrangements induced by these 3 vectors revealed reduced diversity (as shown by lower Shannon index) and higher clonality (as indicated by increased Simpson index; Fig 5, D).
DISCUSSION
In this study we determined the recombination activity of naturally occurring RAG2 mutant proteins and demonstrated that mutations associated with SCID and OS have significantly lower residual activity than mutations detected in patients with less severe clinical presentations (AS and CID-G/AI). Overall, these data confirm that for human RAG2 deficiency, residual activities of the mutations correlate with the severity of the clinical presentation, as previously demonstrated for RAG1 deficiency.6 Although numbers of both patients and mutations analyzed in our current study for RAG2 deficiency was lower than in our previous study of RAG1 deficiency (79 mutations identified in 63 patients with RAG1 deficiency vs 41 mutations identified in 55 patients with RAG2 deficiency), significant differences in average RAG2 recombination activity were observed in patients presenting with different clinical phenotypes. Specifically, mutations associated with CID-G/AI supported a recombination activity that was significantly greater than that in mutations associated with more severe phenotypes (AS, OS, and SCID). In the case of RAG2-mutated patients, the F62L, T77N, and G451A RAG2 mutations were only found in patients with CID-G/AI. In addition, recurrence of the same low-activity mutations (C41W, R229Q, R229W, and C478Y) in multiple patients presenting with either SCID or OS phenotype could be predictive of a severe clinical phenotype in newly diagnosed patients carrying these mutations.
We have also developed a method whereby use of bicistronic vectors allows us to test the recombination activity of compound heterozygous RAG2 mutations and have shown that this approach is particularly useful in cases in which the 2 mutant alleles exert very different levels of recombination activity. Recently, another method based on a multiple plasmid transfection assay has been published that allows simultaneous testing of RAG variants that have been reported as compound heterozygous changes in patients.32 Compared with that method, ours has the advantage of equimolar ratios of the 2 RAG2 cDNA variants introduced. In addition, it is flow cytometry based and thus can be scaled up easily to test the activity of multiple pairs of RAG2 variants in a single experiment and permits investigation of RAG activity in a more physiologic genomic context by analyzing rearrangements at the endogenous Igh locus.
Overall, use of the bicistronic vector is preferable when faced with compound heterozygous mutations because it allows us to study the net effect of the 2 mutations on the recombination activity of the heterotetrameric RAG complex and might lead to improved correlation with the clinical phenotype, as shown in the case of P9 in this study. However, even with this approach, correlation between in vitro recombination activity and in vivo clinical and immunologic phenotypes is not absolute. In particular, environmental factors, including exposure to microorganisms and use of drugs, have been shown to modulate the phenotype of RAG deficiency in both patients and mice.33–36
Nonetheless, functional testing of the recombination activity of newly identified occurring RAG2 variants is important to assess whether these are disease causing. In this study 4 variants (P253R, F386L, N474S, and M502V) were found to support WT levels of recombination activity. One of these variants (M502V) was detected in compound heterozygosity with the S160L variant in patient P12. When tested in combination in a bicistronic vector, the S160L and M502V variants supported robust levels of recombination activity (64.6% of WT).
To further assess possible pathogenicity of the M502V variant, we analyzed its CADD-PHRED score and evaluated its frequency in gnomAD. The latter represents a recent evolution of the Exome Aggregation Consortium database, but compared with that database, it has removed subjects known to be affected by severe pediatric diseases and as such might serve as a useful reference set of allele frequency for severe disease studies. The M502V variant is reported in the gnomAD database with an MAF of 0.0019 (including the presence of 4 homozygous subjects) and has a low CADD-PHRED score of 10.54 (see Table E2). Altogether, these data indicate that the M502V variant is nonpathogenic.
Patient P39 with SCID and maternal T-cell engraftment was homozygous for the N474S variant, which also supported WT levels of recombination activity. Although this variant is reported in the gnomAD database with an MAF of 2.5e-5, its CADD-PHRED score is rather low (8.358). Furthermore, 2 previous studies analyzed in detail both the expression and function of this variant, demonstrating that it supports efficient V(D)J recombination.37,38 Patient P44 was compound heterozygous for the T251I and R229Q variants. Although the latter is a well-known pathogenic variant and supports modest levels of recombination activity (8.9% of WT), the T215I variant was permissive for high levels of recombination (67.2%). The combination of the 2 was also permissive for robust recombination activity (63%) and generation of polyclonal rearrangements at the endogenous Igh locus when introduced into Rag2−/− v-Abl pro-B cells by using a bicistronic vector. Furthermore, although the T215I variant has a relatively high CADD-PHRED score of 21.4, it is reported in the gnomAD database with an MAF of 0.002984, although it was as high as 0.02527 in South Asians, including 11 homozygous subjects (10 of whom are from South Asia). Finally, patient P11 with OS was compound heterozygous for the P253R and K440N variants, with 95.4% and 26.7% activity, respectively. Although the P253R variant is not reported in public databases and is scored as possibly damaging by Polyphen-2,39 the observation that it supports WT levels of recombination activity raises doubts about its possible pathogenicity. According to American College of Medical Genetics and Genomics criteria, both the T215I and the P253R variants are scored as variants of unknown significance, and additional in vitro data and in vivo observations are needed to resolve their pathogenicity.
In summary, we have performed a comprehensive analysis of the recombination activity of naturally occurring RAG2 variants. Our data support the notion that genotype-phenotype correlation exists for this disease, as previously demonstrated for RAG1 deficiency. However, this correlation is not absolute, and other genetic and environmental factors can contribute to determine the clinical phenotype. Notwithstanding this limitation, the flow cytometry–based assay to test the functional activity of RAG1 and RAG2 variants (with use of bicistronic vectors to assess the effects of compound heterozygous variants) might help assess the possible pathogenicity of newly identified RAG variants. This is particularly important because RAG deficiency has emerged as the second most common form of SCID that can be identified by using newborn screening.40,41
Supplementary Material
{kind=link}
Clinical implications: In vitro testing of recombination activity of naturally occurring RAG2 variants might help assess their pathogenicity and reveals correlation with the severity of the clinical phenotype.
Acknowledgments
Supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH; grant 2R01AI100887 to L.D.N.); the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health; and the JSPS Research Fellowship for Japanese Biomedical and Behavioral Research at NIH (to Y.Y.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Abbreviations used
- AS:
Atypical severe combined immunodeficiency
- CADD:
Combined annotation-dependent depletion
- CID-G/AI:
Combined immunodeficiency with granulomas and/or autoimmunity
- GFP:
Green fluorescent protein
- gnomAD:
Genome Aggregation Database
- IRES:
Internal ribosome entry site
- MAF:
Minor allele frequency
- OS:
Omenn syndrome
- RAG:
Recombinase-activating gene
- SCID:
Severe combined immunodeficiency
- v-Abl:
Abelson virus
- WT:
Wild-type
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have relevant no conflicts of interest.
REFERENCES
- 1.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016;16:234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science 1996;274:97–9. [DOI] [PubMed] [Google Scholar]
- 3.Villa A, Sobacchi C, Vezzoni P. Omenn syndrome in the context of other B cell-negative severe combined immunodeficiencies. Isr Med Assoc J 2002;4:218–21. [PubMed] [Google Scholar]
- 4.Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen T, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 2001;97:81–8. [DOI] [PubMed] [Google Scholar]
- 5.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008; 358:2030–8. [DOI] [PubMed] [Google Scholar]
- 6.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol 2014; 133:1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter JE, Lo MS, Kis-Toth K, Tirosh I, Frugoni F, Lee YN, et al. Impaired receptor editing and heterozygous RAG2 mutation in a patient with systemic lupus erythematosus and erosive arthritis. J Allergy Clin Immunol 2015;135:272–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutmer CM, Asturias EJ, Smith C, Dishop MK, Schmid DS, Bellini WJ, et al. Late onset hypomorphic RAG2 deficiency presentation with fatal vaccine-strain VZV infection. J Clin Immunol 2015;35:754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood 2009;114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT® tools for the nucleotide analysis of immunonoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol 2012;882:569–604. [DOI] [PubMed] [Google Scholar]
- 11.Rechavi E, Lev A, Lee YN, Simon AJ, Yinon Y, Lipitz S, et al. Timely and spatially regulated maturation of B and T cell repertoire during human fetal development. Sci Transl Med 2015;7:276ra225. [DOI] [PubMed] [Google Scholar]
- 12.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014;133:1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corneo B, Moshous D, Callebaut I, de Chasseval R, Fischer A, de Villartay JP. Three-dimensional clustering of human RAG2 gene mutations in severe combined immune deficiency. J Biol Chem 2000;275:12672–5. [DOI] [PubMed] [Google Scholar]
- 14.Corneo B, Moshous D, Güngör T, Wulffraat N, Philippet P, Le Deist FL, et al. Identical mutations in RAG1 or RAG2 genes leading to defective V(D)J recombinase activity can cause either T-B-severe combined immune deficiency or Omenn syndrome. Blood 2001;97:2772–6. [DOI] [PubMed] [Google Scholar]
- 15.Noordzij JG, de Bruin-Versteeg S, Verkaik NS, Vossen JM, de Groot R, Bertatowska E, et al. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood 2002;100:2145–52. [PubMed] [Google Scholar]
- 16.Tabori U, Mark Z, Amariglio N, Etzioni A, Golan H, Biloray B, et al. Detection of RAG mutations and prenatal diagnosis in families presenting with either T-B- severe combined immunodeficiency or Omenn’s syndrome. Clin Genet 2004;65: 322–6. [DOI] [PubMed] [Google Scholar]
- 17.Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Hum Mutat 2006;27:1174–84. [DOI] [PubMed] [Google Scholar]
- 18.Alsmadi O, Al-Ghonaium A, Al-Muhsen S, Arnaout R, Al-Dhekri H, Al-Saud B, et al. Molecular analysis of T-B-NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet 2009;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dalal I, Tasher D, Somech R, Etzioni A, Garti BZ, Lev D, et al. Novel mutations in RAG1/2 and ADA genes in Israeli patients presenting with T-B-SCID or Omenn syndrome. Clin Immunol 2011;140:284–90. [DOI] [PubMed] [Google Scholar]
- 20.Meshaal S, El Hawary R, Elsharkawy M, Mousa RK, Farid RJ, Abd Elaziz D, et al. Mutations in Recombination Activating Gene 1 and 2 in patients with severe combined immunodeficiency disorders in Egypt. Clin Immunol 2015;158:167–73. [DOI] [PubMed] [Google Scholar]
- 21.Safaei S, Pourpak Z, Moin M, Houshmand M. IL7R and RAG1/2 genes mutations/polymorphisms in patients with SCID. Iran J Allergy Asthma Immunol 2011;10: 129–32. [PubMed] [Google Scholar]
- 22.Gomez CA, Ptaszek LM, Villa A, Bozzi F, Sobacchi C, Broorks EG, et al. Mutations in conserved regions of the predicted RAG2 kelch repeats block initiation of V(D)J recombination and result in primary immunodeficiencies. Mol Cell Biol 2000;20:5653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ktiouet S, Bertrand Y, Rival-Tringali AL, Kanitakis J, Malcus C, Poitevin F, et al. Omenn syndrome due to mutation of the RAG2 gene. J Eur Acad Dermatol Venereol 2009;23:1449–51. [DOI] [PubMed] [Google Scholar]
- 24.Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood 2009;114:105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asai E, Wada T, Sakakibara Y, Toga A, Toma T, Shimizu T, et al. Analysis of mutations and recombination activity in RAG-deficient patients. Clin Immunol 2011; 138:172–7. [DOI] [PubMed] [Google Scholar]
- 26.Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol 2012;130:1414–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lev A, Simon AJ, Trakhtenbrot L, Goldstein I, Nagar M, Stepensky P, et al. Characterizing T cells in SCID patients presenting with reactive or residual T lymphocytes. Clin Dev Immunol 2012;2012:261470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency [published erratum in J Clin Invest 2016;126:4389]. J Clin Invest 2015;125:4135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawless D, Geier CB, Farmer JR, Allen Lango H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol 2018. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Villartay JP, Lim A, Al-Mousa H, Dupont S, Déchanet-Merville J, Coumau-Gat-bois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest 2005;115:3291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest 2005;115:3140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract 2018. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rigoni R, Fontana E, Guglielmetti S, Fosso B, D’Erchia AM, Maina V, et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med 2016;213:355–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elkin SK, Ivanov D, Ewalt M, Ferguson CG, Hyberts SG, Sun ZY, et al. A PHD finger motif in the C terminus of RAG2 modulates recombination activity. J Biol Chem 2005;280:28701–10. [DOI] [PubMed] [Google Scholar]
- 38.Couëdel C, Roman C, Jones A, Vezzoni P, Villa A, Cortes P. Analysis of mutations from SCID and Omenn syndrome patients reveals the central role of the Rag2 PHD domain in regulating V(D)J recombination. J Clin Invest 2010;120: 1337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014;312:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution an dsurvival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood 2017; 130:2718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.